

Abstract
Loop 181-197 of human thymidylate synthase (hTS) populates two conformational states. In the first state, Cys195, a residue crucial for catalytic activity, is in the active site (active conformer); in the other conformation, it is about 10 Å away, outside the active site (inactive conformer). We have designed and expressed an hTS variant, R163K, in which the inactive conformation is destabilized. The activity of this mutant is 33% higher than that of wt hTS, suggesting that at least one-third of hTS populates the inactive conformer. Crystal structures of R163K in two different crystal forms, with six and two subunits per asymmetric part of the unit cells, have been determined. All subunits of this mutant are in the active conformation while wt hTS crystallizes as the inactive conformer in similar mother liquors. The structures show differences in the environment of catalytic Cys195, which correlate with Cys195 thiol reactivity, as judged by its oxidation state. Calculations show that the molecular electrostatic potential at Cys195 differs between the subunits of the dimer. One of the dimers is asymmetric with a phosphate ion bound in only one of the subunits. In the absence of the phosphate ion, that is in the inhibitor-free enzyme, the tip of loop 47-53 is about 11 Å away from the active site.
Thymidylate synthase (TS)1 catalyzes the reaction that converts the nucleotide deoxyuridylate (dUMP) into thymidylate (TMP) using 5,10-methylenetetrahydrofolate (mTHF) as a cosubstrate (1). TS is a dimer of identical subunits which generate asymmetry upon substrate/ligand binding (2, 3). Substrates bind to the active site in an ordered manner with dUMP binding prior to mTHF. The active site cysteine (Cys195 in human TS) attacks the 6-position of the pyrimidine base of the nucleotide resulting in the formation of a covalent bond between the enzyme and the nucleotide, thus activating the 5-position for subsequent covalent bond formation with the C-11 substituent of mTHF (reviewed in ref 4).
TS is the sole source of de novo thymidylate which is required for DNA synthesis. Cessation of this reaction not only halts cell replication but also leads to apoptosis of rapidly dividing cells, an effect named thymineless death (5). Inhibitors mimicking either the substrate or cosubstrate have been developed. Their binding leads to strained conformations of TS (6, 7).
TS is ubiquitous and highly conserved among species. Human TS (hTS) is different from bacterial TS's in three regions: the N-terminus is extended by 28-29 residues and there are two insertions of 12 and 8 residues at positions 117 and 146, respectively (8). hTS was originally crystallized in high ammonium sulfate conditions, and when its structure was compared to other TS enzymes, it was observed that loop 181-197 was rotated 180° (9). This new conformation, seen so far only in hTS, has the catalytic cysteine out of the active site and must be inactive. With loop 181-197 in the inactive conformation, one of the eukaryotic inserts, loop 107-128, is disordered. In solution, the active and inactive conformations are in equilibrium (10). Substrate or substrate analogue binding shifts the equilibrium toward the active conformer as hTS in an inhibitory complex with dUMP and raltitrexed (11) has loop 107-128 ordered and loop 181-197 in the active conformation. The equilibrium is also affected by the N-terminus as other structural studies of hTS with residues 7-29 deleted showed the active conformation (12). The positions of Trp182 are quite different in the active and inactive conformations, and this allowed Phan et al. (10) to monitor the equilibrium by intrinsic fluorescence studies. The addition of phosphate, which stabilizes the inactive conformer, shows an increase in hTS fluorescence. The addition of dUMP in the presence of phosphate quenches this increase (10).
In general, TS subunits show negative cooperativity for substrates or inhibitor binding. The ratios of ligand dissociation constants, Kd, for the two subunits depend strongly on the ligand studied and on the source of the enzyme (3). The only reported structure of the TS dimer with half of the sites filled by ligands is that of Pneumocystis carinii, for which the Kd ratio is about 8000 (13).
We report here the design and structural analyses of an hTS mutant in which the inactive conformer has been destabilized. The structures show differences between the environments of catalytic Cys195 residues in the dimer which correlate with their propensity to undergo oxidation. Molecular potential calculations link the structural differences with the protonation state of the thiol groups.
MATERIALS AND METHODS
Chemicals
Salts, β-mercaptoethanol, and polyethylene glycols (PEGs) were obtained from Sigma (St. Louis, MO). (6S)-5,6,7,8-tetrahydrofolic acid (H4folate) was prepared from folic acid and converted to (6R)-CH2H4folate as described previously (14). PDPA was obtained from Epsilon-Chimie (Brest, France).
Protein Expression and Crystallization
The R163K mutation of the pTS080 plasmid obtained from W. S. Dallas, Glaxo Wellcome, Research Triangle Park, NC (3), was produced using the QuickChange site-directed mutagenesis kit (Stratagene, LaJolla, CA) following the manufacturer's instructions. Mutagenic primers were purchased from Integrated DNA Technology (IDT, Coralville, IA). The sequence containing the correct mutation was confirmed by LiCor sequencing (The DNA Sequencing Laboratory, University of South Carolina, Columbia, SC). Plasmid containing the mutation was transformed into the Escherichia coli TX61− (thyA−) bacterial strain that does not produce its own thymidylate synthase enzyme (15).
R163K hTS was expressed and purified following the procedures previously developed for hTS (10) with only minor modifications. Briefly, cell-free extract was separated on a Q-Sepharose anion-exchange column using a 0-100% saturated KCl gradient. Active fractions were pooled, dialyzed, and separated on a Blue Sepharose CL-6B column using a similar gradient. Active fractions were again pooled, dialyzed, and separated a second time on the Q-Sepharose anion-exchange column using a similar gradient. Active fractions were then pooled, concentrated to 12 mg/mL, and analyzed for purity on an SDS-PAGE gel. R163K crystals were grown in low-salt conditions, 100 mM Tris, pH 9.0, 20 mM BME, 3 mM KH2PO4, and 10-20% PEG 4K, by hanging drop diffusion at 4 °C.
X-ray Diffraction Data Measurement and Processing
Crystals were transferred to artificial mother liquor containing 20% ethylene glycol and flash frozen in liquid N2 vapors. X-ray diffraction data were collected at the SER-CAT 22-ID or 22-BM beamlines, at APS, Argonne National Laboratory. The data were indexed and processed with the HKL software (16); processing parameters and statistics are summarized in Table 1.
Table 1.
Crystallographic Data and Refinement Statistics for R163K
| form 1 | form 2 | |
|---|---|---|
| X-ray source | APS SER-CAT ID | APS SER-CAT BM |
| wavelength (Å) | 1.00000 | 0.97243 |
| no. of frames high pass (low pass) | 300 (200) | 190 (n/a) |
| oscillation range (deg) | 0.5 (0.5) | 0.5 |
| crystal to detector distance (mm) | 380 (450) | 300 |
| temperature (K) | 100 | 100 |
| space group | P3221 | P3221 |
| unit cell dimensions | ||
| a, b (Å) | 123.74 | 120.74 |
| c (Å) | 284.77 | 129.77 |
| volume (Å3) | 2531628 | 1098404 |
| % solvent, Matthews coeff | 57, 3.0 | 67, 3.9 |
| mosaicity (deg) | 0.33 | 0.52 |
| resolution range (Å) (outer shell) | 50.0-2.7 (2.77-2.70) | 50.0-2.5 (2.59-2.50) |
| average redundancy | 7.1 | 4.7 |
| average I/σ(I) | 15.5 | 7.8 |
| total no. of reflections | 517709 | 142162 |
| no. of unique reflections | 72672 | 30034 |
| completeness (%) (50.0-3.0 Å) | 95.5 (95.1) | 70.4 (97.4) |
| total linear R-merge | 20.7 | 13.8 |
| R-value (%) | 27.8(Refmac5) | 26.8(CNS) |
| Rfree-value (%) | 27.8(Refmac5) | 26.8(CNS) |
| Ramachandran statistics | ||
| residues in most favored regions (%) | 83.1 | 86.6 |
| residues in additionally allowed regions (%) | 16.5 | 13.2 |
| residues in generously allowed regions (%) | 0.4 | 0.2 |
| residues in disallowed regions (%) | 0 | 0.0 |
| average B factors for subunits A-F | 30.5, 24.8, 37.5, 46.9, 68.2, 81.9 | 45.6, 56.3 |
Structure Determination and Refinement
The structure of R163K form 1 was solved by molecular replacement with the CNS software (17) using the hTS/dUMP/raltitrexed structure (PDB code 1HVY) (11) without ligands as the search model. The structure of form 2 was solved by molecular replacement using Amore (18) in the CCP4 suite (19) and the R163K structure as the search model. Structure rebuilding and subsequent refinements were performed using Turbo-Frodo software (20) and CNS, respectively. The final refinement of the form 1 structure was carried out with Refmac5 (19). Figures 1, 5, and 6 were prepared using Turbo-Frodo. Superpositions were obtained using the Lsqkab program in the CCP4 suite. Figure 4 was made using MOLSCRIPT (21) and Raster3D (22). Molecular electrostatic potentials were calculated using DelPhi (23).
Figure 1.

(A) Hydrogen bonding between Arg163 and loop 181-197 (from the other subunit) present in the hTS dimer in the inactive conformation. (B) Bond lengths in a model in which Arg163 was replaced by a lysine while loop 181-197 was kept unchanged.
Figure 5.

Stereoview of the active sites of the dimer in crystal form 2 with 2Fo - Fc electron density, contoured at the 1.0σ level, for subunits A (top) and B (middle). Superposition of subunits A (in yellow) and B (in blue) is shown in the bottom panel. The β-mercaptoethanol molecule is connected to the side chain of Cys195 by a disulfide bond in subunit B while it forms a noncovalent adduct in subunit A. The distances from Cys195 to Ser216 are also different. Different oxidation states of Cys195 reflect its propensity to oxidation.
Figure 6.

Stereoview of electron density, from a 2Fo - Fc map contoured at the 1.0σ level, for the active site of subunit B (top) and subunit A (middle) showing the positions of Cys195 with respect to Ser216. (A) Superposition of subunits A (in blue) and B in yellow (bottom).
Figure 4.
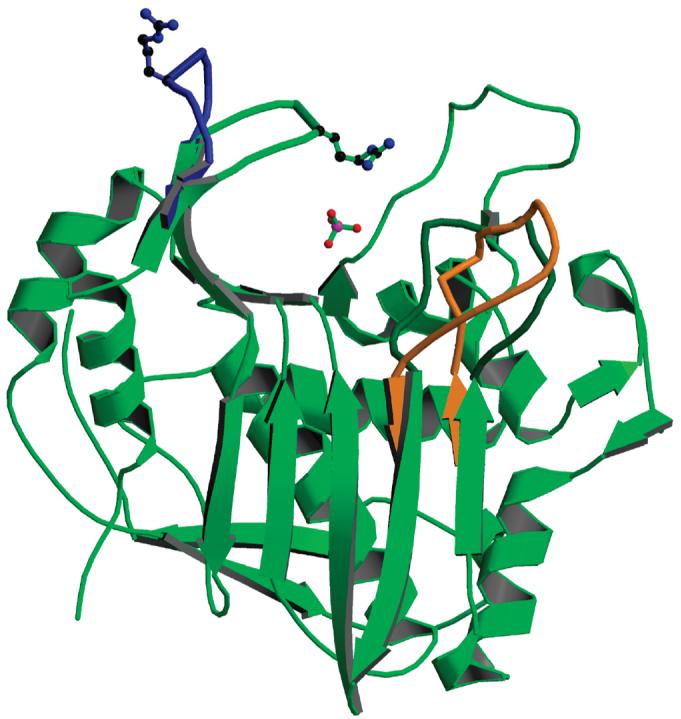
Subunit B, from the form 1 structure (in green), with superposed loop 45-56 (in blue) from subunit A of the same dimer. The side chain of Arg50 is shown for the two conformations. The phosphate ion is present in subunit B but not in subunit A; its position corresponds to the binding site of the phosphate of the dUMP substrate. Also shown is the position of loop 181-197 in the inactive conformation (in orange). Subunit A is the first crystallographic study of inhibitor-free hTS.
Activity Assays
Enzyme concentration was determined by measurement of absorbance at 280 nm, as described previously (24). Enzyme activity was determined spectrophotometrically as previously described (25). Measurements at 340 nM were made at 22 °C using a UV210 spectrophotometer with a temperature-controlled six-cell sample chamber. A typical assay uses 40 nM TS, 100 μM dUMP, and 150 μM mTHF in Morrison buffer [120 mM Tris, 60 mM 2-(N-morpholino)ethanesulfonic acid, 60 mM acetic acid, pH 7.2].
Fluorescence Studies
Intrinsic fluorescence of R163K was measured using an approach similar to that used in phosphate binding studies (26). The samples were excited at 295 nm, and the emission was scanned from 305 to 446 nm at a rate of 3 nm/s. A 2 mL sample containing 1 μM protein in buffer A was titrated by microliter additions of propane-1,3-bisphosphonate (PDPA) followed by mixing. The total volume added during titration was less than 5% of the final volume. All measurements were done in triplicate in order to eliminate errors due to nonhomogeneity of the sample and instrumental drift. The dissociation constant Kd was calculated with SigmaPlot (27) using the best fit to the function ΔF = ΔFmax/(Kb + [PDPA]), where ΔF is the fluorescence change for a given PDPA concentration.
RESULTS
Modeling
Analysis of the structure of hTS (PDB code 1YPV) (28) showed that the inactive conformation of loop 181-197 is stabilized by a pair of hydrogen bonds from the guanidinium group of Arg163 to the main chain carbonyls of Ala191 and Leu192 from the other subunit. The geometry of hydrogen bonds is very favorable with the N—H bonds being nearly collinear with the carbonyls' dipole moments (Figure 1). Arginine in position 163 is so far found only in human TS (8); in rodent TS enzymes the equivalent residue is a lysine, and murine TS (mTS) does not populate the inactive conformer (26). Modeling of lysine in position 163 showed that its side chain is too short to reach the carbonyls (Figure 1) and that even a conformational change that reduces the distance between residues 191-192 and Lys163 would not generate a favorable hydrogen-bonding geometry. Thus “murinization” of hTS through R163K mutation should destabilize the inactive conformation and shift the loop 181-197 conformational equilibrium toward the active conformation.
Protein Yields, Activity, and Fluorescence
The R163K variant was expressed with yields comparable to that of the wt-hTS. It has a higher activity than wt-hTS (133% of wt-hTS activity) and is somewhat less inhibited by PDPA, which is a potent inhibitor of wt-hTS that stabilizes the inactive conformer (26), as shown in Figure 2a. Wt-hTS showed a complex pattern of inhibition by PDPA: uncompetitive at low concentrations while mixed at higher PDPA concentrations (26). The uncompetitive region was interpreted as resulting from the inhibitor binding to the inactive conformer of hTS. In contrast, the R163K enzyme shows simple competitive inhibition as shown in Figure 2b. This is consistent with our postulate that R163K does not populate the inactive conformer and is supporting the previously proposed hypothesis that the uncompetitive/mixed inhibition of wt-hTS is related to the presence of the inactive conformer. The value of Ki calculated assuming the competitive model is 7.9 μM.
Figure 2.

(a) Activity of hTS and R163K hTS in the presence of varying concentrations of PDPA. Values above the bars are in reference to the wt-hTS activity while values in parentheses indicate R163K activity with reference to 100% activity of the mutant. The activity of wild-type hTS which is used as the reference was 1.29. One unit of enzyme activity is defined as the amount of enzyme required to synthesize 1 μmol of dTMP per minute. (b) Inhibition of R163K by PDPA versus increasing concentrations of mTHF presented as a Lineweaver-Burk plot shows competitive character of inhibition.
Fluorescence studies, presented in Figure 3, showed that PDPA has an effect on R163K similar to that on hTS. Since crystallography shows only one high-affinity phosphate ion binding site, which corresponds to the site in which the phosphate moiety of dUMP binds (vide infra), it is likely that PDPA binds in the active site of R163K. This is supported by the kinetic studies that showed competitive inhibition. The PDPA dissociation constant calculated from the data shown in Figure 3 is 2.1(0.3) μM, not far from the Ki value.
Figure 3.
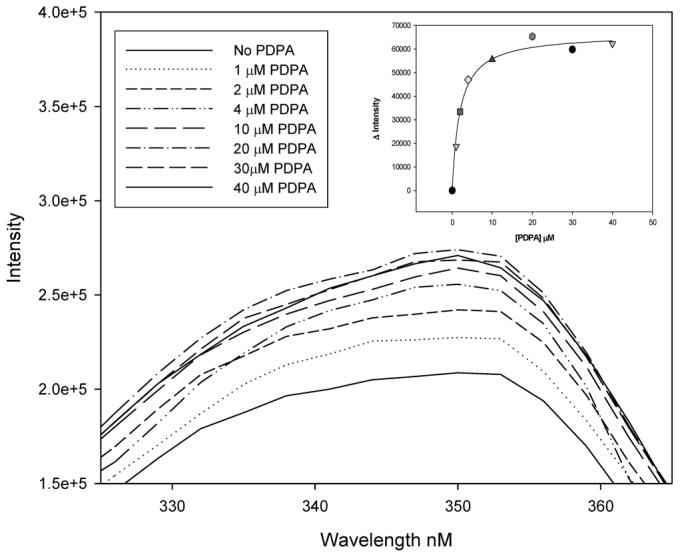
Intrinsic fluorescence of R163K upon titration with PDPA. Changes in peak fluorescence as a function of PDPA concentration are shown in the inset.
Crystallography
Crystallization efforts revealed that the R163K protein has a propensity for polymorphism. Crystals of two forms, 1 and 2, both trigonal, grew in the same drops, form 1 first and then 2-3 days later crystals of form 2 appeared. Form 1 contains two asymmetric dimers (in crystallographic general positions) and two on 2-fold crystallographic axes (six subunits per the asymmetric part of the unit cell). Form 2 has one asymmetric dimer in a crystallographic general position. Somewhat surprisingly, the forms have significantly different solvent content: 57% and 67%, respectively. A factor, which may have contributed to the polymorphism, is the progressing oxidation of protein thiols and formation of S,S-(2-hydroxyethyl)thiocysteines, presumably by reacting with 2-hydroxyethyl disulfide, the product of oxidation of BME. Indeed, one of Cys195 in form 2, which crystallized later, is oxidized while in form 1 cysteines are not oxidized.
Data collection for form 1 was somewhat challenging due to fairly large unit cell dimensions. R-merge was quite high, 20.7%, but with a redundancy of 7.1 the average Fs yielded R-factors in a range typical for a structure at this resolution. The subunits in form 1 have fairly different average B factors (Table 1), and this likely affects the precision of their coordinates. The lowest factors are for subunits A and B that form an asymmetric dimer which in general has excellent electron density in composite omit maps. Indeed, we were able to extend the previous best model by adding another N-terminal residue, Arg25, in subunit A.
Loop Conformation
In all of the independently observed eight subunits loop 181-197 is in the active conformation and loop 107-128 is ordered. The ammonium moieties of Lys163 side chains are in the solvent area and do not form contacts with protein atoms; typically they have poor electron density. The same is true about Arg163 in hTS in the active conformation. Loop 47-53 in subunit A from crystal form 1 has a different conformation than the one observed previously; the position of the Cα atom of Arg50 is shifted 11 Å in respect to that observed in subunit B and in previous models (Figure 4). Loop 47-53 does not form contacts with neighbors; thus it is not an artifact of crystal packing. Rather, all previously determined structures of hTS contain a phosphate/sulfate ion (or dUMP) in the active site, and the Arg50 side chain interacts with the phosphate ion. It is likely that this interaction stabilizes the loop position at the active site. Thus the conformation of subunit A is representative for phosphate-free hTS. The phosphate ion concentration in the crystals is about 3 mM, a value within the physiological range. The presence of a phosphate ion in only one subunit of the dimer indicates different binding constants and negative cooperativity. In crystal form 2 subunit B the density for this region is poor, suggesting that perhaps a fraction of the molecules populates this alternative conformation as well; the density for the phosphate is also fairly weak, suggesting a partial occupancy. Since enzymes in compact conformations tend to be easier to crystallize, the observation of the open conformation for loop 47-53 explains why TSs are more likely to crystallize in the presence of phosphate.
Loop 107-128 is disordered in the inactive conformation (9, 10, 28). In the active conformation this region has a single conformation stabilized by interactions with loop 181-197 (11, 12). This correlation between the loop conformation is confirmed in R163K as loop 107-128 is ordered in all subunits.
Active Site
Two types of Cys195 environment can be distinguished in structures of both forms; in the discussion below we use the distances observed in the dimer of form 2. In the first one (subunit A) the thiol is positioned at a distance of about 3.3 Å from the peptide oxygen of Ser216 and further (4.4 Å) from the guanidinium of Arg215; in the second (subunit B) the thiol is in the solvent area (4.5 Å from the oxygen of Ser216) and closer (4.0 Å) to the guanidinium of Arg215 (Figure 5). These different environments should lead to different pKa values for the Cys195 thiol. To put this analysis in more quantitative terms, the molecular electrostatic potential at Sγ of Cys195 is −225 kT/e in subunit A and −169 kT/e in subunit B. These different potentials lead to different propensity to form thiolate ions, the reactive form of cysteine. Indeed, the thiol of Cys195 in subunit B is modified and forms a disulfide bond with a molecule of BME (Figure 5) while the thiol in subunit A is not modified. Although the active sites in the dimer are different, there is no major conformational shift between the subunits of form 2. In the structure of crystal form 1 the dimers formed by subunits A and B and subunits E and F also have asymmetric environments of Cys195; the distances between the Sγ and O of Ser216 are 4.3, 3.3, 3.9, and 3.5, respectively. No evidence for thiol derivatization is present, indicating that the dimer asymmetry is causing different thiol reactivity not vice versa (Figure 6). Subunits C and D form dimers with subunits equivalent by 2-fold crystallographic symmetry.
DISCUSSION
The R163K variant has indeed stabilized the active conformation as predicted. Since there are eight independent observations, this cannot be an artifact of crystal environment but must reflect the situation in solution. The mutant is a catalyst 1.33 times better than hTS at the assay conditions. One interpretation of this is that the extent to which hTS populates the inactive conformation of loop 181-197 is approximately 1/3. This could be, however, more complicated since the substrate binding stabilizes the active conformer and the enzyme is quite slow, with a turnover rate of about 3 s−1 (25). Thus if the active/inactive conformational equilibrium is faster than catalysis, the stabilization of the active conformation would have a smaller effect on the rate, and the observed population of the inactive conformer of 1/3 should be considered a lower limit. It should be indicated that the replaced residue, Arg163, is not considered to be involved in catalysis and is poorly conserved; for instance, in E. coli TS the corresponding residue is a threonine.
Depopulating the inactive conformer of loop 181-197 by R163K mutation facilitated the enzyme crystallization at a low phosphate/sulfate concentration and helped to observe the conformational state of loop 47-53 which has not been reported before and which likely represents the conformation of inhibitor-free hTS. All previously reported structures have the phosphate binding site occupied by a phosphate ion or a sulfate ion or the phosphate moiety of a nucleotide. Interactions with these ions induce loop 47-53 closure.
Even in the absence of the major conformational differences, minor structural differences between the subunits lead to dimer asymmetry and differences between the environments of the active site cysteines: one forming a short contact with a peptide carbonyl, the other pointing into solvent. Upon longer exposure to oxygen during crystallization the exposed thiol of Cys195 reacts with oxidated exogenous thiols, forming an asymmetric product in which only one subunit is modified. To observe asymmetry in crystallographic studies there has to be a correlation between crystal environment and asymmetry. This always raises a possibility that the asymmetry is an artifact of crystal packing. The observation of three asymmetric dimers of R163K in different crystal environments, each with Sγ Cys195 close to Ser216 carbonyl in one subunit and pointing to solvent in the other, strongly suggests that this asymmetry is a property of TS and not an artifact. Danenberg and co-workers observed that titration of Lactobacillus casei TS with thiol reagents leads to the modification of only one cysteine per dimer and proposed structural asymmetry of the dimer (2). Thus asymmetric reactivity of the catalytic cysteines in the dimer is not limited to the human enzyme but appears to be a conserved property of TS. It may be thought that some conformational states populated by TS represent situations that protect the catalytic cysteine from oxidative events. Indeed, asymmetric Cys195 oxidation during the crystallization process may reflect an arrangement in which one-half of the cysteines is “stashed away for later use”. It was proposed that substrate binding promotes the deprotonation of Cys195 thiols, the main factor in the cysteine reactivity (29). E. coli TS is a half-the-sites activity enzyme (30), and it is likely that hTS is as well. It may be speculated that the partial protection of catalytic thiol is one of the properties generated by this phenomenon.
Another possibility is that the inactive conformer is involved in hTS protein binding to a regulatory region of mRNA molecules coding for hTS and preventing ribosome attachment. This was proposed to be a feedback mechanism regulating translation and, consequently, intracellular protein levels. The inactive conformer has three to four phosphate ion binding sites which appear to have a suitable geometry for interacting with phosphate moieties of mRNA, while the active conformer has only one phosphate ion binding site (10, 31).
Inhibition of hTS by stabilization of the inactive conformer of loop 181-197 rather than by blocking the active site was proposed (9). Inhibitors functioning in such mode may have an advantage of not blocking the autoregulatory TS binding to its own mRNA. This is considered one of the mechanisms leading to resistance by increasing hTS levels in tumors of patients treated with presently used active site-directed hTS inhibitors which prevent mRNA binding. The R163K variant of hTS may be used as a tool in the search for allosteric inhibitors since it should be less sensitive to these inhibitors than hTS. We observed this effect in R163K interactions with PDPA.
ACKNOWLEDGMENT
We thank Sangita Koli for help with constructing the expression system for R163K and Dr. Sondra H. Berger for helpful discussions.
This work was supported by NIH Grant CA 76560. Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Abbreviations: TS, thymidylate synthase; hTS, human TS; FdUMP, 5-fluoro-2′-deoxyuridine monophosphate; dUMP, 2′-deoxyuridine 5′-monophosphate; dTMP, 2′-deoxythymidine 5′-monophosphate; CH2H4folate or mTHF, 5, 10-methylenetetrahydrofolate; EDTA, ethyl-enediaminetetraacetic acid; BME, β-mercaptoethanol; PDPA, 1,3-propanediphosphonic acid; PDB, Protein Data Bank; PEG, polyethylene glycol.
The PDB files with atomic coordinates of R163K structures have been deposited in the Protein Data Bank as entries 2RDA (form 1) and 2RD8 (form 2).
REFERENCES
- 1.Humphreys GK, Greenberg DM. Studies on the conversion of deoxyuridylic acid to thymidylic acid by a soluble extract from rat thymus. Arch. Biochem. Biophys. 1958;78:275–287. doi: 10.1016/0003-9861(58)90351-5. [DOI] [PubMed] [Google Scholar]
- 2.Danenberg KD, Danenberg PV. Evidence for a sequential interaction of the subunits of thymidylate synthase. J. Biol. Chem. 1979;254:4345–4348. [PubMed] [Google Scholar]
- 3.Dev IK, Dallas WS, Ferone R, Hanlon M, McKee DD, Yates BB. Mode of binding of folate analogs to thymidylate synthase. Evidence for two asymmetric but interactive substrate binding sites. J. Biol. Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 4.Stroud RM, Finer-Moore JS. Conformational dynamics along an enzymatic reaction pathway: thymidylate synthase, “the movie”. Biochemistry. 2003;42:239–247. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- 5.Houghton PJ. Thymineless death. In: Jackman AL, editor. Antifolate drugs in cancer chemotherapy. Humana Press Inc.; Totowa, NJ: 1999. pp. 423–436. [Google Scholar]
- 6.Matthews DA, Villafranca JE, Janson CA, Smith WW, Welsh K, Freer S. Stereochemical mechanism of action for thymidylate synthase based on the X-ray structure of the covalent inhibitory ternary complex with 5-fluoro-2′-deoxyuridylate and 5,10-methylenetetrahydrofolate. J. Mol. Biol. 1990;214:937–948. doi: 10.1016/0022-2836(90)90347-O. [DOI] [PubMed] [Google Scholar]
- 7.Montfort WR, Weichsel A. Thymidylate synthase: structure, inhibition, and strained conformations during catalysis. Pharmacol. Ther. 1997;76:29–43. doi: 10.1016/s0163-7258(97)00099-5. [DOI] [PubMed] [Google Scholar]
- 8.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 9.Schiffer CA, Clifton IJ, Davisson VJ, Santi DV, Stroud RM. Crystal structure of thymidylate synthase: a structural mechanism for guiding substrates into the active site. Biochemistry. 1995;34:16279–16287. doi: 10.1021/bi00050a007. [DOI] [PubMed] [Google Scholar]
- 10.Phan J, Steadman DJ, Koli S, Ding WC, Minor W, Dunlap RB, Berger SH, Lebioda L. Structure of human thymidylate synthase suggests advantages of chemotherapy with noncompetitive inhibitors. J. Biol. Chem. 2001;276:14170–14177. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 11.Phan J, Koli S, Minor W, Dunlap RB, Berger SH, Lebioda L. Human thymidylate synthase is in the closed conformation when complexed with dUMP and raltitrexed, an antifolate drug. Biochemistry. 2001;40:1897–1902. doi: 10.1021/bi002413i. [DOI] [PubMed] [Google Scholar]
- 12.Almog R, Waddling CA, Maley F, Maley GF, Van Roey P. Crystal structure of a deletion mutant of human thymidylate synthase Δ(7-29) and its ternary complex with Tomudex and dUMP. Protein Sci. 2001;10:988–996. doi: 10.1110/ps.47601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson AC, O'Neil RH, DeLano WL, Stroud RM. The structural mechanism for half-the-sites reactivity in an enzyme, thymidylate synthase, involves a relay of changes between subunits. Biochemistry. 1999;38:13829–13836. doi: 10.1021/bi991610i. [DOI] [PubMed] [Google Scholar]
- 14.Steadman DJ, Zhao P-S, Spencer HT, Dunlap RB, Berger SH. A structural role for glutamine 214 in human thymidylate synthase. Biochemistry. 1998;37:7089–7095. doi: 10.1021/bi9725428. [DOI] [PubMed] [Google Scholar]
- 15.Dev IK, Yates BB, Leong J, Dallas WS. Functional role of cysteine-146 in Escherichia coli thymidylate synthase. Proc. Natl. Acad. Sci. U.S.A. 1988;85:1472–1476. doi: 10.1073/pnas.85.5.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 18.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. 1994;D50:157–163. [Google Scholar]
- 19.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Roussel A, Cambillau C. “Turbo Frodo” Silicon Graphics Geometry Partners Directory, Silicon Graphics. Vol. 86. Mountain View, CA: 1991. [Google Scholar]
- 21.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 22.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 23.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 2001;105:6507–6514. [Google Scholar]
- 24.Zapf JW, Weir MS, Emerick V, Villafranca JE, Dunlap RB. Substitution of glutamine for glutamic acid-58 in Escherichia coli thymidylate synthase results in pronounced decreases in catalytic activity and ligand binding. Biochemistry. 1993;32:9274–9281. doi: 10.1021/bi00087a003. [DOI] [PubMed] [Google Scholar]
- 25.Steadman DJ, Spencer HT, Dunlap RB, Berger SH. Substitution at residue 214 of human thymidylate synthase alters nucleotide binding and isomerization of ligand-protein complexes. Biochemistry. 1999;38:5582–5587. doi: 10.1021/bi982910n. [DOI] [PubMed] [Google Scholar]
- 26.Lovelace LL, Gibson LM, Lebioda L. Cooperative inhibition of human thymidylate synthase by mixtures of active site binding and allosteric inhibitors. Biochemistry. 2007;46:2823–2830. doi: 10.1021/bi061309j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SigmaPlot. (version 9.0.1) www.systat.com/products/SigmaPlot/
- 28.Lovelace LL, Minor W, Lebioda L. Structure of human thymidylate synthase under low-salt conditions. Acta Crystallogr. 2005;D61:622–627. doi: 10.1107/S0907444905005895. [DOI] [PubMed] [Google Scholar]
- 29.Phan J, Mahdavian E, Nivens C, Minor W, Berger SH, Spencer HT, Dunlap RB, Lebioda L. Catalytic cysteine of TS is activated upon substrate binding. Biochemistry. 2000;39:6969–6978. doi: 10.1021/bi000367g. [DOI] [PubMed] [Google Scholar]
- 30.Maley F, Pedersen-Lane J, Changchien L. Complete restoration of activity to inactive mutants of Escherichia coli thymidylate synthase: evidence that E. coli thymidylate synthase is a half-the-sites activity enzyme. Biochemistry. 1995;34:1469–1474. doi: 10.1021/bi00005a001. [DOI] [PubMed] [Google Scholar]
- 31.Berger SH, Berger FG, Lebioda L. Effects of ligand binding and conformational switching on intracellular stability of human thymidylate synthase. Biochim. Biophys. Acta. 2004;1696:15–22. doi: 10.1016/j.bbapap.2003.09.005. [DOI] [PubMed] [Google Scholar]