

Abstract
Objective
Recent studies have suggested that oxidative stress and DNA damage may play a role in the pathophysiology of bipolar disorder (BD). We investigated the effects of the mood stabilizers lithium and valproate on amphetamine-induced DNA damage in an animal model of mania and their correlation with oxidative stress markers.
Methods
In the first experiment (reversal model), we treated adult male Wistar rats with D-amphetamine (AMPH) or saline for 14 days; between the 8th and 14th days, rats also received lithium, valproate or saline. In the second experiment (prevention model), rats received either lithium, valproate or saline for 14 days; between the 8th and 14th days, we added AMPH or saline. We evaluated DNA damage using single-cell gel electrophoresis (comet assay), and we assessed the mutagenic potential using the micronucleus test. We assessed oxidative stress levels by lipid peroxidation levels (TBARS) and antioxidant enzyme activities (superoxide dismutase and catalase). We assessed DNA damage and oxidative stress markers in blood/plasma and hippocampal samples. We evaluated mutagenesis in fresh lymphocytes.
Results
In both models, we found that AMPH increased peripheral and hippocampal DNA damage. The index of DNA damage correlated positively with lipid peroxidation, whereas lithium and valproate were able to modulate the oxidative balance and prevent recent damage to the DNA. However, lithium and valproate were not able to prevent micronucleus formation.
Conclusion
Our results support the notion that lithium and valproate exert central and peripheral antioxidant-like properties. In addition, the protection to the integrity of DNA conferred by lithium seems to be limited to transient DNA damage and does not alter micronuclei formation.
Medical subject headings: models, animal; bipolar disorder; lithium; DNA damage; dopamine; oxidative stress
Abstract
Objectif
Des études récentes ont indiqué que le stress oxydatif et les dommages causés à l'ADN peuvent jouer un rôle dans la pathophysiologie du trouble bipolaire (TB). Nous avons étudié les effets du lithium et du valproate, agents thymorégulateurs, sur les dommages causés à l'ADN par les amphétamines dans un modèle animal de la manie et leurs liens avec des marqueurs du stress oxydatif.
Méthodes
Au cours de la 1e expérience (modèle d'inversion), nous avons traité des rats Wistar mâles adultes en leur administrant de la D-amphétamine (AMPH) ou une solution physiologique pendant 14 jours. Entre les 8e et 14e jours, les rats ont aussi reçu du lithium, du valproate, ou une solution physiologique. Au cours de la 2e expérience (modèle de prévention), les rats ont reçu du lithium, du valproate ou une solution physiologique pendant 14 jours. Nous avons ajouté de l'amphétamine ou une solution physiologique entre les 8e et 14e jours. Nous avons évalué les dommages causés à l'ADN par électrophorèse sur gel à cellule unique (essai de comet) et nous avons évalué le potentiel mutagène en utilisant le test du micronoyau. Nous avons évalué les niveaux de stress oxydatif en fonction des concentrations de peroxydation des lipides (TBARS) et de l'activité des enzymes anti-oxydantes (superoxyde dismutase et catalase). Nous avons évalué les dommages causés à l'ADN et les marqueurs du stress oxydatif dans des échantillons de sang/plasma et d'hippocampe. Nous avons évalué la mutagenèse dans des lymphocytes frais.
Résultats
Dans les 2 modèles, nous avons constaté que les amphétamines augmentaient les dommages périphériques causés à l'ADN et à l'hippocampe. Il y avait un lien positif entre l'indice des dommages causés à l'ADN et la peroxydation des lipides, tandis que le lithium et le valproate ont pu moduler l'équilibre oxydatif et prévenir des dommages récents à l'ADN. Le lithium et le valproate n'ont toutefois pu prévenir la formation de micronoyaux.
Conclusion
Nos résultats appuient le concept selon lequel le lithium et le valproate ont des propriétés semblables aux antioxydants centraux et périphériques. En outre, la protection de l'intégrité de l'ADN conférée par le lithium semble limitée au dommage transitoire causé à l'ADN et n'altère pas la formation des micronoyaux.
Introduction
Bipolar disorder (BD) is a prevalent, chronic and highly disabling psychiatric disorder.1 It is considered to be one of the leading causes of disability among all medical and psychiatric conditions,2 and untreated BD has been associated with increased morbidity and mortality due to general medical conditions such as vascular disorders and cancer.3 In addition, evidence from postmortem studies suggests that the pathophysiology of BD may involve apoptotic cell death and neuronal and glial cell loss,4 which may in part explain the high rates of morbidity and the persistent interepisode cognitive impairment in patients with BD.4 It has been suggested that increased oxidative stress may be a common mechanism underlying the neurobiologic basis of these conditions.5 Reactive oxygen species (ROS) are known to play an important role in the pathogenesis of several general medical diseases6 and in neurodegenerative and psychiatric disorders,7 including Alzheimer disease,7 schizophrenia6,8 and BD.9,10
The central nervous system is extremely vulnerable to peroxidative damage. The human brain has a high concentration of oxidizable substrates and high oxygen tension because it metabolizes 20% of total body oxygen.11 In addition, the human brain contains a large amount of polysaturated fatty acids, which are very susceptible to lipid peroxidation. Thus under pathologic conditions, levels of ROS can easily exceed antioxidant capacity, which is relatively low in the central nervous system.6 Such excess of ROS may cause direct damage to cellular proteins, DNA and lipids, thereby affecting cellular function.12,13 The amount of DNA damage and the integrity of the repair system would determine whether the damage to DNA strands would be transient (DNA single-and double-strand breaks) or permanent (micronucleus formation). The single-cell gel-electrophoresis technique (comet assay) is used to detect transient DNA single-and double-strand breaks, whereas the DNA damage that persists after the action of different repair systems can be assessed using the micronucleus test.14,15
The understanding of the transient and permanent consequences of oxidative stress to DNA integrity and the protective potential of mood stabilizers have turned into central issues in the study of BD. We have recently shown that patients with BD have increased oxidative stress9,10 and DNA damage10,16 in peripheral blood. Moreover, it seems that increased oxidative stress occurs in proportion with DNA damage in such patients.10 A recent study showed that telomere shortening, which is thought to occur as a consequence of increased oxidative stress, is increased among patients with BD.17 Because the main source of free radicals is the respiratory chain in the mitochondria, attention should be drawn to the fact that abnormalities in respiratory complex activity18 and mitochondrial dysfunction19,20 were also demonstrated in BD. Using an animal model of mania, we have shown that chronic treatment with D-amphetamine (AMPH) increases superoxide dismutase (SOD) and catalase activity21 as well as lipid peroxidation and protein carbonyl formation.22 Chronic AMPH exposure induced a 3- to 6-fold increase of thiobarbituric acid reactive substances (TBARS) and a 1.5- to 2-fold increase in superoxide production in submitochondrial particles of the prefrontal cortex and hippocampus.23 Further, we showed that lithium and valproate exert protective effects against AMPH-induced oxidative stress.24 However, the deleterious effects of oxidative stress on DNA integrity and the protective potential of mood stabilizers have not been examined.
We sought to investigate the effects of lithium and valproate on transient and permanent DNA damage (as assessed with comet and micronucleus assays, respectively) in the peripheral blood and the hippocampus using an animal model of mania (repeated exposure to AMPH). Moreover, we evaluated parameters of oxidative stress, such as lipid peroxidation and antioxidant enzyme activities, to test the correlation between DNA damage and oxidative stress markers.
Methods
Chemicals
The chemicals we used included AMPH (Sigma-Aldrich), low and normal melting point agarose (Gibco), ethylene-diamine-tetracetic acid (EDTA; Labsynth), tris (Labsynth), triton X-100 (Labsynth), dimethyl sulfoxide (DMSO) (Labsynth), ethidium bromide (Sigma-Aldrich), RMPI 1640 medium (Nutricell), cytochalasin-B, valproic acid sodium salt and lithium carbonate (Sigma-Aldrich) and Giemsa (Merck).
Animals
We performed our experiments in male Wistar rats (age 3–4 mo, weight 220–310 g) obtained from our breeding colony. We housed the rats 5 to a cage, on a 12-hour light/dark cycle (lights on between 7 am and 7 pm), with food and water available ad libitum. We carried out all experimental procedures in accordance with the National Institutes of Health (NIH) Guide for the care and use of laboratory animals25 and the Brazilian Society for Neuroscience and Behaviour (SBNeC) recommendations for animal care. The local ethics committee (Universidade do Extremo Sul Catarinense, Criciúma, SC, Brazil) approved our research project.
Reversal model
We designed our first model to reproduce the management of an acute manic episode. Animals (n = 48) received a daily intraperitoneal injection of either AMPH (2 mg/kg) or saline for 14 days. Between the 8th and the 14th day, we divided the animals that received saline or AMPH into 6 experimental groups of 6–8 animals per group: saline+saline (SAL), saline+lithium (Li), saline+valproate (VPA), AMPH+saline, AMPH+lithium (AMPH+Li) and AMPH+valproate (AMPH+VPA). Animals in the Li group received intraperitoneal injections of lithium (47.5 mg/kg) twice a day; those in the VPA group received valproate (200 mg/kg) twice a day. We measured locomotor activity 2 hours after the last injection of AMPH or saline, submitted the rats to intracardiac punction immediately after the open field task and sacrificed them by decapitation after the intracardiac punction. We rapidly dissected the hippocampus and immediately used a slice for the analyses of DNA damage. We stored the remaining hippocampal tissue at –80°C until it was assayed for oxidative stress markers.
Prevention model
We designed the second model to mimic the maintenance phase of the treatment of BD. Animals (n = 48) received intraperitoneal injections of either lithium (47.5 mg/kg) twice a day, valproate (200 mg/kg) twice a day or saline for 14 days. Between the 8th and the 14th day, we divided the animals that received lithium, valproate or saline into 6 experimental groups of 6–8 animals per group: SAL, Li, VPA, AMPH, Li+AMPH and VPA+AMPH. The AMPH group received 1 daily intraperitoneal injection of either AMPH (2 mg/kg) or saline. We measured locomotor activity 2 hours after the last injection, submitted the rats to an intracardiac punction and sacrificed them by decapitation after the open field task. We rapidly dissected the hippocampus, and we immediately used a slice to analyze DNA damage. We stored the remaining hippocampal tissue was at –80°C until it was assayed for oxidative stress markers.
Locomotor activity
We assessed locomotor activity in the open-field task, which we performed in a 40 × 60–cm open field surrounded by walls 50 cm high made of brown plywood with a frontal glass wall. We divided the floor of the open field into 12 equal rectangles with black lines. We placed the animals on the left rear quadrant to freely explore the arena for 5 minutes. We considered the number of crossings of the black lines to be a measure of horizontal locomotor activity and we considered rearings to be a measure of vertical locomotor activity.
Sample preparation
We extracted blood by intracardiac punction with heparin. We used total blood for the micronucleus test and comet assay. For enzyme assays, we used plasma that we obtained by centrifugation at 1800 g for 10 minutes. We homogenized the hippocampus in cold phosphate-buffered saline (PBS) for the comet assay and antioxidant enzymes assay, which we carried out immediately.
Single-cell gel electrophoresis (comet assay)
We adopted a standard protocol for the comet assay preparation and analysis.26 We performed the comet assay under alkaline conditions (pH 12.6), which detect double-and single-strand breaks and alkali-labile sites.26,27 We prepared the slides by mixing 5 μL of whole blood or 20 μL of hippocampus homogenate (homogenized in 20 vol [wt/vol] of STM buffer [sucrose 250 mmol/L, Tris-HCl 50 mmol/L, pH 7.4, MgSO4 5 mmol/L, phenylmethylsulfonyl fluoride 0.5 mmol/L]), with 95 μL of low melting point agarose (0.75%) for blood samples or 80 μL for hippocampus samples. We added the mixture (cells/agarose) to a fully frosted microscope slide coated with a layer of 500 μL of normal melting agarose (1%). After solidification, we removed the cover slip and placed the slides in a lyses solution (2.5 M NaCl, 100 mM EDTA and 10 mM Tris, pH 10.0–10.5, with freshly added 1% Triton X-100 and 10% DMSO) for 1 day. Subsequently, we incubated the slides in freshly made alkaline buffer (300 mM NaOH and 1 mM EDTA, pH 12.6) for 10 minutes. The DNA was electrophoresed for 20 minutes at 25 V (0.90 V/cm) and 300 mA under alkaline conditions (pH 12.6). After that, we neutralized the slides with 0.4 M Tris (pH 7.5). Finally, we stained the DNA with ethidium bromide. We used negative and positive controls for each electrophoresis assay to ensure the reliability of the procedure. We analyzed images of 100 randomly selected cells (50 cells from 2 replicated slides) from each treatment group. Based on the size of the tails, we scored cells visually from 0 to 4, with 0 indicating no tail and 4 indicating the longest tail, resulting in a single DNA damage score for each cell and, consequently, for each study group. We calculated the index of DNA damage by multiplying the number of cells by each index score and then summing the results. Therefore, a group damage index could range from 0 (all cells, no tail; 100 cells × 0) to 400 (all cells, longest tail; 100 cells × 4).27
Cytokinesis-blocked lymphocyte micronucleus test
For each blood sample, we set up duplicate blood cultures in culture flasks by adding 0.3 mL of whole blood to 5 mL of RPMI 1640 medium containing 20% fetal calf serum and 1% (v/v) phytohemaglutinin. We incubated the flasks at 37°C for 44 hours before adding 5 μg/mL of cytochalasin-B,28 and we continued incubation until the total incubation time reached 72 hours. After incubation, we harvested the lymphocytes by centrifugation at 800 revolutions per minute for 8 minutes. The lymphocytes were then recentrifuged, fixed in 3:1 (v/v) methanol/acetic acid, placed onto a clean microscope slide and stained with 5% (v/v) Giemsa. For each blood sample, we scored 1000 binucleated lymphocytes (i.e., 500 from each of the 2 slides prepared from the duplicate lymphocyte cultures) for micronuclei presence, using bright-field optical microscopy (original magnification ×200–1000). Blinded to the treatment groups, we coded all slides.
Lipid peroxidation
We measured the plasma levels of lipid peroxidation using the TBARS method described by Wills.29 Data are expressed in nanomoles per millilitre.
Superoxide dismutase activity
We determined superoxide dismutase activity spectrophotometrically in peripheral plasma and hippocampal homogenates by measuring the inhibition of the ratio of autocatalytic adrenochrome formation at 480 nm in a reaction medium containing 1 mM adrenaline and 50 mM glycine (pH 10.2). We conducted this reaction at a constant temperature of 30°C for 3 minutes. Enzyme activity is expressed as superoxide dismutase units per gram of protein. We define 1 unit as the amount of enzyme that inhibits the ratio of adrenochrome formation to 50%.30
Catalase activity
We assayed catalase activity according to the method of Aebi.31 Briefly, first we added 2 mL quartz cuvette contained in 1.980 mL phosphate buffer (pH 7.4, 50 mM) to 20 mL of peripheral plasma or hippocampal homogenates, and we adjusted the spectrophotometer to auto zero. We then added 2 mL of H2O2 (3 mM freshly diluted). Values are expressed as micromoles of H2O2 consumed per minute per milligram of protein.
Statistical analysis
We determined the differences in oxidative stress parameters, index of DNA damage and micronuclei frequency among experimental groups using one-way analysis of variance (ANOVA). When ANOVA results were significant, we performed the Tukey post-hoc test. We used a Spearman coefficient to calculate the correlation between lipid peroxidation and DNA damage. In all experiments, we considered p < 0.05 to indicate statistical significance.
Results
Locomotor activity
In the first experiment (reversal treatment), we replicated our previous findings24 to confirm that DNA damage was measured in a proper model of mania. We found that AMPH increased crossings (F5–39 = 12.89, p < 0.001) and rearings (F5–39 = 22.91, p < 0.001) in rats treated with saline; lithium and valproate reverted AMPH-related hyperactive behaviour (Fig. 1A). The administration of lithium or valproate in saline-treated animals did not change behavioural measures, indicating that the effects of mood stabilizers in rats treated with AMPH were not associated with sedation. Behavioural measures of the second experiment (prevention treatment) demonstrated that both lithium and valproate pretreatment were also able to prevent AMPH-related hyperactivity (crossings F5–40 = 21.44, p < 0.001) and rearings (F5–40 = 31.56, p < 0.001). Saline administration in rats pretreated with mood stabilizers did not affect locomotor behaviour (Fig. 1B).

Fig. 1: Effects of D-amphetamine (AMPH), lithium (Li) and valproate (VPA) on crossing (horizontal) and rearing (vertical) behaviour in the (A) reversal and (B) prevention models. Values are expressed as means and standard deviation. *Statistically different from control group (SAL), as determined by one-way analysis of variance and Tukey test (p < 0.05).
Transient and permanent DNA damage
The results of transient DNA damage (comet assay) are shown as an index of DNA damage (Table 1), while those of permanent DNA damage are shown by frequency of micronuclei (Fig. 2). Treatment with AMPH increased transient DNA damage in central (hippocampus) samples in both the prevention (F5–40 = 10.14, p < 0.001) and the reversal (F5–39 = 13.57, p < 0.001) models (Table 1). We observed the same results in blood samples (F5–40 = 21.25, p < 0.001 in the prevention model; F5–39 = 19.01, p < 0.001 in the reversal model). Results of the Tukey test showed that treatment with lithium was able to reverse and prevent transient DNA damage in the peripheral blood (p = 0.233 in the reversal model; p = 0.235 in the prevention model) and hippocampal samples (p = 0.345 in the reversal model; p = 0.641 in the prevention model). Valproate did not have any effect. In prevention (F5–40 = 10.01, p < 0.001) and reversal models (F5–39 = 9.55, p < 0.001), animals treated with AMPH showed an enhanced micronucleus frequency. In the prevention model, administration of lithium and valproate diminished the frequency of micronuclei; however, the decrease was not statistically significant (Fig. 2A). In the reversal model, neither lithium nor valproate were able to reverse such damage (Fig. 2B).
Table 1

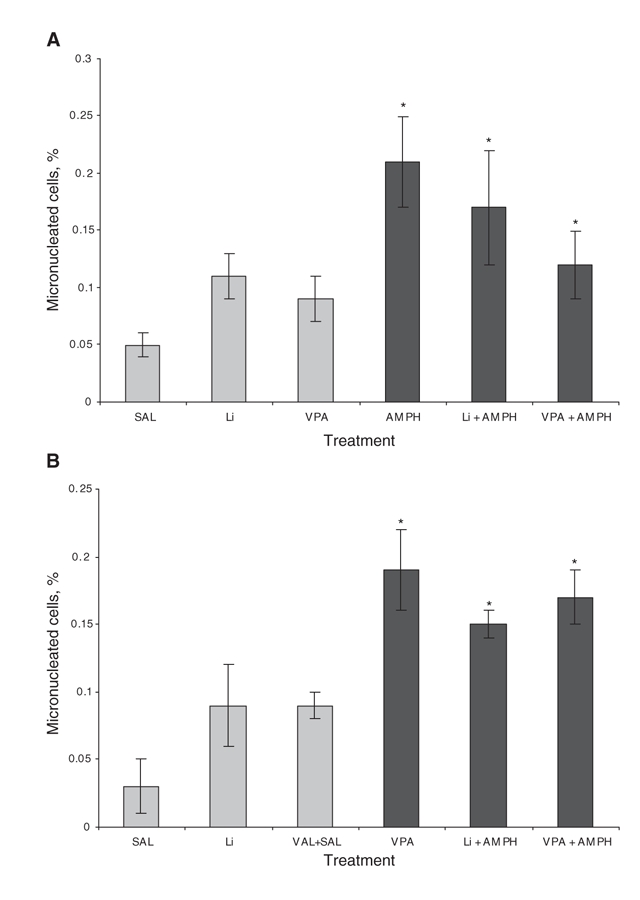
Fig. 2: Frequency of micronucleus in the (A) prevention and (B) reversal models. We carried out the micronucleus analysis in lymphoblastic cells. Values are expressed as means and standard deviation. *Statistically different from control group (SAL), as determined by one-way analysis of variance and Tukey test (p < 0.05). Treatments with D-amphetamine + mood stabilizers did not differ from AMPH + saline in any of the models. AMPH = D-amphetamine; Li = lithium; SAL = saline; VPA = valproate.
Oxidative stress
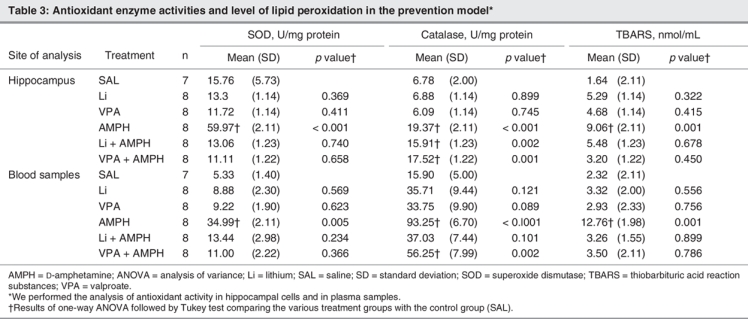
Table 2 and Table 3 show the results of oxidative stress markers in the reversal and the prevention models, respectively. We found that AMPH increased lipid peroxidation in the plasma (F5–40 = 18.91, p < 0.001 in the prevention model; F5–39 = 11.99, p < 0.001 in the reversal model) and in the hippocampus (F5–40 = 11.11, p < 0.001 in the prevention model; F5,39 = 9.81, p < 0.001 in the reversal model), as indicated by increased levels of TBARS in both models. In addition, lithium reversed and prevented hippocampal and peripheral AMPH-induced lipid peroxidation, whereas valproate prevented hippocampal lipid peroxidation, and reverted and prevented peripheral AMPH-induced lipid peroxidation. We found that AMPH lead to about a 3-fold increase of SOD activity in the hippocampus (F5–40 = 10.98, p < 0.001 in the prevention model; F5–39 = 18.45, p < 0.001 in the reversal model) and a 5-fold increase of SOD activity in the plasma (F5–40 = 18.99, p < 0.001 in the prevention model; F5–39 = 21.45, p < 0.001 in the reversal model) in both models. In the reversal model, lithium decreased SOD activity after AMPH exposure in plasma and hippocampal samples, whereas in the prevention model, lithium and valproate prevented the increase of SOD activity in both samples. Treatment with AMPH alone increased catalase activity in the hippocampus (F5–40 = 12.48, p < 0.001 in the prevention model; F5–39 = 19.49, p < 0.001 in the reversal model) and plasma (F5–40 = 15.61, p < 0.001 in the prevention model; F5–39 = 11.95, p < 0.001 in the reversal model). Valproate alone increased catalase and SOD activity in the hippocampus in the reversal model. Lithium and valproate, when administered before AMPH, increased catalase activity in the hippocampus; however, only valproate had the same effect in the plasma samples. Lithium and valproate were able to reverse AMPH-induced increase in catalase activity.
Table 2
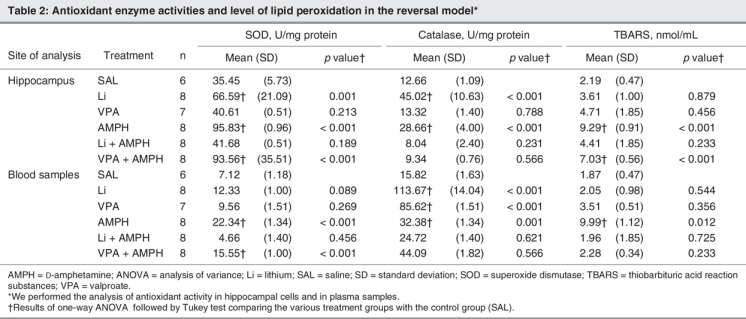
Table 3
Correlation between DNA damage and oxidative stress markers
We found a positive correlation between TBARS and transient DNA damage in plasma (rs = 0.62, no. of pairs = 29, p = 0.003) and in the hippocampus (rs = 0.4, no. of pairs = 29, p = 0.005) in the AMPH group, which indicates that AMPH-induced oxidative stress is associated with increased DNA damage. In addition, we found that the frequency of type-IV DNA damage (highest degree of damage) was positively correlated with the frequency of micronuclei in rats treated with AMPH alone (rs = 0.45, no. of pairs = 16, p = 0.034). We observed no significant correlation between TBARS levels and micronucleus frequency.
Discussion
Our results suggest that our earlier finding of increased oxidative stress in an animal model of mania,24 which we replicated in the present study, is associated with transient and permanent DNA damage. We also found that, along with the increased locomotor activity induced by AMPH, lithium and valproate were able to prevent and/or reverse DNA damage, lipid peroxidation and antioxidant enzyme changes. These results are consistent with recent reports indicating that the neuroprotective effects of lithium and valproate may be related to their antioxidant properties.24,32,33 However, neither lithium nor valproate were able to alter permanent DNA damage (as indicated by the micronuclei frequency), which suggests that these mood stabilizers may protect against transient, but not permanent, DNA damage. It is conceivable that the transient DNA damage reported herein may be a consequence of increased oxidative stress, because we found a positive correlation between the lipid peroxidation markers and the index of transient DNA damage.
Our findings are in line with recent evidence showing that mood stabilizers may regulate numerous pathways involved in the oxidative stress response.34,35 In our experiment, lithium and valproate were able to modulate the antioxidant enzymes, and lithium in particular prevented oxidative DNA damage. De Vasconcelos and colleagues13 showed that lithium decreased free-radical formation and increased the total antioxidant capacity in the hippocampus. Moreover, in a primary culture of cerebral cells from rats, treatment with lithium and valproate prevented glutamate-induced oxidative stress and DNA fragmentation32 and increased glutathione-S-transferase mRNA and protein levels.35 Compared with our previous findings, we found higher activity of catalase and SOD, possibly because in the present study we changed the method to allow the comet assay and micronucleus technique to be performed in the same sample as the oxidative stress markers.
The effects of chronic lithium and valproate treatments seem to take place through multiple pathways, including the induction of the survival signaling systems (e.g., activation of PI3K/Akt and MEK/ERK cascades). These signalling systems upregulate brain-derived neurotrophic factor (BDNF) expression;36,37 increase levels of neuroprotective and antiapoptotic protein bcl-2 in the frontal cortex, hippocampus and striatum of rats;38 inhibit glycogen synthase kinase-3β activity by phosphorilation in serine-9, an apoptotic promoter involved in neurodegenerative and affective diseases;39 and prevent lipid and protein peroxidation.24,26,35 Moreover, valproate increases the expression of heat shock protein 70 (Hsp70) and the binding of Hsp70 to apoptosis-proteases-activating factor 1,40 which prevents this factor from activating procaspase 9.39
Our results showed that valproate did not prevent transient DNA damage, which is in line with previous reports.41,42 For instance, valproate has been reported to function as a histone deacetylase inhibitor, leading to the acetylation of histone tails.42,43 Histone acetylation results in attenuation of the electrostatic charge interactions between histones and DNA, and has been associated with chromatin decondensation.43 Valproate-induced chromatin decondensation may lead to enhanced sensitivity of DNA to nucleases and increased DNA binding by intercalating agents,41 thereby increasing the access of macromolecules to DNA. In addition, it has been suggested that H2O2 can increase histone acetylation and decrease deacetylation.41 Thus an increase in ROS concentration and valproate intake may increase the susceptibility of the cell to DNA damage. Our data corroborate the notion that treatment with lithium may be more protective against oxidative stress and DNA damage than treatment with valproate. However, it is conceivable that valproate and lithium share part of their protective mechanisms. For instance, both lithium and valproate decreased AMPH-induced ROS generation, an effect that may be partly due to a decrease in dopamine levels;38,44 AMPH increases dopamine release, which may suffer auto-oxidation, thereby leading to the formation of highly reactive quinones.45 It has been demonstrated that reactive quinones may cause a direct inhibition of mitochondrial electron transport chain complexes46 and increase glutamate release,47 which may alter the redox status.
Along with the effects of mood stabilizers, the neurochemical effects of AMPH itself should be considered. Repeated exposure to AMPH is known to be associated with neurotoxicity,48,49 and our results showed increased lipid peroxidation, DNA damage and SOD activity in rats treated with AMPH. Importantly, SOD accelerates the dismutation of O2- to H2O2 and thus diminishes ONOO- formation from O2- as well as direct damage caused by O2-.6 Therefore, it seems that the increment of SOD activity after treatment with AMPH that we observed was a response against AMPH-induced oxidative DNA damage.
Limitations
Some limitations should be considered in the interpretation of our results. The animal model of mania using AMPH is restricted to mimic one aspect of BD, which is a far more complex disorder. However, the unique hallmark of bipolar illness is the presence of mania.1 Manic symptoms can be reproduced in normal volunteers after the administration of AMPH, and a number of case reports have suggested that the euphoric effects induced by AMPH in humans can be attenuated by the administration of lithium.50,51 Similar to the changes associated with mania, the administration of AMPH increases the heart rate, blood pressure and cortisol levels.50 Indeed, the magnitude of ventral striatal dopamine release induced by AMPH correlated positively with the hedonic response in humans.52
Another limitation of the present study is that we did not assess apoptosis and the efficiency of the repair system. However, the purpose of this study was to assess the correlation between oxidative stress and DNA damage, and further investigation of the fate of cells after DNA damage was beyond its scope.
Conclusion
The present study suggests that increased oxidative stress in an animal model of mania may be associated with both transient and permanent DNA damage. Further studies are needed to clarify whether this is a consequence of a dysfunctional repair system and whether this damage will ultimately result in neuronal apoptosis. More importantly, data from the treatment models indicate that lithium and, to a lesser extent, valproate exert antioxidant properties, which are likely to be associated with protective effects on DNA. However, the extent of the protection conferred by these drugs seems limited to transient DNA damage (comet assay). Once the cell had undergone micronuclei formation, (considered to be permanent damage) neither lithium nor valproate were able to prevent or reverse the damage. Further investigation is needed to ascertain whether these antioxidant/protective properties are correlated with the ability of these drugs to control the symptoms and negative outcomes in BD. We are tempted to speculate that new drugs targeted to lessen the oxidative stress burden in BD have the potential to move treatment beyond the control of symptoms, moving it closer to the preventive model already consolidated in other disorders.
Acknowledgments
This study was supported in part by the Brazilian Council, CNPQ and FAPERGS, University Federal of Rio Grande do Sul and University of Caxias do Sul and Hospital of Clínicas of Porto Alegre.
Footnotes
Contributors: Drs. Andreazza, Valvassori, Réus, Quevedo and Kapczinski designed the study. Drs. Andreazza, Stertz, Zanotto, Ribeiro, Giasson and Kapczinski acquired the data, which Drs. Andrezza, Kauer-Sant'Anna, Frey, Zanotto, Ribeiro, Salvador, Goncalves and Kapczinski analyzed. Drs. Andreazza, Kauer-Sant'Anna, Frey, Giasson and Kapczinski wrote the article, which Drs. Andreazza, Kauer-Sant'Anna, Frey, Stertz, Zanotto, Valvassori, Réus, Salvador, Quevedo, Goncalves and Kapczinski reviewed. All authors approved publication of the article.
Competing interests: None declared for Drs. Andreazza, Kauer-Sant'Anna, Frey, Stertz, Zanotto, Ribeiro, Giasson, Valvassori, Réus, Salvador, Quevedo and Goncalves. Dr. Kapczinski has received speakers fees and travel assistance from Abbott.
Correspondence to: Dr. F. Kapczinski, Bipolar Disorders Program, Centro de Pesquisas, Hospital de Clínicas de Porto Alegre, Rua Ramiro Barcelos, 2350, zip code 90035-003, Porto Alegre, RS, Brazil; fax 55 51 21018846; kapcz@terra.com.br
References
- 1.Belmaker RH. Bipolar disorder. N Engl J Med 2004;351:476-86. [DOI] [PubMed]
- 2.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997;349:1498-504. [DOI] [PubMed]
- 3.Angst F, Stassen HH, Clayton PJ, et al. Mortality of patients with mood disorders: follow-up over 34–38 years. J Affect Disord 2002;68:167-81. [DOI] [PubMed]
- 4.Rajkowska G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 2000;48:766-77. [DOI] [PubMed]
- 5.Carter CJ. Schizophrenia susceptibility genes converge on interlinked pathways related to glutamatergic transmission and long-term potentiation, oxidative stress and oligodendrocyte viability. Schizophr Res 2006;86:1-14. [DOI] [PubMed]
- 6.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 2001;18:685-716. [DOI] [PubMed]
- 7.Liu Q, Xie F, Rolston R, et al. Prevention and treatment of Alzheimer disease and aging: antioxidants. Mini Rev Med Chem 2007;7:171-80. [DOI] [PubMed]
- 8.Palmieri B, Sblendorio V. Oxidative stress detection: What for? Part I. Eur Rev Med Pharmacol Sci 2006;10:291-317. [PubMed]
- 9.Andreazza AC, Cassini C, Rosa AR, et al. Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res 2007;41:523-9. [DOI] [PubMed]
- 10.Frey BN, Andreazza AC, Kunz M, et al. Increased oxidative stress and DNA damage in bipolar disorder: a twin-case report. Prog Neuropsychopharmacol Biol Psychiatry 2007;31:283-5. [DOI] [PubMed]
- 11.Zecca L, Youdim MB, Riederer P, et al. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 2004;5:863-73. [DOI] [PubMed]
- 12.Cochrane CG. Mechanisms of oxidant injury of cells. Mol Aspects Med 1991;12:137-47. [DOI] [PubMed]
- 13.de Vasconcelos AP, Bouilleret V, Riban V, et al. Role of nitric oxide in cerebral blood flow changes during kainate seizures in mice: genetic and pharmacological approaches. Neurobiol Dis 2005;18:270-81. [DOI] [PubMed]
- 14.Maluf SW, Erdtmann B. Follow-up study of the genetic damage in lymphocytes of pharmacists and nurses handling antineoplastic drugs evaluated by cytokinesis-block micronuclei analysis and single cell gel electrophoresis assay. Mutat Res 2000;471:21-7. [DOI] [PubMed]
- 15.Fenech M, Morley AA. Cytokinesis-block micronucleus method in human lymphocytes: effect of in vivo ageing and low dose X-irradiation. Mutat Res 1986;161:193-8. [DOI] [PubMed]
- 16.Andreazza AC, Frey BN, Rombaldi F, et al. DNA damage in bipolar disorder. Psychiatry Res 2007;153:27-32. [DOI] [PubMed]
- 17.Simon NM, Smoller JW, McNamara KL, et al. Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry 2006;60:432-5. [DOI] [PubMed]
- 18.Calabrese V, Scapagnini G, Giuffrida Stella AM, et al. Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res 2001;26:739-64. [DOI] [PubMed]
- 19.Kato T. The role of mitochondrial dysfunction in bipolar disorder. Drug News Perspect 2006;19:597-602. [DOI] [PubMed]
- 20.Konradi C, Eaton M, MacDonald ML, et al. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 2004;61:300-8. [DOI] [PubMed]
- 21.Frey BN, Valvassori SS, Reus GZ, et al. Changes in antioxidant defense enzymes after d-amphetamine exposure: implications as an animal model of mania. Neurochem Res 2006;31:699-703. [DOI] [PubMed]
- 22.Frey BN, Martins MR, Petronilho FC, et al. Increased oxidative stress after repeated amphetamine exposure: possible relevance as a model of mania. Bipolar Disord 2006;8:275-80. [DOI] [PubMed]
- 23.Frey BN, Valvassori SS, Gomes KM, et al. Increased oxidative stress in submitochondrial particles after chronic amphetamine exposure. Brain Res 2006;1097:224-9. [DOI] [PubMed]
- 24.Frey BN, Valvassori SS, Reus GZ, et al. Effects of lithium and valproate on amphetamine-induced oxidative stress generation in an animal model of mania. J Psychiatry Neurosci 2006;31:326-32. [PMC free article] [PubMed]
- 25.Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Guide for the care and use of laboratory animals. National Academy Press. Washington: National Academy Press; 1996.
- 26.Tice RR, Agurell E, Anderson D, et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 2000;35:206-21. [DOI] [PubMed]
- 27.Collins A, Dusinska M, Franklin M, et al. Comet assay in human biomonitoring studies: reliability, validation, and applications. Environ Mol Mutagen 1997;30:139-46. [DOI] [PubMed]
- 28.Fenech M, Holland N, Chang WP, et al. The HUman MicroNucleus Project–An international collaborative study on the use of the micronucleus technique for measuring DNA damage in humans. Mutat Res 1999;428:271-83. [DOI] [PubMed]
- 29.Wills ED. Mechanisms of lipid peroxide formation in animal tissues. Biochem J 1966;99:667-76. [DOI] [PMC free article] [PubMed]
- 30.Bannister JV, Calabrese L. Assays for superoxide dismutase. Methods Biochem Anal 1987;32:279-312. [DOI] [PubMed]
- 31.Aebi H. Catalase in vitro. Methods Enzymol 1984;105:121-6. [DOI] [PubMed]
- 32.Shao L, Young LT, Wang JF. Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 2005;58:879-84. [DOI] [PubMed]
- 33.Brunello N. Mood stabilizers: protecting the brain. J Affect Disord 2004;79:S15-20. [DOI] [PubMed]
- 34.Manji HK, Moore GJ, Chen G. Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness. J Clin Psychiatry 2000;61(Suppl. 9):82-96. [PubMed]
- 35.Chuang DM, Chen RW, Chalecka-Franaszek E, et al. Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord 2002;4:129-36. [DOI] [PubMed]
- 36.Fukumoto T, Morinobu S, Okamoto Y, et al. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 2001;158:100-6. [DOI] [PubMed]
- 37.Hashimoto R, Hough C, Nakazawa T, et al. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 2002;80:589-97. [DOI] [PubMed]
- 38.Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem 1999;274:6039-42. [DOI] [PubMed]
- 39.Rowe MK, Chuang DM. Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev Mol Med 2004;6:1-18. [DOI] [PubMed]
- 40.Pan T, Li X, Xie W, et al. Valproic acid-mediated Hsp70 induction and anti-apoptotic neuroprotection in SH-SY5Y cells. FEBS Lett 2005;579:6716-20. [DOI] [PubMed]
- 41.Marchion DC, Bicaku E, Daud AI, et al. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res 2005;65:3815-22. [DOI] [PubMed]
- 42.Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001;20:6969-78. [DOI] [PMC free article] [PubMed]
- 43.Phiel CJ, Zhang F, Huang EY, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001;276:36734-41. [DOI] [PubMed]
- 44.Carli M, Morissette M, Hebert C, et al. Effects of a chronic lithium treatment on central dopamine neurotransporters. Biochem Pharmacol 1997;54:391-7. [DOI] [PubMed]
- 45.Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem 1999;73:1127-37. [DOI] [PubMed]
- 46.Burrows KB, Gudelsky G, Yamamoto BK. Rapid and transient inhibition of mitochondrial function following methamphetamine or 3,4-methylenedioxymethamphetamine administration. Eur J Pharmacol 2000;398:11-8. [DOI] [PubMed]
- 47.Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science 1989;243:398-400. [DOI] [PubMed]
- 48.Wagner GC, Preston K, Ricaurte GA, et al. Neurochemical similarities between d,l-cathinone and d-amphetamine. Drug Alcohol Depend 1982;9:279-84. [DOI] [PubMed]
- 49.Armstrong V, Reichel CM, Doti JF, et al. Repeated amphetamine treatment causes a persistent elevation of glial fibrillary acidic protein in the caudate-putamen. Eur J Pharmacol 2004;488:111-5. [DOI] [PubMed]
- 50.Jacobs D, Silverstone T. Dextroamphetamine-induced arousal in human subjects as a model for mania. Psychol Med 1986;16:323-9. [DOI] [PubMed]
- 51.Zacny JP, Bodker BK, de Wit H. Effects of setting on the subjective and behavioral effects of d-amphetamine in humans. Addict Behav 1992;17:27-33. [DOI] [PubMed]
- 52.Anand A, Verhoeff P, Seneca N, et al. Brain SPECT imaging of amphetamine-induced dopamine release in euthymic bipolar disorder patients. Am J Psychiatry 2000;157:1108-14. [DOI] [PubMed]