

Abstract
Humanin (HN) is an anti-apoptotic peptide that suppresses neuronal cell death induced by Alzheimer's disease, prion protein fragments, and serum deprivation. Recently, we demonstrated that Gly14-HN (HNG), a variant of HN in which the 14th amino acid serine is replaced with glycine, can decrease apoptotic neuronal death and reduce infarct volume in a focal cerebral ischemia/reperfusion mouse model. In this study, we postulate that the mechanism of HNG's neuroprotective effect is mediated by the PI3K/Akt pathway. Oxygen-glucose deprivation (OGD) was performed in cultured mouse primary cortical neurons for 60 min. The effect of HNG and PI3K/Akt inhibitors on OGD-induced cell death was examined at 24 h after reperfusion. HNG increased cell viability after OGD in primary cortical neurons, whereas the PI3K/Akt inhibitors wortmannin and Akti-1/2 attenuated the protective effect of HNG. HNG rapidly increased Akt phosphorylation, an effect that was inhibited by wortmannin and Akti-1/2. Mouse brains were injected intraventricularly with HNG before being subjected to middle cerebral artery occlusion (MCAO) for 75 min followed by 24 h reperfusion. HNG treatment significantly elevated p-Akt levels after cerebral I/R injury and decreased infarct volume. The protective effect of HNG on infarct size was attenuated by wortmannin and Akti-1/2. Taken as a whole, these results suggest that PI3K/Akt activation mediates HNG's protective effect against hypoxia/ischemia reperfusion injury.
Keywords: Humanin, PI3K/Akt, OGD, cortical neuron, MCAO
1. Introduction
Humanin (HN) is a recently identified 24-amino acid anti-apoptotic peptide that is best known for its ability to suppress neuronal cell death induced by Alzheimer's disease-related insults, such as amyloid beta (Aβ) toxicity (Tajima et al., 2002). Recent studies have shown that HN not only has neuroprotective effects on Aβ toxicity but also inhibits apoptosis in many different diseases. HN and a highly potent HN variant, Gly14-HN (HNG), prevent apoptotic cell death induced by soluble prion protein fragments in rat cortical neurons (Sponne et al., 2004). HN can reverse memory impairments induced by different toxic agents in vivo (Mamiya et al., 2001; Krejcova et al., 2004). We recently showed that HNG protects against cerebral ischemia/reperfusion (I/R) injury by inhibiting apoptotic cell death, reducing infarct volume, and decreasing neurological deficits in mice (Xu et al., 2006).
Despite the above studies, the protective mechanisms and signaling pathways of HN are not clear. Most importantly, the putative HN receptor on the cell membrane has not been cloned or characterized (Hashimoto et al., 2001). Ying et al. (2004) reported that HN binds to a human G protein-coupled formylpeptide receptor-like-1 (FPRL1), which induces chemotaxis of mononuclear phagocytes and prevents Aβ-induced apoptotic cell death by competitively inhibiting Aβ binding to FPRL1. Harada et al. (2004) also published findings that supported this theory. However, Hashimoto et al. (2005) proposed that the HN receptor might belong to a tyrosine kinase receptor family since a tyrosine kinase inhibitor (genistein) attenuates the neuroprotective activity of HNG against Aβ toxicity in F11 neuronal hybrid cells. These investigators also found that HN-mediated protection is completely blocked by the expression of a dominant-negative STAT3, suggesting that STAT3 is involved in HN's protective mechanism. Our previous studies demonstrated that HNG exerts its neuroprotective effects on cerebral ischemia injury by inhibiting the activation of the extracellular signal regulated kinase (ERK), indicating that HNG's protective effect is at least partially mediated by the tyrosine kinase Ras/MEK/ERK pathway (Xu et al., 2006). Finally, several studies reported that HN inhibits apoptosis by binding intracellularly to Bax and Bid, thus blocking apoptosis independently of receptors (Guo et al., 2003; Zhai et al., 2005).
The PI3K/Akt pathway is a central mediator in signal transduction pathways involved in cell growth, cell survival, and metabolism (Brazil, Yang and Hemmings, 2004). The neuroprotective role of the PI3K/Akt pathway in cerebral ischemia has been widely studied (Janelidze et. al., 2001; Noshita et al., 2001; Shibata et al., 2002). In models of cerebral ischemia, Akt phosphorylation increases at 1 h and 4 h after I/R, but decreases significantly 24 h after reperfusion (Janelidze et al., 2001, Shibata et al., 2002). Akt blocks apoptotic stimuli by inactivating pro-apoptotic proteins such as Bad, caspase-9, and glycogen synthase kinase-3β. Akt also exerts anti-apoptotic effects by activating endothelial nitric oxide synthase (eNOS) (Brazil, Yang and Hemmings, 2004)
Given the neuroprotective role of the PI3K/Akt pathway in cerebral ischemia, we hypothesized that HN's neuroprotective effect involves the activation of the PI3K/Akt pathway after cerebral I/R injury. In this study, we provide evidence that this is indeed the case by using an in vitro oxygen-glucose deprivation (OGD) cell model and a focal cerebral I/R mouse model.
2. Results
2.1. HNG protects against OGD-induced neuronal death
In our study, HNG (0.2 μM) treatment alone had no effect on the viability of the mouse cortical neurons. After 60 min of OGD and 24 h reperfusion, cell viability decreased to 52.9 ± 0.93% of the control. HNG treatment significantly reduced cell death induced by OGD (69.1 ± 0.77% of the control) (Figure 1, P<0.05).
Figure 1.

The effects of HNG and the PI3K/Akt inhibitors wortmannin and Akti-1/2 on OGD-induced cell death in primary cortical neurons. OGD experiments were conducted in cultured mouse cortical neurons at DIV 10. Primary cortical neurons were incubated with 0.2 μM HNG, 0.1 μM wortmannin (W), or 1 μM Akti-1/2 (Akti) in glucose-free HBSS in a hypoxia chamber for 60 min. The plate was then restored to normoxic conditions. Cell viability was assessed by the MTS assay at 24 h of reperfusion. Control culture plates in the presence of HNG, wortmannin, or Akti-1/2 were exposed to oxygenated HBSS containing 5.5 mM glucose in normoxic condition. Bars represent mean ± SEM of 8 samples. * P<0.01 versus the OGD group; #, versus the OGD+HNG group.
2.2. PI3K/Akt inhibitors attenuate HNG's neuroprotective effects
To examine whether the protective effects of HNG are mediated by the PI3K/Akt pathway, OGD-treated cortical neurons were incubated with HNG and a PI3K inhibitor wortmannin (1 μM) or an Akt inhibitor Akti-1/2 (1 μM). In our study, administration of wortmannin or Akti-1/2 alone had no effect on cell survival after OGD. Cells exposed to either HNG and wortmannin or HNG and Akti-1/2 had significantly decreased cell viability compared with the HNG-treated group (P<0.05) (Figure 1).
2.3. HNG activates Akt in primary cortical neurons
We next investigated whether the addition of HNG activates Akt. Treatment of mouse cortical neurons with 0.2 μM HNG led to a time-dependent phosphorylation of Akt, with the maximal level occurring at 10 min (Figure 2A). Wortmannin and Akti-1/2 were used to block PI3K and Akt pathways, respectively. Mouse cortical neurons were pretreated with 100 nM wortmannin for 15 min or 1 μM Akti-1/2 for 30 min before incubating with 0.2 μM HNG for 10 min. Quantification was performed with densitometric analysis of p-Akt and total Akt. As shown in Figure 2 B,C, HNG upregulated p-Akt levels by 1.8-fold (P<0.05); this effect was attenuated with wortmannin or Akti-1/2. A lower concentration of wortmannin was chosen because wortmannin at 1 μM completely blunted p-Akt levels and made densitometric analysis impossible.
Figure 2.
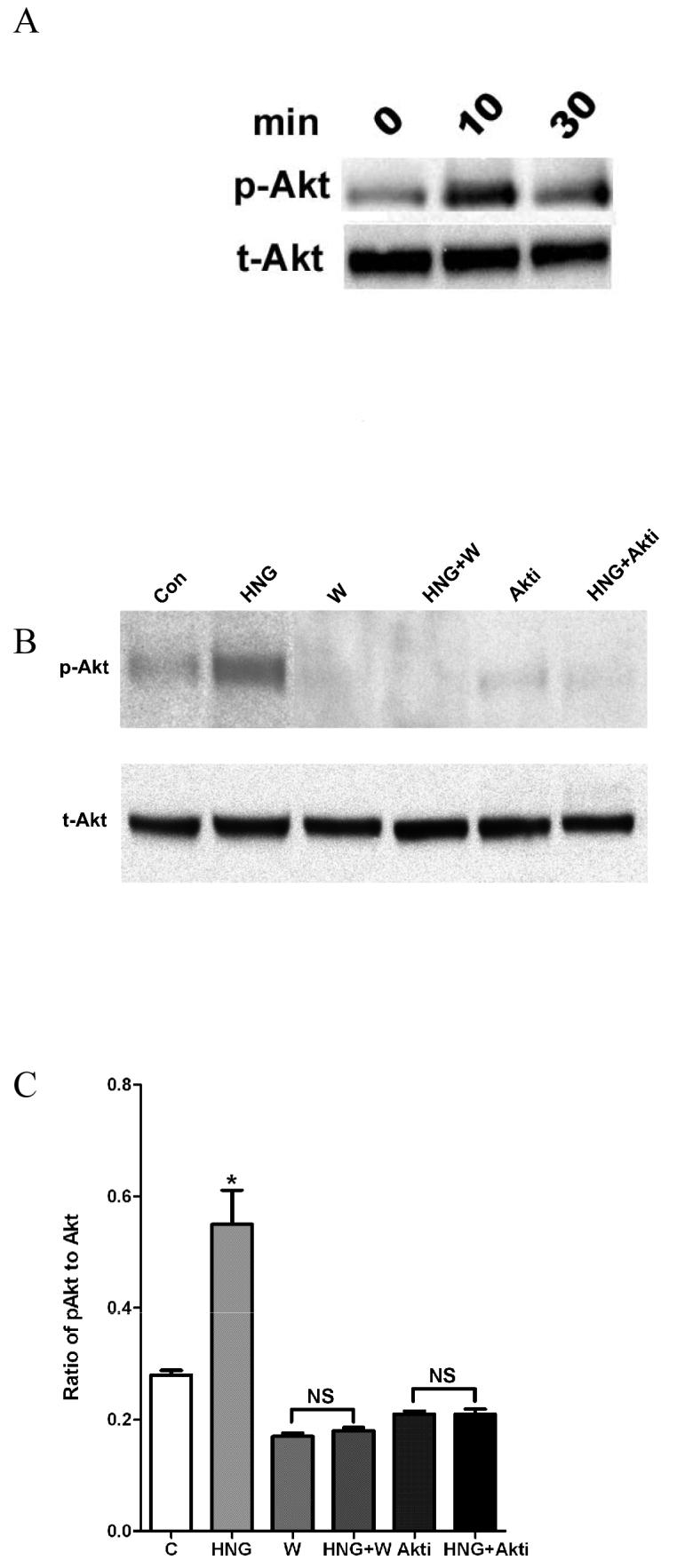
The effect of inhibitors on p-Akt in primary cortical neurons. Primary mouse neurons were treated with 0.2 μM HNG for 10 min or 30 min. Western blot analyses were carried out with 30 μg of homogenate using antibodies against p-Akt (Ser473). A duplicate blot was probed with antibodies against total Akt (A). In B, neurons were pretreated with 0.1 μM wortmannin for 15 min or 1 μM Akti-1/2 for 30 min before treatment with 0.2 μM HNG for 10 min. Total Akt and p-Akt levels were determined by Western blot analysis. Bar graph shows a densitometric analysis of the relative intensity of p-Akt, normalized against total Akt (C). Data represent means ± SEM from 4 samples. *P<0.05 versus control cells.
2.4. HNG activates Akt in vivo
Given the protective effect of HNG in vitro, we tested the effect of HNG in an in vivo mouse model of middle cerebral artery occlusion. As shown in our previous study (Xu et al, 2006), mouse brains subjected to 75 min of ischemia experienced greater than 50% infarction 24 h after ischemia/reperfusion. The heterogeneity of populations of live and dead cells hampers the interpretation of p-Akt result. Therefore, Western blot analysis was performed after a period of 30 min of ischemia and 6 h of reperfusion in the absence of Cell death. HNG treatment increased the p-Akt level by two-fold compared with the saline-treated group (P<0.05, Figure 3).
Figure 3.

The effect of HNG on p-Akt level after cerebral I/R injury. Mice were treated with HNG (0.1 μg, i.c.v.) or a same volume of saline 30 min before ischemia. After 30 min of ischemia and 6 h of reperfusion, brains were removed and the ischemic hemispheres were used for Western blot analysis. (A) Representative photographs of p-Akt and total Akt. (B) Quantitative analysis of the ratio of p-Akt to total Akt after cerebral ischemia in saline (NS) group and HNG group. Bars represent mean ± SEM of 3 brains. *P<0.01 versus NS group.
2.5. Wortmannin reduces the protection of HNG on cerebral infarct volume
We investigated whether the protective effect of HNG on cerebral I/R injury could be blocked by wortmannin, a PI3K inhibitor. The infarct size of wortmannin-treated mice (56.6 ± 2.0%) was comparable to that of vehicle-treated control mice (59.3 ± 2.6%, P>0.05, Figure 4C). The infarct size in the HNG-treated group was significantly decreased compared to other groups (29.2 ± 1.1%, P<0.01 for all other groups). The combination of HNG and wortmannin partially blocked the protective effect of HNG on infarct size (44.2 ± 1.3%).
Figure 4.

The effects of PI3K inhibitor wortmannin on neuroprotection of HNG on cerebral I/R injury. Mice were pretreated with wortmannin (43 ng, i.c.v ) or vehicle (DMSO) 60 min before ischemia. HNG (0.1 μg, i.c.v.) or saline was administered 30 min before ischemia. After 75 min of ischemia and 24 h of reperfusion, cerebral infarct volume was determined by TTC staining. Quantitative analysis of cerebral infarct volume in four groups was shown. Bars represent mean ± SEM of 6-7 brains. *P<0.01; NS, not significant; Con, control group.
2.6. Akti-1/2 reduces the protection of HNG on cerebral infarct volume
We next examined whether the protective effect of HNG on cerebral I/R injury could be blocked by an Akt inhibitor, Akti-1/2 (Cheng et al., 2005). Akti-1/2 alone had no effect after cerebral I/R compared with the vehicle-treated I/R control group (56.9 ± 2.1% vs. 59.3 ± 2.6%, respectively; P>0.05, Figure 5). Infarct volume was significantly lower in the HNG and Akti-1/2-treated I/R groups, compared with the HNG-treated I/R group (29.2 ± 1.1%, P<0.01). The combination of HNG and Akti-1/2 partially blocked the protective effect of HNG on infarct size (47.0 ± 2.0%).
Figure 5.

The effect of Akti-1/2 on neuroprotection of HNG on cerebral I/R injury. Mice were pretreated with Akti-1/2 (55 ng, i.c.v.) or vehicle (DMSO) 60 min before ischemia. HNG (0.1 μg, i.c.v.) or saline was administered 30 min before ischemia. After 75 min of ischemia and 24 h of reperfusion, cerebral infarct volume was determined by TTC staining. Quantitative analysis of cerebral infarct volume in four groups was shown. Bars represent mean ± SEM of 6-7 brains. *P<0.01; NS, not significant; Con, control group.
3. Discussion
This study demonstrated for the first time that HNG protects against hypoxia/ischemia both in vitro and in vivo. The study also provides evidence that the mechanism of this protective effect is mediated by the PI3K/Akt pathway. First, the protective effect of HNG in OGD-treated neurons was attenuated in the presence of wortmannin and Akti-1/2 (Figure 1). Second, HNG significantly increased p-Akt levels in cortical neurons, an increase that was abolished by both wortmannin and Akti-1/2 (Figure 2). Finally, wortmannin and Akti-1/2 attenuated HNG's protective effect on infarct volume (Figures 4, 5).
Activation of Akt has been shown to play an important role in neuronal survival after cerebral I/R injury. One study showed that transgenic mice overexpressing Akt1 had 35% reduced infarct volume after I/R (Ohba et al, 2004). Recently, ischemic post-conditioning by an episode of 10 min of ischemia followed by 10 min of reperfusion has been demonstrated to reduce infarct volume by 50%; the neuroprotective effect is thought to be mediated by the activation of Akt (Pignataro et al, 2008). A number of neuroprotectants, including VEGF (Kaya et al., 2005), GDNF (Jin et al., 2003), and erythropoietin (Kilic et al., 2005), exert their protective effect through the PI3K/Akt pathway. Our study provides evidence that HNG's protective effect also relies on the activation of the PI3K/Akt pathway, which agrees with the results of previous studies.
In the initial step of Akt activation, membrane-bound receptor tyrosine kinase (RTK) is phosphorylated. RTK phosphorylation activates PI3K, which generates phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). 3-Phosphoinositide-dependent protein kinase-1 (PDK1) in turn phosphorylates Akt at Ser 473 or Thr 308. Activated Akt blocks apoptosis by inactivating several targets, including Bad, glycogen synthase kinase-3β, forkhead transcription factors, or caspase-9 (Zhao et al., 2006 for review). In our study, wortmannin and Akti-1/2 lead to an attenuation of HNG-mediated protection, indicating that PI3K/Akt pathway plays a role in HNG's protection. One issue that was not addressed in this study is whether p-Akt is inhibited completely by wortmannin and Akti-1/2 in vivo, although we have shown complete blockade in vitro (Figure 2). It possible that wortmannin and Akti-1/2 only partially reduce Akt phosphorylation in vivo, although it is also possible that HNG's protection is mediated by multiple signaling pathways. This question will need to be addressed in future studies.
Hashimoto et al. (2005) demonstrated that HN can inhibit cell death induced by ASK1/JNK (apoptosis signal-regulating kinase1/c-Jun N-terminal kinase). Wang et al. (2005) found that HN delays serum deprivation-induced apoptosis in K562 cells by downregulating the expression of p38 MAP kinase. Our previous study demonstrated that HNG had no effect on ischemia-induced p38 MAPK activation in vivo (Xu et al., 2006). A recent study indicated that the Jak2/STAT3 pathway is involved in the protective mechanism of HN against Aβ toxicity (Hashimoto et al., 2005). Finally, our previous study showed that HNG can decrease phospho-ERK levels in a focal cerebral I/R model (Xu et al., 2006). In the current study, we found that wortmannin did not change the effect of HNG on ERK activation (data not shown), indirectly supporting the idea of multiple signaling pathways. The full characterization of these other signaling pathways awaits further investigation.
One issue not addressed by the current study is whether HN acts via a receptor or whether it acts independently of a receptor on an intracellular level. The Jak2/STAT3 pathway plays an important role in cerebral ischemia injury (Satriotomo et al., 2006). The possible involvement of this pathway in HNG's neuroprotective effect warrants further investigation. Investigators have argued that both the G protein-coupled receptor FPRL1/FPR2 and tyrosine kinase receptor are related to HN's function (Hashimoto et al., 2001; Ying et al., 2004; Harada et al., 2004; Hashimoto et al., 2005). It is possible that there is a tyrosine kinase in the putative HN receptor that activates the PI3K/Akt pathway. The resolution of this question will require the cloning of the HN receptor.
We have shown previously that intraperitoneal administration of HNG after ischemia/reperfusion reduces infarct volume (Xu et al, 2006), which suggests that HNG could be used as a neuroprotective agent in the treatment of stroke. A complete understanding of the mechanisms of HNG's protective effects will greatly facilitate the realization of its clinical potential.
4. Experimental Procedure
4.1. Materials and Animals
Humanin (HNG) was purchased from Peptide International, Inc. (Lexington, KY). Wortmannin was obtained from Alomone (Israel). Akti-1/2 (Akt inhibitor VIII) was purchased from Calbiochem (San Diego, CA).
Male CD-1 mice, 25-30g, were purchased from Harlan (Indianapolis, IN). All animal procedures were approved by the University Committee on Animal Care and Use of East Tennessee State University.
4.2. Middle cerebral artery occlusion model
The MCAO model was performed as described previously (Xu et al., 2006). Briefly, after a mouse was anesthetized with 7.2% chloral hydrate (400 mg/kg, i.p.), the right common carotid artery (CCA), the right external carotid artery (ECA), and the internal carotid artery (ICA) were exposed through a ventral midline neck incision. A 6-0 nylon monofilament (Ethilon, Ethicon Inc., Somervill, NJ) coated with silicon resin (Heraeus, Kulzer, Germany) was introduced into the right CCA and advanced until faint resistance was felt. Reperfusion was achieved by withdrawing the suture after 75 min of occlusion to restore blood supply to the middle cerebral artery (MCA) territory. Body temperature was maintained at 36.5-37.5°C throughout the procedure from the start of the surgery until the animals recovered from anesthesia. Occlusion and reperfusion of the MCA was monitored by a laser Doppler blood flowmeter (Periflux 5010, PERIMED, Sweden) positioned 1 mm posterior and 3 mm lateral to the bregma bilaterally.
4.3. Animal experimental groups
In each experiment, CD-1 mice were randomly divided into four groups (n=6-7): (1) vehicle-treated I/R control group; (2) HNG-treated I/R group; (3) wortmannin or Akti-1/2-treated I/R group; (4) HNG and wortmannin- or HNG and Akti-1/2-treated I/R groups. HNG-treated groups were administered 0.1 μg HNG intraventricularly (i.c.v.) in 5 μl saline and 1 μl DMSO. This dose was chosen based on results from our previous study (Xu et al., 2006). Wortmanninor Akti-1/2-treated I/R groups were administered 43 ng wortmannin or 55 ng Akti-1/2 in 1 μl DMSO with or without HNG. Saline-treated groups were administered 5 μl saline and 1 μl DMSO. For the injection of HNG or saline into the contralateral ventricle to the ischemic side, a small burr hole was made in the parietal region (0.5 mm posterior and 1.0 mm lateral to the bregma on the left side). A 28G needle on a Hamilton syringe was inserted into the lateral ventricle 2.5 mm in depth.
4.4. Evaluation of infarct volume
Mice were anesthetized and the brains were removed. Brains with subarachnoid hemorrhage and/or clot formation in the MCA were eliminated from the analysis in this study. All brains were sliced into 1 mm sections. Slices were incubated for 30 min in a 0.1% solution of 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO) at 37°C and then fixed in 10% buffered formaldehyde solution. For analysis, the sections were photographed by a high-resolution digital camera (Nikon Coolpix 5700). The cross-sectional area of infarction in the right MCA territory of each brain slice was determined with a computerized image analysis system (AlphaEase Image Analysis Software V 3.1.2). The total mean infarct area of each section was calculated as the average of the area on its rostral and caudal surface. The hemispheric lesion volume was calculated by multiplying the area by the thickness of slices. The areas of the infarcted tissue and the areas of both hemispheres were calculated for each brain slice. The percent hemispheric infarct volume was calculated as described by Giffard and Swanson (2005).
4.5. Primary cortical neuron culture
Embryonic day 16-18 mice were obtained from pregnant CD-1 mice anesthetized with tribromoethanol (350 mg/kg, i.p.). Meninges were carefully removed and cerebral cortices were isolated from the mouse brains. Cerebral cortices were dissociated with 8.2 U/ml papain (Worthington Biochemical, Lakewood, NJ) for 30 min at 37°C. Fetal bovine serum and trypsin inhibitor were used to stop digestion. Tissues were then triturated with a Pasteur pipette. Freshly dissociated cells were seeded at 2×105 cells/cm2 into 96-well plastic plates coated with L-polyornithine (10 μg/ml) and then incubated in Neurobasal medium (Invitrogen, Carlsbad, CA) with 2% B-27 supplement, Glutamax (1:100) (Invitrogen, Calsbad, CA), penicillin, and streptomycin at 37°C with 5% CO2 and 95% air. The medium was changed 24 h after plating, and half of the medium was changed every 3 d. Experiments were conducted at the DIV 10 after culture. Immunocytochemical analysis of neuronal marker protein gene product 9.5 (PGP 9.5) (Chemicon International, Inc., Temecula, CA) was used to confirm the purity of neuronal cells.
4.6. Oxygen-glucose deprivation (OGD)
Culture medium in a 96-well plate was removed and rinsed with 1× Hank's balanced salt solution (HBSS, 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM HEPES, 30 μM glycine, pH 7.4). Cultured cortical neurons were incubated in the pre-gassed HBSS buffer containing 0.2 μM HNG, 0.1 μM wortmannin, or 1 μM Akti-1/2 and then the plate was placed in a Billups-Rothenberg modular incubator chamber (Del Mar, CA), flushed with mixed gas of 5% CO2 and 95% N2 for 10 min. The chamber was then sealed and placed into a humidified CO2 incubator at 37°C. After 60 min in the hypoxic chamber, the OGD treatment was stopped by replacing HBSS with Neurobasal medium supplemented with B27 containing HNG and/or wortmannin or Akti-1/2. The plate was placed back to normoxic conditions and incubated for 24 h for the cell viability determination. Control culture plates in the presence of HNG or inhibitors were exposed to oxygenated HBSS containing 5.5 mM glucose in normoxic conditions during the same period as the OGD cultures.
4.7. Cell viability assay
Cell viability was assessed by the ability of the viable cells to metabolize 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)2-H-tetrazolium, inner salt (MTS), as described previously (Cory et al., 1991). At 24 h of OGD treatment in cultured cortical neurons, 10 μl of MTS solution (5 mg/ml; Promega, Madison, WI) was added to each well and the cells were maintained in growth medium for 3 h at 37°C. Absorbance was subsequently measured at 490 nm. Untreated cells were considered as control and the culture medium without cells in the presence of MTS solution was used as solution background. Cell viability was expressed as the percentage of the untreated control. For the MTS assay, there were eight samples in each group, and the experiment was repeated at least 3 times.
4.8. Western blot analysis
Mouse primary cortical neurons were incubated for 4 h in Neurobasal medium without B27. HNG was added directly to the conditioned medium for 10 min or 30 min at a final concentration of 0.2 μM. This concentration was chosen based on our previous studies on mouse heart-derived endothelial cells and rat myoblast H9c2 cells (unpublished data). In some experiments, cells were pretreated with 0.1 μM wortmannin for 15 min or 1 μM Akti-1/2 for 30 min before HNG treatment. Cells were harvested in a buffer containing 20 mM Tris-HCl (pH 7.8), 137 mM NaCl, 15% glycerol, 1% Triton X-100, 2 μg/ml each of leupeptin, aprotinin, and pepstatin, 2 mM benzamidine, 20 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium vanadate, 25 mM beta-glycerophosphate, and 1 mM phenylmethylsulfonylfluoride and PhosSTOP (Roche, Indianapolis, IN).
To study the activation of Akt by HNG in vivo, HNG-treated mice were sacrificed after 30 min of ischemia followed by 6 h of reperfusion. The right hemisphere was quickly removed and pulverized into powder in liquid nitrogen. Brains were homogenized in the lysis buffer as described above. Protein concentration was determined by the Bio-Rad protein assay (BioRad, Hercules, CA). Aliquots of 30 μg of lysates were electrophoresed on 12% SDS-PAGE and transferred to nitrocellulose membranes. Western blot analysis was carried out with antibodies against p-Akt (Ser473) and total Akt (Cell Signaling Technology). Blots were developed with the ECL chemiluminescence system (GE Healthcare, Piscataway, NJ) and were captured on autoradiographic films (Kodak). Films were scanned, and densitometric analysis of the bands was performed with AlphaEase Image Analysis Software.
4.9. Statistical analysis
All data are expressed as mean ± SEM. Differences between groups were determined with the Student's t test for p-Akt level; differences among groups were compared by one-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison test if there was a significant difference between groups. Differences were deemed statistically significant if P <0.05.
Acknowledgments
We appreciate the help of Dr. Deling Yin, Dr. Meng-Yang Zhu and Jennifer Hoard on culturing the primary mouse neurons. This study was supported by a grant from the Department of Veterans Affairs Merit Review, an NIH grant HL087271, and a grant-in-aid from the American Heart Association Southeast Affiliate to B.H.L.C.
Abbreviations
- HN
humanin
- MCAO
middle cerebral artery occlusion
- OGD
Oxygen-glucose deprivation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section: Cellular and Molecular Biology of Nervous Systems
Literature references
- 1.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase b signalling: Aktion on multiple fronts. Trends Biochem. Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The akt/pkb pathway: Molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 3.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 4.Giffard RG, Swanson RA. Ischemia-induced programmed cell death in astrocytes. Glia. 2005;50:299–306. doi: 10.1002/glia.20167. [DOI] [PubMed] [Google Scholar]
- 5.Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 6.Harada M, Habata Y, Hosoya M, Nishi K, Fujii R, Kobayashi M, Hinuma S. N-formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun. 2004;324:255–261. doi: 10.1016/j.bbrc.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I. A rescue factor abolishing neuronal cell death by a wide spectrum of familial alzheimer's disease genes and abeta. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto Y, Suzuki H, Aiso S, Niikura T, Nishimoto I, Matsuoka M. Involvement of tyrosine kinases and stat3 in humanin-mediated neuroprotection. Life Sci. 2005;17:3092–104. doi: 10.1016/j.lfs.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Janelidze S, Hu BR, Siesjo P, Siesjo BK. Alterations of akt1 (pkbalpha) and p70(s6k) in transient focal ischemia. Neurobiol. Dis. 2001;8:147–154. doi: 10.1006/nbdi.2000.0325. [DOI] [PubMed] [Google Scholar]
- 10.Jin G, Omori N, Li F, Nagano I, Manabe Y, Shoji M, Abe K. Protection against ischemic brain damage by GDNF affecting cell survival and death signals. Neurol. Res. 2003;25:249–253. doi: 10.1179/016164103101201454. [DOI] [PubMed] [Google Scholar]
- 11.Kaya D, Gursoy-Ozdemir Y, Yemisci M, Tuncer N, Aktan S, Dalkara T. VEGF protects brain against focal ischemia without increasing blood-brain permeability when administered intracerebroventricularly. J. Cereb. Blood Flow Metab. 2005;25:1111–1118. doi: 10.1038/sj.jcbfm.9600109. [DOI] [PubMed] [Google Scholar]
- 12.Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- 13.Krejcova G, Patocka J, Slaninova J. Effect of humanin analogues on experimentally induced impairment of spatial memory in rats. J. Pept. Sci. 2004;10:636–639. doi: 10.1002/psc.569. [DOI] [PubMed] [Google Scholar]
- 14.Mamiya T, Ukai M. [gly(14)]-humanin improved the learning and memory impairment induced by scopolamine in vivo. Br J Pharmacol. 2001;134:1597–159. doi: 10.1038/sj.bjp.0704429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noshita N, Lewen A, Sugawara T, Chan PH. Evidence of phosphorylation of akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Ohba N, Kiryu-Seo S, Maeda M, Muraoka M, Ishii M, Kiyama H. Transgenic mouse overexpressing the Akt reduced the volume of infarct area after middle cerebral artery occlusion. Neurosci. Lett. 2004;359:159–62. doi: 10.1016/j.neulet.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 17.Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J. Cereb. Blood Flow Metab. 2008;28:232–41. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- 18.Satriotomo I, Bowen KK, Vemuganti R. Jak2 and stat3 activation contributes to neuronal damage following transient focal cerebral ischemia. J. Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- 19.Shibata M, Yamawaki T, Sasaki T, Hattori H, Hamada J, Fukuuchi Y, Okano H, Miura M. Upregulation of akt phosphorylation at the early stage of middle cerebral artery occlusion in mice. Brain Res. 2002;942:1–10. doi: 10.1016/s0006-8993(02)02474-5. [DOI] [PubMed] [Google Scholar]
- 20.Sponne I, Fifre A, Koziel V, Kriem B, Oster T, Pillot T. Humanin rescues cortical neurons from prion-peptide-induced apoptosis. Mol. Cell Neurosci. 2004;25:95–102. doi: 10.1016/j.mcn.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 21.Tajima H, Niikura T, Hashimoto Y, Ito Y, Kita Y, Terashita K, Yamazaki K, Koto A, Aiso S, Nishimoto I. Evidence for in vivo production of humanin peptide, a neuroprotective factor against alzheimer's disease-related insults. Neurosci. Lett. 2002;324:227–231. doi: 10.1016/s0304-3940(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, Li H, Yuan H, Zheng M, Bai C, Chen L, Pei X. Humanin delays apoptosis in K562 cells by downregulation of P38 MAP kinase. Apoptosis. 2005;10:963–71. doi: 10.1007/s10495-005-1191-x. [DOI] [PubMed] [Google Scholar]
- 23.Xu X, Chua CC, Gao J, Hamdy RC, Chua BH. Humanin is a novel neuroprotective agent against stroke. Stroke. 2006;37:2613–2619. doi: 10.1161/01.STR.0000242772.94277.1f. [DOI] [PubMed] [Google Scholar]
- 24.Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu ZX, Le Y, Cui Y, Wang JM. Humanin, a newly identified neuroprotective factor, uses the g protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004;172:7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- 25.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies bid activity by blocking its activation of bax and bak. J. Biol. Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 26.Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/Akt survival signaling pathways are implicated in neuronal survival after stroke. Molecular Neurobiology. 2006;34:249–269. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]