

Summary
Listeria monocytogenes is an intracellular bacterial pathogen that causes life-threatening disease. The mechanisms used by L. monocytogenes to invade non-professional phagocytic cells are not fully understood. In addition to the requirement of bacterial determinants, host cell conditions profoundly influence infection. Here, we have shown that inhibition of the RhoA/ROCK pathway by pharmacological inhibitors or RNA interference (RNAi) results in increased L. monocytogenes invasion of murine fibroblasts and hepatocytes. InlF, a member of the internalin multigene family with no known function, was identified as a L. monocytogenes-specific factor mediating increased host cell binding and entry. Conversely, activation of RhoA/ROCK activity resulted in decreased L. monocytogenes adhesion and invasion. Furthermore, virulence of wild-type bacteria during infection of mice was significantly increased upon inhibition of ROCK activity, whereas colonization and virulence of an inlF deletion mutant was not affected, thus supporting a role for InlF as a functional virulence determinant in vivo under specific conditions. In addition, inhibition of ROCK activity in human-derived cells enhanced either bacterial adhesion or adhesion and entry in an InlF-independent manner, further suggesting a host species or cell type specific role for InlF and that additional bacterial determinants are involved in mediating ROCK-regulated invasion of human cells.
Keywords: Listeria monocytogenes, ROCK, InlF, invasion, virulence
Introduction
Listeria monocytogenes is a food-borne, intracellular bacterial pathogen that causes listeriosis, a disease characterized by gastroenteritis, meningitis, encephalitis, and maternofetal infections. The frequent contamination of food by L. monocytogenes makes listeriosis a serious public health concern due to the high mortality rate (20-30%) (Lorber, 1997). L. monocytogenes has a broad host range and is able to infect humans and animals. During infection, bacteria disseminate from the intestine to the blood, allowing spread to internal organs and eventually the brain. L. monocytogenes can infect a variety of tissues and cell types, including phagocytic and non-professional phagocytic cells, such as epithelial, endothelial, fibroblasts, hepatocytes and neurons (Vazquez-Boland et al., 2001). Following entry into host cells, bacteria escape the phagocytic vacuole, replicate within the host cell cytosol and spread via actin-based motility to neighboring cells.
To date, several bacterial factors have been shown to be involved in infection. Entry of bacteria into non-professional phagocytic host cells is facilitated by several bacterial surface proteins such as InlA and InlB, both members of the internalin multigene family. InlA interacts with E-cadherin on the surface of host cells and has been shown to be required for invasion of intestinal tissue in vivo using a transgenic mouse model (Lecuit et al., 2001). Furthermore, InlA was shown to play a key role for crossing the human maternofetal barrier, although it was shown not to be essential for this role in the pregnant guinea-pig infection model (Bakardjiev et al., 2004; Lecuit et al., 2004). InlB, important for liver and splenic colonization in vivo (Lecuit et al., 2004; Shen et al., 2000), triggers bacterial entry by interacting with the hepatocyte growth factor receptor (HGF-R or Met) and two other cellular components: gC1q-R and proteoglycans (Braun et al., 2000; Jonquieres et al., 2001; Shen et al., 2000). Less is known about the precise function of the remaining 23 identified members of the internalin multigene family. However, deletion mutants of inlC or the inlGHE gene cluster display reduced virulence in the mouse infection model (Engelbrecht et al., 1996; Raffelsbauer et al., 1998). During tissue culture infections, inlC and the inlGHE gene cluster were shown to have a supportive role in InlA-mediated invasion (Bergmann et al., 2002). Additional studies have determined that InlH and InlJ play a role in L. monocytogenes virulence, since mutant strains are attenuated in colonization of mice (Sabet et al., 2005; Schubert et al., 2001). No roles in infection have been determined thus far for the products of the inlC2, inlD and inlF genes as in vitro and in vivo infection studies with the respective null mutants have not revealed a phenotype (Dramsi et al., 1997). In addition to internalins, several other L. monocytogenes surface proteins have been shown to be involved in infection. Several adhesins and invasion-mediating factors, including FbpA, the autolysin Ami, ActA, Lap and Vip (Bierne and Cossart, 2007; Dramsi et al., 2004; Milohanic et al., 2001) have been characterized.
Whereas numerous bacterial determinants that facilitate L. monocytogenes infection have been characterized, less is known about the host cell factors that are required for infection. Our laboratory has recently performed a genome-wide RNAi screen in Drosophila cells to identify host factors required for the pathogenesis of L. monocytogenes (Agaisse et al., 2005). The identified factors spanned a wide range of associated cellular functions. Interestingly, knockdown of several gene products caused an increase in infection by L. monocytogenes, including the small GTPase Rho1, the Drosophila homolog of mammalian RhoA. Small GTPases of the Rho family act as molecular switches in signal transduction and thereby regulate a variety of cellular processes. The major downstream targets of RhoA are Rho kinases (ROCKs), serine-threonine protein kinases that are involved in many fundamental cellular processes such as cell adhesion, motility, contractility, gene expression and cytokinesis (Noma et al., 2006). ROCKs are important regulators of cell growth, migration, and apoptosis via control of actin cytoskeletal assembly (Riento and Ridley, 2003). By affecting tight and adherens junctions through actin cytoskeletal contractions, ROCKs can also regulate macrophage phagocytic activity and endothelial cell permeability (Wojciak-Stothard et al., 2001; Wojciak-Stothard and Ridley, 2002). In mammalian cells, two ROCK isoforms have been identified: ROCK-I (also known as ROKβ or p160ROCK) and ROCK-II (also known as ROKα or Rho kinase) (Ishizaki et al., 1996; Matsui et al., 1996). ROCKs are comprised of an N-terminal catalytic kinase domain, a central coiled-coil domain that includes the Rho-binding domain, and a C-terminal pleckstrin homology domain that is split by a cysteine-rich region (Nakagawa et al., 1996). ROCK-I and ROCK-II share 65% homology in overall amino acid sequence and 92% homology in their kinase domains. ROCK-I mRNA is preferentially expressed in lung, liver, spleen, kidney, and testes, whereas ROCK-II mRNA is highly expressed in the heart and brain (Di Cunto et al., 2000; Nakagawa et al., 1996).
Several effectors for ROCKs have been proposed. Myosin light chain (MLC) is one of the major downstream targets of ROCKs and phosphorylation of MLC controls the assembly of actin-myosin structures (Kureishi et al., 1997; Li et al., 2006; Samarin et al., 2007; Yoneda et al., 2005). The myosin binding subunit (MBS) on myosin light chain phosphatase (MLCP) is another important downstream target, resulting in the subsequent phosphorylation of MLC by inhibiting the phosphatase activity (Somlyo and Somlyo, 2000). In addition, ROCKs can also phosphorylate LIM kinase, ERM proteins, adducin and the Na-H exchanger NHE1 (Maekawa et al., 1999; Riento and Ridley, 2003). The overall physiological effects of ROCKs are to enhance actin-myosin association through increasing MLC phosphorylation and preventing actin depolymerization. In addition, ROCKs are involved in the regulation of other cellular functions independent of their effects on the actin cytoskeleton, such as inhibition of insulin signaling and regulation of cell size (Farah et al., 1998; Sordella et al., 2002). Although extensively studied, limited information exists about the specific role of each ROCK isoform in the various cellular processes. While previously assumed to be functionally redundant, recent studies suggest isoform specific functions (Coleman et al., 2001; Riento et al., 2003; Riento et al., 2005; Sebbagh et al., 2001; Yoneda et al., 2005; Yoneda et al., 2007).
ROCKs have also become an important therapeutic target since increased ROCK activity has been shown to be involved in various diseases, including hypertension, cancer, and neurological disorders (Mueller et al., 2005; Noma et al., 2006). Recently, the ROCK inhibitor Fasudil (HA-1077) has been approved and marketed in Japan for treatment in humans of systemic and pulmonary arterial hypertension, angina pectoris, acute ischemic stroke and cerebral vasospasm (Fukumoto et al., 2005; Masumoto et al., 2001; Masumoto et al., 2002; Shibuya et al., 2005; Shimokawa et al., 2002).
In this study, we investigated the impact of the RhoA/ROCK pathway on L. monocytogenes infection of mammalian cells. We have shown that depending on the host cell type, inhibition of RhoA and ROCK activity increases bacterial adhesion or both adhesion and entry into host cells. Conversely, increases in RhoA and ROCK activity results in decreased L. monocytogenes adhesion and invasion. Furthermore, we identified InlF as a bacterial factor involved in mediating adhesion and invasion of murine fibroblasts and hepatocytes under ROCK inhibition conditions. Interestingly, inhibition of ROCK activity in human-derived host cells enhanced either bacterial adhesion or adhesion and entry in an InlF-independent manner, suggesting a possible species-specific activity for InlF. Lastly, we have also demonstrated that inhibition of ROCK activity during in vivo infection of mice increased the virulence of L. monocytogenes in an InlF-dependent manner, implying potential effects of clinically used ROCK inhibitors, such as Fasudil, on susceptibility to L. monocytogenes infection.
Results
RhoA and ROCK activity affect L. monocytogenes invasion
Results of a genome-wide RNA interference (RNAi) screen in Drosophila cells indicated that depletion of the small GTPase Rho1 (homolog of human RhoA) increases L. monocytogenes infection efficiency (Agaisse et al., 2005). Using a chemical inhibitor approach, we evaluated the impact of RhoA and its major downstream effector target ROCK on L. monocytogenes infection in mammalian host cells. Treatment of host cells with CT04 (Exoenzyme C3 transferase from Clostridium botulinum) is commonly used to selectively inactivate RhoA, while Y27632 is a specific inhibitor of ROCK activity. Furthermore, lysophosphatidic acid (LPA) is known to activate RhoA and consequently ROCK (Bian et al., 2006; Mills and Moolenaar, 2003; Moolenaar et al., 2004; Riento and Ridley, 2003). To determine a role for RhoA and ROCK activity in the early stages of L. monocytogenes infection, L2 murine fibroblast cell monolayers were incubated with CT04, Y27632 or LPA prior to infection and bacterial host cell association (adhesion and entry) as well as host cell entry (intracellular) were determined by gentamicin protection assay (Fig. 1A). Inhibition of RhoA as well as ROCK activity led to a 2-fold increase in cell-associated bacteria and a >5-fold increase in intracellular bacteria compared to untreated L2 cells. In contrast, treatment with the RhoA/ROCK activator LPA reduced the number of cell-associated bacteria by ∼1.5-fold and the number of intracellular L. monocytogenes by >2-fold. Cell culture medium is known to contain LPA, present in supplemented serum at concentrations ranging from 2 μM to 10 μM, that can function to stimulate RhoA activity (Moolenaar, 1995). Interestingly, incubation of L2 cells in serum-free medium prior to and during infection increased adhesion and entry of L. monocytogenes to a similar level as CT04 or Y27632 treatment (data not shown). To confirm the effect of drug treatment on RhoA/ROCK pathway activity, the phosphorylation level of myosin light chain (MLC), a major downstream target of ROCK, was examined. Western blot analysis of L2 cell lysates showed, respectively, a decrease of 55.6% and 31.3% in MLC phosphorylation upon CT04 and Y27632 treatment, whereas LPA treatment increased phosphorylation of MLC by 39.1 % (Fig. 1B).
Fig. 1. RhoA and ROCK activity affects L. monocytogenes invasion.
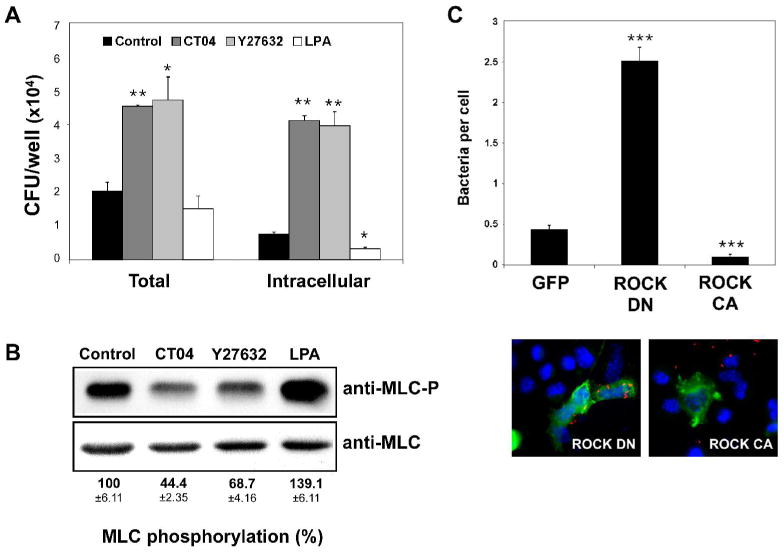
A. L2 cells were treated for 5 h with 2 μg/ml CT04 or for 30 min with 10 μM Y27632, 10 μM LPA or with DMSO (control) prior to infection with wild-type L. monocytogenes (10403S). At 1 h post-infection, cell-associated (total) bacteria were quantified as described in Experimental Procedures. For determination of intracellular bacteria, samples were washed and subsequently incubated with medium containing 50 μg/ml gentamicin for an additional 1 h to kill extracellular bacteria prior to quantitation of intracellular bacteria. Data represents the mean colony forming units (CFU) and standard deviations (s.d.) of one of three experiments performed in triplicate with similar results. * p < 0.05, ** p < 0.01 compared to control.
B. Western blot analysis of L2 cell extracts treated as in (A) with 2 μg/ml CT04, 10 μM Y27632, 10 μM LPA or DMSO (control). Phospho-myosin light chain antibody (MLC-P, upper panel) or myosin light chain 2 antibody (MLC, lower panel) was used. Shown is a representative autoradiograph. MLC phosphorylation (MLC-P) was quantified by densitometry as described in Experimental Procedures and normalized against total MLC protein. Numerical values indicate the means ±s.d. of the percentage of MLC phosphorylation relative to control samples of three independent experiments.
C. L2 cells seeded on glass coverslips were transfected with myc-tagged ROCK constructs or the respective control vector expressing GFP. ROCK DN denotes dominant negative ROCK, whereas ROCK CA denotes constitutively active ROCK. Twenty-four hours post-transfection, cells were infected with wild-type L. monocytogenes for 1 h. Coverslips were washed several times to remove unbound bacteria and then analyzed by immunofluorescence microscopy. Transfected cells were identified by detection of Myc-tag expression (green). Cell-associated bacteria (red) were quantified by counting 100 transfected cells for each sample. Cell nuclei are shown in blue. The upper graph indicates the means ±s.d. of bacteria associated per transfected L2 cell (n=3). *** p < 0.001 compared to GFP.
Although Y27632 is selective for ROCK, when applied at higher concentrations Y27632 has been shown to affect other kinases, such as protein kinase C-related kinase (PRK) 2, citron kinase, MSK1 and protein kinase N (PKN) (Ishizaki et al., 2000). To verify the association of the observed effects on L. monocytogenes infection with inhibition of ROCK activity, we analyzed the effect of two additional ROCK inhibitors, HA-1077 (Fasudil) and HA-1100 (Davies et al., 2000; Shimokawa et al., 1999; Sward et al., 2000). Treatment with all three inhibitors resulted in a similar phenotype (Supplemental Fig. S1). The number of cell-associated (total) as well as intracellular bacteria increased in a dose-dependent manner upon inhibitor treatment. To further confirm the impact of ROCK activity on L. monocytogenes infection, L2 cells were transfected with dominant negative (ROCK DN) or constitutive active ROCK (ROCK CA) and then infected with L. monocytogenes (Fig. 1C). ROCK DN expressing cells contained 4.6-fold more bacteria associated per L2 cell, whereas ROCK CA expression decreased the number of associated bacteria per L2 cell by 5.2-fold compared to transfected control cells (GFP-expressing vector). Taken together, these data indicated that infection of murine L2 fibroblasts by L. monocytogenes is regulated by RhoA and ROCK activity. Inhibition of RhoA or ROCK activity increases bacterial host cell-association and entry. Conversely, activation of RhoA and ROCK results in decreased L. monocytogenes host cell-association and entry.
ROCK activity does not affect intracellular growth or cell-to-cell-spread of L. monocytogenes
Following entry into host cells, L. monocytogenes replicates within the cytosol and spreads cell-to-cell via actin-based motility. To determine whether ROCK activity affects intracellular replication, L2 cells treated with Y27632 or LPA were infected with bacteria and intracellular growth was examined (Fig. 2A). To ensure that Y27632 and LPA are efficiently active over the course of the infection, drug-containing medium was replaced at 3 h intervals. Compared to the untreated control, the initial number of intracellular bacteria at 2 h post-infection was increased by Y27632 treatment and decreased upon LPA treatment, which is consistent with the RhoA/ROCK-regulated effect on bacterial entry observed in Figure 1A. However, the rates of intracellular growth over the 10-h infection period were similar under all conditions examined (Fig. 2A). In addition, cell-to-cell spread was assayed by analyzing plaque formation in L2 cell monolayers treated with Y27632 or LPA. Although the number of plaques per monolayer was increased >7-fold following Y27632 treatment, and decreased >3-fold following LPA treatment, plaque sizes were not significantly altered compared to control samples (Fig. 2B, upper panel). Because Y27632 and LPA activity may be decreased over the infection period for the plaquing assay and the experimental design does not allow for replacement of drug-containing medium, spreading efficiency of bacteria within drug-treated L2 cells was further analyzed by determining the size of foci of infection. As shown in Figure 2B (lower panel) and Supplemental Figure S2, the foci size at 12 h and 24 h post-infection was not significantly altered by Y27632 or LPA treatment compared to control samples. These data indicated that neither intracellular replication nor cell-to-cell spread of L. monocytogenes was affected by alterations in RhoA/ROCK activity.
Fig. 2. ROCK activity does not affect intracellular growth or cell-to-cell-spread of L. monocytogenes.
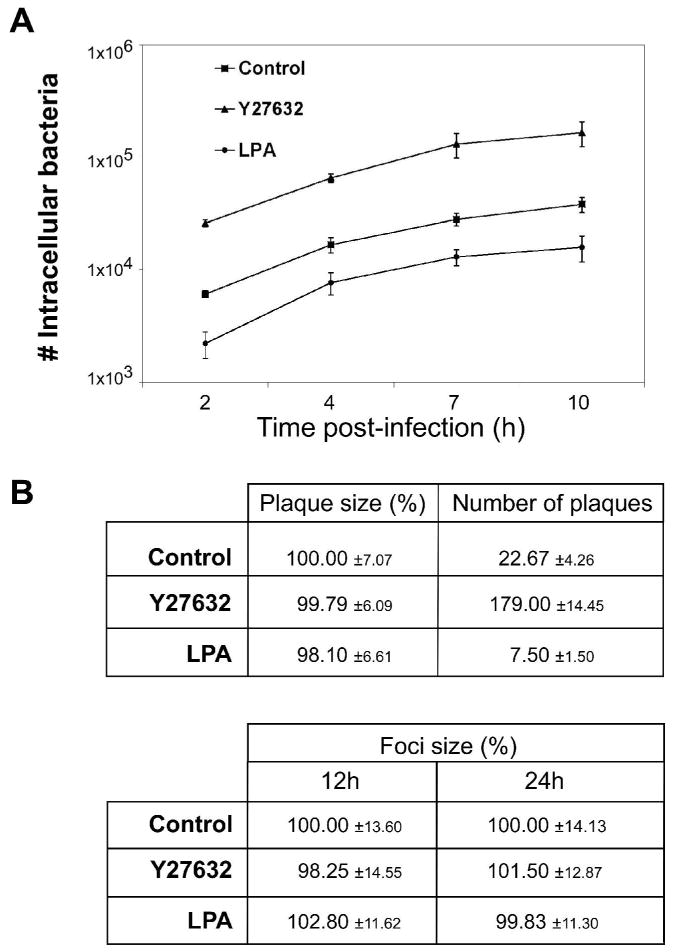
A. Intracellular growth of L. monocytogenes in L2 cells. L2 cell monolayers seeded onto glass coverslips were treated with 10 μM Y27632, 10 μM LPA or DMSO (control) and infected with wild-type L. monocytogenes (10403S). At 1 h post-infection, coverslips were washed and medium containing 10 μM Y27632, 10 μM LPA, or DMSO and gentamicin was added and replaced at 4 h and 7 h post-infection. At the indicated times post-infection, coverslips were removed and the number of intracellular bacteria determined as described in Experimental Procedures. Data shown represents the means ±s.d. of one of three independent experiments performed in triplicate with similar results.
B. Cell-to-cell spread in L2 fibroblasts. For plaque formation assays, L2 cell monolayers seeded in 6-well dishes were treated with 10 μM Y27632, 10 μM LPA, or DMSO (control). Wild-type L. monocytogenes (10403S) were added to monolayers for 1 h. The infected monolayers were washed with PBS, and a medium-agarose overlay containing 10 μM Y27632, 10 μM LPA, or DMSO and gentamicin was added to kill extracellular bacteria. Intracellular growth and cell-to-cell spread of bacteria were visualized after 72 h by the formation of clearing zones (plaques) within the L2 monolayers. The number of plaques/well was determined and the diameters of 10 plaques/sample were measured. Data represent the mean percent diameter of plaques ±s.d. relative to the control and the mean number of plaques formed/well ±s.d. Data are from three independent experiments performed in duplicate. For microscopy analysis of foci of infection, L2 cell monolayers seeded onto glass coverslips were treated with 10 μM Y27632, 10 μM LPA, or DMSO (control) and infected with GFP-expressing wild-type L. monocytogenes (10403S). At 1 h post-infection, coverslips were washed and medium containing 10 μM Y27632, 10 μM LPA, or DMSO and gentamicin was added and replaced at 4 h, 8 h, and 12 h post-infection. At 12 h and 24 h post-infection, coverslips were fixed, nuclei stained and analyzed by fluorescence microscopy. The diameters of 15 foci/sample were measured. Data represent the mean percent diameter of foci ±s.d. relative to the control. Data are from three independent experiments.
Depletion of ROCK-I or ROCK-II isoforms increases L. monocytogenes invasion
There are two isoforms of ROCK, ROCK-I and ROCK-II, which are encoded by separate genes. However, ROCK inhibitors, such as Y27632, HA-1100 and HA-1077, target both isoforms equally. To determine whether one specific isoform of ROCK is involved in regulating L. monocytogenes infection, expression of ROCK-I and ROCK-II was individually down regulated in L2 cells using RNAi. ROCK-I or ROCK-II specific shRNA constructs were used for transfection of L2 cells. An shRNA construct targeting GFP was included as a control. Western blot analysis using antibodies for each ROCK isoform was used to confirm ROCK-specific down-regulation (Fig. 3, bottom panel). Seventy-two hours post-transfection, L2 cells were infected with wild-type L. monocytogenes and host cell-associated (total) as well as intracellular bacteria were quantified using gentamicin protection assays. As shown in Figure 3 (upper graph), down-regulation of either ROCK-I or ROCK-II resulted in increased L2 cell-associated and intracellular bacteria. Compared to cells transfected with shRNA targeting GFP, host cell-associated bacteria were increased by 2-fold while intracellular bacteria were increased 5.7-fold in cells transfected with the shRNA targeting ROCK-I. Down-regulation of ROCK-II expression increased the number of cell-associated bacteria by 2.7-fold and intracellular bacteria by 7.2-fold. Furthermore, concurrent down-regulation of both ROCK isoforms enhanced total cell-associated bacteria by 3.1-fold and intracellular bacteria by 10.1-fold compared to the control. However, co-transfection of shRNAs targeting ROCK-I and ROCK-II resulted in less efficient down-regulation of each isoform (Fig. 3, lower panel). These data demonstrate that decreased expression of both ROCK isoforms resulted in similar increases in L. monocytogenes invasion, suggesting that a common downstream mechanism is responsible.
Fig. 3. Depletion of either ROCK isoform increases L. monocytogenes invasion.
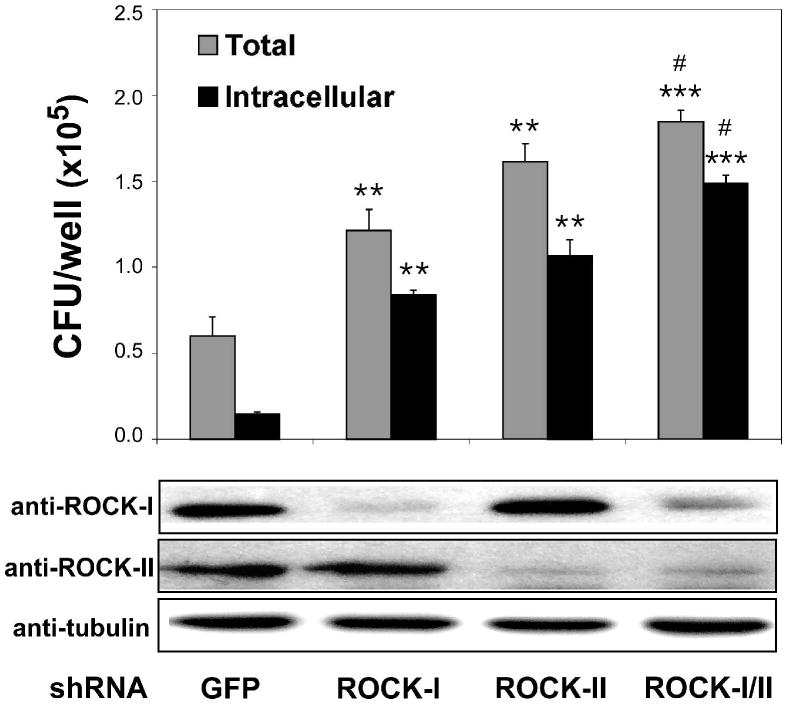
Expression of ROCK-I and ROCK-II was down-regulated in L2 cells by RNAi using shRNA constructs targeting GFP, ROCK-I, ROCK-II or both ROCK-I and ROCK-II. Seventy-two hours post-transfection, L2 cells were infected with wild-type L. monocytogenes (10403S) for 1 h and cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay. The upper graph indicates the means ±s.d. CFU per well for one of three representative experiments performed in triplicate with similar results. ** p < 0.01 and *** p < 0.001 compared to GFP control. # p < 0.05 compared to ROCK-I and ROCK-II samples. Bottom panels: shRNA-transfected L2 cell lysates were analyzed by Western blot using antibodies specific for ROCK-I, ROCK-II, or tubulin.
Impact of myosin II activity on L. monocytogenes infection
ROCKs are important regulators of diverse cellular functions, including cell contraction, actin cytoskeleton organization, cell adhesion, and motility. A principle substrate of ROCKs is myosin light chain (MLC), the regulatory subunit of myosin II. Phosphorylation of MLC results in stimulation of actin-myosin interactions. We used the myosin II-specific inhibitor blebbistatin (Straight et al., 2003) to elucidate a role of actin-myosin interactions for the observed increase in L. monocytogenes host cell-association and entry. Pre-treatment of L2 cells with blebbistatin increased bacterial host cell-association and entry to similar levels observed when L2 cells were treated with Y27632 (Fig. 4). Simultaneous incubation of L2 cells with both Y27632 and blebbistatin did not result in an additive effect, suggesting that the same myosin II-dependent pathway is involved in mediating the increase in cell-associated and intracellular bacteria observed with each inhibitor. These data suggest that actin-myosin structures are involved in regulating L. monocytogenes interaction with host cells, and that disruption of actin-myosin interactions leads to increased host cell-association and entry.
Fig. 4. Inhibition of myosin II activity increases L. monocytogenes invasion.
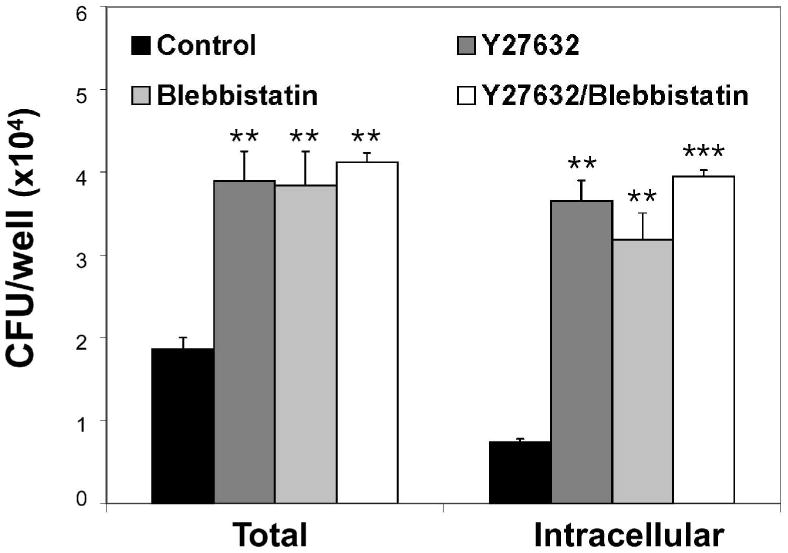
L2 cells were treated for 30 min with DMSO (control), 10 μM Y27632, 10 μM blebbistatin or both Y27632 and blebbistatin prior to infection with wild-type L. monocytogenes (10403S) for 1 h. Cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay. Data represents the means ±s.d. CFU per well for one of three experiments performed in triplicate with similar results. ** p < 0.01, *** p < 0.001 compared to control samples.
The ROCK-dependent effects on L. monocytogenes infection are host cell type specific
To determine whether the impact of ROCK activity on L. monocytogenes invasion is restricted to murine fibroblasts, various murine and human cell lines, including both professional and non-professional phagocytic cells, were treated with the ROCK inhibitor Y27632 and the effect on L. monocytogenes infection assessed. Y27632 induced changes in cell-associated and intracellular bacteria were quantified by gentamicin protection assay (Table 1). For murine fibroblast cell lines (L2, L929 and NIH 3T3), as well as for the murine hepatocyte cell line TIB 75 and the human fibroblast cell line WI38, Y27632 treatment increased host cell-association between 1.88 and 2.61-fold and host cell entry 3.45 to 5.44-fold. Interestingly, Y27632 treatment of HeLa and Hep2 human epithelial cell lines resulted in increased cell-association (∼2.5-fold), but did not significantly affect host cell entry (Table 1). No significant changes in L. monocytogenes host cell-association or entry were observed for cell lines 1308.1 (murine epithelial), HEK293 (human epithelial), HepG2 (human hepatocyte) or for any of the macrophage cell lines examined (murine RAW 264.7, RAW 309 Cr.1; human U937). In addition, treatment of cell lines with higher concentrations of Y27632 (30 μM) gave similar results (data not shown). These data reveal a cell type specific effect of ROCK activity inhibition on L. monocytogenes infection, resulting in two distinct phenotypes. One phenotype, which was observed for murine fibroblasts and hepatocytes, as well as for human fibroblast cells, resulted in increased bacterial host cell-association and entry. The second phenotype, observed with HeLa and Hep2 human epithelial cells, was restricted to increased host cell-association (adhesion) with no affect on bacterial entry into host cells. Furthermore, host cell-association and uptake by the phagocytic cell lines tested was not increased upon Y27632 treatment.
Table 1.
Cell type specific effects of ROCK activity inhibition on L. monocytogenes infection.
Monolayers of cell lines were treated with 10 μM Y27632 or DMSO (control) for 30 min prior to washing of monolayers and infection with wild-type L. monocytogenes 10403S. Cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay as described in Experimental Procedures. Data presented represents the means ±s.d. fold change in CFU upon Y27632 treatment relative to control samples for one of three experiments performed in triplicate with similar results. * p<0.01; bold type indicates changes in CFU greater than 2-fold with p< 0.05.
| Classification | Species | Cell line | Cell type | Total | Intracellular |
|---|---|---|---|---|---|
| non-professional phagocytic | murine | L2 | fibroblast | 2.61 (±0.04) | 5.44 (±0.43) |
| L929 | fibroblast | 1.88 (±0.09)* | 3.88 (±0.17) | ||
| NIH 3T3 | fibroblast | 2.35 (±0.08) | 3.45 (±0.52) | ||
| 1308.1 | epithelial | 0.97 (±0.08) | 1.11 (±0.21) | ||
| TIB 75 | hepatocyte | 2.40 (±0.06) | 4.93 (±0.10) | ||
| human | WI38 | fibroblast | 2.16 (±0.06) | 4.88 (±1.17) | |
| HeLa | epithelial | 2.54 (±0.26) | 1.26 (±0.04) | ||
| Hep2 | epithelial | 2.48 (±0.13) | 1.02 (±0.03) | ||
| HEK-293 | epithelial | 1.04 (±0.05) | 0.65 (±0.17) | ||
| HepG2 | hepatocyte | 1.12 (±0.04) | 0.97 (±0.01) | ||
| phagocytic | murine | RAW 264.7 | macrophage | 0.99 (±0.05) | 1.14 (±0.09) |
| RAW 309 Cr.1 | macrophage | 0.89 (±0.01) | 0.90 (±0.08) | ||
| human | U937 | macrophage | 0.86 (±0.05) | 0.87 (±0.03) |
The ROCK-dependent effects on host cell infection may be L. monocytogenes specific
To determine if the increase in host cell association and bacterial uptake following inhibition of ROCK activity was specific to L. monocytogenes, various bacterial species including non-invasive (Bacillus subtilis, Escherichia coli, and Pseudomonas aeruginosa) and invasive (E. coli inv; Salmonella enterica serovar Typhimurium and Shigella flexneri) were analyzed for alterations in host cell-association and entry into Y27632-treated host cells. Infections of L2 and HeLa cell lines, representative of the two distinct L. monocytogenes invasion phenotypes observed following Y27632 treatment (Table 1), were performed. As seen in Supplemental Figure S3, none of the additional bacterial strains examined were altered for host cell-association or entry in Y27632-treated L2 or HeLa cells. Again, L. monocytogenes was the only bacterial species examined found to be affected in host cell interactions by inhibition of ROCK activity, suggesting that alterations in interactions with host cells following inhibition of ROCK activity may be L. monocytogenes specific.
To investigate the potential species specificity of the ROCK-dependent effects on bacteria/host cell interactions, various Listeria strains were analyzed for infection of Y27632-treated L2 and HeLa cells. Bacterial strains examined included another pathogenic Listeria species, L. ivanovii, a non-pathogenic member of the Listeria genus, L. innocua, and an additional L. monocytogenes strain, EGDe (Fig. 5A). As observed for L. monocytogenes 10403S, infection of host cells with EGDe was similarly affected by Y27632 treatment. Host cell-association and bacterial uptake were increased in L2 cells, whereas only the number of cell-associated (adhered) bacteria was observed to increase during infection of HeLa cells. L. ivanovii and L. innocua did not show alternations in cell-association or uptake upon Y27632 treatment of host cells. Based upon these data, we speculate that interactions between a L. monocytogenes specific factor and a host cell receptor(s) mediate the ROCK-dependent effects on bacterial adhesion and uptake.
Fig. 5. Increased L. monocytogenes infection upon inhibition of ROCK activity is independent of InlA, InlB, and PrfA.
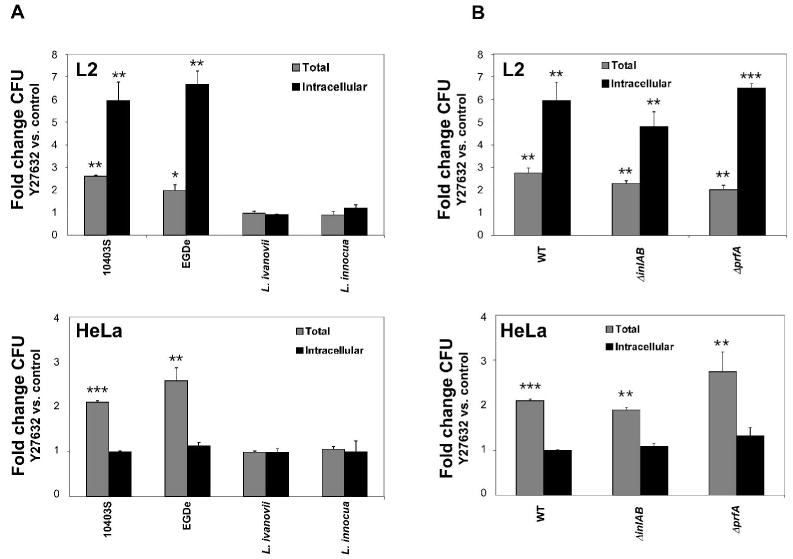
L2 or HeLa cell monolayers were treated with 10 μM Y27632 or DMSO (control) and subsequently incubated with Listeria strains for 1 h. Cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay. Data represent the means ±s.d. fold change in CFU upon Y27632 treatment relative to control samples for one of three experiments performed in triplicate with similar results.
A. L. monocytogenes strains 10403S or EGDe, L. ivanovii, or L. innocua were used in infections.
B. L. monocytogenes 10403S deletion mutants in inlAB or prfA were used in infections. * p < 0.05, ** p < 0.01, *** p < 0.001 compared to the control (control fold change =1).
To gain insight into the identity of the putative L. monocytogenes specific invasion determinant, L. monocytogenes deletion mutants were analyzed in gentamicin protection assays. Two principal L. monocytogenes invasion factors are InlA and InlB. To determine whether these determinants were involved in mediating the Y27632-dependent increase in infection, an inlAB deletion mutant (10403S ΔinlAB) was analyzed. Although the ΔinlAB strain showed decreased infection of both L2 and HeLa cells (50% of 10403S; data not shown), a similar increase in host cell-association (HeLa) or cell-association and entry (L2) was observed upon Y27632 treatment (Fig. 5B). Furthermore, the PrfA transcriptional activator positively regulates expression of many L. monocytogenes virulence determinants (Dussurget et al., 2002; Milohanic et al., 2003). To determine whether PrfA regulates expression of the putative invasion factor, a prfA deletion mutant (10403S ΔprfA) was analyzed. The increase in Y27632-dependent host cell-association and uptake was not abolished during infection by the ΔprfA strain (Fig. 5B). Taken together, these data suggest that the increase in L. monocytogenes infection upon inhibition of ROCK activity is not mediated by InlA or InlB nor is dependent upon a PrfA-regulated factor.
InlF mediates L. monocytogenes invasion of Y27632-treated host cells
We next used a genetic screening approach to identify the L. monocytogenes factor(s) mediating the ROCK-dependent increase in infection. L. innocua does not show significant invasion of untreated or Y27632-treated L2 cells (Fig. 5A). Therefore, a L. monocytogenes genomic expression library was transformed into L. innocua and bacteria were used to infect Y27632-treated L2 cells. L. innocua transformants demonstrating at least a 3–fold increase in invasion of Y27632-treated L2 cells were isolated and further analyzed by DNA sequencing as described in Experimental Procedures. A transforming clone (YInv-4A) harboring a 2.2 kb L. monocytogenes DNA insert was found to encode InlF (Lmo0409), a member of the internalin multigene family with unknown function. Compared to mock-treated host cells, L. innocua YInv-4A showed a 2(±0.22)-fold increase in L2 cell-associated bacteria and a 3.75(±0.28)-fold increase in bacterial entry into Y27632-treated L2 cells.
Role of InlF in L. monocytogenes infection
To further investigate the role of InlF in host cell infection, an inlF in-frame deletion mutant in 10403S (ΔinlF) was constructed. Infection of L2, TIB 75, WI38 and HeLa cells by ΔinlF bacteria was analyzed by gentamicin protection assay (Fig. 6). During infection of L2 and TIB 75 cells under mock-treated conditions (control), ΔinlF displayed similar host cell-association and entry efficiencies as wild-type bacteria (Fig. 6). However, following infection of Y27632-treated cells, ΔinlF did not show a significant increase in host cell-association or uptake (Fig. 6). Furthermore, during infection of WI38 and HeLa cells, no differences were observed between wild-type and ΔinlF bacteria under untreated or Y27632-treated conditions. These data indicated that InlF mediates the Y27632-dependent increase in infection of L2 murine fibroblasts and TIB 75 murine hepatocytes, but that an additional L. monocytogenes factor(s) is required to facilitate Y27632-dependent increased adherence and/or entry of human-derived WI38 fibroblasts and HeLa epithelial cells.
Fig. 6. Increased infection of L2 and TIB 75 cells following inhibition of ROCK activity is InlF-dependent.
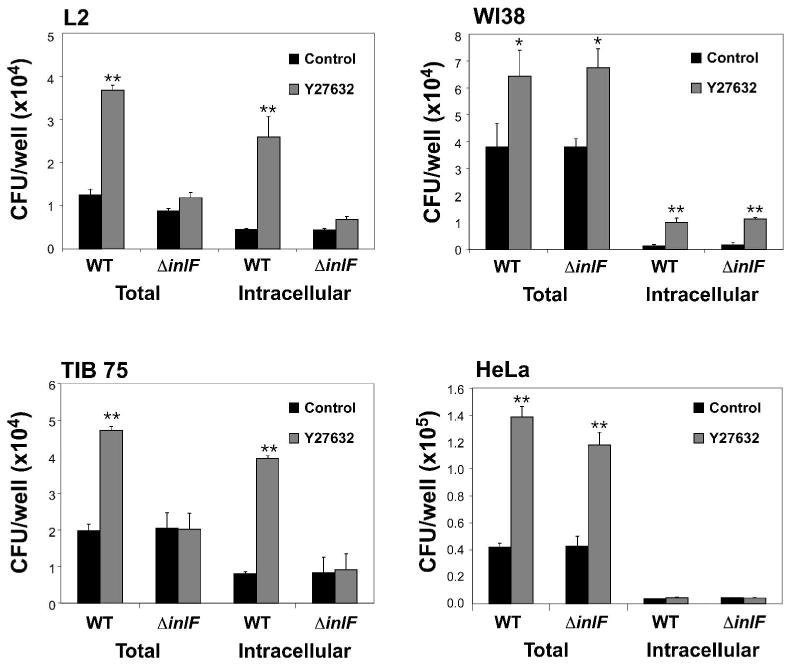
L2, TIB 75, WI38, or HeLa cells were treated with DMSO (control), or 10 μM Y27632 prior to infection with wild-type (WT) L. monocytogenes 10403S or an isogenic inlF in-frame deletion mutant (ΔinlF) for 1 h. Cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay. Data represents the means ±s.d. CFU per well for one of three experiments performed in triplicate with similar results. * p < 0.05, ** p < 0.01 compared to control samples.
Based on phylogenetic and serological studies, L. monocytogenes strains have been divided into three evolutionary groups, Lineages I, II and III. Attempts have been made to correlate lineage affiliation with pathogenic potential, including host specificity and virulence. Interestingly, Lineage I strains have been described as lacking inlF (Jia et al., 2007; Tsai et al., 2006). We analyzed infection of L2 cells by various L. monocytogenes Lineage I strains, FSL J1-194 (Jia et al., 2007; Tsai et al., 2006), FSL R2-503 (Borucki et al., 2004; Ducey et al., 2007) and FSL N1-017 (Gray et al., 2004) (Fig. 7A). Compared to 10403S (Lineage II) none of the Lineage I strains examined showed a significant increase in L2 cell-association or entry upon Y27632 treatment, further implicating a role of InlF for increased infection following inhibition of ROCK activity. Moreover, a Y27632-dependent increase in adherence to HeLa cells was still observed following infection with Lineage I strains (data not shown), further indicating that a L. monocytogenes factor(s) besides InlF can mediate Y27632-dependent increased adherence to HeLa cells.
Fig. 7. Expression of InlF in Listeria mediates increased infection of Y27632-treated L2 cells.
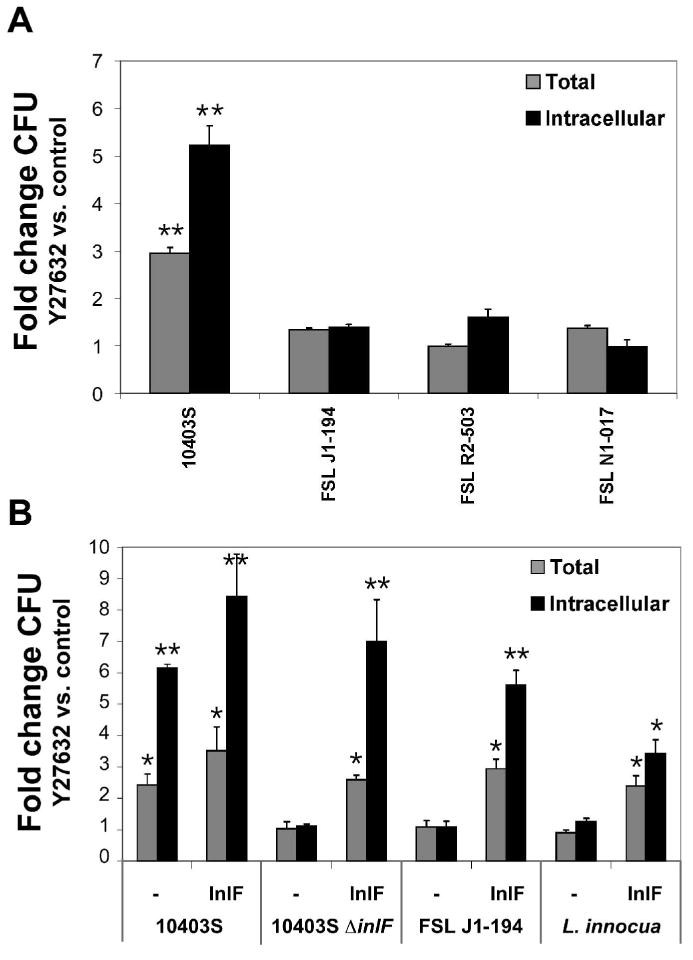
A. L2 cells were treated with 10 μM Y27632 or DMSO (control) and infected with L. monocytogenes (10403S) or Lineage I strains FSL J1-194, FSL R2-503, or FSL N1-017 for 1 h. Cell-associated (total) and intracellular bacteria were quantified by gentamicin protection assay.
B. L. monocytogenes 10403S, ΔinlF, FSL J1-194 or L. innocua were transformed with plasmid pAM-inlF (InlF) or pAM401spacOid-BamHI (-) and analyzed for infection of L2 cells treated with 10 μM Y27632 or DMSO (control) as in (A). Data represent the means ±s.d. fold change in CFU upon Y27632 treatment relative to control samples for one of three experiments performed in triplicate with similar results. * p < 0.05, ** p < 0.01 compared to the control (control fold change =1).
As additional confirmation for InlF mediating increased invasion following inhibition of ROCK activity, we analyzed infection of L2 cells with L. monocytogenes strains complemented for expression of InlF. L. monocytogenes 10403S-derived ΔinlF, Lineage I strain FSL J1-194 and L. innocua harboring a plasmid encoding InlF demonstrated an increase in host cell-association and entry upon Y27632 treatment (Fig. 7B). Infection of L2 cells with Listeria strains lacking InlF and harboring the cloning vector alone was not affected by Y27632 treatment. Taken together, the data shown in Figures 6 and 7 indicate a role of InlF for increased infection of L2 cells following inhibition of ROCK activity.
Inhibition of ROCK activity in vivo increases L. monocytogenes virulence and is dependent on InlF
Infection of BALB/c mice was used to determine the impact of inhibiting ROCK activity and a role of InlF for L. monocytogenes virulence during in vivo infection. Since HA-1077 (Fasudil) is an approved drug for clinical use, HA-1077 was used for ROCK activity inhibition studies in mice. Animals were treated with HA-1077 followed by intravenous injection (i.v.) of wild-type or ΔinlF bacteria. The number of bacteria present in the liver and spleen was determined 72 h post-infection (Fig. 8). The wild-type bacterial burden in organs was significantly increased when mice were treated with HA-1077 prior to infection, yielding a 30-fold increase in bacterial numbers in both the livers and spleens of infected mice. In addition, a 4-fold increase in colonization of liver and spleen was observed in HA-1077 treated mice at 24 h post-infection (data not shown), suggesting early events in infection were involved in mediating the increase in bacterial burden. In the absence of inhibitor treatment, ΔinlF colonized organs similar to wild-type bacteria. In contrast to infection with wild type, the increase in bacterial burden observed in liver or spleen following treatment with HA-1077 was completely abolished during infection with ΔinlF bacteria. These results suggest that inhibition of ROCK activity in mice can increase L. monocytogenes infection in vivo and is dependent on expression of InlF.
Fig. 8. Inhibition of ROCK activity increases virulence in mice and is dependent on expression of InlF.
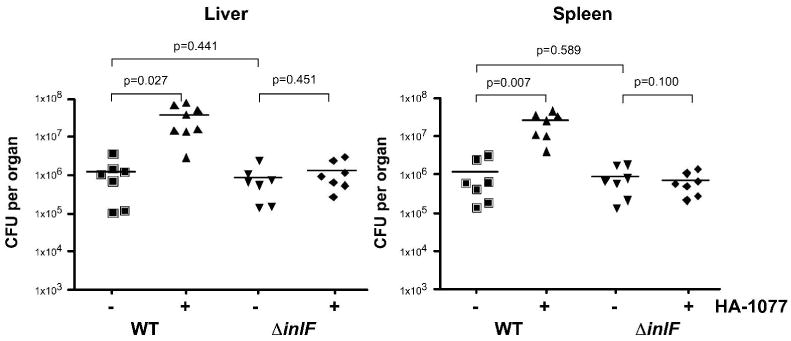
Groups of seven BALB/c mice were injected intravenously (i.v.) with 10 mg/kg HA-1077 (+) or PBS (-) and 5 h later infected via i.v. injection with wild-type 10403S (WT) or ΔinlF bacteria (1×104 bacteria/animal). At 72 h post-infection, the bacterial burden in the liver and spleen was determined. Each symbol represents the CFU per organ of one mouse. Horizontal bar indicates the mean value per group. p, represents the statistical significance between bracketed groups.
Discussion
We recently determined that RNAi knockdown of the small GTPase Rho1 in Drosophila cells resulted in increased infection by L. monocytogenes (Agaisse et al., 2005). In this report, we investigated the impact of the homologous RhoA/ROCK pathway for L. monocytogenes infection of mammalian host cells. Inhibition of RhoA/ROCK activity increased host cell-association and entry into murine fibroblast and hepatocyte cell lines. Conversely, activation of RhoA/ROCK activity resulted in decreased L. monocytogenes invasion. Interestingly, down-regulation of expression of either ROCK isoform resulted in increased L. monocytogenes infection (Fig. 3). The mechanism by which inhibition of ROCK activity leads to increased infection is currently unknown. However, ROCKs affect a wide range of cellular functions and recent studies report isoform specific activities (Coleman et al., 2001; Riento et al., 2003; Sebbagh et al., 2001; Yoneda et al., 2005; Yoneda et al., 2007) and distinct isoform distribution within cells (Riento et al., 2003; Yoneda et al., 2005). Therefore, it is unclear whether a common pathway or distinct isoform specific functions are involved in the regulation of L. monocytogenes infection.
Studies with the myosin II specific inhibitor blebbistatin strongly suggest that the assembly stage of myosin-actin structures has an impact on L. monocytogenes infection. Similar to inhibition of ROCK activity, disassembly of myosin-actin filaments resulted in increased host cell-association and bacterial uptake (Fig. 4). Myosin-actin assembly is regulated by phosphorylation of MLC, a major substrate for ROCKs (Kureishi et al., 1997). Both ROCK-I and ROCK-II isoforms were shown to phosphorylate MLC in vitro and in vivo. Furthermore, siRNA-mediated ROCK depletion resulted in a substantial reduction of MLC phosphorylation (Li et al., 2006; Samarin et al., 2007; Yoneda et al., 2005). Therefore, it is possible that a common function of ROCK-I and ROCK-II, such as regulation of myosin-actin assembly, accounts for the increase in L. monocytogenes infection. The mechanism by which myosin-actin structures may contribute to the regulation of L. monocytogenes infection is being further investigated.
Several bacterial species were examined for ROCK-dependent interactions with host cells, including invasive and non-invasive bacteria. Within the spectrum of bacteria tested in this study, an increase in host cell association or entry following inhibition of ROCK activity was only observed with L. monocytogenes (Supplemental Fig. S3). In addition, interaction of L. monocytogenes with phagocytic host cells was not affected by inhibition of ROCK activity (Table 1). Therefore, our studies suggest a specific receptor-ligand interaction leading to invasion of host cells. Given that increased infection was also observed following blebbistatin treatment, we hypothesize that alterations in myosin-actin structures may lead to surface exposure of a putative host cell receptor as has been described for integrins (Carragher et al., 2006). Another possibility is that receptor conformation and/or membrane localization is affected by alteration in myosin-actin structures (Charrasse et al., 2006; Lawler et al., 2006; Rodriguez-Fernandez et al., 2001; Shewan et al., 2005; Wojciak-Stothard et al., 1999). As a consequence, a putative receptor could be made more accessible resulting in bacterial adhesion and uptake. An impact of RhoA and ROCK activity in infection has been shown for some pathogenic organisms. Binding of the fungal pathogen Cryptococcus neoformans to endothelial cells was increased upon Y27632 treatment (Chen et al., 2003) and expression of a dominant negative RhoA enhanced host cell invasion by Vibrio parahaemolyticus (Akeda et al., 2002). Nonetheless, the mechanisms governing these interactions are not known. In addition, Vaccinia virus has been shown to directly inhibit RhoA/ROCK signaling to facilitate virus morphogenesis and motility (Valderrama et al., 2006). Our ongoing studies are aimed at determining if L. monocytogenes can actively interfere with RhoA or ROCK activity during infection.
We have also identified InlF as a L. monocytogenes-specific factor that mediates increased invasion of murine-derived cell lines, such as fibroblasts and hepatocytes, following inhibition of ROCK activity (Fig. 6). Although a role for fibroblasts in naturally occurring L. monocytogenes infection has not been described thus far, hepatocytes represent a cell type crucial for host colonization in vivo (Vazquez-Boland et al., 2001). InlF is one of 25 members of the internalin multigene family present in Listeria species (Hamon et al., 2006). However, a role for only a few internalin family members has been described during infection. InlA has been shown to promote invasion of human non-phagocytic cells expressing E-cadherin (Gaillard et al., 1991; Mengaud et al., 1996), while InlB mediates entry into several host cell types by interacting with multiple cellular receptors, including hepatocyte growth factor receptor (Met) and gC1q-R (Braun et al., 2000; Shen et al., 2000). Furthermore, interactions between host-cell receptors and InlA or InlB are species specific (Khelef et al., 2006; Lecuit et al., 1999). Transcription of inlF has been shown to be PrfA- and σB-independent and up-regulated by high salt (McGann et al., 2007). While inlF is present in L. monocytogenes, the gene is absent in L. innocua and L. ivanovii strains. This is consistent with our results indicating that increased infection following inhibition of ROCK activity was not observed with L. innocua and L. ivanovii or abolished upon deletion of prfA in L. monocytogenes (Fig. 5). Prior studies have shown that deletion of inlF did not affect invasion of Caco-2 and HepG2 cells or virulence in mice (Dramsi et al., 1997). Nonetheless, we have identified a condition where host cells are susceptible to InlF-mediated infection by L. monocytogenes both in vitro and in vivo. Consistent with previous reports (Dramsi et al., 1997), an inlF mutant did not show any defect in host cell-association or entry under standard cell culture conditions (Fig. 6).
Based on genotypic and epidemiological characteristics, L. monocytogenes isolates can be grouped into three evolutionary divisions (Lineages, I, II and III). Only Lineage I and II strains have been isolated from humans, although all three lineages are represented in animal isolates. Interestingly, inlF is present in all Lineage II isolates, including 10403S and EGDe, but was not found in Lineage I strains (Dramsi et al., 1997; Tsai et al., 2006). Consistent with a role of InlF in L. monocytogenes infection, none of the Lineage I strains examined in this study demonstrated increased infection following inhibition of ROCK activity (Fig. 7A). Previous attempts to correlate lineages with pathogenic potential suggest that in animals, Lineage I strains represent a narrower spectrum for pathogenesis than Lineage II strains (Pohl et al., 2006). The absence of inlF in Lineage I strains may contribute to this outcome. However, no such correlation was found in humans as epidemiological studies do not indicate a higher or specific pathogenic potential of Lineage II strains in humans (Jeffers et al., 2001). The fact that inlF is highly conserved within Lineage II strains may support its potential importance and suggests that the presence of inlF in Lineage II strains may represent an advantage for infection of hosts other than humans.
In vitro infection of various host cell lines demonstrated that inhibition of ROCK activity resulted in two host cell type-specific phenotypes. For murine fibroblasts and hepatocytes, as well as for human fibroblasts, increased host cell-association and entry was observed, whereas for the human epithelial cell lines HeLa and Hep2, an increase in adhesion but not in entry was observed (Table 1). It is possible that a different or altered host cell receptor-bacterial ligand interaction is responsible for increased adhesion, but not entry into epithelial cells. This is consistent with our results indicating that InlF is not involved in mediating increased adhesion to HeLa cells (Fig. 6). In addition, InlF was not involved in increased infection of WI38 human fibroblasts (Fig. 6), suggesting a possible species-specific interaction of InlF with murine cells. Future studies will be aimed at identifying L. monocytogenes factors mediating increased infection of human-derived cells upon inhibition of ROCK activity.
In addition to in vitro studies, we determined that inhibition of ROCK activity in vivo using HA-1077 (Fasudil) treatment of mice enhanced L. monocytogenes infection leading to increased colonization of the liver and spleen (Fig. 8). Importantly, with ΔinlF bacteria the increase in bacterial burden in organs upon infection of HA-1077 treated mice was completely abolished, supporting a functional role of InlF during infection. Although consistent with in vitro infection data, we can only speculate on the mechanism supporting an InlF-mediated increase in colonization following inhibition of ROCK activity in vivo. In tissue culture infection models, host cell-association and entry of L. monocytogenes into murine fibroblasts and hepatocytes was affected by ROCK activity dependent on expression of InlF. Whether ROCK activity has the same impact on host cell invasion in the in vivo mouse model needs to be investigated. In addition, colonization of both liver and spleen was similarly affected by inhibitor treatment, suggesting a systemic effect. Although less pronounced, an increase in colonization of organs was detectable at 24 h post-infection in inhibitor-treated mice. Therefore, the increase in bacterial burden observed at 72 h post-infection (Fig. 8) may be due to alterations in initial colonization events. Currently, it is unclear at which stage of infection or which host cell types are involved in mediating enhanced colonization following inhibition of ROCK activity in mice. Our in vitro infection studies with murine TIB 75 hepatocytes (Fig. 6) suggest that enhanced bacterial entry of host tissue may account for increased liver colonization in inhibitor-treated mice. Consequently, other organs or host cell types may be similarly affected, as inhibition of ROCK activity appears to systemically affect L. monocytogenes infection in mice. Previous studies have shown that inhibition of ROCK activity can attenuate innate immune responses in vivo, such as leukocyte infiltration, neutrophil migration and the production of pro-inflammatory cytokines and chemokines (Bao et al., 2004; Tasaka et al., 2005; Thorlacius et al., 2006). However, in this study, a repression of general innate immune responses is unlikely to be the cause of increased colonization in mice as infection with ΔinlF bacteria was not altered by drug treatment. Detailed investigation of colonization events during the course of in vivo infection is currently being performed to elucidate the mechanism of enhanced L. monocytogenes pathogenesis following inhibition of ROCK activity.
In conclusion, we have shown that inactivation of the host cell kinase ROCK leads to increased L. monocytogenes infection in vitro and in vivo. Furthermore, although a role in infection has not been defined for the majority of internalin family members, we have identified InlF as a L. monocytogenes-specific factor involved in mediating enhanced infection in a host cell type/species specific manner following inhibition of ROCK activity. Besides the expression of specific bacterial invasive determinants, the susceptibility of host cells to invasion will determine which tissues L. monocytogenes can infect. Inhibition of ROCK activity in cultured murine host cells provides an in vitro model where the function of InlF as an invasin becomes apparent. Current studies are aimed at identifying the host receptor(s) for InlF. Taken together, this study represents a valuable approach to reveal the function of a bacterial virulence factor that may be relevant under certain host cell conditions or during infection of specific host species. Several host conditions can influence the success of infection, primarily through alterations in the function of the immune system. Individuals who are immunocompromised are more susceptible to L. monocytogenes infection (Vazquez-Boland et al., 2001). Nonetheless, there are also adult patients for whom no obvious predisposing condition has been identified. The potential relevance of ROCK activity in L. monocytogenes infection is not readily apparent as information regarding in vivo ROCK activity is limited. However, animal studies have shown that under certain physiological conditions ROCK activity can be down-regulated, as demonstrated for aorta and myometrium tissues in pregnant rats (Cario-Toumaniantz et al., 2003; Katoue et al., 2006). More importantly, the ROCK inhibitor Fasudil is an approved drug already therapeutically used in patients with cardiovascular diseases, and the range of applications is likely to expand, as ROCKs have become an attractive drug target for treatment of cancer and neurological disorders (Mueller et al., 2005; Noma et al., 2006). Since adequate epidemiological data are not yet available, whether treatment of humans with ROCK inhibitors enhances susceptibility to L. monocytogenes infection is unknown, but may now potentially be considered as a risk factor.
Experimental procedures
Bacteria strains and media
Bacterial strains and plasmids used in this study are listed in Table 2. L. monocytogenes, L. innocua and L. ivanovii strains were grown in Brain Heart Infusion (BHI) medium (Difco, Detroit, MI). Salmonella enterica serovar Typhimurium IR715 (ATCC 14028), Pseudomonas aeruginosa PAK1 and Bacillus subtilis were grown in Luria-Bertani (LB) broth or on LB agar. Shigella flexneri 2457T, containing the large Shigella virulence plasmid, was grown in Tryptic Soy Broth (TSB). E. coli strains were grown in LB broth at 37°C with shaking. Antibiotics were used at the following concentrations: chloramphenicol at 20 μg/ml for selection of pAM401 and 100 μg/ml carbenicillin for pCON1 derivatives in E. coli; chloramphenicol 7.5 μg/ml for selection of pAM401 and pCON1 derivatives in L. monocytogenes and L. innocua; kanamycin at 30 μg/ml for maintaining pJP2 in E. coli.
Table 2.
Bacterial strains used in this study
| Strain | Species | Description | Source or Reference |
|---|---|---|---|
| 10403S | L. monocytogenes | Wild-type strain | (Bishop and Hinrichs, 1987) |
| DH-L1252 | L. monocytogenes | 10403S expressing GFP | This study |
| DH-L368 | L. monocytogenes | 10403S ΔinlAB | H. Shen |
| DH-L371 | L. monocytogenes | 10403S ΔprfA | H. Shen |
| DH-L478 | L. monocytogenes | Wild-type strain EGDe | M. Loessner |
| DH-L1681 | L. monocytogenes | 10403S with pAM401spacOid-BamHI | This study |
| DH-L1676 | L. monocytogenes | 10403S with pAM-inlF | This study |
| DH-L1674 | L. monocytogenes | 10403S ΔinlF | This study |
| DH-L1682 | L. monocytogenes | 10403SΔinlF with pAM401spacOid-BamHI | This study |
| DH-L1677 | L. monocytogenes | 10403SΔinlF with pAM-inlF | This study |
| DH-L1668 | L. monocytogenes | Lineage I strain FSL J1-194 (DUP-1042B) | (Tsai et al., 2006) |
| DH-L1669 | L. monocytogenes | Lineage I strain FSL R2-503 (G6054) | (Ducey et al., 2007) |
| DH-L1671 | L. monocytogenes | Lineage I strain FSL N1-017 | (Gray et al., 2004) |
| DH-L1683 | L. monocytogenes | DH-L1668 with pAM401spacOid-BamHI | This study |
| DH-L1678 | L. monocytogenes | DH-L1668 with pAM-inlF | This study |
| DH-L1353 | L. ivanovii | Wild-type strain FSL C2-010 | ATCC C2-010 |
| DH-L657 | L. innocua | Wild-type L. innocua | N. Freitag |
| DH-L1684 | L. innocua | DH-L657 with pAM401spacOid-BamHI | This study |
| DH-L1679 | L. innocua | DH-L657 with pAM-inlF | This study |
| DH-1664 | Salmonella typhimurium | Wild-type strain IR715 | ATCC 14028 |
| DH-P1665 | Pseudomonas aeruginosa | Wild-type strain PAK1 | (Takeya and Amako, 1966) |
| DH-B1666 | Bacillus subtilis | Wild-type strain PY79 | (Youngman et al., 1983) |
| DH-1667 | Shigella flexneri | Wild-type strain 2457T, serotype 2a | (Labrec et al., 1964) |
| DH-E112 | E. coli | JM109(DE3) | Promega Corp. |
| DH-E182 | E. coli | XL1-Blue | Stratagene Corp. |
| DH-E123 | E. coli | pCON1 in JM109 | (Freitag, 2000) |
| DH-E652 | E. coli | pJP2 (pACYC184 with Y. pseudotuberculosis invasin) in XL1-Blue | J. Pratt (Isberg et al., 1987) |
| DH-E659 | E. coli | pAM401spacOid in XL1-Blue | (Gründling et al., 2003) |
| DH-E969 | E. coli | pAM401spacOid-BamHI in XL1-Blue | This study |
| DH-E1673 | E. coli | pCON1/ΔinlF in XL1-Blue | This study |
| DH-E1675 | E. coli | pAM-inlF in XL1-Blue | This study |
Cell culture
The human-derived epithelial cell lines HeLa (ATCC CCL-2) and HEp-2 (ATCC CCL-23), the murine-derived epithelial cell line 1308.1, the murine-derived fibroblast cell lines L2 and L929, the murine-derived macrophage cell lines RAW 264.7 and RAW 309 Cr.1, and the human-derived macrophage cell line U937 (ATCC CRL-1593.2) were cultured in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% FBS (HyClone, Logan, UT), 55 μM 2-β-mercaptoethanol, 1 mM sodium pyruvate, and 2 mM glutamine. The human kidney epithelial cell line HEK293, the murine fibroblast cell line NIH 3T3 and the murine hepatocyte cell line TIB 75 (ATCC TIB 75) were propagated in DMEM medium (Mediatech, Herndon, VA) supplemented with 10% FBS, 55 μM 2-β-mercaptoethanol, 1 mM sodium pyruvate, and 2 mM glutamine. The human hepatocyte cell line HepG2 was cultured in DMEM medium (Mediatech, Herndon, VA) supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM glutamine, and 1X non-essential amino acids. The human fibroblast cell line WI38 was grown in MEM medium (Mediatech, Herndon, VA) supplemented with 10% FBS, 2 mM glutamine and Earle's BSS adjusted to contain 1.5 g/L sodium bicarbonate, 0.1 mM non-essential amino acids and 1 mM sodium pyruvate. All cell lines were maintained at 37°C in a 5% CO2-air atmosphere.
DNA constructs and transfection
pCAG-myc-based plasmids expressing dominant negative (ROCK-I DN) and constitutive active (ROCK-I CA) ROCK were a gift from Dr. L. Luo (Stanford University, Stanford, CA). The control vector pCAG-GFP (Matsuda and Cepko, 2004) was purchased from Addgene (Plasmid 11150, Addgene, Cambridge, MA). Transfection was performed using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Cells were used for experiments 24 h post-transfection. pLKO.1-based shRNA constructs targeting GFP, murine ROCK-I and ROCK-II were kindly provided by Dr. William Hahn (Dana-Farber Cancer Institute, Boston, MA). The gene-specific hairpin sequences are as follows: GFP: 5′–CGCAAGCTGACCCTGAGTTC–3′; ROCK-I: 5′–GACATTTGAAGTTAGCAGA–3′; ROCK-II: 5′–CTCGTCAACCTTATGAGTA –3′. Transfection of shRNA constructs was performed using Lipofectamine 2000 transfection reagent (Invitrogen) according to manufacturer's instructions. L2 cells were transfected twice at approximately 24 h intervals with 4 μg of DNA per 1×106 cells. Transfected cells were used for experiments 72 h after the first transfection.
Antibodies and reagents
The antibodies used were: rabbit polyclonal anti-phospho-myosin light chain (Ser19) (#3671, Cell Signaling, Danvers, MA); rabbit polyclonal anti-myosin light chain 2 (FL-172), goat polyclonal anti-ROCK-I (C-19), goat polyclonal anti-ROCK-II (C-20) and rabbit polyclonal anti-tubulin (H-235) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); mouse monoclonal anti-Myc (M4439, Sigma, St. Louis, MO); and rabbit polyclonal anti-L. monocytogenes (Difco, Detroit, MI). For microscopy, all secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit (BioRad, Hercules, CA) or anti-goat (Santa Cruz Biotechnology, Inc.) secondary antibodies were used for Western blot analyses. Hoechst stain was purchased from Sigma. Reagents used were: Rho inhibitor CT04 (Cytoskeleton, Inc., Denver, CO); Y27632 (Biomol, Plymouth Meeting, PA); HA-1077 (Fasudil; LC-Laboratories, Woburn, MA); HA-1100 (Hydroxyfasudil; Calbiochem, San Diego, CA); Blebbistatin and Lysophosphatidic acid (LPA, Sigma, St. Louis, MO).
Western blot analyses
Total L2 cell lysates were resolved by 7% (ROCK detection) or 12% (MLC detection) sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with Tris-buffered saline containing 0.1% Tween 20 and 4% bovine serum albumin, membranes were probed with specific antibodies. Proteins were visualized with peroxidase-coupled secondary antibody using the ECL system (Amersham, Piscataway, NJ). Autoradiographs were imaged using the Typhoon 9400 scanner (Amersham). Quantitative analysis was performed using the ImageQuant TL software program (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). Mean values and standard deviation were calculated from three independent experiments. The control value was set as 100% and all other values were calculated with respect to the control.
Microscopy
L2 cell monolayers seeded on glass coverslips were infected with L. monocytogenes at a multiplicity of infection (MOI) of 50:1. At 1 h post-infection, monolayers were washed three times with PBS and fixed with 3.2% paraformaldehyde for 15 min at room temperature. Subsequently, the monolayers were permeabilized with 0.1% Triton X-100 in PBS for 15 min and then blocked with 1% bovine serum albumin in PBS for 30 min at room temperature. For immunodetection, coverslips were co-incubated with rabbit polyclonal antibody specific for L. monocytogenes and mouse monoclonal antibody against Myc, washed with PBS, followed by treatment with Rhodamine Red X-conjugated donkey anti-rabbit IgG, FITC-conjugated goat anti-mouse IgG and Hoechst stain. Coverslips were mounted and analyzed by fluorescence microscopy. Images were acquired using MetaMorph imaging software (Molecular Devices, Downingtown, PA). For each acquisition, a Z-series was obtained and the collected Z-stack was merged into one plane using the ‘Stack Arithmetic: Maximum’ command of MetaMorph. Appropriate colors were assigned to each fluorescent image (blue for Hoechst, green for FITC and red for Rhodamine Red X). The color images were scaled and ultimately overlaid using the ‘Overlay’ command of MetaMorph.
Gentamicin protection assay
Host cells were grown in 24-well cell culture dishes to 70–80% confluency. On the day of infection, monolayers were pre-incubated with or without inhibitors for the indicated time periods and then incubated with bacteria from 14-16 h cultures that had been pelleted, washed in PBS and resuspended in cell culture medium. Infected monolayers were incubated for 1 h or 30 min (macrophage cell lines) in a 5% CO2-air atmosphere at 37°C. Cultures were washed three times with medium and processed further to determine cell-associated and intracellular colony-forming units. To quantitate total cell-associated bacteria, washed monolayers were immediately lysed with 1% Triton X-100 in PBS. Bacteria were suspended by vigorous pipetting and colony-forming units in lysates were determined by plating dilutions on agar plates. To determine bacterial internalization, extracellular bacteria were selectively killed by incubating washed monolayers for 1 h in culture medium containing gentamicin followed by Triton X-100 lysis and plating for viable intracellular bacteria. For macrophage cell lines, a concentration of 10 μg/ml gentamicin was used. For all other cell lines, 50 μg/ml gentamicin was applied. The following multiplicities of infection (MOI) were used for infections with Listeria strains: MOI 50:1 for L2, L929, NIH3T3, WI38, 1308.1, HeLa, Hep2 and HepG2; MOI 5:1 for HEK293; MOI 1:1 for RAW 264.7, RAW 309 Cr.1 and U937. L2 and HeLa cell infections with B. subtilis, E. coli, E. coli inv, S. enterica serovar Typhimurium, P. aeruginosa, and S. flexneri, were performed using a MOI of 50:1.
Intracellular growth assay
A total of 2.0 × 106 host cells were seeded 1 day prior to infection in 60-mm-diameter culture dishes containing 12-mm-diameter round glass coverslips. 10 μM Y27632, 10 μM LPA or DMSO was added to host cells 30 min prior to infection. Wild-type L. monocytogenes (10403S) was grown 14-16 h in 3 ml of BHI medium at 30°C without shaking. The bacterial culture was washed once with PBS and used for infection of host cells at a multiplicity of infection (MOI) of 50:1. At 1 h post-infection, monolayers were washed three times with RPMI medium, and RPMI-10% FBS medium containing DMSO, 10 μM Y27632 or 10 μM LPA and 50 μg/ml gentamicin was added and replaced at 4 h and 7 h post-infection. The number of CFU per coverslip were determined at the indicated time points by separately placing coverslips, in triplicate, into 15-ml conical tubes containing 5 ml of sterile water. Following vortexing of conical tubes, dilutions of lysates were plated on LB agar plates.
Plaque assay in L2 cells
Twenty-four hours prior to infection, 2 × 106 L2 cells were seeded in each well of a six-well dish. 10 μM Y27632, 10 μM LPA or DMSO (control) was added to L2 cells 30 min prior to infection. Fourteen to sixteen hour L. monocytogenes cultures were washed with PBS and 2 × 105 bacteria in culture medium were added to each well. At 1 h post-infection, monolayers were washed twice with PBS and a 0.7% agarose-medium overlay in DMEM, 5% FBS, 30 μg/ml gentamicin, and either 10 μM Y27632, 10 μM LPA or DMSO was applied. At 48 h post-infection, a second agarose-medium overlay was applied that contained 187 μg/ml of neutral red and 30 μg/ml gentamicin. At 72 h post-infection, plates were scanned to digital images, and the diameters of 10 plaques per well were determined using Adobe Photoshop 6.0 software.
Analysis of foci of infection
L2 cell monolayers seeded onto glass coverslips were treated with 10 μM Y27632, 10 μM LPA, or DMSO (control) and infected with strain DH-L1252, a variant of GFP-expressing wild-type 10403S L. monocytogenes DH-L1039 (Agaisse et al., 2005), at a multiplicity of infection (MOI) of 1:1. After 1 h of infection, coverslips were washed three times with PBS and medium containing 10 μM Y27632, 10 μM LPA, or DMSO and 50 μg/ml gentamicin was added. At 4 h, 8 h, and 12 h post-infection the medium was replaced with fresh medium containing 10 μM Y27632, 10 μM LPA, or DMSO and 10 μg/ml gentamicin. At 12 h and 24 h post-infection, coverslips were removed, fixed and stained with Hoechst. Samples were analyzed by fluorescence microscopy using a 20X objective. The diameters of 15 foci per sample were determined using Adobe Photoshop 6.0 software.
in vivo virulence studies
Six- to eight-week-old female BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were injected intravenously (i.v.) with HA-1077 in PBS (10 mg/kg; approximately 250 μg/animal) or PBS only (Rikitake et al., 2005; Shin et al., 2007). After 5 h, mice were infected i.v. with 1 × 104 L. monocytogenes. At 24 h or 72 h post-infection, mice were humanely euthanized and livers and spleens were sterilely dissected and homogenized in 5 ml PBS. The number of CFU/organ was determined by plating dilutions of the organ homogenates on LB agar.
Expression library construction and selection
Using primer pair 256 and 257 (Table 3), the SPAC/lacOid promoter/operator region was PCR amplified from plasmid pAMspacOid (Gründling et al., 2003), digested with NruI and BamHI and cloned into the multicopy plasmid vector pAM401 (Wirth et al., 1986), which had been digested with the same enzymes, resulting in the plasmid pAMspacOid-BamHI (DH-E969). A L. monocytogenes 10403S expression library was constructed by digesting chromosomal DNA with Sau3AI. One to 4 kb fragments were ligated into pAMspacOid-BamHI that had been digested with BamHI. After transformation into E. coli XL1-Blue, plasmid DNA from approximately 2 × 104 pooled single colonies was isolated and transformed into L. innocua. A total of 1.1 × 104 independent colonies were generated by selective growth on BHI agar plates containing 7.5 μg/ml chloramphenicol. Colonies were pooled and amplified by growth in BHI broth with 7.5 μg/ml chloramphenicol for 4 h at 37°C. The library was enriched for invasion-mediating clones by infecting Y27632 treated L2 cells at an MOI of 100:1 for 1 h. After an additional hour of infection in the presence of 50 μg/ml gentamicin, intracellular clones were isolated by lysing infected L2 cells with 1% Triton X-100 in PBS. Clones were amplified by 14-16 h growth at 37°C and used for additional infection of L2 cells. This enrichment was repeated twice. After the third round of infection, isolated intracellular clones were plated directly on BHI plates containing 7.5 μg/ml chloramphenicol. Single colonies were tested individually for invasion (see gentamicin protection assay) and putative clones were analyzed by plasmid isolation and sequencing of DNA inserts.
Table 3.
Primer sequences used in this study
| Number | Sequence | Restriction sitea |
|---|---|---|
| 256 | 5′-ACGCTCGCGACTAACAGCACAAGAGCGGAAAGATG-3′ | NruI |
| 257 | 5′-CGGGATCCGTCGACCAGATAAAATATTTCTAGAACACCTCC-3′ | BamHI |
| 660 | 5′-GCTCTAGATCGAGCCGGTCAACGGAAAT-3′ | XbaI |
| 661 | 5′-CTCCAAATATAAAACGCGGATGAACGGTCATTATGGTGGTGATTT-3′ | – |
| 662 | 5′-AAATCACCACCATAATGACCGTTCATCCGCGTTTTATATTTGGAG-3′ | – |
| 663 | 5′-GGGGATCCTTGCTACTTTGGATGGTGGTG-3′ | BamHI |
| 664 | 5′-CGCGGATCCAGGAGGAAAAATATGAAATCTAAAAATAATTATTTCAAAC-3′ | BamHI |
| 665 | 5′-AAGTCGACTTATGCTTTTTTTCTCCAAATATAAAA-3′ | – |
the indicated restriction endonuclease site is underlined within each oligonucleotide sequence.
Mutagenesis and cloning of inlF
Primer pair 660 and 661 was used with wild-type 10403S genomic DNA to amplify ∼1.0 kB of the region upstream of inlF including DNA sequence encoding the first 10 amino acids of InlF. Primers 662 and 663 were used to amplify ∼1 kB of DNA sequence downstream of and encoding the last 10 amino of InlF. The 5′ and 3′ PCR products were gel purified using the QIAquick gel extraction kit (Qiagen, Valencia, CA) and used as templates for a splicing by overlap extension (SOE) PCR reaction (Horton et al., 1989). The flanking primers, 660 and 663, were used to amplify a ∼2.0 kB PCR product containing an in-frame deletion of sequences encoding amino acids 11 to 810 of InlF. The SOE PCR product was gel-purified, digested with XbaI and BamHI, ligated to plasmid pCON1 digested with the same restriction enzymes, and transformed into XL1-Blue to create strain DH-E1673. The resulting plasmid, pCON1/ΔinlF was introduced into wild-type 10403S by electroporation, and allelic exchange was performed (Camilli et al., 1993) to generate strain DH-L1674. To create a complementing InlF construct, the inlF gene was cloned into pAMspacOid-BamHI (DH-E969). inlF carrying an optimized ermC RBS was amplified from 10403S genomic DNA using primers 664 and 665. The resulting PCR product was gel purified, digested with BamHI, ligated into pAMspacOid-BamHI digested with BamHI and EcoRV, and transformed into XL1-Blue, creating strain DH-E1675. The resulting plasmid pAM-inlF was sequenced and transformed into 10403S, 10403S ΔinlF, L. innocua and FSL J1-194 by electroporation, creating stains DH-L1676, DH-L1677, DH-L1678 and DH-L1679. As a control, the empty pAMspacOid-BamHI vector was also transformed in the same strains, creating strains DH-L1681, DH-L1682, DH-L1683 and DH-L1684, respectively.
Statistical analysis
Statistical analysis was performed using the Student's t-test (two-tailed, unpaired). Differences were considered significant at p < 0.05.
Supplementary Material
Acknowledgments
We would like to thank Wade Harper for providing shRNA constructs, Connie Cepko for construction of the pCAG-GFP plasmid, Liqun Luo for the gift of the ROCK expression plasmids, Wendy Loomis for training in mice infection procedures and Linda Lieberman for critical reading of the manuscript. This work was supported by US Public Health Service Grant AI53669 from the National Institutes of Health (D.E.H.). M. K. is a recipient of a fellowship from the Deutsche Forschungsgemeinschaft (KI 1088/1-1).
References
- Agaisse H, Burrack LS, Philips JA, Rubin EJ, Perrimon N, Higgins DE. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science. 2005;309:1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- Akeda Y, Kodama T, Kashimoto T, Cantarelli V, Horiguchi Y, Nagayama K, Iida T, Honda T. Dominant-negative Rho, Rac, and Cdc42 facilitate the invasion process of Vibrio parahaemolyticus into Caco-2 cells. Infect Immun. 2002;70:970–973. doi: 10.1128/IAI.70.2.970-973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakardjiev AI, Stacy BA, Fisher SJ, Portnoy DA. Listeriosis in the pregnant guinea pig: a model of vertical transmission. Infect Immun. 2004;72:489–497. doi: 10.1128/IAI.72.1.489-497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–558. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Bergmann B, Raffelsbauer D, Kuhn M, Goetz M, Hom S, Goebel W. InlA- but not InlB-mediated internalization of Listeria monocytogenes by non-phagocytic mammalian cells needs the support of other internalins. Mol Microbiol. 2002;43:557–570. doi: 10.1046/j.1365-2958.2002.02767.x. [DOI] [PubMed] [Google Scholar]
- Bian D, Mahanivong C, Yu J, Frisch SM, Pan ZK, Ye RD, Huang S. The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene. 2006;25:2234–2244. doi: 10.1038/sj.onc.1209261. [DOI] [PubMed] [Google Scholar]
- Bierne H, Cossart P. Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol Mol Biol Rev. 2007;71:377–397. doi: 10.1128/MMBR.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Borucki MK, Kim SH, Call DR, Smole SC, Pagotto F. Selective discrimination of Listeria monocytogenes epidemic strains by a mixed-genome DNA microarray compared to discrimination by pulsed-field gel electrophoresis, ribotyping, and multilocus sequence typing. J Clin Microbiol. 2004;42:5270–5276. doi: 10.1128/JCM.42.11.5270-5276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun L, Ghebrehiwet B, Cossart P. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. Embo J. 2000;19:1458–1466. doi: 10.1093/emboj/19.7.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cario-Toumaniantz C, Reillaudoux G, Sauzeau V, Heutte F, Vaillant N, Finet M, Chardin P, Loirand G, Pacaud P. Modulation of RhoA-Rho kinase-mediated Ca2+ sensitization of rabbit myometrium during pregnancy - role of Rnd3. J Physiol. 2003;552:403–413. doi: 10.1113/jphysiol.2003.047738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher NO, Walker SM, Scott Carragher LA, Harris F, Sawyer TK, Brunton VG, Ozanne BW, Frame MC. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene. 2006;25:5726–5740. doi: 10.1038/sj.onc.1209582. [DOI] [PubMed] [Google Scholar]
- Charrasse S, Comunale F, Grumbach Y, Poulat F, Blangy A, Gauthier-Rouviere C. RhoA GTPase regulates M-cadherin activity and myoblast fusion. Mol Biol Cell. 2006;17:749–759. doi: 10.1091/mbc.E05-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Stins MF, Huang SH, Chen YH, Kwon-Chung KJ, Chang Y, Kim KS, Suzuki K, Jong AY. Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol. 2003;52:961–970. doi: 10.1099/jmm.0.05230-0. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–127. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Dramsi S, Dehoux P, Lebrun M, Goossens PL, Cossart P. Identification of four new members of the internalin multigene family of Listeria monocytogenes EGD. Infect Immun. 1997;65:1615–1625. doi: 10.1128/iai.65.5.1615-1625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S, Bourdichon F, Cabanes D, Lecuit M, Fsihi H, Cossart P. FbpA, a novel multifunctional Listeria monocytogenes virulence factor. Mol Microbiol. 2004;53:639–649. doi: 10.1111/j.1365-2958.2004.04138.x. [DOI] [PubMed] [Google Scholar]
- Ducey TF, Page B, Usgaard T, Borucki MK, Pupedis K, Ward TJ. A single-nucleotide-polymorphism-based multilocus genotyping assay for subtyping lineage I isolates of Listeria monocytogenes. Appl Environ Microbiol. 2007;73:133–147. doi: 10.1128/AEM.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussurget O, Cabanes D, Dehoux P, Lecuit M, Buchrieser C, Glaser P, Cossart P. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol Microbiol. 2002;45:1095–1106. doi: 10.1046/j.1365-2958.2002.03080.x. [DOI] [PubMed] [Google Scholar]
- Engelbrecht F, Chun SK, Ochs C, Hess J, Lottspeich F, Goebel W, Sokolovic Z. A new PrfA-regulated gene of Listeria monocytogenes encoding a small, secreted protein which belongs to the family of internalins. Mol Microbiol. 1996;21:823–837. doi: 10.1046/j.1365-2958.1996.541414.x. [DOI] [PubMed] [Google Scholar]
- Farah S, Agazie Y, Ohan N, Ngsee JK, Liu XJ. A rho-associated protein kinase, ROKalpha, binds insulin receptor substrate-1 and modulates insulin signaling. J Biol Chem. 1998;273:4740–4746. doi: 10.1074/jbc.273.8.4740. [DOI] [PubMed] [Google Scholar]
- Freitag NE. Genetic tools for use with Listeria monocytogenes. In: Fischetti VA, Novivk RP, Ferretti JJ, Portnoy DA, Rood JI, editors. Gram-positive pathogens. Washinghton, D.C.: ASM press; 2000. pp. 488–498. [Google Scholar]
- Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91:391–392. doi: 10.1136/hrt.2003.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell. 1991;65:1127–1141. doi: 10.1016/0092-8674(91)90009-n. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Zadoks RN, Fortes ED, Dogan B, Cai S, Chen Y, Scott VN, Gombas DE, Boor KJ, Wiedmann M. Listeria monocytogenes isolates from foods and humans form distinct but overlapping populations. Appl Environ Microbiol. 2004;70:5833–5841. doi: 10.1128/AEM.70.10.5833-5841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Gonzalez MD, Higgins DE. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J Bacteriol. 2003;185:6295–6307. doi: 10.1128/JB.185.21.6295-6307.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Isberg RR, Voorhis DL, Falkow S. Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell. 1987;50:769–778. doi: 10.1016/0092-8674(87)90335-7. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. Embo J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- Jeffers GT, Bruce JL, McDonough PL, Scarlett J, Boor KJ, Wiedmann M. Comparative genetic characterization of Listeria monocytogenes isolates from human and animal listeriosis cases. Microbiology. 2001;147:1095–1104. doi: 10.1099/00221287-147-5-1095. [DOI] [PubMed] [Google Scholar]
- Jia Y, Nightingale KK, Boor KJ, Ho A, Wiedmann M, McGann P. Distribution of internalin gene profiles of Listeria monocytogenes isolates from different sources associated with phylogenetic lineages. Foodborne Pathog Dis. 2007;4:222–232. doi: 10.1089/fpd.2006.0081. [DOI] [PubMed] [Google Scholar]
- Jonquieres R, Pizarro-Cerda J, Cossart P. Synergy between the N- and C-terminal domains of InlB for efficient invasion of non-phagocytic cells by Listeria monocytogenes. Mol Microbiol. 2001;42:955–965. doi: 10.1046/j.1365-2958.2001.02704.x. [DOI] [PubMed] [Google Scholar]
- Katoue MG, Khan I, Oriowo MA. Pregnancy-induced modulation of calcium mobilization and down-regulation of Rho-kinase expression contribute to attenuated vasopressin-induced contraction of the rat aorta. Vascul Pharmacol. 2006;44:170–176. doi: 10.1016/j.vph.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Khelef N, Lecuit M, Bierne H, Cossart P. Species specificity of the Listeria monocytogenes InlB protein. Cell Microbiol. 2006;8:457–470. doi: 10.1111/j.1462-5822.2005.00634.x. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1997;272:12257–12260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- Labrec EH, Schneider H, Magnani TJ, Formal SB. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. J Bacteriol. 1964;88:1503–1518. doi: 10.1128/jb.88.5.1503-1518.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler K, Foran E, O'Sullivan G, Long A, Kenny D. Mobility and invasiveness of metastatic esophageal cancer are potentiated by shear stress in a ROCK- and Ras-dependent manner. Am J Physiol Cell Physiol. 2006;291:C668–677. doi: 10.1152/ajpcell.00626.2005. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. Embo J. 1999;18:3956–3963. doi: 10.1093/emboj/18.14.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M, Vandormael-Pournin S, Lefort J, Huerre M, Gounon P, Dupuy C, Babinet C, Cossart P. A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science. 2001;292:1722–1725. doi: 10.1126/science.1059852. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Nelson DM, Smith SD, Khun H, Huerre M, Vacher-Lavenu MC, Gordon JI, Cossart P. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E-cadherin. Proc Natl Acad Sci U S A. 2004;101:6152–6157. doi: 10.1073/pnas.0401434101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, Zhang X, Li A, Burbelo P, Stanley ER, Pestell RG. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol. 2006;26:4240–4256. doi: 10.1128/MCB.02124-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber B. Listeriosis. Clin Infect Dis. 1997;24:1–9. doi: 10.1093/clinids/24.1.1. quiz 10-11. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–1310. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105:1545–1547. doi: 10.1161/hc1002.105938. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. Embo J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- McGann P, Ivanek R, Wiedmann M, Boor KJ. Temperature-dependent expression of Listeria monocytogenes internalin and internalin-like genes suggests functional diversity of these proteins among the listeriae. Appl Environ Microbiol. 2007;73:2806–2814. doi: 10.1128/AEM.02923-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Lecuit M, Lebrun M, Nato F, Mazie JC, Cossart P. Antibodies to the leucine-rich repeat region of internalin block entry of Listeria monocytogenes into cells expressing E-cadherin. Infect Immun. 1996;64:5430–5433. doi: 10.1128/iai.64.12.5430-5433.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- Milohanic E, Jonquieres R, Cossart P, Berche P, Gaillard JL. The autolysin Ami contributes to the adhesion of Listeria monocytogenes to eukaryotic cells via its cell wall anchor. Mol Microbiol. 2001;39:1212–1224. doi: 10.1111/j.1365-2958.2001.02208.x. [DOI] [PubMed] [Google Scholar]
- Milohanic E, Glaser P, Coppee JY, Frangeul L, Vega Y, Vazquez-Boland JA, Kunst F, Cossart P, Buchrieser C. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol Microbiol. 2003;47:1613–1625. doi: 10.1046/j.1365-2958.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. J Biol Chem. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–398. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol. 2006;290:C661–668. doi: 10.1152/ajpcell.00459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl MA, Wiedmann M, Nightingale KK. Associations among Listeria monocytogenes genotypes and distinct clinical manifestations of listeriosis in cattle. Am J Vet Res. 2006;67:616–626. doi: 10.2460/ajvr.67.4.616. [DOI] [PubMed] [Google Scholar]
- Raffelsbauer D, Bubert A, Engelbrecht F, Scheinpflug J, Simm A, Hess J, Kaufmann SH, Goebel W. The gene cluster inlC2DE of Listeria monocytogenes contains additional new internalin genes and is important for virulence in mice. Mol Gen Genet. 1998;260:144–158. doi: 10.1007/s004380050880. [DOI] [PubMed] [Google Scholar]
- Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. Embo J. 2005;24:1170–1180. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fernandez JL, Sanchez-Martin L, Rey M, Vicente-Manzanares M, Narumiya S, Teixido J, Sanchez-Madrid F, Cabanas C. Rho and Rho-associated kinase modulate the tyrosine kinase PYK2 in T-cells through regulation of the activity of the integrin LFA-1. J Biol Chem. 2001;276:40518–40527. doi: 10.1074/jbc.M102896200. [DOI] [PubMed] [Google Scholar]
- Sabet C, Lecuit M, Cabanes D, Cossart P, Bierne H. LPXTG protein InlJ, a newly identified internalin involved in Listeria monocytogenes virulence. Infect Immun. 2005;73:6912–6922. doi: 10.1128/IAI.73.10.6912-6922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarin SN, Ivanov AI, Flatau G, Parkos CA, Nusrat A. Rho/ROCK-II signaling mediates disassembly of epithelial apical junctions. Mol Biol Cell. 2007;18:3429–3439. doi: 10.1091/mbc.E07-04-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert WD, Gobel G, Diepholz M, Darji A, Kloer D, Hain T, Chakraborty T, Wehland J, Domann E, Heinz DW. Internalins from the human pathogen Listeria monocytogenes combine three distinct folds into a contiguous internalin domain. J Mol Biol. 2001;312:783–794. doi: 10.1006/jmbi.2001.4989. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- Shen Y, Naujokas M, Park M, Ireton K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 2000;103:501–510. doi: 10.1016/s0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, Kovacs EM, Yap AS. Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol Biol Cell. 2005;16:4531–4542. doi: 10.1091/mbc.E05-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E. Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci. 2005;238:31–39. doi: 10.1016/j.jns.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Seto M, Katsumata N, Amano M, Kozai T, Yamawaki T, Kuwata K, Kandabashi T, Egashira K, Ikegaki I, Asano T, Kaibuchi K, Takeshita A. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc Res. 1999;43:1029–1039. doi: 10.1016/s0008-6363(99)00144-3. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Hiramori K, Iinuma H, Hosoda S, Kishida H, Osada H, Katagiri T, Yamauchi K, Yui Y, Minamino T, Nakashima M, Kato K. Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol. 2002;40:751–761. doi: 10.1097/00005344-200211000-00013. [DOI] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, Huang PL, Boas DA, Liao JK, Moskowitz MA, Ayata C. Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab. 2007;27:998–1009. doi: 10.1038/sj.jcbfm.9600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522(Pt 2):177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, Zhang L, Takami H, Yamada Y, Settleman J. Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Dev Cell. 2002;2:553–565. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol. 2000;522(Pt 1):33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeya K, Amako K. A rod-shaped Pseudomonas phage. Virology. 1966;28:163–165. doi: 10.1016/0042-6822(66)90317-5. [DOI] [PubMed] [Google Scholar]
- Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, Ishizaka A. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol. 2005;32:504–510. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- Thorlacius K, Slotta JE, Laschke MW, Wang Y, Menger MD, Jeppsson B, Thorlacius H. Protective effect of fasudil, a Rho-kinase inhibitor, on chemokine expression, leukocyte recruitment, and hepatocellular apoptosis in septic liver injury. J Leukoc Biol. 2006;79:923–931. doi: 10.1189/jlb.0705406. [DOI] [PubMed] [Google Scholar]
- Tsai YH, Orsi RH, Nightingale KK, Wiedmann M. Listeria monocytogenes internalins are highly diverse and evolved by recombination and positive selection. Infect Genet Evol. 2006;6:378–389. doi: 10.1016/j.meegid.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Valderrama F, Cordeiro JV, Schleich S, Frischknecht F, Way M. Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science. 2006;311:377–381. doi: 10.1126/science.1122411. [DOI] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth R, An FY, Clewell DB. Highly efficient protoplast transformation system for Streptococcus faecalis and a new Escherichia coli-S. faecalis shuttle vector. J Bacteriol. 1986;165:831–836. doi: 10.1128/jb.165.3.831-836.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–453. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Ushakov D, Multhaupt HA, Couchman JR. Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II. Mol Biol Cell. 2007;18:66–75. doi: 10.1091/mbc.E06-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman PJ, Perkins JB, Losick R. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc Natl Acad Sci U S A. 1983;80:2305–2309. doi: 10.1073/pnas.80.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.