

Abstract
Activation of soluble guanylate cyclase by nitric oxide (NO) controls signaling pathways that play critical roles in normal vascular physiology and in the pathogenesis of cardiovascular disease. We have identified the secreted protein thrombospondin-1 as a key regulator of NO signaling. Thrombospondin-1 limits the angiogenic activity of NO in endothelial cells, its vasodilator activity in vascular smooth muscle, and its antithrombotic activity in platelets. Loss of either thrombospondin-1 or its receptor CD47 in transgenic mice results in hyperdynamic responses to NO and reveals the importance of this pathway in normal physiology. Thrombospondin-1 and CD47 null mice show improved abilities to respond to ischemic stress, suggesting that therapeutic targeting of this pathway could benefit patients with a variety of ischemic conditions. We review the preclinical development of therapeutics targeting thrombospondin-1 or CD47 for improving survival of fixed ischemia, ischemia due to aging and peripheral vascular disease, and skin grafting.
Keywords: ischemic injury, tissue perfusion, angiogenesis, hemostasis, blood flow, nitric oxide, vascular smooth muscle, platelets
A. Introduction
Cardiovascular disease is the single greatest source of morbidity and mortality in Western societies, affecting more than 80% of people over the age of 65 [1, 2]. Manifestations include stroke, macular degeneration, peripheral vascular disease, hypertension, heart and other vital organ failure, heart attack, and poor wound healing. The etiology of cardiovascular disease involves either acute or subacute decreases in blood flow with concurrent under-perfusion of tissue [3]. These flow alterations are associated with changes in the structure of arteries and the contractile function of vascular smooth muscle cells (VSMC) [4, 5]. Arteries, the major conduits of blood flow to peripheral tissues, dynamically alter their luminal diameter in response to changing regional tissue metabolic needs. Control of blood flow and arterial diameter is achieved through an integrated system of local and central modifiers that stimulate VSMC relaxation or contraction. Intrinsic regulation of vascular contractility and blood flow is an area of great research interest. Among the currently known locally acting vasodilators, nitric oxide (NO) is perhaps the most critical to cardiovascular health [6].
B. Nitric Oxide
NO is a gaseous diatomic radical that is produced through the conversion of L-arginine to L-citrulline by three nitric oxide synthases (nNOS/NOS1, iNOS/NOS2 and eNOS/NOS3) [7, 8]. eNOS is the predominant NOS isoform expressed in endothelial cells and platelets. The activity of eNOS is extensively regulated by its subcellular localization, calcium/calmodulin and Hsp90 binding, cofactor levels, and several phosphorylation sites [9]. NO is also generated in low oxygen environments through the nitrite reductase activities of several proteins including hemoglobin, myoglobin, and xanthine oxidase [10-12]. Among its several bioactivities, NO is one of the most potent antagonists of VSMC contraction known [13]. Treatment with acetylcholine relaxes intact arterial segments but not those lacking endothelium. These observations led to a search for the endothelial derived relaxing factor that controls smooth muscle contractility, which was found to be NO [14-17]. In its target cells, NO binds to the heme moiety of soluble guanylate cyclase (sGC), increasing its enzymatic activity and leading to accumulation of intracellular cGMP [18]. Elevated cGMP in turn activates a number of downstream targets controlling signaling pathways that modulate the contractility of VSMC [19] (Fig. 1). In this manner NO relaxes VSMC, dilates blood vessels, and enhances tissue perfusion [20]. Therapeutics such as nitroglycerine and sodium nitroprusside have capitalized upon this property of NO and for generations have been a mainstay in the treatment of myocardial ischemic vascular disease [21, 22].
Fig. 1. Thrombospondin-1 regulation of vascular smooth muscle contractility.

VSMC contraction requires interaction between myosin and actin. This process is stimulated by myosin light chain kinase (MLCK)-mediated phosphorylation of serine-19 on myosin light chain-2 (MLC2). Vasoconstrictors promote VSMC contraction by activating MLCK and inactivating MLC phosphatase. NO relaxes VSMC through increasing cellular cGMP, which activates cGK and, via cyclic nucleotide gated ion channels, reduces the amount of Ca available for activation of MLCK. cGK phosphorylates MYP1 on serine-695 to simultaneously activate myosin light chain phosphatase (MLCP). Engagement of CD36 or CD47 at the cell membrane by the type 1 repeats or the C-terminal domains of TSP1 inhibits NO-stimulation of sGC and thereby prevents intracellular cGMP accumulation.
Normal physiologic levels of NO (≤10 nM) exert pro-survival effects on endothelial and VSMC by promoting their proliferation, adhesion, and migration [23, 24]. Such levels of NO are produced by eNOS and nNOS and serve to provide tonic pro-survival support to vascular cells [25]. The availability of pro-survival NO concentrations in vivo in vascular cells depends on levels of the substrate arginine, regulatory phosphorylation of eNOS, and the availability of necessary NOS cofactors such as NADPH and BH4. However, at higher concentrations (>100 nM) NO is cytostatic or cytotoxic for vascular cells [26]. The latter amounts of NO are found in the context of inflammatory cell activation, are produced by iNOS, and induce phosphorylation of p53 and dephosphorylation of MAP kinases via induction of MKP1 [27]. Although high levels of NO are pro-inflammatory, physiologic concentrations of NO produced by eNOS can promote tissue survival under inflammatory stress by limiting the effects of inflammatory cytokines. This is particularly true in terms of VSMC contractility and inflammation at the endothelium. Physiologic NO tempers endothelial activation by inflammatory agents, suppresses expression of adhesion molecules and minimizes inflammatory cell interactions with endothelial cells [28, 29]. All of these effects support the maintenance of tissue perfusion by limiting thrombosis and leukocyte diapedesis.
Sustained low doses of NO enhance endothelial cell proliferation, adhesion and migration and thereby stimulate new blood vessel formation [23, 30, 31]. An ex vivo model of wound healing using muscle tissue incorporated into a type I collagen matrix can be used to quantify vascular cell invasion and migration through the collagen matrix. Vascular outgrowth in this assay is significantly increased by exogenous NO or L-arginine. In contrast, vascular outgrowth from muscle explants is inhibited by the nonspecific NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME) [23]. Consistent with these results, in vivo models of wound healing have shown positive roles for NO and L-arginine in promoting wound healing [32-34].
C. Physiologic Regulation of NO Signaling
NO diffuses rapidly through tissue and can engage targets both at the site of production and in neighboring cells. The major route for elimination of NO in circulation is its reaction with oxygenated hemoglobin (Hb) to form MetHb and biologically inactive NO3−. Some NO is consumed following conversion to reactive metabolites that modify free thiols on proteins and glutathione, forming S-nitrosothiols on albumin and other proteins [35]. In inflammatory environments, superoxide scavenging of NO can limit its ability to signal via sGC [36]. Although these pathways are important for limiting the duration of NO-signaling within the vascular lumen and in pathologic states, they do not provide an explanation for the dynamic regulation of NO signaling within the arterial wall. Hydrolysis of cGMP, the product of NO-activated sGC, by several phosphodiesterases and feedback control of eNOS activation through substrate and byproduct modifications and availability provide some limits for NO/cGMP signaling [37, 38]. However, we recently discovered that the secreted matricellular protein thrombospondin-1 (TSP1) provides another major physiological control for this pathway [23].
D. Inhibition of Nitric Oxide Signaling by Thrombospondin-1
We found that NO-stimulated ex vivo angiogenesis is significantly enhanced in the absence of endogenous TSP1 [23]. Furthermore, addition of picomolar concentrations of exogenous TSP1 is sufficient to potently block NO-driven angiogenic responses. TSP1 is produced by all vascular cell types and modifies cellular response from an extracellular position by engaging a number of cell surface receptors [39, 40]. Though defined roles for TSP1 in tumor angiogenesis and wound healing were known and inhibition of cGMP synthesis had been reported in a melanoma cell line [41], a role for the protein to regulate vascular NO signaling was not appreciated. Notably, prior studies had shown that TSP1 expression in endothelial, VSMC, and kidney mesangial cells is itself a target of NO/cGMP signaling [42-45]. We found that exogenous TSP1 potently blocks NO-stimulated increases in endothelial cell proliferation, adhesion and migration [23]. Primary murine endothelial cells from TSP1 null animals demonstrate significantly greater increases in proliferation and adhesion when compared to wild type cells. The physiologic importance of these findings was emphasized by the finding that TSP1 null endothelial cells consistently demonstrate increased basal levels of cGMP compared to wild type cells. Thus, endogenous TSP1 functions as a tonic antagonist of NO signaling in endothelial cells. NO-stimulated accumulation of cGMP is also profoundly sensitive to exogenous TSP1, with picomolar concentrations being sufficient to completely block NO stimulation of sGC. A recombinant fragment of TSP1 derived from its type 1 repeats that interacts with the cell receptor CD36 is equally effective at inhibiting NO-driven angiogenic responses and cGMP flux in vascular cells. Peptides derived from this area of the protein that bind to CD36 and certain CD36 antibodies also block NO signaling in vascular cells.
Endothelial cells sense changes in blood flow and shear but cannot directly regulate vascular diameter to maintain vascular homeostasis. Endothelial cells alter their NO production in response to these physiological stimuli, and the NO diffuses into the vessel wall to control the contractile state of the underlying VSMC [46-49]. Like endothelial cells, VSMC contain sGC and express several TSP1 receptors. Because TSP1 potently inhibits NO-stimulated VSMC proliferation, adhesion, migration and cGMP accumulation [24], it is reasonable then to consider a role for TSP1 in regulating VSMC contractility.
At the molecular level, VSMC contracture involves interactions between myosin and the actin cytoskeleton. Agents that promote VSMC contraction enhance actin/myosin cross-bridging. NO-induced cGMP accumulation decreases intracellular calcium and activates myosin light chain phosphatase to dephosphorylate the myosin regulatory subunit myosin light chain-2 (MLC2), leading to reduced myosin-actin interactions and relaxation of VSMC [50, 51] (Fig. 1). We found that NO-stimulated relaxation of VSMC is directly blocked by TSP1 at the level of MLC2 by preventing its dephosphorylation [52]. Exogenous TSP1 prevents NO-driven relaxation of pre-contracted VSMC. Conversely, TSP1 null VSMC contract to a known agonist much less than wild type cells. Therefore, TSP1 also antagonizes this acute response of VSMC to NO.
E. CD47 is Necessary for TSP1 Inhibition of NO Signaling in Vascular Cells
In vitro experiments using human cells and recombinant domains of TSP1 demonstrated that the type 1 repeats and the C-terminus of TSP1 effectively block NO-stimulated events. A recombinant N-terminal domain that engages integrin receptors and calreticulin/LRP did not alter or moderately enhanced the NO response. The receptors for the active domains of TSP1 are CD36 and CD47 (Fig. 1). Peptides derived from the amino acid sequence of the C-terminus that bind to CD47 also block NO signaling. Targeting CD36 using peptides derived from the type 1 repeats of TSP1 or recombinant type 1 repeats also blocks NO-stimulated cGMP signaling in vascular cells. Therefore, ligation of CD47 or CD36 is sufficient to block NO-driven vascular cell responses. However, TSP1 inhibits NO/cGMP signaling in vascular cells and tissue explants from CD36 null but not those from CD47 null mice. Thus, CD47 is necessary for TSP1 blockade of NO signaling, whereas CD36 is only sufficient in cells that also express CD47. CD47 is expressed on the surface of virtually all cells due to its role in immune cell modulation and self-recognition [53].
F. CD36 as a Target for TSP1 Inhibition of NO Signaling
Although CD36 is not the necessary receptor for TSP1 signaling, it remains a potential target for therapeutic control of this pathway. CD36 functions not only as a receptor for TSP1 but also as a translocase for free fatty acids (reviewed in [54]). The free fatty acid myristate was recently reported to stimulate eNOS in a CD36-dependent manner, resulting in increased endogenous NO production [55]. We in turn discovered that TSP1 and TSP1-based agents block myristate uptake into vascular cells and thereby block eNOS-mediated production of NO and NO-stimulated events including sGC stimulation [56]. More than 100 cytoplasmic proteins, including a number of important signaling proteins, are regulated by acylation of subterminal Gly residues with myristate [57]. We demonstrated that myristoylation and membrane translocation of Fyn, a Src family kinase, is regulated in a CD36-dependent manner. Blocking myristate uptake in vascular cells using a CD36-binding peptide from TSP1 prevented Fyn translocation and tyrosine phosphorylation of Src family kinases. eNOS is also a myristoylated protein, but membrane translocation of eNOS was not CD36-dependent. Therefore, the myristate/CD36-dependent activation of eNOS must occur at a later step. Interestingly, Src kinase-mediated phosphorylation of eNOS at Tyr-83 in endothelial cells and intact vessels was recently shown to play a critical role its functional activation [58]. This finding provides a mechanism by which TSP1 could prevent eNOS activation by preventing Src family kinase activation via CD36. Therefore, these CD36-binding peptides could be lead compounds for designing inhibitors of NO signaling that act by inhibiting myristate uptake via CD36.
A drug targeting CD36 is currently in clinical trails as an anti-angiogenic agent for treating cancer [59, 60]. ABT-510 was designed based on a CD36-binding peptide from the type 1 repeats of TSP1 [61]. Therefore, we examined its efficacy as an inhibitor of NO/cGMP signaling in vascular cells. Although the TSP1-based mimetic Gly-Asp-Gly-Val-(dIle)-Thr-Arg-Ile-Arg potently inhibits myristate uptake, the synthetic derivative ABT-510 (NAc-Sar-Gly-Val-(d-allo-Ile)-Thr-Nva-Ile-Arg-ProNEt) is less potent than the former peptide or whole TSP1 [62]. ABT-510 is also less active than TSP1 or Gly-Asp-Gly-Val-(dIle)-Thr-Arg-Ile-Arg for inhibiting myristate-stimulated vascular cell adhesion to collagen and cGMP accumulation [62]. Consistent with its ability to modulate NO signaling in vascular cells, ABT-510 moderately stimulates thrombin-driven platelet aggregation, suggesting a potential pro-thrombotic side effect of this anti-angiogenic agent. Although ABT-510 was less potent than expected for inhibiting NO/cGMP signaling via CD36, it is a potent inducer of endothelial and VSMC apoptosis via an as yet undefined mechanism. Therefore, ABT-510 remains an interesting candidate for inhibiting tumor angiogenesis, but this therapeutic activity is probably not mediated by inhibition of NO/cGMP signaling.
G. TSP1 Regulation of NO Signaling in Platelets
Nitric oxide also plays a critical role in maintaining blood flow by minimizing platelet activation and platelet adhesion to endothelium and matrix [63]. This is particularly important in the microcirculation where aggregation of activated platelets could rapidly obstruct blood flow. TSP1 comprises the major protein of platelet α-granules, and on activation at the site of hemorrhage platelets degranulate, resulting in a local burst of TSP1. Concurrently, alterations in shear at the point of hemorrhage drives increased eNOS activity and a local burst in endogenous NO [64]. The later stimulates sGC in the adjacent vascular cells including platelets and VSMC. Yet, the precise function TSP1 plays in modifying platelets has remained controversial, with studies demonstrating a pro- as well as anti-thrombotic roles [65-68]. Most surprisingly mice with a disrupted Thbs1 gene, whose platelets lacked any detectable thrombospondins, failed to show increased bleeding times on a tail snip assay [69, 70]. The confusion in these contradictory results becomes understandable when re-interpreted in terms of TSP1 inhibition of NO signaling. In vitro, NO delays thrombin-stimulated aggregation and platelet adhesion to collagen. TSP1 and recombinant domains and peptides derived from TSP1 potently inhibit NO-stimulated delay in platelet aggregation, adhesion and cGMP accumulation [71]. Platelets lacking either TSP1 or CD47 require several times more thrombin to initiate aggregation as compared to wild type cells and, in the presence of exogenous NO, were rendered refractory to thrombin.
As in other vascular cells, TSP1 inhibits activation of sGC by NO in platelets, but further studies identified an additional downstream target of TSP1 for inhibiting NO-signaling. Our previous studies using endothelial and VSMC showed that TSP1 inhibits several functional responses of vascular cells stimulated by a cell-permeable cGMP analog [23, 24]. This indicated that a second target must exist downstream of sGC. NO is known to inhibit platelet adhesion by limiting αIIb/β3 integrin activation (Fig. 2). NO-stimulated cGMP in platelets activates cGK-I, which in turn blocks activation of Rap1 by phosphorylation of the Rap1 GTPase activator protein Rap1GAP2, preventing Rap1-mediated activation of the integrin αIIb/β3 [72, 73]. TSP1 restores GTP loading of Rap1 and also inhibits NO- and cGMP-stimulated phosphorylation of a second cGK-I target VASP in platelets, suggesting that cGK-I is a downstream target of TSP1/CD47 signaling. This was confirmed using a defined peptide substrate of cGK-I, phosphorylation of which was blocked in an in vitro kinase assay when the platelets were previously exposed to TSP1.
Fig. 2. Thrombospondin-1 regulation of platelet aggregation.

Activated platelets rapidly secrete the TSP1 from their α-granules. Extracellular TSP1 can then engage CD36 and CD47 located on the platelet membrane and inhibit NO stimulation of sGC or downstream cGMP-driven activation of cGK-I. Decreased activation of cGK-I decreases Rap1GAP2-mediated unloading of thrombin-activated GTP-Rap1, thereby increasing platelet αIIb/β3 integrin activation and enhancing platelet adhesion and aggregation. TSP1 also prevents cGK-I-mediated phosphorylation of VASP and other targets to further stimulate platelet aggregation and adhesion.
H. TSP1 and Wound Healing
Transgenic mice have proven to be a very powerful tool for asking fundamental questions about the roles of particular proteins in development and physiology. The TSP1 null mouse at first appeared quite unremarkable, lacking obvious developmental defects and having minimal irregularities as an adult [74]. In a dermal punch biopsy model of wound repair TSP1 null animals had slower healing rates, a finding supported by studies in similar wounds treated with TSP1 antisense oligomers, which also healed at a slower rate [75, 76]. These findings were unexpected given the known inhibitory role of TSP1 in controlling neovascularization and the accelerated wound healing observed in TSP2 null mice [75]. The differences in wound repair between these two null mice may reflect a distinct role of TSP1 in stimulating macrophage recruitment. This excisional wound model reveals a limiting role of TSP1 in the macrophage recruitment for formation of granulation tissue [75]. In light of our recent findings concerning regulation of NO-stimulated vasodilation, we searched for additional roles of TSP1 utilizing wound repair models that are more appropriate for assessing the role of TSP1 in controlling tissue blood flow.
I. TSP1-CD47 Regulation of Blood Flow and Tissue Survival
Maintenance of tissue blood flow and perfusion is central to survival. Acute or chronic decreases in flow are a key factor in many diseases that affect adults in Western societies. Some form of vasculopathy can be demonstrated in the vast majority of elderly persons. Yet the ability to therapeutically enhance tissue blood flow remains elusive with minimal clinical developments beyond those obtained at the level of macro-vascular surgery [77, 78]. Experimental therapies have attempted to capitalize upon the known role of NO/cGMP signaling in increasing blood vessel diameter [79]. Therapies employing L-arginine, cGMP phosphodiesterase inhibitors, or NO donors can increase tissue survival [80-87]. The finding that TSP1 via CD47 limits NO-stimulated VSMC relaxation in vitro suggested a role in controlling blood flow in vivo. Using real time functional magnetic resonance imaging (MRI) analysis of blood flow, we found that NO-stimulated increases in muscle and soft tissue blood flow are approximately 2-fold greater in TSP1 null mice than in wild type [52]. The enhanced blood flow in TSP1 null mice is evident immediately following NO administration, too fast for de novo synthesis or secretion of TSP1. Therefore, endogenous TSP1 in the extracellular matrix surrounding arterial smooth muscle must tonically function to temper acute vascular responses to NO. These experiments were performed using resting and otherwise unstressed mice with intact autonomic systems. Hence, TSP1 regulation of blood flow via NO signaling supersedes the homeostatic role of autonomic input.
The central role that TSP1 plays in controlling blood flow extends beyond normal physiologic conditions. The soft tissue cutaneous flap model creates a fixed ischemic challenge. Tissue blood flow and perfusion in this injury model were dramatically increased in the absence of TSP1 or CD47 [52]. Null animals experienced close to 100% tissue survival, whereas wild type animals subjected to comparable ischemic challenge underwent close to 50% tissue necrosis Functional imaging demonstrated that tissue oxygen levels were preserved in ischemic tissues in the absence of TSP1 or CD47, confirming the maintenance of tissue perfusion [88].
An identical protective effect was found following major arterial interruption in an ischemic hindlimb model. Mice subjected to femoral artery ligation at the inguinal ligament showed rapid loss of perfusion with eventual tissue necrosis. In contrast, TSP1 and CD47 null mice experienced a transient decrease in limb flow following femoral artery ligation with rapid restoration of limb perfusion. As documented by laser Doppler imaging, restoration of hindlimb perfusion in null animals occurs within minutes via collateral remodeling, presumably stimulated by enhanced NO signaling. Though comparable vascular collateral networks should be present in the wild type, dynamic remodeling appears to be prevented by TSP1/CD47 inhibition of NO-signaling. These in vivo findings provide genetic evidence for the dynamic regulation of tissue blood flow by TSP1/CD47 under vasoactive challenge or fixed ischemia.
Random soft tissue flaps and hindlimbs with proximal arterial occlusion represent fixed ischemic injury models. They replicate clinical ischemic conditions encountered in surgery, trauma, and vascular disease. However, these models retain some residual perfusion. In contrast, full thickness skin grafts (FTSG) are completely anoxic at the time of grafting, and circulation is restored only following neovascularization from the underlying wound bed that requires 48 to 76 hours [89]. FTSG experience profound ischemia and in wild type mice undergo essentially complete necrosis. In contrast, FTSG in TSP1 or CD47 null mice demonstrate over 80% survival. FTSG wound beds in null animals demonstrate dramatic vascular remodeling in the early post-graft period, whereas wild type wound beds do not [90]. Based on transplant experiments between null and wild type mice, the presence or absence of TSP1 or CD47 in the wound bed rather than in the skin graft is critical for graft survival. Wild type FTSG on null wound beds enjoy significantly increased survival and perfusion, whereas null grafts on wild type wound beds experience more necrosis. Thus, TSP1/CD47 signaling in the underlying wound bed appears to limit its ability to increase perfusion to provide nutrients and oxygen needed to maintain the overlying avascular graft until revascularization can occur.
J. Loss of TSP1/CD47 Signaling Increases Blood Flow and Tissue Survival Despite Advanced Age or Peripheral Vascular Disease
Trauma and elective surgical procedures are the most common etiologies behind tissue under-perfusion in the young. With advancing age, however, a majority of persons in Western societies develop varying degrees of cardiovascular pathology [91-94]. Clinical manifestations include peripheral vascular disease, atherosclerosis, myocardial infarction, hypertension, stroke and macular degeneration. All these diseases involve varying degrees of tissue under-perfusion. NO insufficiency is increasingly recognized as a primary feature of cardiovascular disease in aging [95]. NO production and eNOS expression in blood vessels markedly decrease with age [96, 97]. Concurrently, vascular TSP1 expression increases with age in humans [98] and rodents [99, 100]. Consistent with these findings, aged wild type mice (14 to 18 months old) demonstrate greater degrees of tissue necrosis in response to an ischemic insult than young animals, with some animals experiencing complete flap loss [101]. In contrast, TSP1 or CD47 null mice of similar age mice demonstrate near complete tissue survival. Blood flow responses to a vasoactive challenge analyzed using functional MRI are significantly greater in aged null mice compared to both aged and juvenile wild type animals. At the tissue level, cGMP levels fall with age in wild type mice but are preserved in TSP1 null mice. Despite advanced age, the dynamic potential of NO-stimulated cardiovascular events persists in the absence of TSP1.
Atherosclerotic plaque formation is pathoneumonic of peripheral vascular disease in the subendothelial layers of the arterial vasculature. These lesions contribute directly to decreased tissue blood flow and perfusion. Apolipoprotein-E null mice fed a high fat diet experience an aggressive form of this vasculopathy [102]. Under ischemic stress, these animals experience near complete tissue necrosis. Importantly, ischemic tissue survival is significantly enhanced through suppressing CD47 in the tissue unit [101].
K. Enhanced Cardiovascular Responses and Tissue Survival by Therapeutic Targeting of TSP1/CD47
Selective enhancement of tissue blood flow and perfusion has tremendous therapeutic potential to treat the ubiquitous vascular pathology in aging populations. Current approaches have had limited clinical impact. Given the preeminent role that NO plays in controlling vascular tone and cardiac function, we predicted that targeting TSP1/CD47 would enhance the beneficial effects of physiologic NO on blood flow and vascular remodeling. Such a strategy could synergize with current therapies that provide an exogenous NO source to increase blood flow [103, 104].
We employed several approaches to therapeutically block TSP1/CD47 signaling (Fig. 3). Because preformed TSP1 can be released from granules in platelets and perhaps other cells at sites of ischemic injury, rather than blocking TSP1 expression we attempted to prevent TSP1 from interacting with CD47 using antibodies. Random dorsal soft tissue flaps in wild type mice that were injected per-operatively with a monoclonal antibody that recognizes murine TSP1 demonstrated increased tissue survival [88]. The same TSP1 antibody was effective for enhancing tissue preservation in wild type hindlimbs following proximal femoral artery ligation. In these experiments the antibody was injected into the medial and lateral thigh musculature at the time of arterial flow interruption. Because therapeutic antibodies typically have long half lives, a single injection appears to be sufficient to maintain blood flow and permit tissue repair.
Fig. 3. Therapeutic targets for overcoming restriction of blood flow in ischemic injuries by thrombospondin-1.
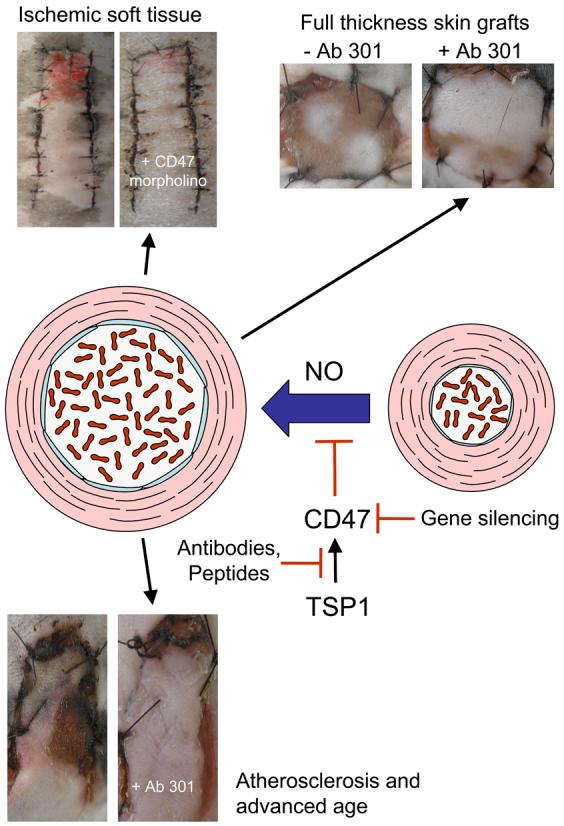
TSP1 expression in ischemic injuries limits the ability of NO to restore and maintain tissue perfusion by vasodilation. Enhanced tissue blood flow and survival can be achieved through antibody engagement of either TSP1 (e.g. clone A6.1) or CD47 (e.g clone 301) under conditions of a fixed ischemic insult in a range of composite tissue units including skin grafts, soft tissue flaps, and hindlimbs. Antisense suppression of CD47 leads to similar benefits in tissue survival. These therapeutic advantages are realized in both young and aged animals and in the presence of atherosclerotic vasculopathy. Beneficial responses to regulation of TSP1/CD47 signaling have been demonstrated in injury models employing mice and pigs and are conserved in vascular cells from multiple species (murine, bovine, porcine, and human).
Engaging CD47 is necessary for TSP1 to inhibit an NO-mediated enhancement of blood flow and tissue survival, so we also targeted CD47 using an antibody. Treatment of fixed tissue ischemia in random myocutaneous flaps and FTSG several models using a CD47 monoclonal antibody resulted in dramatic increases in tissue survival [88, 90]
Alternatively, one can block TSP1/CD47 signaling by suppression of CD47 at the mRNA level using antisense agents such as morpholino oligonucleotides. A morpholino oligonucleotide was designed complementary to a conserved sequence in the 5' region of murine and human CD47 mRNA and verified by western blotting to prevent translation of this mRNA in vascular cells in vitro [88]. Direct injection of this agent into FTSG, soft tissue flaps, and ischemic hindlimbs greatly enhanced tissue survival [88, 90, 101]. Morpholinos can suppress expression of their targets for several days, so this approach should be well suited to further applications involving acute ischemic injury.
Inhibition of NO/cGMP signaling by TSP1/CD47 based agents has now been shown in murine, porcine, bovine and human vascular cells. In mice, targeting TSP1-CD47 enhances tissue blood flow and survival. However, murine cutaneous vascular anatomy differs from that of humans [105]. For this reason, porcine models are often employed in pre-clinical testing of therapeutics. To characterize our agents in a cutaneous ischemic injury that better represents human anatomy, we studied a porcine model of tissue ischemia. We first determined the sequence of CD47 in this species using VSMC obtained from the femoral artery of Yucatan white hairless miniature pigs. The nucleotide sequence differed slightly from the murine sequence, which our CD47 morpholino was designed to complement, but the CD47 morpholino dramatically and specifically suppressed CD47 protein levels in porcine cells [106]. Similarly, treatment of porcine VSMC with the morpholino but not a mismatched control ablated TSP1 inhibition of NO-stimulated sGC activation. Random cutaneous flaps in Yucatan mini-pigs treated with the morpholino showed increased tissue survival and decreased CD47 expression on immunohistology. Treatment with a CD47 antibody also enhanced the survival of ischemic porcine flaps. Thus, suppressing CD47 by either approach can improve tissue survival of ischemic injury in an animal having similar vascular anatomy as humans.
These studies establish that targeting TSP1 or CD47 can enhance tissue survival in several murine and porcine models of partial and total fixed ischemia. We propose that targeting TSP1 may be less efficient given the constant potential for platelet release of TSP1 in areas of injury or inflammation and the accumulating evidence that TSP1 expression can be stimulated in ischemic injuries [107-109]. Targeting the necessary receptor CD47 may more efficiently restore the protective NO/cGMP signaling and protect ischemic tissue from endogenous TSP1 and that induced secondary to injury.
L. Clinical Implications and Future Directions
NO is a central regulator of the cardiovascular system in vertebrates. With aging, the vasculature and tissues experience a progressive loss of the pro-survival and vascular protective signals provided by physiologic NO. This may result from both decreased endogenous NO synthesis and increased expression of TSP1. The net result is a loss of vascular plasticity, decreased tissue blood flow, and loss of perfusion. Genetic or pharmacologic ablation of TSP1/CD47 signaling enhances the beneficial effects of NO on cardiovascular dynamics following a vascular challenge in aged animals to a level superior to that of an untreated juvenile. This results in increased blood flow and improved tissue survival of ischemic injuries. Given the protean beneficial physiologic NO exerts effects of in mammalian cells, the clinical applications of such therapeutics may extend far beyond enhancing tissue blood flow.
Heart disease and stroke remain the number 1 and number 3 causes of death in the United States. In 2004 close to 475,000 men and 400,000 women died from complications related to peripheral vascular disease. Cardiovascular disease accounted for some 6 million hospital admissions in 2005 [110]. More than 70% of men and women over the age of 65 have manifestations of cardiovascular disease, and targeting TSP1/CD47 may benefit all of these patient populations. Cardiovascular disease in aging populations involves both acute and chronic ischemia, so further work is needed to assess the utility of these agents for treating chronic ischemia and, if effective, the potential side effects that may result from chronic suppression of this signaling pathway.
The agent with the most immediate potential for use in people is the CD47 morpholino, which is known to suppress CD47 expression in human cells and so could be used in a human clinical trail without further modification. Antibody A6.1 recognizes human TSP1 but would need to be humanized to permit repeated administration during clinical testing. The CD47 antibody is specific for murine CD47, so further research is needed to identify anti-human CD47 antibodies that protect ischemic tissues. The TSP1/CD47 interaction could also be targeted by inhibitory small molecules. However, appropriate screening assays must be established to identify such inhibitors.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (D.A.W, M.C.K., D.D.R.) and NIH grants HL54390 and GM57573 (W.A.F.).
References
- 1.Daviglus ML, Lloyd-Jones DM, Pirzada A. Preventing cardiovascular disease in the 21st century: therapeutic and preventive implications of current evidence. Am J Cardiovasc Drugs. 2006;6:87–101. doi: 10.2165/00129784-200606020-00003. [DOI] [PubMed] [Google Scholar]
- 2.Hanna IR, Wenger NK. Secondary prevention of coronary heart disease in elderly patients. Am Fam Physician. 2005;71:2289–2296. [PubMed] [Google Scholar]
- 3.Glasser SP. On arterial physiology, pathophysiology of vascular compliance, and cardiovascular disease. Heart Dis. 2000;2:375–379. [PubMed] [Google Scholar]
- 4.Bajwa TK, Shalev YA, Gupta A, Khalid MA, Moussavi N. Peripheral vascular disease, Part 1. Curr Probl Cardiol. 1998;23:245–297. doi: 10.1016/s0146-2806(98)80014-5. [DOI] [PubMed] [Google Scholar]
- 5.Bajwa TK, Shalev YA, Gupta A, Khalid MA. Peripheral vascular disease, Part 2. Curr Probl Cardiol. 1998;23:305–348. doi: 10.1016/s0146-2806(98)80013-3. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson IB, McEniery CM. Arterial stiffness, endothelial function and novel pharmacological approaches. Clin Exp Pharmacol Physiol. 2004;31:795–799. doi: 10.1111/j.1440-1681.2004.04074.x. [DOI] [PubMed] [Google Scholar]
- 7.Ignarro LJ. Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;16:477–483. doi: 10.1161/01.hyp.16.5.477. [DOI] [PubMed] [Google Scholar]
- 8.Ignarro LJ. Physiology and pathophysiology of nitric oxide. Kidney Int Suppl. 1996;55:S2–5. [PubMed] [Google Scholar]
- 9.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2026–2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 11.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- 12.Rassaf T, Flogel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res. 2007;100:1749–1754. doi: 10.1161/CIRCRESAHA.107.152488. [DOI] [PubMed] [Google Scholar]
- 13.Knowles RG, Moncada S. Nitric oxide as a signal in blood vessels. Trends Biochem Sci. 1992;17:399–402. doi: 10.1016/0968-0004(92)90008-w. [DOI] [PubMed] [Google Scholar]
- 14.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, Gruetter CA. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 15.Gruetter CA, Gruetter DY, Lyon JE, Kadowitz PJ, Ignarro LJ. Relationship between cyclic guanosine 3′:5′-monophosphate formation and relaxation of coronary arterial smooth muscle by glyceryl trinitrate, nitroprusside, nitrite and nitric oxide: effects of methylene blue and methemoglobin. J Pharmacol Exp Ther. 1981;219:181–186. [PubMed] [Google Scholar]
- 16.Ignarro LJ, Harbison RG, Wood KS, Kadowitz PJ. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther. 1986;237:893–900. [PubMed] [Google Scholar]
- 17.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol: 2006:213–254. doi: 10.1007/3-540-32967-6_7. [DOI] [PubMed] [Google Scholar]
- 19.Boerth NJ, Dey NB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res. 1997;34:245–259. doi: 10.1159/000159231. [DOI] [PubMed] [Google Scholar]
- 20.Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science. 1999;286:1583–1587. doi: 10.1126/science.286.5444.1583. [DOI] [PubMed] [Google Scholar]
- 21.Kim-Shapiro DB, Schechter AN, Gladwin MT. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arterioscler Thromb Vasc Biol. 2006;26:697–705. doi: 10.1161/01.ATV.0000204350.44226.9a. [DOI] [PubMed] [Google Scholar]
- 22.Warren JB, Pons F, Brady AJ. Nitric oxide biology: implications for cardiovascular therapeutics. Cardiovasc Res. 1994;28:25–30. doi: 10.1093/cvr/28.1.25. [DOI] [PubMed] [Google Scholar]
- 23.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isenberg JS, Wink DA, Roberts DD. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc Res. 2006;71:785–793. doi: 10.1016/j.cardiores.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsui M. Neuronal nitric oxide synthase as a novel anti-atherogenic factor. J Atheroscler Thromb. 2004;11:41–48. doi: 10.5551/jat.11.41. [DOI] [PubMed] [Google Scholar]
- 26.Lau HK. Cytotoxicity of nitric oxide donors in smooth muscle cells is dependent on phenotype, and mainly due to apoptosis. Atherosclerosis. 2003;166:223–232. doi: 10.1016/s0021-9150(02)00333-7. [DOI] [PubMed] [Google Scholar]
- 27.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laroux FS, Pavlick KP, Hines IN, Kawachi S, Harada H, Bharwani S, Hoffman JM, Grisham MB. Role of nitric oxide in inflammation. Acta Physiol Scand. 2001;173:113–118. doi: 10.1046/j.1365-201X.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- 29.Laroux FS, Lefer DJ, Kawachi S, Scalia R, Cockrell AS, Gray L, Van der Heyde H, Hoffman JM, Grisham MB. Role of nitric oxide in the regulation of acute and chronic inflammation. Antioxid Redox Signal. 2000;2:391–396. doi: 10.1089/15230860050192161. [DOI] [PubMed] [Google Scholar]
- 30.Isenberg JS, Ridnour LA, Thomas DD, Wink DA, Roberts DD, Espey MG. Guanylyl cyclase-dependent chemotaxis of endothelial cells in response to nitric oxide gradients. Free Radic Biol Med. 2006;40:1028–1033. doi: 10.1016/j.freeradbiomed.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 31.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281:26069–26080. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 32.Shabani M, Pulfer SK, Bulgrin JP, Smith DJ. Enhancement of wound repair with a topically applied nitric oxide-releasing polymer. Wound Repair Regen. 1996;4:353–362. doi: 10.1046/j.1524-475X.1996.40312.x. [DOI] [PubMed] [Google Scholar]
- 33.Frank S, Kampfer H, Wetzler C, Pfeilschifter J. Nitric oxide drives skin repair: novel functions of an established mediator. Kidney Int. 2002;61:882–888. doi: 10.1046/j.1523-1755.2002.00237.x. [DOI] [PubMed] [Google Scholar]
- 34.Isenberg JS, Ridnour LA, Espey MG, Wink DA, Roberts DD. Nitric oxide in wound-healing. Microsurgery. 2005;25:442–451. doi: 10.1002/micr.20168. [DOI] [PubMed] [Google Scholar]
- 35.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 36.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreopoulos S, Papapetropoulos A. Molecular aspects of soluble guanylyl cyclase regulation. Gen Pharmacol. 2000;34:147–157. doi: 10.1016/s0306-3623(00)00062-8. [DOI] [PubMed] [Google Scholar]
- 38.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 39.Lawler J. The functions of thrombospondin-1 and-2. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 40.Calzada MJ, Roberts DD. Novel integrin antagonists derived from thrombospondins. Curr Pharm Des. 2005;11:849–866. doi: 10.2174/1381612053381792. [DOI] [PubMed] [Google Scholar]
- 41.Guo N, Zabrenetzky VS, Chandrasekaran L, Sipes JM, Lawler J, Krutzsch HC, Roberts DD. Differential roles of protein kinase C and pertussis toxin-sensitive G-binding proteins in modulation of melanoma cell proliferation and motility by thrombospondin 1. Cancer Res. 1998;58:3154–3162. [PubMed] [Google Scholar]
- 42.Lincoln TM, Dey NB, Boerth NJ, Cornwell TL, Soff GA. Nitric oxide--cyclic GMP pathway regulates vascular smooth muscle cell phenotypic modulation: implications in vascular diseases. Acta Physiol Scand. 1998;164:507–515. doi: 10.1111/j.1365-201x.1998.tb10700.x. [DOI] [PubMed] [Google Scholar]
- 43.Phelan MW, Forman LW, Perrine SP, Faller DV. Hypoxia increases thrombospondin-1 transcript and protein in cultured endothelial cells. J Lab Clin Med. 1998;132:519–529. doi: 10.1016/s0022-2143(98)90131-7. [DOI] [PubMed] [Google Scholar]
- 44.Wang S, Wu X, Lincoln TM, Murphy-Ullrich JE. Expression of constitutively active cGMP-dependent protein kinase prevents glucose stimulation of thrombospondin 1 expression and TGF-beta activity. Diabetes. 2003;52:2144–2150. doi: 10.2337/diabetes.52.8.2144. [DOI] [PubMed] [Google Scholar]
- 45.Wang S, Shiva S, Poczatek MH, Darley-Usmar V, Murphy-Ullrich JE. Nitric oxide and cGMP-dependent protein kinase regulation of glucose-mediated thrombospondin 1-dependent transforming growth factor-beta activation in mesangial cells. J Biol Chem. 2002;277:9880–9888. doi: 10.1074/jbc.M108360200. [DOI] [PubMed] [Google Scholar]
- 46.Kono T, Saito M, Kinoshita Y, Satoh I, Shinbori C, Satoh K. Real-time monitoring of nitric oxide and blood flow during ischemia-reperfusion in the rat testis. Mol Cell Biochem. 2006;286:139–145. doi: 10.1007/s11010-005-9105-3. [DOI] [PubMed] [Google Scholar]
- 47.Dursun N, Arifoglu C, Suer C, Keskinol L. Blood pressure relationship to nitric oxide, lipid peroxidation, renal function, and renal blood flow in rats exposed to low lead levels. Biol Trace Elem Res. 2005;104:141–149. doi: 10.1385/BTER:104:2:141. [DOI] [PubMed] [Google Scholar]
- 48.Kudej RK, Vatner SF. Nitric oxide-dependent vasodilation maintains blood flow in true hibernating myocardium. J Mol Cell Cardiol. 2003;35:931–935. doi: 10.1016/s0022-2828(03)00174-3. [DOI] [PubMed] [Google Scholar]
- 49.Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698. doi: 10.1113/jphysiol.2002.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rapoport RM, Murad F. Endothelium-dependent and nitrovasodilator-induced relaxation of vascular smooth muscle: role of cyclic GMP. J Cyclic Nucleotide Protein Phosphor Res. 1983;9:281–296. [PubMed] [Google Scholar]
- 51.Murad F, Waldman S, Molina C, Bennett B, Leitman D. Regulation and role of guanylate cyclase-cyclic GMP in vascular relaxation. Prog Clin Biol Res. 1987;249:65–76. [PubMed] [Google Scholar]
- 52.Isenberg JS, Hyodo F, Matsumoto K, Romeo MJ, Abu-Asab M, Tsokos M, Kuppusamy P, Wink DA, Krishna MC, Roberts DD. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood. 2007;109:1945–1952. doi: 10.1182/blood-2006-08-041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 54.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu W, Smart EJ. Myristic acid stimulates endothelial nitric-oxide synthase in a CD36- and an AMP kinase-dependent manner. J Biol Chem. 2005;280:29543–29550. doi: 10.1074/jbc.M501238200. [DOI] [PubMed] [Google Scholar]
- 56.Isenberg JS, Jia Y, Fukuyama J, Switzer CH, Wink DA, Roberts DD. Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J Biol Chem. 2007;282:15404–15415. doi: 10.1074/jbc.M701638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maurer-Stroh S, Eisenhaber B, Eisenhaber F. N-terminal N-myristoylation of proteins: prediction of substrate proteins from amino acid sequence. J Mol Biol. 2002;317:541–557. doi: 10.1006/jmbi.2002.5426. [DOI] [PubMed] [Google Scholar]
- 58.Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC. Agonist-Stimulated Endothelial Nitric Oxide Synthase Activation and Vascular Relaxation. Role of eNOS Phosphorylation at Tyr83. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.107.162933. [DOI] [PubMed] [Google Scholar]
- 59.Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2006;28:185–206. [PubMed] [Google Scholar]
- 60.Westphal JR. Technology evaluation: ABT-510, Abbott. Curr Opin Mol Ther. 2004;6:451–457. [PubMed] [Google Scholar]
- 61.Haviv F, Bradley MF, Kalvin DM, Schneider AJ, Davidson DJ, Majest SM, McKay LM, Haskell CJ, Bell RL, Nguyen B, Marsh KC, Surber BW, Uchic JT, Ferrero J, Wang YC, Leal J, Record RD, Hodde J, Badylak SF, Lesniewski RR, Henkin J. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: design, synthesis, and optimization of pharmacokinetics and biological activities. J Med Chem. 2005;48:2838–2846. doi: 10.1021/jm0401560. [DOI] [PubMed] [Google Scholar]
- 62.Isenberg JS, Yu C, Roberts DD. Differential effects of ABT-510 and a CD36-binding peptide derived from the type 1 repeats of thrombospondin-1 on fatty acid uptake, nitric oxide signaling, and caspase activation in vascular cells. Biochem Pharmacol. 2008;75:875–882. doi: 10.1016/j.bcp.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schafer A, Wiesmann F, Neubauer S, Eigenthaler M, Bauersachs J, Channon KM. Rapid regulation of platelet activation in vivo by nitric oxide. Circulation. 2004;109:1819–1822. doi: 10.1161/01.CIR.0000126837.88743.DD. [DOI] [PubMed] [Google Scholar]
- 64.Boo YC. Shear stress stimulates phosphorylation of protein kinase A substrate proteins including endothelial nitric oxide synthase in endothelial cells. Exp Mol Med. 2006;38:63–71. doi: 10.1038/emm.2006.8. [DOI] [PubMed] [Google Scholar]
- 65.Leung LL. Role of thrombospondin in platelet aggregation. J Clin Invest. 1984;74:1764–1772. doi: 10.1172/JCI111595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chung J, Wang XQ, Lindberg FP, Frazier WA. Thrombospondin-1 acts via IAP/CD47 to synergize with collagen in alpha2beta1-mediated platelet activation. Blood. 1999;94:642–648. [PubMed] [Google Scholar]
- 67.Dorahy DJ, Thorne RF, Fecondo JV, Burns GF. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J Biol Chem. 1997;272:1323–1330. doi: 10.1074/jbc.272.2.1323. [DOI] [PubMed] [Google Scholar]
- 68.Fujimoto TT, Katsutani S, Shimomura T, Fujimura K. Thrombospondin-bound integrin-associated protein (CD47) physically and functionally modifies integrin alphaIIbbeta3 by its extracellular domain. J Biol Chem. 2003;278:26655–26665. doi: 10.1074/jbc.M302194200. [DOI] [PubMed] [Google Scholar]
- 69.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonnefoy A, Daenens K, Feys HB, De Vos R, Vandervoort P, Vermylen J, Lawler J, Hoylaerts MF. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. 2006;107:955–964. doi: 10.1182/blood-2004-12-4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, Rick ME, Wink DA, Frazier WA, Roberts DD. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood. 2008;111:613–623. doi: 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schultess J, Danielewski O, Smolenski AP. Rap1GAP2 is a new GTPase-activating protein of Rap1 expressed in human platelets. Blood. 2005;105:3185–3192. doi: 10.1182/blood-2004-09-3605. [DOI] [PubMed] [Google Scholar]
- 73.Danielewski O, Schultess J, Smolenski A. The NO/cGMP pathway inhibits Rap 1 activation in human platelets via cGMP-dependent protein kinase I. Thromb Haemost. 2005;93:319–325. doi: 10.1160/TH04-09-0582. [DOI] [PubMed] [Google Scholar]
- 74.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 75.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–839. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DiPietro LA, Nissen NN, Gamelli RL, Koch AE, Pyle JM, Polverini PJ. Thrombospondin 1 synthesis and function in wound repair. Am J Pathol. 1996;148:1851–1860. [PMC free article] [PubMed] [Google Scholar]
- 77.Yla-Herttuala S. An update on angiogenic gene therapy: vascular endothelial growth factor and other directions. Curr Opin Mol Ther. 2006;8:295–300. [PubMed] [Google Scholar]
- 78.Kastrup J. Therapeutic angiogenesis in ischemic heart disease: gene or recombinant vascular growth factor protein therapy? Curr Gene Ther. 2003;3:197–206. doi: 10.2174/1566523034578366. [DOI] [PubMed] [Google Scholar]
- 79.Mendoza MG, Robles HV, Romo E, Rios A, Escalante B. Nitric oxide-dependent neovascularization role in the lower extremity disease. Curr Pharm Des. 2007;13:3591–3596. doi: 10.2174/138161207782794103. [DOI] [PubMed] [Google Scholar]
- 80.Komorowska-Timek E, Timek TA, Brevetti LS, Zhang F, Lineaweaver WC, Buncke HJ. The effect of single administration of vascular endothelial growth factor or L-arginine on necrosis and vasculature of the epigastric flap in the rat model. Br J Plast Surg. 2004;57:317–325. doi: 10.1016/j.bjps.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 81.Cordeiro PG, Santamaria E, Hu QY. Use of a nitric oxide precursor to protect pig myocutaneous flaps from ischemia-reperfusion injury. Plast Reconstr Surg. 1998;102:2040–2048. doi: 10.1097/00006534-199811000-00035. discussion 2049-2051. [DOI] [PubMed] [Google Scholar]
- 82.Ercocen AR, Apaydin I, Emiroglu M, Gultan SM, Ergun H, Yormuk E. The effects of L-arginine and iloprost on the viability of random skin flaps in rats. Scand J Plast Reconstr Surg Hand Surg. 1998;32:19–25. doi: 10.1080/02844319850158912. [DOI] [PubMed] [Google Scholar]
- 83.Um SC, Suzuki S, Toyokuni S, Kim BM, Tanaka T, Hiai H, Nishimura Y. Involvement of nitric oxide in survival of random pattern skin flap. Plast Reconstr Surg. 1998;101:785–792. doi: 10.1097/00006534-199803000-00030. [DOI] [PubMed] [Google Scholar]
- 84.Cordeiro PG, Mastorakos DP, Hu QY, Kirschner RE. The protective effect of L-arginine on ischemia-reperfusion injury in rat skin flaps. Plast Reconstr Surg. 1997;100:1227–1233. doi: 10.1097/00006534-199710000-00023. [DOI] [PubMed] [Google Scholar]
- 85.Ayyildiz A, Uysal A, Kocer U, Karaaslan O, Huri E, Germiyanoglu C, Caydere M. Effect of sildenafil citrate on viability of flaps: an experimental study in rats. Scand J Plast Reconstr Surg Hand Surg. 2005;39:204–208. doi: 10.1080/02844310510006268. [DOI] [PubMed] [Google Scholar]
- 86.Sarifakioglu N, Gokrem S, Ates L, Akbuga UB, Aslan G. The influence of sildenafil on random skin flap survival in rats: an experimental study. Br J Plast Surg. 2004;57:769–772. doi: 10.1016/j.bjps.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 87.Hart K, Baur D, Hodam J, Lesoon-Wood L, Parham M, Keith K, Vazquez R, Ager E, Pizarro J. Short- and long-term effects of sildenafil on skin flap survival in rats. Laryngoscope. 2006;116:522–528. doi: 10.1097/01.mlg.0000200792.67802.3b. [DOI] [PubMed] [Google Scholar]
- 88.Isenberg JS, Romeo MJ, Abu-Asab M, Tsokos M, Oldenborg A, Pappan L, Wink DA, Frazier WA, Roberts DD. Increasing survival of ischemic tissue by targeting CD47. Circ Res. 2007;100:712–720. doi: 10.1161/01.RES.0000259579.35787.4e. [DOI] [PubMed] [Google Scholar]
- 89.Andreassi A, Bilenchi R, Biagioli M, D'Aniello C. Classification and pathophysiology of skin grafts. Clin Dermatol. 2005;23:332–337. doi: 10.1016/j.clindermatol.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 90.Isenberg JS, Pappan LK, Romeo MJ, Abu-Asab M, Tsokos M, Wink DA, Frazier WA, Roberts DD. Blockade of thrombospondin-1-CD47 interactions prevents necrosis of full thickness skin grafts. Ann Surg. 2008;247:180–190. doi: 10.1097/SLA.0b013e31815685dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rectenwald JE, Upchurch GR., Jr. Impact of outcomes research on the management of vascular surgery patients. J Vasc Surg. 2007;45(Suppl A):A131–140. doi: 10.1016/j.jvs.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 92.Sigvant B, Wiberg-Hedman K, Bergqvist D, Rolandsson O, Andersson B, Persson E, Wahlberg E. A population-based study of peripheral arterial disease prevalence with special focus on critical limb ischemia and sex differences. J Vasc Surg. 2007;45:1185–1191. doi: 10.1016/j.jvs.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 93.Roger VL. Epidemiology of Myocardial Infarction. Med Clin North Am. 2007;91:537–552. doi: 10.1016/j.mcna.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Poredos P, Jug B. The prevalence of peripheral arterial disease in high risk subjects and coronary or cerebrovascular patients. Angiology. 2007;58:309–315. doi: 10.1177/0003319707302494. [DOI] [PubMed] [Google Scholar]
- 95.Soucy KG, Ryoo S, Benjo A, Lim HK, Gupta G, Sohi JS, Elser J, Aon MA, Nyhan D, Shoukas AA, Berkowitz DE. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol. 2006;101:1751–1759. doi: 10.1152/japplphysiol.00138.2006. [DOI] [PubMed] [Google Scholar]
- 96.Bach MH, Sadoun E, Reed MJ. Defects in activation of nitric oxide synthases occur during delayed angiogenesis in aging. Mech Ageing Dev. 2005;126:467–473. doi: 10.1016/j.mad.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 97.Smith AR, Visioli F, Hagen TM. Plasma membrane-associated endothelial nitric oxide synthase and activity in aging rat aortic vascular endothelia markedly decline with age. Arch Biochem Biophys. 2006;454:100–105. doi: 10.1016/j.abb.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 98.Favier J, Germain S, Emmerich J, Corvol P, Gasc JM. Critical overexpression of thrombospondin 1 in chronic leg ischaemia. J Pathol. 2005;207:358–366. doi: 10.1002/path.1833. [DOI] [PubMed] [Google Scholar]
- 99.Riessen R, Kearney M, Lawler J, Isner JM. Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries. Am Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x. [DOI] [PubMed] [Google Scholar]
- 100.Roth JJ, Gahtan V, Brown JL, Gerhard C, Swami VK, Rothman VL, Tulenko TN, Tuszynski GP. Thrombospondin-1 is elevated with both intimal hyperplasia and hypercholesterolemia. J Surg Res. 1998;74:11–16. doi: 10.1006/jsre.1997.5209. [DOI] [PubMed] [Google Scholar]
- 101.Isenberg JS, Hyodo F, Pappan LK, Abu-Asab M, Tsokos M, Krishna MC, Frazier WA, Roberts DD. Blocking thrombospondin-1/CD47 signaling alleviates deleterious effects of aging on tissue responses to ischemia. Arterioscler Thromb Vasc Biol. 2007;27:2582–2588. doi: 10.1161/ATVBAHA.107.155390. [DOI] [PubMed] [Google Scholar]
- 102.Kincer JF, Uittenbogaard A, Dressman J, Guerin TM, Febbraio M, Guo L, Smart EJ. Hypercholesterolemia promotes a CD36-dependent and endothelial nitric-oxide synthase-mediated vascular dysfunction. J Biol Chem. 2002;277:23525–23533. doi: 10.1074/jbc.M202465200. [DOI] [PubMed] [Google Scholar]
- 103.Giles TD. Aspects of nitric oxide in health and disease: a focus on hypertension and cardiovascular disease. J Clin Hypertens (Greenwich) 2006;8:2–16. doi: 10.1111/j.1524-6175.2006.06023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thatcher GR. An introduction to NO-related therapeutic agents. Curr Top Med Chem. 2005;5:597–601. doi: 10.2174/1568026054679281. [DOI] [PubMed] [Google Scholar]
- 105.Zhang HM, Yan YP, Sun GC, Hum HX, Liu ZF, Feng YJ. Cutaneous blood vessels in scent pigs. Plast Reconstr Surg. 2000;106:1555–1565. doi: 10.1097/00006534-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 106.Isenberg J, Romeo J, Maxhimer J, Smedley J, Frazier W, Roberts D. Gene Silencing of CD47 and Antibody Ligation of Thrombospondin-1 Enhance Ischemic Tissue Survival in a Porcine Model: Implications for Human Disease. Ann Surg. 2008 doi: 10.1097/SLA.0b013e31816c4006. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sage E, Mercier O, Van den Eyden F, de Perrot M, Barlier-Mur AM, Dartevelle P, Eddahibi S, Herve P, Fadel E. Endothelial cell apoptosis in chronically obstructed and reperfused pulmonary artery. Respir Res. 2008;9:19. doi: 10.1186/1465-9921-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lario S, Bescos M, Campos B, Mur C, Luque P, Alvarez R, Campistol JM. Thrombospondin-1 mRNA expression in experimental kidney transplantation with heartbeating and non-heart-beating donors. J Nephrol. 2007;20:588–595. [PubMed] [Google Scholar]
- 109.Thakar CV, Zahedi K, Revelo MP, Wang Z, Burnham CE, Barone S, Bevans S, Lentsch AB, Rabb H, Soleimani M. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J Clin Invest. 2005;115:3451–3459. doi: 10.1172/JCI25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.American Heart Association 2008 http://www.americanheart.org/presenter.jhtml?identifier=4478.