

Abstract
A commercial quadrupole/time-of-flight (QqTOF) tandem mass spectrometer has been adapted for ion/ion reaction studies. To enable mutual storage of oppositely charged ions in a linear ion trap, the oscillating quadrupole field of the second quadrupole of the system (Q2) serves to store ions in the radial dimension while auxiliary RF is superposed on the end lenses of Q2 during the reaction period to create barriers in the axial dimension. A pulsed dual electrospray (ESI) source is directly coupled to the instrument interface for the purpose of proton transfer reactions. Singly and doubly charged protein ions as high in mass as 66 kDa are readily formed and observed after proton transfer reactions. For the modified instrument, the mass resolving power is about 8000 for a wide m/z range and the mass accuracy is ~20 ppm for external calibration and ~5 ppm for internal calibration after ion/ion reactions. Parallel ion parking is demonstrated with a six-component protein mixture, which shows the potential application of reducing spectral complexity and concentrating certain charge states. The current system has high flexibility with respect to defining MSn experiments involving collision-induced dissociation (CID) and ion/ion reactions. Protein precursor and CID product masses can be determined with good accuracy, providing an attractive platform for top-down proteomics. Electron transfer dissociation (ETD) ion/ion reactions are implemented by using a pulsed nano-ESI/atmospheric pressure chemical ionization (APCI) dual source for ionization. The reaction between protonated peptide ions and radical anions of 1,3-dinitrobenzene formed exclusively c- and z- type fragment ions.
Keywords: quadrupole/time-of-flight mass spectrometry, ion/ion reactions, top-down proteomics, collision-induced dissociation, electron transfer dissociation
INTRODUCTION
The nature of an ionized biomolecule (e.g., magnitude1 and polarity of charge,2 the identities of cationizing agents,3 etc.) plays an important role in determining characteristics of an ion in the gas phase, including its reactivity and three-dimensional structure. Proton transfer ion/ion reactions provide an effective means for altering the nature of gas phase ions after their initial formation via manipulation of ion charge and polarity.4 The utility of ion/ion reactions has been further broadened as a probe of primary structure with the development of electron transfer dissociation ion/ion reactions,5–12 where structurally informative dissociation is induced, giving rise to cleavages analogous to those noted in electron capture dissociation (ECD).13, 14
Ion/ion reactions have been conducted both outside and inside a mass spectrometer. A desirable characteristic of the former approach is that it allows ion/ion reactions to be coupled with any type of mass analyzer. For example, a time-of-flight (TOF) mass spectrometer has been used to study the charge reduced ion/ion reaction products of proteins formed outside the mass spectrometer.15–17 Ion/ion reactions within the vacuum system of a mass spectrometer have usually taken place in electrodynamic ion traps. Ion traps have the desirable capability of employing ion/ion reactions within the context of multi-stage mass spectrometry (MSn) experiments. In this regard, ion traps have served as reaction vessels18 with ion isolation and MSn functionalities,19 including the mass analysis of the product ions. To date, ion/ion reactions performed in an ion trap have been applied to mixture analysis20–23 including the use of the “ion parking” technique for gas-phase concentration and purification,24 the formation of ions that cannot be directly produced by ESI for subsequent tandem mass spectrometry studies,25 the reduction of the product ion charge states to singly and doubly charged species so as to simplify the interpretation of product ion spectra26–31 and protein/peptide sequencing by electron transfer dissociation.8 The above applications have proven to be particularly useful in extending the capabilities of an ion trap mass spectrometer to top-down protein analysis. For example, interpretable MS/MS spectra of proteins up to 25.9 kDa have been obtained on ion trap instruments, albeit with limited mass resolution and mass measurement accuracy.30 In addition, an a priori unknown protein which is not present in an annotated protein data base has been identified using a 3D ion trap via whole protein dissociation and subsequent proton transfer ion/ion reactions.32 While ion/ion chemistry provides a solution to address the relative low mass resolving power of ion traps by charge reduction of ions mostly to +1, other mass analysis characteristics of ion traps, such as limited mass accuracy, narrow mass-to-charge ratio (m/z) range available on most commercial instruments, and relatively long mass analysis time are factors limiting the application of ion/ion reaction studies in proteomics. One obvious strategy to address the mass analysis problems associated with ion traps is to spatially separate the ion manipulation and ion/ion reaction steps from the final mass analysis step and to employ a different form of mass analysis. Ion/ion reactions can then be conducted in ion traps with all the ion manipulation capabilities preserved, while mass analyzers with performance characteristics superior to those of ion traps are employed for the product analysis. Some of the logical instrument platforms that can be envisioned include hybrid mass spectrometers with a combination of an electrodynamic ion trap with i) a time-offlight (TOF) mass analyzer,33 ii) an orbitrap mass analyzer34 and iii) a Fourier transform ion cyclotron resonance (FTICR) mass analyzer.35 The high mass measurement accuracy, high speed, wide m/z range, and moderate mass resolving power of TOF makes a quadrupole/time-of-flight combination particularly attractive for ion/ion reaction applications.
In this work, a commercial quadrupole/time-of-flight tandem mass spectrometer,36 which comprises three quadrupole arrays and a reflectron TOF, abbreviated herein as QqTOF, has been adapted for ion/ion reaction studies. The hardware modifications to the QqTOF instrument have been minimized with the use of a pulsed dual electrospray37 or a nano-ESI/atmospheric pressure chemical ionization (APCI)38 source configuration. With such a dual source, no interface modification is needed to accommodate distinct ion sources and each polarity of ions can be sequentially formed and injected through the same ion path. Flexible software control over the voltages of all ion optical elements during the course of an ion/ion reaction experiment is key to the ability to use a common ion path. The only hardware change needed herein is the superposition of auxiliary RF on the end lenses of the quadrupole array that serves as a linear ion trap for mutual simultaneous ion polarity storage. The modified instrument has been characterized in terms of its utility for conducting common ion/ion reaction experiments involving proton transfer reactions and electron transfer reactions. Illustrations and a summary of the capabilities of the instrument are provided in this report.
EXPERIMENTAL SECTION
Materials
Proteins were purchased from Sigma-Aldrich (St. Louis, MO). The peptide KGAILKGAILR was synthesized by SynPep (Dublin, CA). Perfluoro-1-octanol (PFO) and 1,3-dinitrobenzene were obtained from Sigma-Aldrich (Milwaukee, WI). Samples were used without further purification. Solutions of peptides or proteins were dissolved to 5 µM in 50/50/1 (v/v/v) methanol/water/acetic acid solution. PFO, with a final concentration of 200 µM, was prepared as a methanol solution containing 1% ammonium hydroxide.
Ionization Methods
For proton transfer ion/ion reactions, ionization was accomplished via a home-built pulsed dual ESI source,37 comprising a nano-ESI emitter for the formation of positive ions ([M+nH]n+) of proteins and peptides and an ESI emitter for the formation of deprotonated anions from PFO. To induce electron transfer dissociation ion/ion reactions, a nano-ESI/APCI source was employed, as fully described elsewhere.38 These ion sources, which can be directly coupled to the nanospray interface of a commercial QSTAR instrument, share a common atmosphere/vacuum interface as well as a common ion path into the instrument.
Mass Spectrometry
All experiments were performed using a modified QqTOF tandem mass spectrometer36 (QSTAR XL, Applied Biosystems/MDS SCIEX, Concord, ON, Canada). The instrument consists of three quadrupoles (ion guide Q0, mass filter Q1, collision cell Q2) and a reflectron time-of-flight (TOF) analyzer with orthogonal injection of ions, as shown schematically in Figure 1. The key hardware modification of the instrument was the superposition of auxiliary RF signals to the containment lenses of the Q2 quadrupole array, which enables the mutual storage of ions of opposite polarity. The auxiliary RF signals were optimized and set at 80 kHz, 150 V for each ion/ion reaction experiment. Another necessary modification is the change of the instrument software from Analyst to Daetalyst as well as the associated instrument control and data acquisition system. Daetalyst is a research version of software developed by MDS SCIEX, which provides full control for initiating each ion source and for applying the appropriate potentials and their timing to the ion path.
Figure 1.

Schematic drawing of the modified quadrupole time-of-flight tandem mass spectrometer (QSTAR XL). The plots below show the potential along the instrument axis at different steps for single ion/ion reaction experiments.
A typical scan function involving ion/ion reactions consisted of steps such as positive ion injection to Q2 LIT (50 ms), anion injection to Q2 LIT (50 ms), mutual cation/anion storage (100–200 ms), and mass analysis by TOF (20–50 ms). The potentials along the instrument axis during such major steps are plotted in Figure 1. Briefly, during the cation injection step the positive high potential applied to the nanospray tip was programmed to be around 1500 V for ionization. The subsequently produced analyte ions were isolated by Q1 in mass-resolving mode and injected axially into the Q2 LIT (3–8 mTorr, nitrogen) with a kinetic energy of roughly 10 zeV, where z represents the unit charge of the ion (Figure1, cation injection). The ions were cooled in Q2 for 50 ms by raising the DC barrier on IQ2 and IQ3 lenses, during which time the high voltage on the nanospray emitter was turned off. After the cooling step, the negative high potential applied to the other ESI tip or the APCI wire was triggered on (~2.5 kV) and the potentials applied to the ion optical elements before Q2 were adjusted to allow injection of anions of interest at relatively low kinetic energies (Figure 1, anion injection). To prevent ions from leaking out of the Q2 cell, an auxiliary RF signal was applied to the IQ3 lens during this anion injection step. For the mutual ion storage step, the negative high potential was turned off and the auxiliary RF signals were applied to both containment lenses of Q2 to store ions axially. After a defined period of mutual storage, the residual reagent ions were ejected from Q2 by applying attractive DC potentials to the IQ2 and IQ3 lenses while the auxiliary RF signals were terminated. Finally, the product ions of interest were released from Q2 to the TOF section for mass analysis, where they acquired an energy of 4 keV per charge. The LINAC39 function in Q2 was turned off for ion/ion reaction periods and turned on for the ion injection steps or release steps. TOF mass spectra were recorded with a four-channel time-to-digital converter. The spectra shown here were typically the averages of 50–200 individual scans.
RESULTS AND DISCUSSION
Proton Transfer Ion/Ion Reactions and Mass Analysis Performance for Ion/Ion Products
Deprotonated perfluoro-1-octanol ([PFO-H]−) has been demonstrated to be an efficient proton transfer reagent that does not give rise to adduct formation or fragmentation of peptide/protein cations.37 When a concentrated PFO solution (200 µM) was used, abundant PFO dimer anions ([2PFO-H]−) were produced (shown in Figure 2a), which were also found to be effective reagents for proton transfer reactions. From the standpoint of the reduction of the charges of whole protein ions, the dimer anions are advantageous because they allow for the use of higher Q2 RF levels than can the monomer anions. The ability to employ higher Q2 RF levels translates to improved storage of high m/z ions. Therefore, the PFO dimer anions were isolated as shown in Figure 2b and used as the proton transfer reagent in the proton transfer ion/ion reaction experiments described below.
Figure 2.

Mass spectra derived from (a) negative electrospray of 200 µM PFO solution and (b) isolation of the PFO dimer anions ([2PFO-H]−).
Proton transfer reactions were carried on in the Q2 LIT (4 mTorr, nitrogen) with proteins ranging in mass from 5 kDa to 66 kDa. Figure 3 shows the post-ion/ion mass spectra of (a) bovine insulin, (b) bovine ubiquitin, (c) horse heart myoglobin and (d) bovine serum albumin (BSA). Singly and doubly charged ions were readily formed via sequential proton transfer reactions with an excess of PFO anions. A resolving power (M/ΔMFWHM) of 8300 was observed for [M+H]+ of insulin (inset of Figure 3a). For heavier protein ions (>8,000 Da), this resolution is insufficient to distinguish isotopic peaks. An estimate of the resolving power under such conditions can be made by comparing the experimental isotopic envelope with that obtained theoretically. We found that the experimental data were adequately reproduced by the simulated isotopic envelopes with a resolution setting of around 8,000 (data not shown) for the ions of most of the proteins with mass greater than 8,000 Da. However, if the isotopic envelope is broadened due to chemical heterogeneity, a misleading estimate of resolving power obtained by comparison to a predicted peak shape in the absence of heterogeneity can result. A likely example of a case in which heterogeneity is to contribute to the peak shape can be found in Figure 3d, where the unusually broad peak of +1 of BSA is seen.
Figure 3.

Mass spectra derived from ion/ion proton transfer reactions between [2PFOH]− and the protein ions: (a) +4 insulin, (b) +10 ubiqutin, (c) +14 myoglobin and (d) the entire charge state distribution of bovine serum albumin.
The effective upper m/z limit of the current system is also of interest. The +1 ions of BSA (m/z ~66,000) were the highest m/z ions seen after ion/ion reactions, as shown in Figure 3d. As expected, the signal levels were found to decrease as the m/z ratio of the ions increased. For example, an average of 1000 scans was required to achieve the signal-to-noise ratio associated with the m/z 66,000 ions of Figure 3d, while much less averaging was needed for lower m/z ions, despite similar signals in the pre-ion/ion reaction mass spectra. Two factors can give rise to an upper m/z limit for ion detection in this instrument. One is the magnitudes of the RF amplitudes (both in radial and axial dimension) applied to the Q2 LIT during ion/ion reactions, which define the m/z range over which ions of opposite polarity can be stored simultaneously.40 The other limiting factor is the detector’s response to high m/z ions.41 In previous studies on a triple quadrupole/linear ion trap instrument, we have noted that the highest m/z ions seen are about m/z 33 k.42 Under those conditions, the RF amplitude (Vr) applied to the rods of the linear ion trap during mutual storage was 185 V and the q (Mathieu parameter) value for m/z 33 k was 0.00748 (RF angular frequency Ω:650 kHz, field radius r0:4.17 mm). A trapping potential of 0.35 V for the m/z 33 k ions can be calculated according to equation 1, which predicts the pseudo-potential well depth, Dr, in a linear ion trap.43
| (1) |
In the QqTOF instrument, the low mass cut-off (LMCO) for Q2 during the ion/ion reaction period was set to m/z 350, which corresponds to a Vr of 1426 V (Ω:1.8 MHz, r0:4.17 mm). If Dr =0.35 V is used as a minimum barrier for trapping ions on the QqTOF instrument, then an upper trapping limit of ions about m/z 250 k can be estimated for the radial dimension. The estimation of the trapping limit in the axial direction was based on equation 2,44 where Da is the trapping potential in the axial direction , c is a constant, Va is the amplitude of the auxiliary RF applied on the end lenses and F is the frequency.
| (2) |
Ions of roughly m/z 290,000 can be trapped in the axial direction with the current experimental settings and the criteria of a minimum trapping potential of 0.35 V. Based on these estimates, the experimentally observed m/z 66,000 limit appears to be well below the upper m/z limit to ion storage in the Q2 LIT under the conditions used in this work. Detector response therefore appears to be the limiting factor for the analysis of high m/z ions. It is well known that the response of the microchannel plate (MCP) detector used in most TOF MS instruments is dependent upon ion m/z for a given accelerating potential. Several groups have shown that the secondary electron yield decreases significantly as the velocity of the ion decreases.41, 45–48 Since the velocity of the ions decreases as the inverse square root of the mass in a TOF analyzer, large ions move more slowly and thus have a lower probability of detection. Loboda et al. showed that the sensitivity of a very similar QqTOF instrument (coupled with MALDI source) as used in our studies decreased rapidly above m/z 20,000, with the highest m/z ions observed being around 66,000.49 Given that a 10 kV acceleration voltage was used in their studies compared to the 4 kV acceleration voltage in the present QqTOF instrument, the observation of an upper m/z limit around 66,000 herein can be expected. The above considerations suggest that detector efficiency is the factor that limits the upper m/z limit in the present instrument.
Obtaining accurate mass measurement of intact proteins is of great importance in identifying unknown proteins via a “top-down” approach and is a major motivation for the use of TOF rather than ion trap mass analysis.50, 51 Table 1 shows the molecular masses of eight proteins, separately subjected to analysis, deduced from m/z measurement of the singly protonated species formed via ion/ion reactions and via deconvolution from the nanospray (MS) spectra. The two approaches show comparable mass measurement accuracy. The ion/ion reaction approach is most attractive in application to the analysis of complex protein mixtures via electrospray, when the resolution of a TOF instrument is insufficient to resolve isotopes of the protein ions and significant overlapping of peaks makes deconvolution difficult, especially for the low abundance components.52 For example, the molecular mass of lysozyme could not be retrieved from deconvoluting the nanospray spectrum of a six-component protein mixture if the charge states were not manually selected (see the discussion below related to Figure 6).
Table 1.
Comparisons of protein masses deduced from molecular formula, ion/ion proton transfer reactions and deconvolution of the MS sepctra.
| Calculated Mw (Da) | Measured Mw from Reaction (Da) | Measured Mw from MS (Da) | |
|---|---|---|---|
| insulin | 5733.5 | 5733.5 | 5733.5 |
| ubiquitin | 8564.8 | 8564.4 | 8564.1 |
| bovine cytocrhome c | 12231.0 | 12231.5 | 12231.1 |
| equine cytocrhome c | 12360.1 | 12359.6 | 12360.4 |
| α-lactalbumin | 14178.1 | 14178.5 | 14179.5 |
| lysozyme | 14305.2 | 14306.2 | 14306.3 |
| myoglobin | 16950.5 | 16950.5 | 16952.0 |
| carbonic anhydrase | 29024.7 | 29024.6 | 29024.8 |
Figure 6.

(a) MS of six-component protein mixture. (b) Parallel ion parking of protein mixture by applying auxiliary ac on one pair of Q2 rods at 17 kHz and 10 V. (c) Post-ion/ ion reaction of the protein mixture by reducing the charge states mostly to +1.
The utility of the modified QqTOF instrument in the analysis of whole proteins also depends upon its performance in applying ion/ion reactions to the dissociation products of intact protein ions. In the MS/MS mode, parent ions are selected in Q1 in mass resolving mode, then collisionally activated in Q2 either by accelerating into Q2 (low energy beam-type CID) or resonant excitation (ion-trap CID). Ion/ion reactions can then be applied to the dissociation products and the charge reduced products are measured in the TOF analyzer. Figure 4 shows the charge reduced ion trap CID products of bovine ubiquitin ions ([M+8H]8+). Mass resolving power of 6000–8000 was observed over the entire mass range, which is indicated by the product ions shown in the insets of Figure 4.
Figure 4.

Post-ion/ion ion-trap CID spectrum of the [M+8H]8+ ion of bovine ubiquitin via activation at 121.45 kHz, 600 mV for 300 ms, collision gas: nitrogen at 4 mTorr.
The mass accuracy of the instrument for the dissociation products can be illustrated by plotting the mass accuracy of each product ion against its mass. Figure 5 shows such a plot for the major product ions in Figure 4 under external calibration (filled dots) or internal calibration (empty circles). When external mass calibration was done with (CsI)Cs+ ions (m/z 392.7153) and (CsI)7Cs+ ions (m/z 1951.5748), mass accuracy of ~10 ppm was obtained for products below m/z 2000 and mass accuracy of ~20 ppm was obtained for ions beyond the calibration bracket. When the same spectrum (Figure 4) was calibrated with the theoretical masses of b5 and b16 ions, mass accuracy of ~5 ppm can be achieved for products ions falling close to that range.
Figure 5.

Plot for the mass accuracy, under external calibration (filled dots) or internal calibration (empty circles), of the major product ions shown in Figure 4.
Parallel Ion Parking for Protein Mixtures
The multiple charging phenomenon resulting from electrospray ionization of proteins can lead to highly complex spectra when mixtures are analyzed. Extensive charge reduction of the protein ions mostly to the +1 charge state was shown to be an effective way of reducing spectral complexity.53 However, reducing the charge to +1 can greatly reduce the detector response, potentially leading to reduced instrument sensitivity. Recently, Chrisman et al. have shown that instead of charge reduction to mostly the +1 charge state, ion/ion reaction rates can be inhibited over a broad m/z range, resulting in a less complex spectrum with good sensitivity since signal originally spread among multiple charge states can be condensed into only one or two charge states.54 To distinguish this approach from the selective inhibition of ion/ion reaction rates for ions within a narrow m/z range (“ion parking”),24 this technique is referred to as “parallel ion parking”. For the modified QqTOF instrument, parallel ion parking can be implemented in the Q2 LIT by applying a relatively high amplitude, low frequency auxiliary ac signal on one pair of Q2 rods during the ion/ion reaction period. An example of extensive charge reduction and parallel ion parking is demonstrated in Figure 6 for a six-component protein mixture (myoglobin 2 µM, lysozyme 2 µM, insulin 1 µM, α-lactalbumin 2 µM, bovine cytocrhome c 2 µM, equine cytochrome c 2 µM). Figure 6a shows the initial distribution of ions produced from nanospray of the protein mixture. After parallel ion parking is applied to the ion population shown in Figure 6a (auxiliary ac: 17 kHz, 10 V), the signal for each component is largely concentrated into one or two charge states as shown in Figure 6b. Figure 6c shows the result when ion/ion reactions are allowed to proceed in the absence of ion parking for a period sufficiently long to convert the proteins largely to the +1 charge state. The three spectra of Figure 6 represent operation of the instrument under conditions that provide three different peak capacities. Figure 6a and Figure 6c represent the smallest and largest peak capacities, respectively. The multiple charging phenomenon tends to concentrate analyte signals into a narrow m/z range, which minimizes the ability of a mass spectrometer to resolve components. The situation is further complicated by the fact that multiple peaks per component are present. The ion/ion reaction experiments disperse the ions over a wider m/z range and also reduce the number of peaks per component. Reduction of protein charge largely to +1 provides the largest peak capacity but with a sacrifice in signal levels due to reduced detector efficiency for the higher m/z ions. The parallel parking experiment allows for a compromise operating condition that reduces the number of components per peak and increases the m/z range over which the analyte ions are dispersed but minimizes charge state reduction and the concomitant loss of detector efficiency.
Top-down Identification of Unknown Proteins
The relatively wide m/z range, high mass measurement accuracy, and moderately high resolving power of the TOF in the modified QqTOF are useful performance characteristics for the top-down identification and characterization of unknown proteins, in conjunction with ion/ion reactions. During the course of characterizing the instrument, it was noted that a solution containing carbonic anhydrase also contained significant levels of several other species of unknown identity. The positive nanospray spectrum of this solution is shown in Figure 7a. Charge reduction of the initial ion population (Figure 7b) shows there are the two major unknown components, one has a molecular weight of 8565.2 Da (denoted as unknown protein A) and another one has a molecular weight of 15592.1 Da (denoted as unknown protein B).
Figure 7.

(a) MS derived from positive nanospray of 5 µM bovine carbonic anhydrase solution and (b) post-ion/ion spectrum of charge reducing the ions shown in (a).
Both low energy beam-type CID and ion-trap CID were applied to several isolated charge states of the two unknown proteins, while ion/ion proton transfer reactions were used to simplify product ion interpretation. For example, the [M+12H]12+ ions of unknown protein B were collisionally activated at 136.16 kHz, 1.6 V in Q2 for 200 ms. The product ions were subjected to ion/ion proton transfer reactions with [2PFO-H]− anions for 100 ms and then to TOF mass analysis. The post-ion/ion MS/MS spectrum is shown in Figure 8. The database search of the uninterpreted post-ion/ion product ion spectrum was done by comparing the singly charged product ions masses with predicted product ions from in silico fragmentation of the proteins in the SwissProt (v. 45.0) database via software developed in-house.20 A precursor mass of 15592 Da was used in the database search with a precursor mass tolerance of ±200 Da and a fragment mass tolerance of ±2 Da. Only the theoretical b- and y-type ions were searched since they are most common for ion trap CID of whole protein cations. Two scoring algorithms were used here. One is a probability-based approach used by Prosight PTM55 and the other is a product ion abundance-based approach developed in-house.20 The highest ranked protein for both the probability based score (3.61e-11) and the intensity based score (130.7), is SODC_BOVIN (bovine superoxide dismutase, Mw 15592.4), which has one disulfide bond (Cys55-Cys144) and is N-terminally acetylated. Of the 30 fragment ions observed, 25 were found to match with the in silico fragments of this protein, with the other 5 fragments manually determined to be the loss of NH3 or H2O from some b- and y- type ions. The product ions in Figure 8 can then be interpreted on the basis of the protein being bovine superoxide dismutase. Sequence ions such as b5–8, b27–34 and y117–124 are observed as prominent products. The bold letters in the protein sequence indicate where the sequence tag can be retrieved if a de novo sequencing approach is employed. The assignments associated with Figure 8 are consistent with typical positive protein ion dissociation, based on the previous observations in the CID of model protein ions.26–30, 56 In a similar way, the unknown protein A was identified with high confidence as bovine ubiquitin. The above example of identification of two a priori unknown proteins demonstrates that the modified QqTOF instrument provides a good platform in top-down proteomics studies with the advantage of acquiring accurate masses and the direct linkage between molecular mass and the structural information derived from tandem mass spectrometry.
Figure 8.
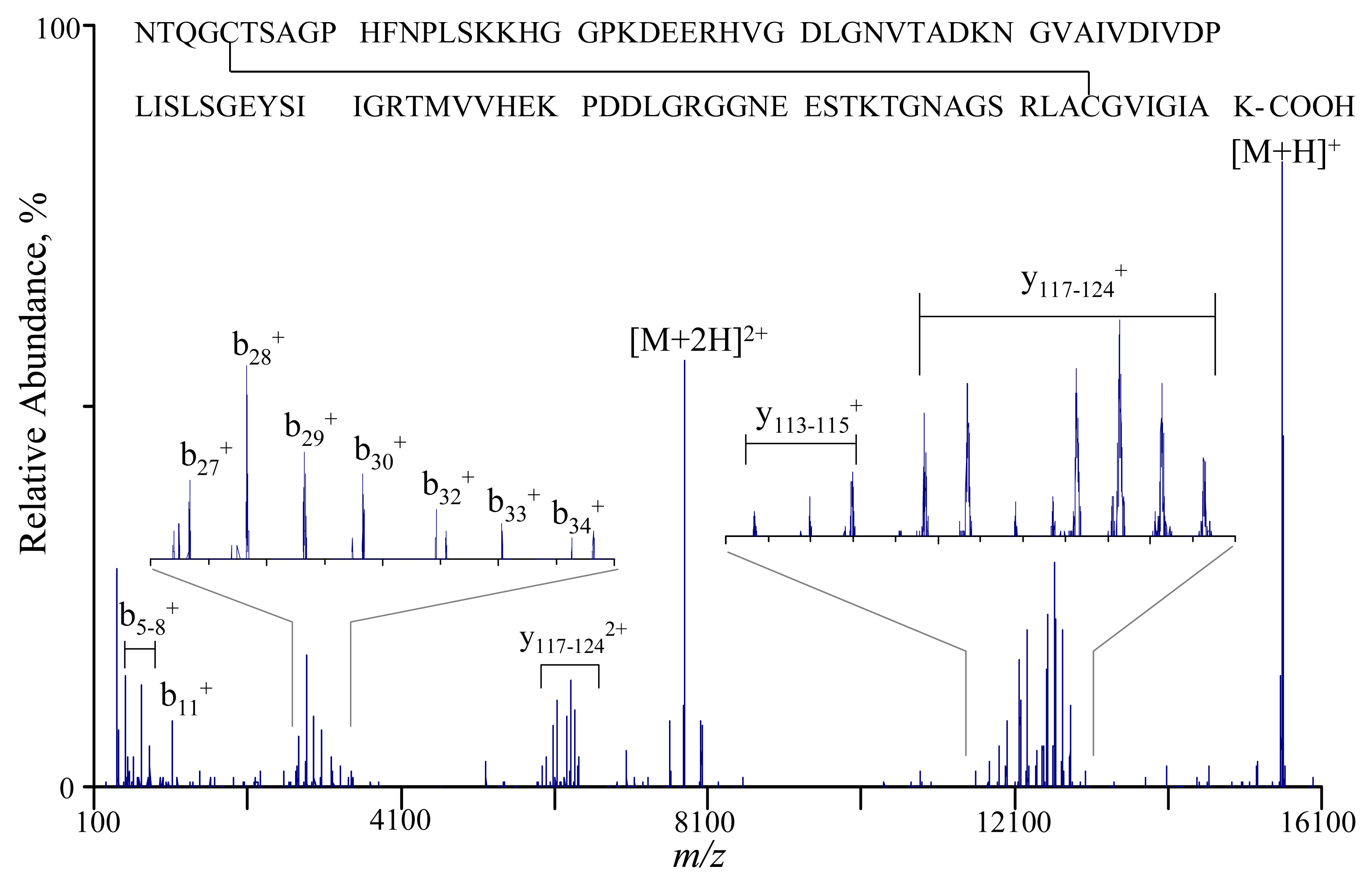
Post-ion/ion ion-trap CID spectrum of the [M+12H]12+ ions of unknown protein B via activation at 136.16 kHz, 1.6 V for 200 ms, collision gas: nitrogen at 4 mTorr. The identified protein sequence (bovine superoxide dismutase) is listed above the spectrum with observed fragmentation labeled.
Electron Transfer Ion/Ion Reactions
It is desirable to be able to implement both proton transfer and electron transfer reactions in the QqTOF. The latter reaction type holds analogies to electron capture dissociation (ECD) in that electron transfer from an anion to multiply protonated polypeptides or proteins typically results in fragmentation of N-Cα bond along peptide/protein backbone, which is less sensitive to the sequence or the presence of labile post-translational modifications.5, 6, 8, 10, 12 We have demonstrated in a previous publication that electron transfer dissociation (ETD) can be implemented on a triple quadrupole/linear ion trap instrument by using a sequentially pulsed ESI/APCI dual ionization source.38 A similar ESI/APCI source configuration was employed on the QqTOF instrument. Figure 9 shows the product ion spectrum derived from ETD of the [M+3H]3+ ions of a synthetic peptide (KGAILKGAILR) with 1,3-dinitrobenzene radical anions for 200 ms. Complete c- and z- type sequence ions were observed. Interestingly, almost all z-type ions are in their adduct form (labeled *zn+ in the spectrum), except z6+· and z10+· ions. Comparisons of the mass measurement of *zn + ions to the calculated z+· ions show that there is a 32 Da mass increase with each of the z+· ions, which clarifies the uncertainty of the adduct mass (31–32 Da) in our previous observations using 3D ion traps.10 The mechanism for the *zn + ions formation is currently under investigation, the results of which will be reported elsewhere. Nevertheless, preliminary results show that the adducts are most likely the product of ion-molecule reactions with the originally formed z+· ions.
Figure 9.

Post-ion/ion spectrum from the reaction of [M + 3H]3+ ions of synthetic peptide KGAILKGAILR with 1,3-dinitrobenzene radical anions for 200 ms. *zn+ denotes the z adducts with 32 Da mass increase compared to zn+· ions. zn+ stands for the even electron z ion species, which is one mass unit higher than zn+· ions. The insets show the theoretical isotopic distribution for the [M+H]+ ions and the experimental result.
Electron transfer and proton transfer are competitive processes for any multiply protonated peptide or protein.9 It is therefore of interest to differentiate the two processes in the ion/ion reactions. Blowup of the +1 charge state of the parent ion (inset of Figure 9) shows that masses and abundances of the isotopic peaks of the intact +1 ions match well the theoretical distribution for [M+H]+ ions, indicating: i) proton transfer takes place in the two step process leading from [M+3H]3+ to [M+H]+ and ii) most of the electron transfer products dissociate under the current reaction conditions. The %ETD (i.e. sum of ion signal due to ETD divided by the sum of all ion signals other than residual peptide cation reactant) with the use of 1,3-dinitrobenzene as ETD reagent on the instrument described herein is about 30–40%. Note that the %ETD is the percentage of ion/ion reactions that lead to ETD, which may differ from the overall ETD efficiency. The overall efficiency, which is the fraction of parent ions that yield ETD products, is a strong function of the reaction time, particularly at times where the residual precursor ion population is high. The %ETD represents the maximum ETD efficiency that can be obtained for a given reactant ion combination (i.e., when all precursor ions undergo ion/ion reactions).
CONCLUSIONS
A commercial quadrupole/linear ion trap/TOF mass spectrometer (QqTOF) has been modified to allow for mutual storage ion/ion reactions (proton transfer and electron transfer) in a linear ion trap and the subsequent product ions were analyzed by an orthogonal acceleration reflectron TOF. The hardware modification for adapting ion/ion reactions on this system involved the application of radio-frequency voltages to the end-plates of the linear ion trap. A pulsed dual ESI source or ESI/APCI source has been used to produce the oppositely charged reactant ions. A key to the ability to take advantage of the pulsed dual ion sources is instrument control software capable of coordination of the ion source pulsing and the establishment of appropriate conditions for transmitting sequentially the ions of opposite polarity. The advantage of adapting ion/ion reactions on a hybrid instrument like the QqTOF, compared to the conventional ion/ion studies in an ion trap, is that the ion/ion reaction process is spatially separated from the mass analysis process, so that the form of mass analysis is independent of the reaction vessel (i.e., the linear ion trap). The reflectron TOF analyzer employed in the current study has several performance characteristics that are attractive for use in peptide and protein analyses involving ion/ion reactions, particularly compared with the mass analysis characteristics of an ion trap. These include higher mass resolving power (~8,000), good mass accuracy (20 ppm for external calibration, 5 ppm for internal calibration), and an upper m/z limit of roughly 66,000. Key ion/ion proton transfer reaction applications in protein and peptide analysis have been demonstrated with the modified instrument. These include the analysis of protein mixtures with and without use of parallel ion parking and the simplification of a protein product ion spectrum to facilitate mass measurement of product ions. Two a priori unknown proteins (15.6 kDa, 8.6 kDa) were identified with high confidence from a protein mixture using a top-down approach based on the use of ion/ion reactions for the simplification of the product ion spectra. Electron transfer dissociation ion/ion reactions have also been implemented on the current instrument, providing means for dissociating peptide and protein ions in addition to beam-type and ion trap CID. The studies described herein demonstrate that the QqTOF instrument with ion/ion reaction capabilities is a flexible tool both for studying ion/ion reactions and for applying ion/ion reactions in peptide and protein analysis.
ACKNOWLEDGMENTS
This research was sponsored by the National Institutes of Health, under Grant GM 45372 and MDS SCIEX, an Industrial Associate of the Department of Chemistry. The authors acknowledge Bruce Thomson, Igor Chernushevich, Mahmoud Rishehri and Bruce Collings for helpful discussions. Y. X. acknowledges support from a Merck Research Laboratories Fellowship during the project.
REFERENCES
- 1.Wysocki HV, Tsaprailis G, Smith LL, Breci LA. J. Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 2.Brinkworth CS, Dua S, McAnoy AM, Bowie JH. Rapid Commun. Mass Spectrom. 2001;15:1965–1973. doi: 10.1002/rcm.457. [DOI] [PubMed] [Google Scholar]
- 3.Lin T, Payne AH, Glish GL. J. Am. Soc. Mass Spectrom. 2001;12:497–504. doi: 10.1016/S1044-0305(01)00234-3. [DOI] [PubMed] [Google Scholar]
- 4.Pitteri SJ, McLuckey SA. Mass Spectrom. Rev. 2005;24:931–958. doi: 10.1002/mas.20048. [DOI] [PubMed] [Google Scholar]
- 5.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Int. J. Mass Spectrom. 2004;236:33–42. [Google Scholar]
- 6.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc. Natl. Acad. Sci. USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. J. Am. Soc. Mass Spectrom. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Proc. Natl. Acad. Sci. USA. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunawardena HP, He M, Chrisman PA, Pitteri SJ, Hogan JM, Hodges BDM, McLuckey SA. J. Am. Chem. Soc. 2005;127:12627–12639. doi: 10.1021/ja0526057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. J. Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Anal. Chem. 2005;77:1831–1839. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunawardena HP, Emory JF, McLuckey SA. Anal. Chem. 2006 doi: 10.1021/ac060164j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zubarev RA, Kelleher NL, McLafferty FW. J. Am. Chem. Soc. 1998;120:3265–3266. [Google Scholar]
- 14.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Anal. Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 15.Scalf M, Westphall MS, Smith LM. Anal. Chem. 2000;72:52–60. doi: 10.1021/ac990878c. [DOI] [PubMed] [Google Scholar]
- 16.Ebeling DD, Westphall MS, Scalf M, Smith LM. Anal. Chem. 2000;72:5158–5161. doi: 10.1021/ac000559h. [DOI] [PubMed] [Google Scholar]
- 17.Scalf M, Westphall MS, Krause J, Kaufman SL, Smith LM. Science. 1999;283:194–197. doi: 10.1126/science.283.5399.194. [DOI] [PubMed] [Google Scholar]
- 18.Mather RE, Todd JF. J. Int. J. Mass Spectrom. Ion Phys. 1980;33:159–165. [Google Scholar]
- 19.McLuckey SA, Glish GL, Van Berkel GJ. Int. J. Mass Spectrom. Ion Processes. 1991;106:213–235. [Google Scholar]
- 20.Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. J. Am. Chem. Soc. 2002;124:7353–7362. doi: 10.1021/ja025966k. [DOI] [PubMed] [Google Scholar]
- 21.McLuckey SA, Stephenson JL, Jr, Asano KG. Anal. Chem. 1998;70:1198–1202. doi: 10.1021/ac9710137. [DOI] [PubMed] [Google Scholar]
- 22.Stephenson JL, Jr, McLuckey SA. Anal. Chem. 1996;68:4026–4032. doi: 10.1021/ac9605657. [DOI] [PubMed] [Google Scholar]
- 23.McLuckey SA, Wu J, Bundy JL, Stephenson JL, Jr, Hurst GB. Anal. Chem. 2002;74:976–984. doi: 10.1021/ac011015y. [DOI] [PubMed] [Google Scholar]
- 24.McLuckey SA, Reid GE, Wells JM. Anal. Chem. 2002;74:336–346. doi: 10.1021/ac0109671. [DOI] [PubMed] [Google Scholar]
- 25.Stephenson JL, Jr, McLuckey SA. J. Am. Chem. Soc. 1996;118:7390–7397. [Google Scholar]
- 26.Engel BJ, Pan P, Reid GE, Wells JM, McLuckey SA. Int. J. Mass Spectrom. 2002;219:171–187. [Google Scholar]
- 27.Newton KA, Chrisman PA, Reid GE, Wells JM, McLuckey SA. Int. J. Mass Spectrom. 2001;212:359–376. doi: 10.1002/rcm.512. [DOI] [PubMed] [Google Scholar]
- 28.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Anal. Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 29.Wells JM, Stephenson JL, Jr, McLuckey SA. Int. J. Mass Spectrom. 2000;203:A1–A9. [Google Scholar]
- 30.Hogan JM, McLuckey SA. J. Mass Spectrom. 2003;38:245–256. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 31.Chrisman PA, McLuckey SA. J. Proteome Res. 2002;1:549–557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 32.Amunugama R, Hogan JM, Newton KA, McLuckey SA. Anal. Chem. 2004;76:720–727. doi: 10.1021/ac034900k. [DOI] [PubMed] [Google Scholar]
- 33.Chernushevich IV, Loboda AV, Thomson BA. J. Mass. Spectrom. 2001;36:849–865. doi: 10.1002/jms.207. [DOI] [PubMed] [Google Scholar]
- 34.Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, Strupat K, Horning S. Anal. Chem. 2006;78:2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 35.Syka JEP, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF. J. Proteome Res. 2004;3:621–626. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- 36.Shevchenko A, Chernushevich I, Ens W, Standing KG, Thomson B, Wilm M, Mann M. Rapid Commun. Mass Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 37.Xia Y, Liang XR, McLuckey SA. J. Am. Soc. Mass Spectrom. 2005;16:1750–1756. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 38.Liang XR, Xia Y, McLuckey SA. Anal. Chem. 2006. in press. [Google Scholar]
- 39.Thomson BA, Jolliffe CL. 5,847,386 U.S. Patent.
- 40.Stephenson JL, Jr, McLuckey SA. Anal. Chem. 1997;69:3760–3766. doi: 10.1021/ac961119m. [DOI] [PubMed] [Google Scholar]
- 41.Chen XY, Westphall MS, Smith LM. Anal. Chem. 2003;75:5944–5952. doi: 10.1021/ac030127h. [DOI] [PubMed] [Google Scholar]
- 42.Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW. J. Am. Soc. Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 43.Dehmelt HG. Adv. Atom. Mol. Phys. 1967;3:53–72. [Google Scholar]
- 44.Hager JW, Londry FA. 2005/0263697 A1 U.S. Patent.
- 45.Hedin A, Hakansson P, Sundqvist BUR. International Journal of Mass Spectrometry and Ion Processes. 1987;75:275–289. [Google Scholar]
- 46.Fitzgerald MC, Zhu L, Smith LM. Rapid Commun. Mass Spectrom. 1993;7:895–897. [Google Scholar]
- 47.Wu KJ, Shaler TA, Becker CH. Anal. Chem. 1994;66:1637–1645. doi: 10.1021/ac00082a007. [DOI] [PubMed] [Google Scholar]
- 48.Frank M, Mears CA, Labov SE, Benner WH, Horn D, Jaklevic JM, Barfknecht AT. Rapid Commun. Mass Spectrom. 1996;10:1946–1950. [Google Scholar]
- 49.Loboda AV, Krutchinsky AN, Bromirski M, Ens W, Standing KG. Rapid Commun. Mass Spectrom. 2000;14:1047–1057. doi: 10.1002/1097-0231(20000630)14:12<1047::AID-RCM990>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 50.Kelleher NL. Anal. Chem. 2004;76:196a–203a. [PubMed] [Google Scholar]
- 51.Nemeth-Cawley JF, Rouse JC. J. Mass. Spectrom. 2002;37:270–282. doi: 10.1002/jms.281. [DOI] [PubMed] [Google Scholar]
- 52.Stephenson JL, Jr, McLuckey SA. J. Mass Spectrom. 1998;33:664–672. doi: 10.1002/(SICI)1096-9888(199807)33:7<664::AID-JMS663>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 53.Stephenson JL, Jr, McLuckey SA. J. Am. Soc. Mass Spectrom. 1998;9:585–596. doi: 10.1016/S1044-0305(98)00025-7. [DOI] [PubMed] [Google Scholar]
- 54.Chrisman PA, Pitteri SJ, McLuckey SA. Anal. Chem. 2006;78:310–316. doi: 10.1021/ac0515778. [DOI] [PubMed] [Google Scholar]
- 55.Meng FY, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Nat. Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 56.Chrisman PA, Newton KA, Reid GE, Wells JM, McLuckey SA. Rapid Commun. Mass Spectrom. 2001;15:2334–2340. doi: 10.1002/rcm.512. [DOI] [PubMed] [Google Scholar]