

Abstract
Free radical-induced macromolecular damage has been studied extensively as a mechanism of oxidative stress, but large-scale intervention trials with free radical scavenging antioxidant supplements show little benefit in humans. The present review summarizes data supporting a complementary hypothesis for oxidative stress in disease that can occur without free radicals. This hypothesis, which is termed the “redox hypothesis,” is that oxidative stress occurs as a consequence of disruption of thiol redox circuits, which normally function in cell signaling and physiological regulation. The redox states of thiol systems are sensitive to two-electron oxidants and controlled by the thioredoxins (Trx), glutathione (GSH), and cysteine (Cys). Trx and GSH systems are maintained under stable, but nonequilibrium conditions, due to a continuous oxidation of cell thiols at a rate of about 0.5% of the total thiol pool per minute. Redox-sensitive thiols are critical for signal transduction (e.g., H-Ras, PTP-1B), transcription factor binding to DNA (e.g., Nrf-2, nuclear factor-κB), receptor activation (e.g., αIIbβ3 integrin in platelet activation), and other processes. Nonradical oxidants, including peroxides, aldehydes, quinones, and epoxides, are generated enzymatically from both endogenous and exogenous precursors and do not require free radicals as intermediates to oxidize or modify these thiols. Because of the nonequilibrium conditions in the thiol pathways, aberrant generation of nonradical oxidants at rates comparable to normal oxidation may be sufficient to disrupt function. Considerable opportunity exists to elucidate specific thiol control pathways and develop interventional strategies to restore normal redox control and protect against oxidative stress in aging and age-related disease.
Keywords: thioredoxin, glutathione, cysteine, hydrogen peroxide, redox signaling, protein thiol
one of the great redox biologists of the past century, Howard S. Mason, professed that to advance science, a scientist must interpret observations at the limit of their meaning. The present review of the redox biology of thiol systems addresses the possibility that disruption of the function and homeostasis of thiol systems is the most central feature of oxidative stress that contributes to mechanisms of aging and age-related disease. I have termed this the “redox hypothesis” to facilitate distinction from free radical hypotheses.
Many proteins contain redox-sensitive thiols, and reactions of thiol systems occur largely by nonradical two-electron transfers. Accumulating data show that central thiol-disulfide couples are maintained under nonequilibrium conditions in biological systems. This presents a condition wherein changes in abundance and distribution of redox catalysts and changes in rates of generation of relevant oxidants (e.g., peroxides) and precursors for NADPH supply can account for pathological effects of oxidative stress through altered functions of enzymes, receptors, transporters, transcription factors, and structural elements, without free radicals.
In this review, I summarize free radical and nonradical mechanisms in the redefinition of oxidative stress (82, 163) and develop the redox hypothesis based on recent data showing that major thiol systems exist under stable nonequilibrium conditions. In this development, I focus on H2O2 as a nonradical oxidant (see Table 1) and do not address details of radical and nonradical oxidants because they have been addressed previously (95, 177). An overview of distribution and function of thiol systems, with a focus on GSH- and thioredoxin (Trx), is presented to illustrate that reversible oxidation/reduction of thiols controls enzymes, receptors, and transcription factors and regulates cell signaling. Evidence is also available to show that protein trafficking, protein synthesis and degradation, and cytoskeletal structure are redox sensitive, but these are not addressed due to space limitations. Accumulating experimental evidence for nonequilibrium conditions of thiol-disulfide couples (88) is summarized, with discussion of the methods and potential artifacts. Finally, available evidence to support nonradical mechanisms of oxidative stress is reviewed. These data indicate that two-electron oxidants are likely to be quantitatively important determinants of oxidative stress. The results identify important opportunities for cell physiology research to define critical thiol targets and novel strategies for therapeutic interventions to prevent nonradical mechanisms of oxidative stress in aging and disease.
Table 1.
Free radicals, nonradical oxidants, and nonradical thiol-reactive species
| Free radicals |
| Superoxide anion radical |
| Nitric oxide |
| Hydroxyl radical |
| ·CCl3 |
| Nonradical oxidants |
| Hydrogen peroxide |
| Hydroperoxyfatty acids |
| Aldehydes |
| Quinones |
| Peroxynitrite |
| Disulfides |
| Nonradical thiol-reactive chemicals |
| Conjugated aldehydes, e.g., acrolein |
| 4-Hydroxynonenal, malondialdehyde |
| Quinones |
| Epoxides |
| Zn2+, Hg2+, other metal ions |
Definition of Oxidative Stress
The contemporary definition of oxidative stress has been refined (82, 163) to account for two different mechanistic outcomes, macromolecular damage, and disruption of thiol redox circuits, which leads to aberrant cell signaling and dysfunctional redox control (Fig. 1). Macromolecular damage is usually considered in terms of oxidative mechanisms linked to free radicals. Free radicals are small, diffusible molecules that differ from most biological molecules in that they have an unpaired electron. Free radicals tend to be reactive and can participate in chain reactions in which a single free radical initiation event can be propagated to damage multiple molecules. Polyunsaturated fatty acids, which are abundant in cell membranes, are oxidatively damaged by free radical chain reactions when exposed to O2 in the presence of trace metal ions. This process is known as lipid peroxidation. Studies of chain reactions in purified chemical systems show that a single initiation event can oxidatively damage 200 to 400 lipid molecules before two radicals react to eliminate the unpaired electrons and terminate the reaction sequence. Early research established that carbon tetrachloride is bioactivated to a free radical, causes lipid peroxidation, and results in hepatotoxicity by a free radical mechanism.
Fig. 1.

Two mechanisms of oxidative stress. Oxidative stress is defined as an imbalance in prooxidants and antioxidants, which results in macromolecular damage and disruption of redox signaling and control (163). Considerable research has focused on free radical (1-electron) mechanisms in macromolecular damage (left). The present review focuses on an alternative mechanism that involves 2-electron oxidants (right). Based on the xanthine oxidase kinetics, which show that univalent reduction of O2 to superoxide anion radical is always a minor fraction of the bivalent reduction to H2O2 (45), one can anticipate that 2-electron oxidants predominate during oxidative stress. Thus for an arbitrary rate of oxidant generation equal to 1,000, perhaps 900 would go directly to 2-electron oxidants and only 100 would be 1-electron oxidants. Free radical scavenging mechanisms are efficient and convert free radicals into nonradical oxidants. The rate of oxidant production would be partitioned; if the radical scavengers are >99% effective, then there would be a rate of macromolecular damage of <1 and a rate of 2-electron oxidant production equal to >999. Thus if nonradical oxidants contribute to disease pathology by disruption of redox signaling and control mechanisms, the pathology can correlate with macromolecular damage and yet be relatively insensitive to free radical scavengers. Nonradical oxidants (e.g., conjugated aldehydes) also cause macromolecular damage, which can disrupt redox signaling and control pathways (not shown).
Four decades of research have clarified the chemistry of free radical processes in biological systems and yielded considerable information on antioxidant defenses. These include free radical scavenging enzymes, such as superoxide dismutases, and abundant radical-scavenging chemicals, such as vitamin E and C. In the presence of these systems and with the very high protein concentration in biological systems, free radical chain reactions are almost completely prevented. Thus most free radical-mediated oxidative damage in biological systems appears to be directly linked to free radical initiation events and not a consequence of chain reactions.
This distinction between free radical chemistry in purified systems and in biological systems may be critical in understanding oxidative stress because biological systems generate more nonradical oxidants than free radicals (Fig. 1). For instance, in the well-studied xanthine oxidase reaction, which produces the free radical superoxide anion, the predominant product is H2O2 (45). A comparison of the univalent flux to the divalent flux showed that under optimal conditions, superoxide represented only 30% of the flux (45); under most conditions the univalent flux was considerably less, consistent with partitioning of radical and nonradical oxidant delivery in cells as depicted in Fig. 1. Sequential univalent transfer occurs with many redox proteins that generate superoxide so that H2O2 is a common product of these reactions. Moreover, dismutation of superoxide anion to H2O2 and O2 occurs at a rapid rate (46), and reaction of radicals with free radical scavengers can result in a highly efficient conversion of free radicals to nonradical oxidants. The free radicals nitric oxide and superoxide react to generate the nonradical oxidant peroxynitrite (179, 180). The two-electron, nonradical oxidant H2O2 is produced at 1 to 4% of the rate of O2 consumption and therefore represents a major oxidant burden. Importantly, if oxidative stress produces radicals and nonradical oxidants as described in Fig. 1 and nonradical oxidants are quantitatively important in disease, then free radical-dependent macromolecular damage can correlate with pathological insult but, none the less, be largely irrelevant to the disease process. Hence, the impetus of the current review is the need to consider two-electron oxidation as a component of oxidative stress that is distinct from free radical-mediated macromolecular damage.
In certain regards, this parallels and overlaps concepts elaborated as the “mitohormesis hypothesis” (176). In mitohormesis, sublethal mitochondrial stress is proposed to produce beneficial outcomes through mitochondrial generation of reactive oxygen species, which serve as signaling elements for cytoprotection. The concept is supported by studies in Caenorhabditis elegans, which show that glucose restriction stimulated mitochondrial respiration and 2,7-dichlorodihydrofluoroscein diacetate oxidation and increased life span (155). A recent review of these concepts proposes novel therapeutic approaches to enhance tolerance to cardiac ischemia by activating the relevant signaling mechanisms (156). However, the mitohormesis hypothesis does not discriminate between free radical and nonradical mechanisms, and a better understanding of the distinct signaling and toxicological mechanisms of the radical and nonradical species will facilitate development of effective therapies.
Thiol-containing proteins that function in redox signaling and physiological regulation are susceptible to two-electron oxidation by nonradical oxidants, including H2O2, lipid hydroperoxides, aldehydes, quinones, and disulfides. Either abnormal oxidation or irreversible modification can interfere with reversible oxidation-reduction reactions of thiols that physiologically function in receptor signaling, transcriptional regulation, cell proliferation, angiogenesis, and apoptosis. Thus the contemporary refinement in definition of oxidative stress (82, 163) represents a shift to include both nonradical oxidants and reversible oxidative reactions of redox signaling and control as key components of oxidative stress.
The Redox Hypothesis
In an attempt to clearly delineate the nonradical complement to free radical theories that have dominated oxidative stress research, I have formulated a “redox hypothesis” with four postulates: 1) All biological systems contain redox elements [e.g., redox-sensitive cysteine, Cys, residues] that function in cell signaling, macromolecular trafficking, and physiological regulation. 2) Organization and coordination of the redox activity of these elements occurs through redox circuits dependent on common control nodes (e.g., thioredoxin, GSH). 3) The redox-sensitive elements are spatially and kinetically insulated so that “gated” redox circuits can be activated by translocation/aggregation and/or catalytic mechanisms. 4) Oxidative stress is a disruption of the function of these redox circuits caused by specific reaction with the redox-sensitive thiol elements, altered pathways of electron transfer, or interruption of the gating mechanisms controlling the flux through these pathways.
The subsequent sections of this review address the experimental basis for these postulates and discuss their potential relevance to oxidative stress as a causal mechanism in disease.
Thiols as critical components in redox systems biology.
Proteins contain two common functional groups [thiol of Cys and thioether of methionine, Met] that undergo reversible oxidation-reduction. The present review focuses on Cys because of the more abundant data on Cys oxidation. The relevant oxidation states are the thiol (-SH), disulfide (-SS-), sulfenate (-SO−), sulfinate (-SO2−), and sulfonate (-SO3−), of which the thiol and disulfide are most common. Thiyl radicals (-RS·) generated from thiols in the presence of oxygen-centered radicals (197), as well as other reactive sulfur species (50), have also been considered as toxic species involved in oxidative stress. Thiyl radicals rapidly react to form disulfides (171), and this may serve as a convergence point between one-electron and two-electron events. Sulfenates are relatively unstable and are converted to disulfides in the presence of thiols. In certain protein structures, they can be stabilized as sulfenamides (150, 185). The higher oxidation states (sulfinates and sulfonates) are typically not reversible in mammalian systems, although a yeast enzyme (sulfiredoxin) was recently discovered, which reduces sulfinate in peroxiredoxin (15).
Reversible oxidations of Met residues and the less common amino acid selenocysteine (Sec) also occur. Oxidation of Met to methionine sulfoxide occurs in association with oxidative stress and aging (169, 170). Such oxidation affects biological activity as shown by loss of α1-antitrypsin inhibitor activity upon Met oxidation (22). Two types of methionine sulfoxide reductases act on the structurally distinct S- and R-sulfoxides (65, 89, 196); methionine sulfoxide reductases activities are dependent on thioredoxins (Trx) (196) and have been associated with longevity (123, 130, 147, 152). The less common selenol of Sec undergoes reversible oxidation-reduction during catalytic functions of Trx reductases and Se-dependent GSH peroxidases (52, 96). These Se-dependent enzymes are present at key positions in both Trx and GSH pathways. Although the present review is focused on Cys oxidation, both Met and Sec oxidation are relevant to the redox hypothesis because of their relationships to the thiol systems and because of their individual functions in control of protein structure and activity.
Regulation of biological functions by thiols.
Reversible oxidation of thiols to disulfides or sulfenic acid residues controls biological functions in three general ways, by chemically altering active site cysteines, by altering macromolecular interactions, and by regulating activity through modification of allosteric Cys (Fig. 2). Although these are not mutually exclusive, classification by these criteria provides an easy way to view the different aspects of redox regulation. Reversible chemical modification of active site Cys residues yields an “on-off” mechanism for control. Crosslinking of proteins through disulfides provides a mechanism for aggregation and trafficking. Oxidation of Cys distal to active sites provides an allosteric mechanism. Because proteins often contain multiple Cys residues that can undergo reversible modification to intramolecular and intermolecular disulfides, sulfenic acids, sulfonic acid, S-glutathione derivatives, and S-nitroso derivatives, a broad range of thiol-dependent regulation, can occur.
Fig. 2.

Sulfur switches (SH) provide flexible control mechanisms for protein function. Cysteine (Cys) is found in the active site of many proteins, and reversible oxidation or modification provides an “on-off” mechanism for protein function (left). Cys residues are also important regulatory elements that control the macromolecular interaction of proteins (center). Reversible modification of nonactive site Cys residues also provides an allosteric type mechanism to regulate activity.
ACTIVE SITE-DEPENDENT “ON-OFF” REGULATION.
Early enzymology research showed that half of all enzyme activities are sensitive to either oxidation, reaction with electrophiles, or interaction with metal ions (190–194). Enzymes with active-site Cys include caspases, kinases, phosphatases, and proteases. Reversible oxidation of the active site of protein tyrosine phosphatase-1B has been associated with control of biological function in redox signaling (39). Oxidation of Cys by S-glutathionylation (GS-ylation) regulates glyceraldehyde 3-phosphate dehydrogenase (125) and caspase-3 (173). Cys residues are present in active sites of detoxification enzymes such as glutathione transferases, cytochromes P-450, Trxs, and peroxiredoxins. Cys is a component of active sites of iron-sulfur clusters of electron transfer proteins. Cys is a component of zinc fingers in transcription factors and zinc-binding domains of metallothioneins. Cys residues are conserved in structural proteins such as actin and docking proteins such as 14-3-3. Oxidation of Cys residues in αIIbβ3 integrin controls platelet activation (43, 99, 118). Cys-rich regions are present in plasma membrane receptors and ion channels, including the NMDA receptors, EGF receptor, and others. Thus reversible oxidation of active site thiols can provide a common and central “on-off” mechanism for control of cell functions.
REGULATION OF MACROMOLECULAR INTERACTIONS.
Thiol oxidation also controls macromolecular interactions of proteins. Early research showed that thiols prevented aggregation during protein purification, and subsequent studies of protein structure revealed that formation of specific disulfide crosslinks between Cys residues was important for correct three-dimensional structure and activity of proteins. The mechanisms for introduction of disulfides have been studied extensively in protein processing and contribute to stability of secondary, tertiary and quaternary structure. β-Actin contains a conserved Cys, which results in reversible binding of proteins, S-GS-ylation, and crosslinking of actin filaments upon oxidation (33, 112, 140). Oxidation functions in glucocorticoid receptor translocation into nuclei (116, 136), and oxidation controls export of yeast AP-1 (Yap-1) from nuclei (38, 98). Disulfide crosslinks control fluidity of mucus (165). Such changes in protein structure and interaction due to reversible oxidation can provide a central mechanism for specificity in redox signaling (34).
ALLOSTERIC REGULATION.
In addition to containing active site and/or structural thiols, many proteins contain Cys which regulate activity by an allosteric mechanism. This type of regulation can provide a “rheostat” rather than an “on-off” switch (88), thereby providing a means to throttle processes by GS-ylation or S-nitrosylation (Fig. 2). Interaction with a metal ion, especially the non-oxidizable Zn2+, provides a further level of control through allosteric Cys residues. GS-ylation regulates activity of NADH dehydrogenase (178). S-Nitrosylation of Cys69 in Trx-1 (62) and Cys118 of Ras (102) regulate respective activities. NMDA-receptor activity is modulated by Zn2+ ions which bind Cys residues and change receptor activity (31).
Orthogonal regulation in proteins with multiple Cys residues.
Individual proteins often contain multiple Cys residues. This allows for a single protein to be controlled in different ways by selective redox mechanisms. These different types of Cys residues can be viewed as orthogonal control elements because they allow a single protein-dependent biological function to be simultaneously regulated by multiple independent mechanisms. Orthogonal regulation is illustrated by known modifications of thioredoxin-1 (Trx-1) (Fig. 3). The active site dithiol motif undergoes oxidation-reduction during the reaction cycle and is important as a regulatory factor through structural interactions with apoptosis signal-regulating kinase-1 (Ask-1). Binding of the C32,C35 dithiol form of Trx-1 inhibits Ask-1 activity. Upon oxidation, release and activation of Ask-1 activates apoptosis. Oxidation of the active site therefore functions as an “on-off” switch for apoptosis. In addition to these active-site thiols, the three nonactive site Cys residues are also subject to redox regulation and provide orthogonal mechanisms for control. Modification of these residues can result in allosteric changes that affect the active site or structural changes, which affect macromolecular interactions. Irreversible modification of C73 with acrolein or 4-hydroxynonenal creates a form that inhibits Trx reductase-1 (TrxR1) and induces pathogenic phenotype (54). This Cys is also a site for GS-ylation and protein-protein disulfide formation, suggesting that physiological regulation could occur through this Cys (23). The remaining Cys residues, C62 and C69, form a second disulfide under oxidative stress conditions (189). Formation of this C62-C69 disulfide inhibits TrxR1 (189), perhaps by disrupting a surface α-helix.
Fig. 3.

Orthogonal regulation can occur through multiple Cys residues on a single protein. Thioredoxin-1 (Trx-1) illustrates how different redox-sensitive elements can be used to simultaneously transmit independent redox signals. The active site (C32,35) undergoes reversible oxidation during catalytic turnover. Oxidation of the active site dithiol results in loss of binding to Ask1 and activation of apoptosis. C73 is an oxidizable amino acid which can undergo glutathionylation (GS-ylation) and also can bind to other cell proteins. C62 undergoes S-nitrosylation and also can form an intramolecular disulfide between C62 and C69 which blocks reduction by TR1.
Cys residues as an organizational feature of cell function.
The finding that the steady-state redox potential of the GSH/GSSG couple becomes progressively oxidized throughout the life cycle of cells (91, 92, 135) suggests that these three types of reversible redox control could organize metabolism in association with different functional demands (92, 153). This can be considered hypothetically in terms of the total Cys content of the proteome. Some proteins have no Cys, whereas others have 20 or more Cys residues. A recent update of the GenBank protein file indicates that there are 406,500 Cys residues in protein entries; with about 1.9 entries per gene. This indicates that there are about 214,000 Cys unique residues encoded in the human genome (Scott Devine, personal communication). A similar calculation yields about 172,000 Met (excluding about 19,800 first Met in sequences), which probably includes some reversibly oxidized residues. The Cys residues vary in their rates of oxidation with many buried within protein structures, present as disulfides or zinc complexes, or otherwise inaccessible. In an experiment to determine rates of protein thiol oxidation in nuclei incubated in air at 37°C, a rapid oxidation of about 10% of the thiols occurred during the first hour, including complete oxidation of Trx1, and then a second 10% of protein thiols was oxidized over the next 7 h (Y.-M. Go, D. P. Jones, unpublished). This suggests that perhaps 80–90% of protein thiols are relatively inert to redox reactions. Extrapolation of the 10–20% readily oxidized thiols to the full 214,000 thiols in the Cys proteome indicates that 21,000–42,000 Cys could be readily oxidized. These are likely to include enzyme, transporter, receptor, or transcription factor “active sites” as well as allosteric and macromolecular interaction sites. Accumulating evidence indicates that dozens, and perhaps hundreds, of proteins undergo S-nitrosylation and GS-ylation (44, 73, 93, 120, 121). The potential significance of a large number of redox-sensitive sites is apparent when one considers the nonequilibrium conditions of thiol-disulfide couples within cells and subcellular compartments as described below.
Together, the data show that there are a large number of redox-sensitive thiols within the Cys proteome. These are widely distributed among signaling, structural, and regulatory proteins. Thus there is a secure basis for the first postulate of the redox hypothesis.
GSH and Trxs as Common Control Nodes for Protein Thiol Redox Circuits
The second postulate is that the redox-sensitive elements are organized into redox pathways and control networks. Two major thiol-containing systems function in redox control of protein thiols. GSH, the most abundant thiol in mammals, was discovered over a century ago, and its central function in detoxification and protection against oxidants was recognized 50 years ago (124). Trx was discovered 45 years ago as a reductant for ribonucleotide reductase (106, 126), but its central protective function has only become recognized over the past two decades (74). Both GSH and Trx support enzyme systems for elimination of peroxides, but each system has other distinct functions.
GSH pathways.
GSH is a high abundance component present at millimolar concentrations in cells; it serves as a short-term storage form of the amino acid Cys, as a nucleophile for efficient detoxification of reactive electrophiles, and as an antioxidant. It is well suited for redox control of monothiols in proteins (153); as described above, GS-ylation can serve as an “on-off” or a “rheostat” mechanism for control of protein function. GS-ylation reactions are catalyzed by thiol transferases known as glutaredoxins (Grx). These reactions are reversible so that a highly reduced GSH/GSSG redox potential drives reduction of protein disulfides while an oxidizing potential drives protein GS-ylation (171, 187). GSH is also used to protect against two-electron oxidants, such as peroxide reduction catalyzed by GSH peroxidases, and aldehydes and quinones catalyzed by GSH transferases.
An overview of the reactions of the GSH-dependent redox network is illustrated in Fig. 4. An important goal of redox systems biology (88) is to integrate these reactions into quantitative models to describe and test biological activities. However, the number and diversity of processes involving GSH present a challenge for development of such models, especially given the heterogeneous distribution of the proteins and difficulty in study at the subcellular level in intact cells and tissues. Furthermore, the wealth of knowledge on GSH systems under toxicological conditions may have obscured a general function of GSH in physiological regulation. There is a long history of research on GS-ylation as a covalent modification (49, 93, 181, 208), but the facile formation of protein oxidation products during oxidative stress and cell fractionation has led to perhaps an overly conservative point of view concerning the biological importance of regulation by thiol modifications.
Fig. 4.

Glutathionine (GSH) redox network. A partial list of GSH-dependent proteins illustrates the need for research to understand the integrated function of these redox systems. 1) GSH is synthesized by a two-step pathway in which abundance of two enzymes, glutamate cysteine ligase (GSH0, GSH1) and GSH synthetase (GSHB), determine synthesis rate (97). GSH is degraded by γ-glutamyltransferase (GGT) at the surface of the brush border of the kidney, small intestine, and a number of other tissues, and probably also in the cisternae of the secretory pathway (142). 2) GSH is transported out of cells by several multidrug resistance proteins (MRP) (12). The chloride channel, which is mutated in cystic fibrosis (CFTR), also transports GSH (113), and GSH is transported into mitochondria by the dicarboxylate carrier (DIC) and a monocarboxylate carrier (OGCP) (103). GSH is transported into the cisternae of the endoplasmic reticulum (13), but the molecular nature of the transporter is not known. 3) GSH is used by a number of GSH transferases (GST), which include microsomal and nonmicrosomal locations, to modify electrophilic chemicals (9). These are thought to largely function in detoxification, but some also act on biosynthetic intermediates for prostaglandins and leukotrienes. A fraction of GSH is present as S-nitroso-GSH, a transnitrosylating agent generated from nitric oxide or its metabolites (168). 4) GSH functions in metabolism as a coenzyme for formaldehyde dehydrogenase, glyoxylase, and other metabolic reactions (4, 168). In these reactions, GSH is cyclically removed by one reaction and regenerated in a second reaction. 5) Several thiol transferases, also known as glutaredoxins, catalyze introduction and removal of GSH (110, 114). 5a) Several proteins are regulated by GS-ylation, and many others undergo GS-ylation under oxidative stress conditions (44, 93). 6) GSH is used as a reductant for selenium-dependent GSH peroxidases (GPX) and selenium-independent peroxiredoxin-6 (PRX6) and some GSH transferases (GST). 6a) The product of these oxidative reactions, GSSG, is reduced back to GSH by GSSG reductase (GSHR) in most tissues. In sperm, thioredoxin reductase-3 (TRXR3) has activity toward both Trx and GSH. The proteins included in this figure are present in multiple cellular compartments and are differentially expressed in cells so that development of functional maps will require tissue-specific measurements of individual reaction rates. Protein designations and common names are from the UniProtKB/Swiss-Prot database. Abbreviations are as follows: GSH0, Glu-Cys ligase, regulatory; GSH1, Glu-Cys ligase, catalytic; GSHB, GSH synthetase; GGT1,4, 5, 6, γ-glutamyltransferase; DIC, mitochondrial dicarboxylate carrier (SLC25A10); OGCP, mitochondrial 2-oxoglutarate/malate carrier; CFTR, cystic fibrosis transmembrane conductance protein; MRP, multidrug resistance-associated protein; MRP2, canalicular multispecific organic anion transporter 1; GST, GSH transferase; ADHX, alcohol dehydrogenase class-3; ESTD, S-formyl-GSH hydrolase; GLO2, Glyoxalase II; HAGHL, hydroxyacylGSH hydrolase-like; LGUL, lactoylGSH lyase; MAAI , maleylacetoacetate isomerase; PTGD2, GSH-requiring prostaglandin D synthase; PTGDS, prostaglandin-H2 D-isomerase; PTGES, prostaglandin E synthase; RBP1, RalA-binding protein 1 (RalBP1); GLRX, glutaredoxin and glutaredoxin-related proteins; YD286, glutaredoxin-like protein; GPX, GSH peroxidase; GSHR, GSSG reductase; TRXR3, thioredoxin reductase 3.
During recent years, data have accumulated for GS-ylation and S-nitrosylation of many proteins (93, 158). PTP-1B and glyceraldehyde 3-phosphate dehydrogenase are inactivated by GS-ylation (39, 125). GS-ylation controls activity of H-Ras at the same C118 residue as S-nitrosylation (3, 120, 121). Both S-nitrosylation and GS-ylation control activity of caspase-3 (138, 173). Actin function is regulated by GS-ylation (33, 187), and both HIV-1 protease (35) and GSH transferases (GST) are activated by GS-ylation (158). GS-ylation of p65-NF-κB potentiates hypoxic apoptosis in murine embryonic fibroblasts (143). Furthermore, the discovery that GST can bind to S-glutathionyl groups on proteins (30) and that GSTπ can transfer GSH to restore Prx6 activity (117) suggests that GST could catalyze GS-ylation. If so, different GST proteins could regulate cell functions by GS-ylation of different subsets of proteins. Thus the number of identified proteins that undergo GS-ylation and S-nitrosylation indicate that there could be a large, currently poorly defined, network of proteins regulated by these mechanisms.
Trx pathways.
Trxs differ from GSH in that they are small proteins and present at several orders of magnitude lower concentrations. GSH is ideally suited to react with monothiols (at the sulfenic acid level) to generate S-glutathione derivatives (mixed disulfides) of proteins and then with S-glutathionyl derivatives to generate GSSG and reduced protein. In contrast, Trx is a two-electron reductant that can directly reduce sulfenic acids in proteins and also control redox state of dithiol-disulfide motifs. The active site dithiol of Trx is highly conserved in evolution and widely distributed among Trx superfamily proteins.
Similar to GSH, Trx supports a large network of redox reactions (Fig. 5), including those catalyzed by ribonucleotide reductase, peroxiredoxins, redox factor-1 (1), and methionine sulfoxide reductases. A global antioxidant function of Trx has been inferred from studies showing that Trx can reduce many oxidized proteins. Kinetic studies with different proteins, however, show that there is an underlying specificity for target proteins in terms of the rates of catalysis (14, 74, 110). C35 mutants of Trx function as dominant-negative forms when expressed in cells and have been used as bait to trap putative targets as stable disulfides (119). Such experiments suggest that Trxs could support individual redox pathways based upon this specificity. In addition, other Trx-like proteins probably exist in networks whereby changes in abundance or oxidation of one component regulates function of multiple target proteins (Fig. 5).
Fig. 5.

A partial list of proteins in the Trx redox network. 1) Thioredoxins are reduced by thioredoxin reductases (TRXR1,2,3). 2) Trx functions as a reductant for ribonucleotide reductase (RNR). 3) Reduced Trx-1 or Trx-2 binds to apoptosis signal-regulating kinase-1 (ASK-1), inhibiting its function. 4) Trx-1 also binds to other proteins, including the vitamin D3-binding protein (VDUP1; TXNIP) (133). Interaction with such proteins regulates activity and may determine distribution between cytoplasm and nuclei. 5) In cell nuclei, Trx-1 reduces redox factor-1 (REF-1), which is the DNA repair/redox enzyme, AP endonuclease (200). REF-1 maintains conserved Cys residues of transcription factors in the reduced form required for DNA binding. These transcription factors include nuclear factor (NF)-κB, Nrf-2, AP-1, P53, glucocorticoid receptor (GR), estrogen receptor (ER), and HIF-1α (1, 17, 72, 75, 116, 182, 184). 6) Trx-1 is a reductant for methionine sulfoxide reductases (MSR) and also for 7) a pathway for MSRB2 reduction mediated by metallothionein in the metal-free thionein form (148). 8) Trx supports six peroxiredoxins (PDRX) that have heterogeneous subcellular distributions but a common activity to eliminate peroxides (146). Redox functions can be provided by Trx-related proteins (TRP14, TRP32, nucleeoredoxin and Trx-related transmembrane protein), which contain a Trx motif and may have evolved to recognize distinctive groups of proteins from those recognized by Trx-1 (105, 107, 122, 198). A large number of Trx-like proteins are known (below dotted line), and many of these are likely to be redox active, either in pathways dependent on Trx or in parallel pathways, which evolved to provide additional specificity beyond that provided by the Trx proteins. Protein designations and common names are from the UniProtKB/Swiss-Prot database. TXN4A, Trx-like protein 4A (spliceosomal U5 snRNP 15 kDa; Dim-1); TXN4B, Trx-like protein 4B (Dim1-like protein); TXND9, ATP-binding protein associated with cell differentiation (APACD); TXNL1, Trx-like protein 1 (32 kDa); NXNL1, nucleoredoxin-like protein 1 (Trx-like protein 6); GLRX3, glutaredoxin-3, PKC-theta-interacting protein; TXND3, spermatid-specific Trx-2; TXND8, spermatid-specific thioredoxin-3 (Trx-6); PDIA6 Protein disulfide-isomerase A6; TXD10, Trx-related transmembrane protein 3 (PDI); TXND1, transmembrane Trx-related protein; DJC10, ER-resident protein; ERdj5, microthioredoxin; TXND4, endoplasmic reticulum protein ERp44; TXND5, endoplasmic reticulum protein ERp46; TXD12, Trx domain-containing protein 12 (ERp18); TXND, Trx domain-containing proteins.
Distribution of electron flow in GSH and Trx pathways.
The number and diversity of the protein interactions in Figs. 4 and 5 illustrate the complexity of antioxidant responses involving GSH and Trx systems. Unfortunately, the Trx systems have mostly been considered independently of GSH in experimental studies; consequently, little is known about the functional coordination of the Trx and GSH systems. In both mitochondrial and nonmitochondrial compartments, the GSH and Trx systems would appear to compete with each other for electrons because both GSSG reductase and Trx reductases utilize NADPH as the electron donor. Similarly the GSH peroxidases and Trx-dependent peroxiredoxins would appear to compete with each other for reduction of peroxides. However, differences in kinetics of the reductases, subcellular localization, or Michaelis constant (KM) for peroxides could provide specificity (34). A study of tumor necrosis factor (TNF)-α-induced cell death showed that Trx-2 and GSH systems function in parallel and are nonredundant in mitochondria (205). In cells with increased Nox-1, H2O2-dependent signaling occurred without detectable oxidation of either cellular GSH/GSSG or Trx-1 (53). In contrast, epidermal growth factor (EGF) signaling oxidized cytoplasmic Trx-1 without detectable oxidation of GSH/GSSG (64). Thus the data suggest that there is an underlying specificity and organization of the GSH- and Trx-dependent redox-signaling events.
Critical issues for integrating Trx and GSH systems into redox network models have been identified (88). Effective control requires both insulation of redox elements and mechanisms to coordinate functions. In this context, an accurate definition of redox pathways is difficult because the pathways are dependent on rate constants for individual redox elements and sensitive to abundance, distribution, and steady-state redox potentials. With a large number of redox-sensitive elements and central GSH- and Trx-dependent systems, it appears likely that there is an underlying organization, i.e., a “redox code” that allows efficient coordination of biological redox functions (88). However, organizing principles for GS-ylation, S-nitrosylation, thiol-disulfide control, and other functions are not yet fully defined (121). Moreover, the concept of a pathway, as applied to glycolysis or other metabolic pathway, may be inaccurate for a redox pathway. In metabolism, a metabolic pathway is defined by the product of one catalytic step being a substrate for a subsequent catalytic step. In redox pathways, specificity may be defined, at least in part, by the proximity of thiols that undergo oxidation. Thus the spatial distribution may determine the substrate-product relationship so that the meaning of “redox pathway” has both spatial and kinetic components.
To develop redox system models, quantification of electron flow is needed for the overall systems and for individual components. Relatively little information is available on physiological reaction rates for any redox signaling or control mechanism. However, the abundant knowledge of GSH-dependent and Trx-dependent control of signaling, structural, and physiological processes supports the postulate that redox elements are organized through pathways dependent on these central redox couples. Initial information described below further indicates that the networks supported by the GSH and Trx systems are kinetically and functionally distinct.
Nonequilibrium Conditions of Thiol-Disulfide Couples in Biological Systems
The third postulate is that redox-sensitive elements are spatially and kinetically insulated so that “gated” redox circuits can be activated by translocation/aggregation and/or catalytic mechanisms. This represents a departure from traditional views that Cys residues in cellular proteins are fully reduced and that GSH provides a “redox buffer” to protect against oxidation of protein thiols. Accumulating data show that critical Cys residues, which function in redox signaling and control, are not in equilibrium with each other or with the NADPH/NADP+ couple. Some Cys residues are present in proximity to cationic amino acids, which lowers their pKa values and provides an autocatalytic means to facilitate oxidation-reduction reactions (166). Kinetic control of the redox-sensitive Cys residues allows the systems to be rapidly responsive and dynamically regulated. At the same time, specificity in redox signaling mechanisms requires that individual signaling elements be insulated from other redox-active components. Such specificity is possible because noncatalyzed oxidation and thiol-disulfide exchange reactions are slow at prevailing conditions in cells. This can occur through spatial and kinetic insulation of redox elements, allowing “gated” redox circuits to be activated by translocation/aggregation of redox components and/or regulated activation of redox catalysts. Importantly, as outlined below, the current through such circuits only needs to be a small fraction of total electron transfer in cells. A low rate of oxidation is often observed during protein purification, and this raises the possibility that what has been traditionally considered nonphysiological “autooxidation” of proteins during purification may be characteristics of unrecognized regulatory mechanisms.
Several lines of evidence support the interpretation that critical thiol-disulfide redox couples are maintained under stable, nonequilibrium conditions in biological systems. One of the most unequivocal pieces of evidence is the disequilibrium of the cysteine/cystine (Cys/CySS) and GSH/GSSG couples in human plasma (84, 87). Steady-state redox potential (Eh) values provide a convenient way to compare the tendency of redox couples to accept or donate electrons (83). The difference in Eh values (ΔEh) between two couples is proportional to the free energy for electron transfer (ΔG = nFΔEh, where n is the number of electrons transferred and F is Faraday's constant). Eh is dependent on both the inherent tendency of the chemical to accept or donate electrons, expressed in the standard potential (Eo), and also the concentrations of the acceptor (oxidized) and donor (reduced) species of the couple, as defined by the Nernst equation {Eh = Eo + RT/nF ln([oxidized]/[reduced])}. The mean Eh of GSH/GSSG in the plasma of 24 healthy, young adults was −137 ± 9 mV, whereas the concurrent value for the Eh of Cys/CySS was −80 ± 9 mV (84). These values are displaced from equilibrium by approximately two orders of magnitude. Because 1) human plasma is a homogeneous fluid with no subcellular compartments, 2) no significant noncovalent binding occurs with proteins, and 3) the same analytic procedures were used for simultaneous measurements of all of the chemical species involved; the results show that the major low molecular weight thiol-disulfide couples in human plasma are maintained in a nonequilibrium state (83, 84).
This supports an earlier conclusion that plasma thiol-disulfide couples are not at equilibrium based on reactions of Cys and GSH with plasma proteins (104). The conclusion is further supported by calculations using the second-order rate constant for thiol-disulfide exchange reactions [20 M−1min−1; (49)]. At prevailing concentrations of GSH (2 μM) and CySS (40 μM) in plasma (84), the calculated rate of reaction of GSH with CySS is 0.0016 μmol·l−1·min−1. This is much lower than the rate of Cys turnover in humans, which was determined by stable isotopic tracer methods to be about 1 μmol·kg−1·min−1 (47, 115, 144). Thus the calculated rates are consistent with measured steady-state redox values showing that the rates of thiol-disulfide exchange are too slow to allow redox couples to equilibrate in human plasma.
Steady-state redox potential of the GSH/GSSG couple in cells and tissues.
Earlier calculations of the Eh in the liver (49) indicated that this value (−255 mV) is highly displaced from the Eh value for NADPH/NADP+ [−380 to −405 mV (161)]. Sies and Summer (164) and a later systematic review by Gilbert (49) concluded that the kinetic characteristics of GSSG reductase were incapable of maintaining GSH/GSSG in equilibrium with NADPH/NADP+ because of a relatively high KM for GSSG. Studies with a number of cell lines and tissues are consistent with the Eh for GSH/GSSG being in the range of −260 to −200 mV (82), i.e., highly displaced from equilibrium with NADPH/NADP+.
Studies performed with HT29 cells evaluated possible artifacts in estimation of Eh due to pH, concentration measurements, and inhomogeneous distribution of GSH and GSSG between compartments. Cell volume was measured by distribution of 3H2O with extracellular volume determined by distribution of [14C]inulin (92). Cytoplasmic pH was determined by distribution of the pH indicator [14C]dimethadione (92). Errors due to sequestration by mitochondrial and secretory compartments were estimated by digitonin fractionation (83, 86). The results showed that the Eh of the cytoplasmic GSH/GSSG couple was about −260 mV under proliferative conditions, about −220 mV under differentiated conditions, transiently oxidized and then reduced following treatment with a GSH synthesis inducer (6), and oxidized to about −170 mV during apoptosis (21, 82, 91). In contrast, Eh of the Cys/CySS couple was −145 ± 3 mV under proliferative conditions and oxidized by about 30 mV by either the differentiation agent, sodium butyrate, or the GSH synthesis inducer benzyl isothiocyanate (92). Results showed that errors due to volume and pH were negligible (92). Contamination due to contents of subcellular compartments was estimated to be no more than 10 mV (82, 86). These results confirmed that Eh for cellular GSH/GSSG is in the range of −260 to −200 mV, i.e., considerably oxidized relative to the NADPH/NADP+ couple (82).
Steady-state Eh of Cys residues in proteins.
Development of methods to evaluate redox potential of individual thiol-disulfide couples in proteins (see Perspectives) has allowed determination of Eh values for Trx-1 and Trx-2 in cells and tissues (64, 69, 70, 188, 189). Results showed that the steady-state Eh for Trx-1 was −300 to −270 mV, a range that is more negative (reduced) than Eh for GSH/GSSG in CaCo-2 cells (135), THP-1 cells (188), HeLa cells (57), and keratinocytes (64). Thus the Eh for cytoplasmic Trx-1 is not in redox equilibrium with Eh for cellular GSH/GSSG. Studies of the mitochondrial compartment are more limited, but in HeLa, HT29, THP-1 cells, and isolated liver mitochondria, Eh for Trx-2 was −360 to −330 mV, whereas estimates for Eh GSH/GSSG were approximately −300 mV (21, 57, 64, 66, 70). Together, these results show that major thiol-disulfide redox components within subcellular and extracellular compartments are not equilibrated.
Trx and GSH redox systems are functionally distinct.
In addition to the biochemical evidence as illustrated in Figs. 4 and 5, several studies show that the Trx and GSH systems are functionally distinct in cells. For instance, mitochondrial Trx-2 was found to be more sensitive to oxidation than cytoplasmic Trx-1 by exogenously added peroxides (27), certain metals (arsenic and cadmium) (69), and TNF-α (70). The nuclear compartment was more resistant to oxidation than cytoplasm as indicated by Trx-1 oxidation (57) and the extent of nuclear protein GS-ylation (57, 63) in cells with targeted nuclear generation of H2O2 or limited NADPH generation. The secretory pathway has also been shown to be relatively oxidized compared with the cytoplasm (76).
The nonequilibrium steady-state Eh values for Trx-1, GSH/GSSG, and Cys/CySS couples show that in biological systems, proteins that rapidly interact with Trx-1 do not rapidly interact with GSH/GSSG or Cys/CySS (88). This explicitly means that Trx-1 and glutaredoxin (Grx-1) do not function as reversible reductants/oxidants for the same protein substrates because such reactivity would equilibrate the couples. Thus the nonequilibrium conditions support the concept that GSH and Trx function as control nodes for different redox networks.
Electron transfer within a redox pathway is kinetically limited.
In a study of electron flow within a thiol-disulfide pathway, Go et al. (57) used redox Western blot analysis to measure Trx-1 redox state and BIAM-blot to measure fractional reduction of TrxR1, Ref-1 (redox factor-1), and the p50 subunit of NF-κB. Under control conditions in HT29 cells, the fractional reduction in the cytosol was consistent with a redox gradient for the sequence NADPH→TrxR1→Trx-1→Ref1→NF-κB(p50) (Fig. 6). In contrast, the fractional reduction in the nuclei indicated that the transfer of electrons was blocked between Trx-1 and Ref-1 (Fig. 6). When cells were transferred to glucose- and glutamine-deficient media, the cellular NADPH pool became oxidized. Under this condition, each component in the cytoplasmic pathway became oxidized while the nuclear pathway was more uniformly reduced (Fig. 6). The results showed that even without an oxidative challenge, this pathway was kinetically limited both in the cytoplasm and in the nuclei, thereby supporting the interpretation that thiol-disulfide redox circuits are kinetically limited.
Fig. 6.

Kinetic limitation in a thiol-disulfide electron transfer pathway. Measurement of the fractional reduction of proteins in the pathway from NADPH through TrxR1, Trx-1, and redox factor-1 (Ref-1) to NF-κB p50 subunit under control conditions showed that the pathway is kinetically limited in both the cytoplasmic and nuclear compartments. In the nuclear compartment, the characteristics suggested that electron transfer was blocked between Trx-1 and Ref-1. Under conditions where NADPH supply was limited by removal of Glc and Gln from the culture media, the cytoplasmic pathway was selectively oxidized while the nuclear pathway showed a loss of the kinetic limitation. The results show that metabolic conditions determine sites of rate limitation, supporting the concept that rate control in redox pathways is important in cell signaling and regulation. Scheme is based upon data of Go et al. (57).
Estimates of tissue oxidant load in terms of H2O2 production rates.
Improved methods and additional study are needed to identify and understand the regulation of redox signaling and control circuits. The quantitative aspects are critically important, and a simple calculation illustrates some of the challenges to development of redox systems biology. The O2 consumption rate in humans is about 0.4 l/min and can be increased to a maximum value about 4 l/min. The latter value is 2,500 μmol·kg−1·min−1 (or 5,000 μmol·kg−1·min−1 if considered as 2-electron transfers) when averaged for the entire body (Table 2). About 1 to 4% of O2 consumption by mitochondria is normally converted to H2O2. At the 1% rate for 5,000 μmol·kg−1·min−1, the oxidant load for reversible two-electron thiol oxidation is 50 μmol·kg−1·min−1. The actual rate of H2O2 generation is greater than this because redox signaling involves cytoplasmic H2O2 generation in addition to that produced by mitochondria, and H2O2 is also produced by metabolic oxidases. At the upper limit, H2O2 production in liver cells in response to exogenously added substrates for peroxisomal glycolate oxidase, outer mitochondrial membrane monoamine oxidase, or microsomal cytochrome P450 showed that 10% of the O2 can be converted to H2O2. Thus H2O2 generation is probably in the range of 50 μmol·kg−1·min−1 but may be lower under some physiological conditions and increased to 500 μmol·kg−1·min−1 upon stimulation (Table 2).
Table 2.
H2O2 generation and redox turnover of protein thiols
| O2 consumption rate (maximal) | 5,000 μmol·kg−1·min−1 |
| H2O2 production | |
| At 1% of maximal O2 consumption rate | 50 μmol·kg−1·min−1 |
| At 10% of O2 consumption rate | 500 μmol·kg−1·min−1 |
| Thiols oxidized by H2O2 | |
| At 1% of O2 consumption | 0.5% of total thiols/min 1,070 of 214,000 protein Cys |
| At 10% of O2 consumption | 5% of total thiols/min |
| Max NADPH supply rate | 500 μmol·kg−1·min−1 |
Rates are estimated based on 2-electron transfer rates, averaged for the entire body. Each value is subject to variation and error in estimation. H2O2 production rates by mitochondria have been measured to be 1 to 4%, and nonmitochondrial H2O2 production rates are similar to the mitochondrial rates, so rates could be higher than indicated. Average O2 consumption rate is about 10% of the maximal O2 consumption rate, so average rates could be lower than the values indicated. Thiol content is about 20,000 μmol/kg; calculations are based on 2 thiol oxidized to disulfide per H2O2. This value could be in error by a factor of 2 in either direction. Most H2O2 metabolism appears to occur by thiol-dependent mechanisms, so calculations of thiol turnover per minute assumes 2-SH oxidized to disulfide per H2O2 generated. NADPH supply rate is estimated from the maximal NADPH supply rate in liver assuming that 50% of the NADPH supply is available for disulfide reduction.
Estimates of global thiol oxidation rate based on H2O2 metabolism.
The thiol content in tissues is about 100 nmol·mg protein−1. If one assumes that tissues are 20% protein, then each kilogram of tissue contains about 200 g of protein. This means that thiol content is probably about 20,000 μmol/kg body mass. Although some tissues contain up to 10 mM GSH, most cells contain only about 1 mM so that the GSH content averaged over the whole body is ∼1,000 μmol/kg. Considered in terms of two-electron oxidation to disulfides, the total dithiol content represents about 10,000 μmol/kg (Table 2). Comparison of this value with the H2O2 generation rate from above shows that the rate of oxidant generation by mitochondrial O2 consumption (50 μmol·kg−1·min−1) is sufficient to oxidize 0.5% of the cellular thiols (2 thiols/H2O2) per minute, assuming that catalase contributes minimally at physiological H2O2 generation rates (32, 85, 164) (Table 2). If 10% of the maximal O2 consumption were converted to H2O2, 5% of the total thiol content could be oxidized per minute. The maximal rate of NADPH supply in liver cells is about 5 nmol·mg protein−1·min−1 (183). If one assumes that 50% of the NADPH supply were available for disulfide reduction, about 500 μmol·kg−1·min−1 could be available for peroxide reduction (Table 2). This value is roughly equivalent to the capacity to reduce 5% of the protein thiols (2 thiols/NADPH) per minute. Although these quantitative estimates could be in error (see legend to Table 2), the results indicate that ongoing thiol oxidation occurs at a rate that is about 0.5% of the cellular thiol content per minute. Thus, in contrast to the view that thiols are present in the reduced form and that GSH buffers against oxidation, the data indicate that a relatively rapid continuous oxidation of thiols occurs.
Partitioning of H2O2 metabolism between enzyme systems.
The knowledge of peroxide metabolizing systems has progressed with sequential discovery of catalase, Sel-dependent GSH peroxidase, Sel-independent GSH peroxidases (glutathione transferases), and peroxiredoxins. Several lines of evidence indicate that in mammalian cells, catalase has little contribution to peroxide metabolism outside the peroxisomes (32, 85, 164). Before the recognition of peroxiredoxins, peroxide metabolism was assumed to be largely catalyzed by the Se-dependent and Se-independent GSH peroxidases (20). Partitioning of metabolism between these systems was inferred from GSSG efflux in perfused liver and heart preparations (20, 201). Protein thiol oxidation was not measured in these studies, and it is unclear how efficiently GSSG was reduced back to GSH, as opposed to being transported out of cells. A similar study with steady-state infusion of diamide in hepatocytes showed that protein thiol oxidation constituted 73% of the net thiol oxidation while GSH oxidation was only 27% of the total (183). Thus substantial protein oxidation can occur without depletion of GSH, indicating that protein and GSH oxidation occur concurrently.
If metabolism of endogenously produced H2O2 is partitioned among all 214,000 Cys residues in proteins, this could mean that about 1,070 specific protein thiols (i.e., 0.5% of total) undergo reversible oxidation/reduction at a rate of 1 per minute (see Table 2). Because thiols are present in redox pathways where thiols of one protein donate electrons to disulfides of other proteins [e.g., NADPH-TrxR1-Trx-1-Ref1-NF-κB(p50)], the actual number could be three- to fivefold higher. Thus the emergent picture is that there are hundreds to thousands of dynamic redox control elements (out of the estimated 21,000 to 42,000 readily oxidizable thiols) that support redox signaling and integration of metabolic functions through low-current redox circuits. In other words, the classes of redox elements described in Fig. 2 can be controlled by redox systems as described in Figs. 4 and 5 to coordinate and regulate cell functions through kinetically controlled pathways.
ΔEh values are sufficient to provide an energetic driving force for low current redox control pathways.
The Eh values for thiol-disulfide couples range from −360 mV for Trx-2 in mitochondria to a mean value of −60 mV for Cys/CySS in plasma. According to the Nernst equation where n = 2, Eh becomes 30 mV more positive with a 10-fold increase in the ratio of oxidized:reduced forms. This means that a ΔEh of 60 mV between two couples is sufficient to drive a 100-fold change in dithiol-to-disulfide ratio. The 300-mV span between mitochondrial Trx-2 and Cys/CySS is therefore sufficient to control different redox functions through relatively shallow redox gradients provided that spatial or catalytic mechanisms are available to control rates. Because H2O2 and O2 are always present under aerobic conditions, coupling of electron transfer to peroxidase or oxidase reactions can further provide an energetic driving force to maintain function of low-current redox control pathways.
The span of redox potentials for known thiol-disulfide couples in different subcellular compartments is summarized in Fig. 7 to illustrate the magnitude of possible effects on protein thiol-disulfide redox state. If a hypothetical protein with a dithiol-disulfide motif with Eo value of −210 mV were equilibrated with cytoplasmic GSH/GSSG at −210 mV, the dithiol-to-disulfide ratio would be 1. If the same protein were equilibrated with cytoplasmic Trx-1 at −270 mV, the ratio would be 100:1. If the protein were equilibrated with mitochondrial Trx-2 at −360 mV, the ratio would be 100,000:1. If the protein were equilibrated with cytoplasmic Cys/CySS at −150 mV, the ratio would be 1:100. Equilibration with plasma Cys/CySS at −60 mV would yield 1:100,000. Thus, if catalysts or autocatalytic structures are present to allow selective interaction of thiols with the central redox control nodes, the range of redox potentials is sufficient to provide a considerable organization of protein structures and functions.
Fig. 7.

Steady-state redox potential (Eh) values for thiol-disulfide couples within different subcellular compartments. Approximate steady-state Eh values for mitochondrial Trx-2, mitochondrial GSH/GSSG, nuclear Trx-1, cytoplasmic Trx-1, cytoplasmic GSH/GSSG, cytoplasmic cysteine/cystine (Cys/CySS), endoplasmic reticular GSH/GSSG, plasma GSH/GSSG, and plasma Cys/CySS are listed along with different dithiol-disulfide ratios (PrSH/PrSS) for a hypothetical protein couple with Eo equal to −210 mV. This comparison shows that the range of Eh values is sufficient for relatively shallow redox gradients between couples to control protein functions based on catalyzed redox circuits between redox couples or between the listed couples and the H2O2/H2O couple (not shown).
Redox pathways control different switchable elements within signal transduction pathways.
On the basis of the knowledge that Trx and GSH have differential activities toward reduction of protein substrates, Trx and GSH could function as control nodes for redox networks that integrate and coordinate cellular processes through series of parallel redox elements (Fig. 8). There are many different reaction pathways that could be considered based on existing knowledge. A general scheme with parallel switches controlling redox state of individual thiols can be written as:
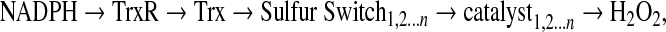 |
where Trx controls a series of sulfur switches. Each switch has an autocatalytic character due to proximal cationic residues or has a specific enzyme catalyst such as one of the peroxiredoxins or other Trx family members. Thus the oxidation of the switch is determined by either proximity of the elements to the H2O2 generation or activation of the catalyst. Similar pathways can be written for GSH-dependent pathways (Fig. 8) in which GS-ylation can be catalyzed by glutathione transferases in the presence of H2O2 and countered by Grx-dependent removal of the GSH moiety. Alternatively, GS-ylation could occur as a consequence of local Gpx-dependent generation of GSSG in the presence of H2O2. In this case, a transient burst of H2O2 could generate a local GS-ylation with an autorecovery after the peroxide burst. Other proteins, such as the Trx reductases (10), thioneins (148), and redox factor-1 [Ref. 1; (200)] may also serve as control nodes supporting additional redox control networks.
Fig. 8.

Scheme depicting possible organization of Trx- and GSH-dependent pathways into redox control networks. Both Trx and glutaredoxins (Grx) catalyze oxidation-reduction reactions with multiple protein substrates. Both are dependent on electron transport pathways with NADPH as the electron donor. Left, Trx is reduced by TrxR and in turn reduces sulfur switches, which are protein disulfide or sulfenic acids [designated Pr1(SS), Pr2(SS), etc.]. The sulfur switches are depicted as either 1) cysteine residues present in autocatalytic structures that are directly oxidized by H2O2 or 2) substrates for catalysts that are oxidized by H2O2. Right, Grx uses reducing equivalents from GSH, which is maintained by GSSG reductase, to reduce sulfur switches designated as PrA, PrB, etc. The precise mechanism for introduction of GSH moieties into proteins is not clear. In this figure, GS-ylation of these sulfur switches is depicted (hypothetically) as being catalyzed by 3) glutathione transferase (GST), in the presence of H2O2. Alternatively, 4) local generation of H2O2 can generate GSSG by Gpx, and the local high GSSG concentration could be used by Grx to form the S-glutathionyl derivative of the sulfur switch. Protein substrates for Trx and Grx are discussed in the text, but no studies are available to test interactions of pathways in functional Trx or GSH/Grx networks.
Integration of redox mechanisms with cell signaling.
In systems biological models, control by redox networks would be superimposed upon other signaling and control processes. To illustrate this point, redox-sensitive sites have been identified in a hypothetical pathway based on the known proinflammatory signaling events in endothelial cells (Fig. 9A). Signaling in this pathway is responsive to 1) extracellular Eh of Cys/CySS through 2) a cell sensor that can be inactivated by pretreatment with a cell-impermeant alkylating agent (55). 3) Intracellular signaling through mitogen-activated protein kinases is opposed by redox-sensitive phosphatases, e.g., PTP-1B (39). H2O2 is generated by 4) Nox enzymes and mitochondria and 5) eliminated by peroxidases [as shown by sensitivity to polyethylene glycol-catalase (55)]. Dependence on mitochondrial oxidant production is indicated by sensitivity to increased Trx-2 (Y. -M. Go and D. P. Jones, unpublished observation). 6) Oxidant activation of NF-κB through enhanced IκB kinase degradation is blocked by both GSH and Trx systems (195). 7) Both p50 and p65 subunits of NF-κB undergo GS-ylation at conserved Cys residues, and NF-κB binding to DNA and transcriptional activation are sensitive to nuclear Trx-1 and changes in GSH/GSSG redox states (143). 8) Translation is sensitive to redox conditions through Trx interactions and Trx-like domains in elongation initiation factors. 9) Processing of proteins destined for secretion or insertion in the plasma membrane, such as VCAM-1, ICAM-1, and E-selectin often require redox-dependent processing in the secretory pathway (7, 24). 10) Function of cell surface receptors is dependent on redox-sensitive Cys in integrins, cytoskeletal proteins (e.g., actin, cofilins), and Cys-rich regions in the receptors. Thus the cumulative knowledge concerning the components of the proinflammatory signaling pathway shows that multiple sites are redox sensitive and suggests that the redox events must have some underlying organization.
Fig. 9.

Redox-sensitive steps in hypothetical redox signaling pathways. Accumulated research from multiple investigators provides evidence for complex spatial and temporal redox events that can be combined in hypothetical signaling pathways with multiple redox sensitive steps. Evidence is collected from experiments using different cell lines and under different conditions so that the combined reactions should not be considered validated reaction pathways, but rather, as evidence that redox signaling involves so many redox-sensitive sites that the regulation is likely to have evolved a functional design that is coordinated through a smaller number of control nodes. Sites sensitive to changes in thiol-disulfide redox state are indicated by an “-SH/-SS-” balance. A: in early proinflammatory signaling in endothelial cells, redox-dependent signaling includes 1) extracellular Cys/CySS redox potential (55); 2) cell surface thiol sensor (55); 3) kinase/phosphatase regulation (39); 4) mitochondrial oxidant generation from mitochondrial electron transport (Mito ET) (Go and Jones, unpublished); 5) H2O2 (55); 6) NF-κB activation involving IκB phosphorylation and degradation (182); 7) NF-κB binding to DNA (160); 8) translation (109); 9) processing of proteins in the secretory pathway (7, 24); and 10) cytoskeletal/surface structure (33, 140, 187). B: in receptor-mediated signaling, redox-sensitive steps include 1) metalloprotease-sensitive growth factor release (134); 2) metalloprotease-sensitive degradation of growth factor inhibitor (51); 3) redox-dependent activation of receptors (31, 94); 4) H2O2-dependent Ca2+ influx and Nox-5 activation (40); 5) active-site Cys residues required for phosphatase activity [PTP1B, SHP2, PTen; (39)]; 6) Ras activity (3); 7) Src activity (48); 8) H2O2 metabolism; 9) lipoxygenase activity (42); 10) LPS activation of cytoplasmic and mitochondrial H2O2 production through Toll-like receptor 4 [TLR4 (77, 139)] (42, 77, 139).
Other biological processes similarly have multiple redox-sensitive steps. To illustrate this point, a composite of receptor activation pathways with identification of redox-sensitive sites is given in Fig. 9B. 1) In this composite, extracellular redox-dependent steps have been identified for metalloproteinase-dependent TGF-α release from the cell surface and activation of signaling by the EGF receptor (134). 2) Redox-dependent growth control can also be mediated by a metalloproteinase-dependent release of IGF-1 from IGF-1 binding protein (51). 3) Conserved Cys-rich domains are present in some receptors, and these are potential redox-sensitive elements (31, 94). 4) Ca2+ influx and activation of Nox-5 production of H2O2 is dependent on an oxidant-mediated activation (40). 5) A number of phosphatases that function in control of receptor tyrosine kinase signaling, including PTP-1B, SHP2, and PTen, are redox sensitive (39). 6) As previously described, Ras activity is dependent on GS-ylation and nitrosylation at Cys118 (3). 7) Src activity is also redox dependent (48). 8) GSH- and Trx-dependent systems control H2O2 concentrations within different subcellular regions (67), potentially differentially controlling signaling steps. 9) Production of other peroxides, such as through Rac-dependent activation of 5-lipoxygenase, can also provide specific oxidant signaling (42). 10) Similarly, mitochondrial and nonmitochondrial production of H2O2 in response to LPS activation of Toll-like receptor-4 provides additional sites of redox sensitivity (77, 139).
The extensive literature on redox signaling clearly supports the concept that multiple redox elements are present. Although integrated models with experimental support for simultaneous involvement of multiple redox sites are not available, the large number of redox-sensitive steps identified indicates that underlying organizational principles must exist and that these probably involve GSH- and Trx-dependent redox networks. Thus there is a considerable need to extend research efforts beyond experimental models with nonspecific reagents [such as N-acetylcysteine (NAC) and high concentrations of H2O2] to ones in which the specificity of redox signaling and control can be elucidated. The available evidence supports the third postulate of the redox hypothesis, namely, that thiol redox pathways are kinetically limited. Studies to identify kinetically limiting sites of redox control will provide sites that are subject to disruption during oxidative stress.
Oxidative Stress as a Disruption of Redox Signaling and Control
The above conceptualization that redox mechanisms contribute to structural and functional organization of cells through a continuous, reversible oxidation/reduction of about 0.5% of the Cys proteome provides a basis to consider the fourth postulate of the redox hypothesis, namely that oxidative stress is a condition where there is a disruption in the normal function of the redox networks. Because the networks involve reversible two-electron oxidations, disruption can occur with or without concomitant free radical-mediated macromolecular damage. Organization of redox elements in insulated redox circuits allows for specificity in oxidative disruption at low levels of oxidants due to type and source of oxidant. In contrast, at high levels, a more global disruption would occur. Thus, in the context of the first three postulates, the fourth postulate predicts specificity in oxidant-dependent mechanisms in disease under usual in vivo conditions and a loss of such specificity when excessive oxidants are introduced.
H2O2 as a constitutively produced nonradical oxidant in cells.
H2O2 is a normal cellular metabolite that is continuously generated and maintained at low concentrations by a number of peroxidases. Techniques based on the spectral and kinetic properties of catalase (137, 162) provided the first reasonable estimates of H2O2 concentration in mammalian cells. Importantly, these studies showed that steady-state H2O2 production occurs at rates that maintain nanomolar concentrations; during detoxification, rates can be stimulated in the liver to >10% of the total O2 consumption rate (25, 85, 164). Measurements by this approach largely reflect concentrations and metabolism in peroxisomes. Peroxisomes are abundant only in some organs, e.g., liver and kidney, and may contribute little to H2O2 metabolism in other tissues (201). Thus quantitative evaluation of peroxide metabolism in other compartments is indirect.
Mitochondria continuously release peroxide at rates that are estimated to be 1% to 4% of the mitochondrial O2 consumption rate (25). Consequently, mitochondria are a major constitutive source of H2O2 outside of peroxisomes (25). Other sources include H2O2-producing enzymes distributed in subcellular compartments, e.g., Nox family enzymes (cytoplasm, plasma membrane, nuclei), xanthine oxidase (cytoplasm, extracellular), endoplasmic reticulum oxidases, sulfhydryl oxidases (nuclei, extracellular), thiol oxidases (plasma membrane, plasma), and monoamine oxidases (mitochondria outer membrane). Data are available, therefore, to show that H2O2 is produced throughout the cell. Furthermore, increased H2O2 has been implicated in disease and/or toxicity from peroxisomal proliferators (100), mitochondrial respiratory inhibitors (186), activators of Nox (59, 60), ER stress (71), precursors of xanthine oxidase (58), and substrates of monoamine oxidases (167). While previously considered in terms of a global imbalance of pro-oxidants and anti-oxidants that cause macromolecular damage, these data could alternatively mean that there is a constitutive peroxide tone in subcellular compartments that provides a counterbalance to the Trx and GSH systems to maintain nonequilibrium steady states for redox control circuits (88).
In vitro studies support the interpretation that H2O2 is a signaling molecule.
The discoveries that low levels of peroxides stimulate cell growth (19), that cancer cells produce increased H2O2 (174), and that a family of NADPH oxidases (Nox) is associated with cell proliferation (101, 172), led to the general recognition that redox signaling pathways have an important function in growth control. Superoxide is an immediate product of Nox, but sequential one-electron transfer and/or rapid dismutation of superoxide result in substantial H2O2 generation. Importantly, catalase and peroxidases affect redox signaling (55, 67, 101) showing that H2O2 is a quantitatively important signaling species.
Studies with targeted increases in abundance of Trx, peroxiredoxins, enzymes of GSH biosynthesis, and GSH peroxidases provide evidence for compartment-specific redox regulation involving H2O2. Cytoplasm- and nuclei-specific redox control in transcription factor activation (1) has been supported by recent evidence that H2O2 functions in both cytoplasmic activation of NF-κB and Nrf-2 and nuclear inhibition of transcription factor binding to DNA (67, 68). For instance, a nuclear export signal (NES)-containing fusion protein (NES-Prx-1) of the antioxidant Prx-1 blocked oxidant-induced activation of NF-κB in the cytoplasm (67). A different fusion protein (NLS-Prx-1) containing Prx1 linked to a nuclear localization sequence (NLS) enhanced nuclear NF-κB activity (67). Thus the data suggest that a constitutive H2O2 concentration exists, which can be decreased in the cytoplasm to block activation while a similar decrease in the nuclei results in increased transcription.
In vitro studies support interpretation that H2O2 is a toxic species.
Elevated peroxide concentrations can contribute to death signaling without macromolecular damage because oxidation of either Trx-1 or Trx-2 stimulates release of Ask-1 and activation of apoptosis (149, 206). Protection against oxidant-induced apoptosis was observed in 143B osteosarcoma cells and SH-SY5Y human neuroblastoma cells with the nonradical reductant mitochondrial Trx-2 (28, 29). Decreased mitochondrial Trx-2 in DT40 chicken B cells increased oxidants and caused spontaneous apoptosis (175). Similarly, decrease in the Trx-2-dependent peroxidase Prx3 increased H2O2 and sensitized HeLa cells to staurosporine- or TNF-α-induced apoptosis (26). Increased abundance of Prx3 also protected against apoptosis caused by H2O2 (205). Depletion of mitochondrial GSH also potentiated peroxide accumulation and sensitized cells to cell death (8, 157). Small interferring RNA (siRNA) for Grx-2 increased sensitivity to doxorubicin, whereas overexpression of mitochondrial Grx-2 inhibited cardiolipin loss and prevented apoptosis induced by doxorubicin (41, 111). Because Trx and GSH protect against two-electron oxidants, this in vitro research points to the plausibility of oxidant-induce cell death mechanisms signaled by nonradical reactions, which disrupt redox control.
In vivo mouse models support nonradical mechanisms of oxidative stress.
Genetic mouse models with altered peroxide-metabolizing enzymes also support the interpretation that two-electron oxidants have a central causative role in oxidative stress. Expression of catalase in mitochondria increased longevity in transgenic mice (154). Decreased Trx-2 in hemizygous Trx-2+/− mice had increased sensitivity to oxidants (141), and Trx-1 transgenic mice were protected against cardiotoxicity from doxorubicin (159). Gpx1 knockout mice demonstrated increased susceptibility to H2O2 (36). Gpx4 (−/−) knockouts are embryonic lethal, but cells isolated from Gpx4-deficient mice (+/−) showed an increased susceptibility to peroxides (202). Similarly, the GSH synthesis inhibitor buthionine sulfoximine (BSO) potentiated MPTP toxicity in mice (199). Furthermore, MPTP treatment depleted brainstem GSH (204), and mice deficient in Gpx-1 exhibited increased vulnerability to MPTP (37) .
Other nonradical oxidative chemicals are produced in tissues.
In this review, I limited discussion to H2O2, but other nonradical oxidants are also likely to contribute to disease. These include lipid hydroperoxides and epoxides generated from polyunsaturated fatty acids by cyclooxygenases, lipoxygenases, and cytochromes P450, aldehydes generated from oxidation of ethanol and other alcohols, and quinones generated from catechols and polyphenols. However, some of these chemicals, such as conjugated aldehydes, are also electrophilic and can disrupt redox circuits by chemical modification of protein thiols. Importantly, thiols are among the most sensitive functional groups for modification by reactive chemicals. Acrolein and 4-hydroxynonenal selectively modify C73 of Trx-1 when added at low concentrations (54). Microinjection of C73-Acr-Trx-1 into endothelial cells showed that this single modification was sufficient to activate pro-inflammatory signaling and monocyte adhesion (54). Specificity in quinone toxicity has also been implicated in the opposing roles of NADPH:quinone reductase (NQO1) and the related quinone reductase-2 (QR2) in quinone toxicity in the retina (18). Metal ions, such as mercury, cadmium, lead, and arsenic also can cause selective disruption of redox circuits based on the specificity of thiol interactions. For instance, 10 μM Fe3+ or Cu2+ oxidized GSH/GSSG but not Trx-1 or Trx-2, whereas 10 μM As3+ or Hg2+ oxidized Trx-1 and Trx-2 without oxidation of GSH/GSSG (69).
Nonradical mechanisms of oxidative stress in human disease.
Disruption of thiol-disulfide redox pathways as a contributing mechanism to human disease has not been explicitly tested, although several correlative studies support this possibility. The most direct studies are based on cellular research showing that signaling pathways that contribute to disease mechanisms are dependent on extracellular thiol-disulfide redox state. For instance, microinjection of C-73-modified Trx-1 into endothelial cells activated signaling for monocyte adhesion (54). This elicitation of pathogenic signaling by modification of a single amino acid on a protein shows that specificity occurs in oxidative stress effects on redox signaling pathways.
Another example involves the death receptor Fas, which has been found in regions of the retina affected by age-related macular degeneration (AMD). Fas ligand stimulates apoptosis in retinal pigment epithelial (RPE) cells, the initial cells to be lost in AMD (78, 80). Cell studies showed that peroxide-induced cell death in RPE is stimulated by oxidized extracellular Eh (79). Extracellular Eh was oxidized in association with age in AMD patients (128), and soluble Fas ligand increased in human plasma with increasing age (78). A relevant double-blind interventional trial with antioxidants (vitamins C and E; β-carotene) and Zn2+ showed that the antioxidants and Zn2+ protected against progression of AMD and also against age-related increase in plasma CySS and oxidation of plasma Cys/CySS redox state (128, 129). Thus the results suggest a causal pathway in which antioxidants and Zn2+ control plasma Cys/CySS redox state and plasma Cys/CySS redox state controls redox-dependent signaling of apoptosis in RPE, which contributes to AMD.
Other studies relevant to cardiovascular disease show that monocyte adhesion to endothelial cells, an early proinflammatory step in atherogenesis, is stimulated by an oxidized Cys/CySS redox state. The specific mechanism involves intracellular generation of H2O2, activation of NF-κB, and transcriptional activation and increased cell surface expression of cell adhesion molecules (55). Both GSH/GSSG and Cys/CySS redox states are oxidized in association with cardiovascular disease risk factors, including cigarette smoking (127) and chronic alcohol abuse (203). Oxidized plasma GSH/GSSG redox state has also been associated with Type 2 diabetes (151) and increased carotid intima media thickness, an early risk factor for cardiovascular disease (11). Other in vivo studies show that an oxidized Cys/CySS redox state is associated with increased persistent atrial fibrillation (132) and with myocardial perfusion defects (2). Together, with in vitro mechanistic studies, these clinical studies indicate that redox-dependent changes in peroxide are likely to be important components of CVD risk.
Other in vitro studies also support mechanistic links between thiol-disulfide redox signaling at cell membranes and human disease. For instance, profibrotic signaling by TGF-β1 in pulmonary fibroblasts is stimulated by oxidized extracellular Eh (145), and this suggests that conditions which oxidize plasma Cys/CySS could contribute to fibrotic disease. Platelet activation has an optimal extracellular redox potential mediated through redox changes of Cys residues in αIIIbβ3 (43) and could be susceptible to aberrant function due to altered plasma redox states. Proliferative signaling in a colon cancer (CaCo-2) cell line is dependent on redox activation by a metalloproteinase, apparently providing redox-dependent growth factor release (134) or cleavage of growth factor inhibitors (51). Plasma Eh values for both GSH/GSSG and Cys/CySS are oxidized following chemotherapy (81), suggesting that antiproliferative effects may, in part, be mediated by altered signaling dependent on extracellular thiol-disulfide couples.
These circumstantial data point to the need for clinical studies to determine whether disruption of thiol-disulfide redox pathways by nonradical oxidants represents a quantitatively important mechanism for oxidative stress in human disease. Reanalysis of data from previous trials could provide information, but experimental designs developed to observe free radical-derived products may be inadequate to measure nonradical perturbation of redox pathways or discriminate relative importance of one- and two-electron reaction schemes. The numerous trials with NAC, a precursor to increase GSH, have provided inconsistent evidence for benefit in vivo (5) and evidence for potential toxicity in vitro (131). However, the extent to which NAC affects tissue GSH in humans is difficult to assess. Recent data showing that GSH/GSSG and Cys/CySS redox states undergo a diurnal variation linked to dietary intake of sulfur amino acids (16) suggest careful timing and dosing may be needed because NAC absorption and clearance are rapid. Furthermore, homeostatic regulation of Cys and GSH declines with age, and this occurs at a younger age in men than in women (16). Thus dosing regimens may be critical in development of antioxidant strategies designed to protect or correct disruption of thiol redox systems by nonradical oxidants.
Perspectives
The available evidence supporting the redox hypothesis indicates both opportunities and challenges for future development. There is considerable need to elucidate the thiol redox signaling and control pathways and identify critical sites where interventions could protect against disruption of function and/or restore normal signaling and control. The recent development of methods to measure redox states of specific proteins in different organelles (56) will allow more complete delineation of compartment-specific disruption of redox pathways in disease. These methods include detection of reduced and oxidized forms of proteins by redox Western blot analysis (64, 189). The redox Western blot analyses of Trx-1 and Trx-2 have recently been found to be suitable for use with human biopsy samples. Results with this approach (Y. Mannery, L. Hao, D. P. Jones, T. R. Ziegler, unpublished observations) show that redox states in normal intestinal biopsies are similar to values obtained in cell culture and that redox states are oxidized in inflamed tissues. An alternate BIAM blot uses an initial labeling of thiols with biotinylated maleimide, followed by immunoprecipitation and Western blot with detection by streptavidin (90).
New discovery-based redox proteomic methods with mass spectrometry use Isotope-Coded Affinity Tag (ICAT) reagent to label thiols with heavy (13C) and light (12C) forms of biotin-labeled iodoacetamide to quantify redox states of specific Cys residues in proteins (56, 108). Sequential addition of the heavy isotopomer before reduction and then addition of light isotopomer after reduction provides a measure of the reduced fraction of specific Cys residues in terms of the ratio of labeling. This method is also sensitive enough to allow measurement of redox states in proteins in blood cells and biopsies (Y.-M. Go, D. P. Jones, unpublished observations). Application of this approach could considerably enhance understanding of physiological and pathological variations in redox signaling and control in humans. Imaging approaches using magnetic resonance spectroscopy for GSH measurement in the brain (207), fluorescence imaging of GSH/GSSG redox potential (61), and thiol-dependent redox-sensitive contrast agents are also being developed and provide means to evaluate redox signaling and control in vivo.
In summary, research literature on thiol-disulfide systems provides the basis for four postulates in the development of a redox hypothesis of oxidative stress. This hypothesis is that critical mechanisms of oxidative stress that contribute to human aging and disease involve nonradical two-electron oxidations rather than free radical reactions. A central observation is that redox-sensitive Cys residues, which undergo reversible two-electron oxidation-reduction reactions, are common in proteins. These Cys residues have important functions as active sites residues, as regulators of macromolecular interactions and as allosteric regulators. The number of distinct redox-sensitive elements, which includes sites undergoing oxidation to disulfides and sulfenic acids, S-nitrosylation, and GS-ylation, appears to be in the range of a few hundred to several thousand distinct Cys residues, with total oxidative turnover of about 0.5% of cell thiols per minute.
Arguments are developed to suggest that these elements exist in redox pathways dependent on controlled generation of oxidant, such as H2O2, balanced by NADPH-dependent reduction pathways. Available evidence indicates that these pathways exist in networks controlled by Trxs and GSH. However, these networks currently are poorly defined and could include other control nodes as well as unidentified catalysts for transnitrosylation, GS-ylation, or other redox reactions.
In the limited number of pathways studied to date, oxidation and reduction of thiol elements are kinetically limited. Noncatalyzed thiol-disulfide exchange rates are slow at prevailing concentrations of thiols and disulfides in cells. This provides a kinetic insulation that is essential for dynamic control of the low-current redox circuits. Under these conditions, efficient control of reactive thiols is obtained by spatial reorganization and regulation of activity of redox catalysts.
Available evidence indicates that redox-sensitive thiol elements function in signaling and control of virtually all aspects of life, including energy regulation, cytoskeletal structure, transport, proliferation, differentiation, and apoptosis. Consequently, disruption of thiol redox signaling and control by nonradical, two-electron oxidants could underlie much of the pathology linked to oxidative stress. To test this hypothesis, research is needed to identify critical redox elements and understand their physiology. Such knowledge can be expected to provide the basis for novel therapeutic approaches to restore normal signaling and control and, thereby, prevent pathological and toxicological consequences of oxidative stress.
GRANTS
Support for research in the author's laboratory was provided by National Institute of Environmental Health Sciences Grants ES-009047 and ES-011195.
Acknowledgments
The author gratefully acknowledges the estimation of the number of unique Cys and methionine residues in the human proteome provided by Scott Devine and the critical review of the manuscript by Young-Mi Go, Jennifer M. Johnson, and Lou Ann S. Brown.
REFERENCES
- 1.Abate C, Patel L, Rauscher FJ, 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 249: 1157–1161, 1990. [DOI] [PubMed] [Google Scholar]
- 2.Abramson JLJD, Bremner JD, Quyyumi AA, Goldberg J, Vaccarino V. Oxidative stress, as indicated by the redox states of plasma thiol/disulfide couples, is associated with myocardial perfusion defects in men without clinical CAD (Abstract). JACC 45: 401A, 2005. [Google Scholar]
- 3.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem 279: 29857–29862, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed U, Dobler D, Larkin SJ, Rabbani N, Thornalley PJ. Reversal of hyperglycemia-induced angiogenesis deficit of human endothelial cells by overexpression of glyoxalase 1 in vitro. Ann NY Acad Sci 1126: 262–264, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aitio ML N-acetylcysteine–passe-partout or much ado about nothing? Br J Clin Pharmacol 61: 5–15, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson CL Effect of Glutathione Synthesis Inhibitor Buthionine Sulfoximine on the Redox State of Cellular and Extracellular Glutathione and Cysteine (Master's Degree). Atlanta, GA: Emory University, 2002.
- 7.Anelli T, Ceppi S, Bergamelli L, Cortini M, Masciarelli S, Valetti C, Sitia R. Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO J 26: 4177–4188, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. FASEB J 16: 1263–1265, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong RN Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol 10: 2–18, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 267: 6102–6109, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Harrison DG, Quyyumi AA. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J Am Coll Cardiol 47: 1005–1011, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol 204: 238–255, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Banhegyi G, Lusini L, Puskas F, Rossi R, Fulceri R, Braun L, Mile V, di Simplicio P, Mandl J, Benedetti A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J Biol Chem 274: 12213–12216, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol 292: H1227–H1236, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425: 980–984, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Blanco RA, Ziegler TR, Carlson BA, Cheng PY, Park Y, Cotsonis GA, Accardi CJ, Jones DP. Diurnal variation in glutathione and cysteine redox states in human plasma. Am J Clin Nutr 86: 1016–1023, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Bloom D, Dhakshinamoorthy S, Jaiswal AK. Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 21: 2191–2200, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Boutin JA, Chatelain-Egger F, Vella F, Delagrange P, Ferry G. Quinone reductase 2 substrate specificity and inhibition pharmacology. Chem Biol Interact 151: 213–228, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Burdon RH Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med 18: 775–794, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Burk RF, Nishiki K, Lawrence RA, Chance B. Peroxide removal by selenium-dependent and selenium-independent glutathione peroxidases in hemoglobin-free perfused rat liver. J Biol Chem 253: 43–46, 1978. [PubMed] [Google Scholar]
- 21.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 273: 11401–11404, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Carp H, Miller F, Hoidal JR, Janoff A. Potential mechanism of emphysema: alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci USA 79: 2041–2045, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci USA 99: 9745–9749, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cenci S, Sitia R. Managing and exploiting stress in the antibody factory. FEBS Lett 581: 3652–3657, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527–605, 1979. [DOI] [PubMed] [Google Scholar]
- 26.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem 279: 41975–41984, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett 580: 6596–6602, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Cai J, Murphy TJ, Jones DP. Overexpressed human mitochondrial thioredoxin confers resistance to oxidant-induced apoptosis in human osteosarcoma cells. J Biol Chem 277: 33242–33248, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Yu M, Jones DP, Greenamyre JT, Cai J. Protection against oxidant-induced apoptosis by mitochondrial thioredoxin in SH-SY5Y neuroblastoma cells. Toxicol Appl Pharmacol 216: 256–262, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Cheng G, Ikeda Y, Iuchi Y, Fujii J. Detection of S-glutathionylated proteins by glutathione S-transferase overlay. Arch Biochem Biophys 435: 42–49, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J Neurosci 21: 392–400, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen G, Hochstein P. Glutathione peroxidase: the primary agent for the elimination of hydrogen peroxide in erythrocytes. Biochemistry 2: 1420–1428, 1963. [DOI] [PubMed] [Google Scholar]
- 33.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. Actin S-glutathionylation: evidence against a thiol-disulphide exchange mechanism. Free Radic Biol Med 35: 1185–1193, 2003. [DOI] [PubMed] [Google Scholar]
- 34.D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Davis DA, Newcomb FM, Starke DW, Ott DE, Mieyal JJ, Yarchoan R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J Biol Chem 272: 25935–25940, 1997. [DOI] [PubMed] [Google Scholar]
- 36.de Haan JB, Bladier C, Griffiths P, Kelner M, O'Shea RD, Cheung NS, Bronson RT, Silvestro MJ, Wild S, Zheng SS, Beart PM, Hertzog PJ, Kola I. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 273: 22528–22536, 1998. [DOI] [PubMed] [Google Scholar]
- 37.De Haan JB, Crack PJ, Flentjar N, Iannello RC, Hertzog PJ, Kola I. An imbalance in antioxidant defense affects cellular function: the pathophysiological consequences of a reduction in antioxidant defense in the glutathione peroxidase-1 (Gpx1) knockout mouse. Redox Rep 8: 69–79, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Delaunay A, Isnard AD, Toledano MB. H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J 19: 5157–5166, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.den Hertog J, Groen A, van der Wijk T. Redox regulation of protein-tyrosine phosphatases. Arch Biochem Biophys 434: 11–15, 2005. [DOI] [PubMed] [Google Scholar]
- 40.El Jamali A, Valente AJ, Lechleiter JD, Gamez MJ, Pearson DW, Nauseef WM, Clark RA. Novel redox-dependent regulation of NOX5 by the tyrosine kinase c-Abl. Free Radic Biol Med 44: 868–881, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enoksson M, Fernandes AP, Prast S, Lillig CH, Holmgren A, Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun 327: 774–779, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Eom YW, Cho SH, Hwang JS, Yoon SB, Na DS, Kang IJ, Kang SS, Song WK, Kim JH. Rac and p38 kinase mediate 5-lipoxygenase translocation and cell death. Biochem Biophys Res Commun 284: 126–132, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Essex DW, Li M. Redox control of platelet aggregation. Biochemistry 42: 129–136, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA 99: 3505–3510, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fridovich I Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J Biol Chem 245: 4053–4057, 1970. [PubMed] [Google Scholar]
- 46.Fridovich I Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem 272: 18515–18517, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Fukagawa NK, Ajami AM, Young VR. Plasma methionine and cysteine kinetics in response to an intravenous glutathione infusion in adult humans. Am J Physiol Endocrinol Metab 270: E209–E214, 1996. [DOI] [PubMed] [Google Scholar]
- 48.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol 25: 6391–6403, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilbert HF Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol 63: 69–172, 1990. [DOI] [PubMed] [Google Scholar]
- 50.Giles GI, Tasker KM, Jacob C. Hypothesis: the role of reactive sulfur species in oxidative stress. Free Radic Biol Med 31: 1279–1283, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Giudice LC, Conover CA, Bale L, Faessen GH, Ilg K, Sun I, Imani B, Suen LF, Irwin JC, Christiansen M, Overgaard MT, Oxvig C. Identification and regulation of the IGFBP-4 protease and its physiological inhibitor in human trophoblasts and endometrial stroma: evidence for paracrine regulation of IGF-II bioavailability in the placental bed during human implantation. J Clin Endocrinol Metab 87: 2359–2366, 2002. [DOI] [PubMed] [Google Scholar]
- 52.Gladyshev VN, Hatfield DL. Selenocysteine-containing proteins in mammals. J Biomed Sci 6: 151–160, 1999. [DOI] [PubMed] [Google Scholar]
- 53.Go YM, Gipp JJ, Mulcahy RT, Jones DP. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J Biol Chem 279: 5837–5845, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Go YM, Halvey PJ, Hansen JM, Reed M, Pohl J, Jones DP. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am J Pathol 171: 1670–1681, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Go YM, Jones DP. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 111: 2973–2980, 2005. [DOI] [PubMed] [Google Scholar]
- 56.Go YM, Pohl J, Jones DP. Quantification of redox conditions in the nucleus. In: Methods in Molecular Biology: The Nucleus, edited by Hancock R. Totawa, NJ: Humana Press, 2008. [DOI] [PubMed]
- 57.Go YM, Ziegler TR, Johnson JM, Gu L, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic Biol Med 42: 363–370, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Granger DN Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 255: H1269–H1275, 1988. [DOI] [PubMed] [Google Scholar]
- 59.Griendling KK NADPH oxidases: new regulators of old functions. Antioxid Redox Signal 8: 1443–1445, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148, 1994. [DOI] [PubMed] [Google Scholar]
- 61.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559, 2008. [DOI] [PubMed] [Google Scholar]
- 62.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol 4: 743–749, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Halvey PJ, Hansen JM, Johnson JM, Go YM, Samali A, Jones DP. Selective oxidative stress in cell nuclei by nuclear-targeted d-amino acid oxidase. Antioxid Redox Signal 9: 807–816, 2007. [DOI] [PubMed] [Google Scholar]
- 64.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J 386: 215–219, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hansel A, Kuschel L, Hehl S, Lemke C, Agricola HJ, Hoshi T, Heinemann SH. Mitochondrial targeting of the human peptide methionine sulfoxide reductase (MSRA), an enzyme involved in the repair of oxidized proteins. FASEB J 16: 911–913, 2002. [DOI] [PubMed] [Google Scholar]
- 66.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol 46: 215–234, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Hansen JM, Moriarty-Craige S, Jones DP. Nuclear and cytoplasmic peroxiredoxin-1 differentially regulate NF-kappaB activities. Free Radic Biol Med 43: 282–288, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci 82: 308–317, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic Biol Med 40: 138–145, 2006. [DOI] [PubMed] [Google Scholar]
- 70.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci 91: 643–650, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003. [DOI] [PubMed] [Google Scholar]
- 72.Hayashi S, Hajiro-Nakanishi K, Makino Y, Eguchi H, Yodoi J, Tanaka H. Functional modulation of estrogen receptor by redox state with reference to thioredoxin as a mediator. Nucleic Acids Res 25: 4035–4040, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hisabori T, Hara S, Fujii T, Yamazaki D, Hosoya-Matsuda N, Motohashi K. Thioredoxin affinity chromatography: a useful method for further understanding the thioredoxin network. J Exp Bot 56: 1463–1468, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Holmgren A Thioredoxin. Annu Rev Biochem 54: 237–271, 1985. [DOI] [PubMed] [Google Scholar]
- 75.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem 271: 32253–32259, 1996. [DOI] [PubMed] [Google Scholar]
- 76.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257: 1496–1502, 1992. [DOI] [PubMed] [Google Scholar]
- 77.Ishida I, Kubo H, Suzuki S, Suzuki T, Akashi S, Inoue K, Maeda S, Kikuchi H, Sasaki H, Kondo T. Hypoxia diminishes toll-like receptor 4 expression through reactive oxygen species generated by mitochondria in endothelial cells. J Immunol 169: 2069–2075, 2002. [DOI] [PubMed] [Google Scholar]
- 78.Jiang S, Moriarty-Craige SE, Li C, Lynn MJ, Cai J, Jones DP, Sternberg P. Associations of plasma-soluble Fas ligand with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci 49: 1345–1349, 2008. [DOI] [PubMed] [Google Scholar]
- 79.Jiang S, Moriarty-Craige SE, Orr M, Cai J, Sternberg P Jr, and Jones DP. Oxidant-induced apoptosis in human retinal pigment epithelial cells: dependence on extracellular redox state. Invest Ophthalmol Vis Sci 46: 1054–1061, 2005. [DOI] [PubMed] [Google Scholar]
- 80.Jiang S, Wu MW, Sternberg P, Jones DP. Fas mediates apoptosis and oxidant-induced cell death in cultured hRPE cells. Invest Ophthalmol Vis Sci 41: 645–655, 2000. [PubMed] [Google Scholar]
- 81.Jonas CR, Puckett AB, Jones DP, Griffith DP, Szeszycki EE, Bergman GF, Furr CE, Tyre C, Carlson JL, Galloway JR, Blumberg JB, Ziegler TR. Plasma antioxidant status after high-dose chemotherapy: a randomized trial of parenteral nutrition in bone marrow transplantation patients. Am J Clin Nutr 72: 181–189, 2000. [DOI] [PubMed] [Google Scholar]
- 82.Jones DP Redefining oxidative stress. Antioxid Redox Signal 8: 1865–1879, 2006. [DOI] [PubMed] [Google Scholar]
- 83.Jones DP Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 348: 93–112, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med 28: 625–635, 2000. [DOI] [PubMed] [Google Scholar]
- 85.Jones DP, Eklow L, Thor H, Orrenius S. Metabolism of hydrogen peroxide in isolated hepatocytes: relative contributions of catalase and glutathione peroxidase in decomposition of endogenously generated H2O2. Arch Biochem Biophys 210: 505–516, 1981. [DOI] [PubMed] [Google Scholar]
- 86.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM Jr, and Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J 18: 1246–1248, 2004. [DOI] [PubMed] [Google Scholar]
- 87.Jones DP, Mody VC Jr, Carlson JL, Lynn MJ, Sternberg P Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med 33: 1290–1300, 2002. [DOI] [PubMed] [Google Scholar]
- 88.Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med 44: 921–937, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim HY, Gladyshev VN. Methionine sulfoxide reduction in mammals: characterization of methionine-R-sulfoxide reductases. Mol Biol Cell 15: 1055–1064, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim JR, Lee SM, Cho SH, Kim JH, Kim BH, Kwon J, Choi CY, Kim YD, Lee SR. Oxidation of thioredoxin reductase in HeLa cells stimulated with tumor necrosis factor-alpha. FEBS Lett 567: 189–196, 2004. [DOI] [PubMed] [Google Scholar]
- 91.Kirlin WG, Cai J, DeLong MJ, Patten EJ, Jones DP. Dietary compounds that induce cancer preventive phase 2 enzymes activate apoptosis at comparable doses in HT29 colon carcinoma cells. J Nutr 129: 1827–1835, 1999. [DOI] [PubMed] [Google Scholar]
- 92.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med 27: 1208–1218, 1999. [DOI] [PubMed] [Google Scholar]
- 93.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem 267: 4928–4944, 2000. [DOI] [PubMed] [Google Scholar]
- 94.Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron 12: 1031–1040, 1994. [DOI] [PubMed] [Google Scholar]
- 95.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol 5: 834–842, 1992. [DOI] [PubMed] [Google Scholar]
- 96.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science 300: 1439–1443, 2003. [DOI] [PubMed] [Google Scholar]
- 97.Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys 423: 116–125, 2004. [DOI] [PubMed] [Google Scholar]
- 98.Kuge S, Toda T, Iizuka N, Nomoto A. Crm1 (XpoI) dependent nuclear export of the budding yeast transcription factor yAP-1 is sensitive to oxidative stress. Genes Cells 3: 521–532, 1998. [DOI] [PubMed] [Google Scholar]
- 99.Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, Knight CG, Essex DW, Farndale RW. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood 102: 2085–2092, 2003. [DOI] [PubMed] [Google Scholar]
- 100.Lake BG Mechanisms of hepatocarcinogenicity of peroxisome-proliferating drugs and chemicals. Annu Rev Pharmacol Toxicol 35: 483–507, 1995. [DOI] [PubMed] [Google Scholar]
- 101.Lambeth JD NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004. [DOI] [PubMed] [Google Scholar]
- 102.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA. Redox regulation of cell signalling. Nature 381: 380–381, 1996. [DOI] [PubMed] [Google Scholar]
- 103.Lash LH Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact 163: 54–67, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lash LH, Jones DP. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch Biochem Biophys 240: 583–592, 1985. [DOI] [PubMed] [Google Scholar]
- 105.Laughner BJ, Sehnke PC, Ferl RJ. A novel nuclear member of the thioredoxin superfamily. Plant Physiol 118: 987–996, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J Biol Chem 239: 3436–3444, 1964. [PubMed] [Google Scholar]
- 107.Lee KK, Murakawa M, Takahashi S, Tsubuki S, Kawashima S, Sakamaki K, Yonehara S. Purification, molecular cloning, and characterization of TRP32, a novel thioredoxin-related mammalian protein of 32 kDa. J Biol Chem 273: 19160–19166, 1998. [DOI] [PubMed] [Google Scholar]
- 108.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci USA, 2008. [DOI] [PMC free article] [PubMed]
- 109.Levings CS, 3rd, Siedow JN. Regulation by redox poise in chloroplasts. Science 268: 695–696, 1995. [DOI] [PubMed] [Google Scholar]
- 110.Lillig CH, Holmgren A. Thioredoxin and related molecules–from biology to health and disease. Antioxid Redox Signal 9: 25–47, 2007. [DOI] [PubMed] [Google Scholar]
- 111.Lillig CH, Lonn ME, Enoksson M, Fernandes AP, Holmgren A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci USA 101: 13227–13232, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys 406: 229–240, 2002. [DOI] [PubMed] [Google Scholar]
- 113.Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol Cell Physiol 275: C323–C326, 1998. [DOI] [PubMed] [Google Scholar]
- 114.Lofgren S, Fernando MR, Xing KY, Wang Y, Kuszynski CA, Ho YS, Lou MF. Thioltransferase (glutaredoxin) knockout increases cellular sensitivity to oxidative stress and decreases cell proliferation in mouse lens epithelial cells. Invest Ophthalmol Vis Sci. 2008, June 27. [Epub ahead of print] [DOI] [PubMed]
- 115.Lyons J, Rauh-Pfeiffer A, Yu YM, Lu XM, Zurakowski D, Tompkins RG, Ajami AM, Young VR, Castillo L. Blood glutathione synthesis rates in healthy adults receiving a sulfur amino acid-free diet. Proc Natl Acad Sci USA 97: 5071–5076, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J, Makino I, Tanaka H. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J Biol Chem 274: 3182–3188, 1999. [DOI] [PubMed] [Google Scholar]
- 117.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med 38: 1422–1432, 2005. [DOI] [PubMed] [Google Scholar]
- 118.Manickam N, Sun X, Li M, Gazitt Y, Essex DW. Protein disulphide isomerase in platelet function. Br J Haematol 140: 223–229, 2008. [DOI] [PubMed] [Google Scholar]
- 119.Marchand C, Le Marechal P, Meyer Y, Decottignies P. Comparative proteomic approaches for the isolation of proteins interacting with thioredoxin. Proteomics 6: 6528–6537, 2006. [DOI] [PubMed] [Google Scholar]
- 120.Martinez-Ruiz A, Lamas S. Proteomic identification of S-nitrosylated proteins in endothelial cells. Methods Mol Biol 357: 215–223, 2007. [DOI] [PubMed] [Google Scholar]
- 121.Martinez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res 75: 220–228, 2007. [DOI] [PubMed] [Google Scholar]
- 122.Matsuo Y, Akiyama N, Nakamura H, Yodoi J, Noda M, and Kizaka-Kondoh S. Identification of a novel thioredoxin-related transmembrane protein. J Biol Chem 276: 10032–10038, 2001. [DOI] [PubMed] [Google Scholar]
- 123.Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, and Smith-Wheelock M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4: 119–125, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mills CG Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem 229: 189–197, 1957. [PubMed] [Google Scholar]
- 125.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem 274: 9427–9430, 1999. [DOI] [PubMed] [Google Scholar]
- 126.Moore EC, Reichard P, Thelander L. Enzymatic synthesis of deoxyribonucleotides. V. purification and properties of thioredoxin reductase from Escherichia Coli B. J Biol Chem 239: 3445–3452, 1964. [PubMed] [Google Scholar]
- 127.Moriarty SE, Shah JH, Lynn M, Jiang S, Openo K, Jones DP, Sternberg P. Oxidation of glutathione and cysteine in human plasma associated with smoking. Free Radic Biol Med 35: 1582–1588, 2003. [DOI] [PubMed] [Google Scholar]
- 128.Moriarty-Craige SE, Adkison J, Lynn M, Gensler G, Bressler S, Jones DP, Sternberg P Jr. Antioxidant supplements prevent oxidation of cysteine/cystine redox in patients with age-related macular degeneration. Am J Ophthalmol 140: 1020–1026, 2005. [DOI] [PubMed] [Google Scholar]
- 129.Moriarty-Craige SE, Ha KN, Sternberg P Jr, Lynn M, Bressler S, Gensler G, and Jones DP. Effects of long-term zinc supplementation on plasma thiol metabolites and redox status in patients with age-related macular degeneration. Am J Ophthalmol 143: 206–211, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci USA 98: 12920–12925, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mukherjee TK, Mishra AK, Mukhopadhyay S, Hoidal JR. High concentration of antioxidants N-acetylcysteine and mitoquinone-Q induces intercellular adhesion molecule 1 and oxidative stress by increasing intracellular glutathione. J Immunol 178: 1835–1844, 2007. [DOI] [PubMed] [Google Scholar]
- 132.Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, Dudley SC Jr. Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem 53: 1652–1657, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem 274: 21645–21650, 1999. [DOI] [PubMed] [Google Scholar]
- 134.Nkabyo YS, Go YM, Ziegler TR, Jones DP. Extracellular cysteine/cystine redox regulates the p44/p42 MAPK pathway by metalloproteinase-dependent epidermal growth factor receptor signaling. Am J Physiol Gastrointest Liver Physiol 289: G70–G78, 2005. [DOI] [PubMed] [Google Scholar]
- 135.Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol 283: G1352–G1359, 2002. [DOI] [PubMed] [Google Scholar]
- 136.Okamoto K, Tanaka H, Ogawa H, Makino Y, Eguchi H, Hayashi S, Yoshikawa N, Poellinger L, Umesono K, Makino I. Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J Biol Chem 274: 10363–10371, 1999. [DOI] [PubMed] [Google Scholar]
- 137.Oshino N, Chance B, Sies H, Bucher T. The role of H2O2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Arch Biochem Biophys 154: 117–131, 1973. [DOI] [PubMed] [Google Scholar]
- 138.Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res 100: 213–219, 2007. [DOI] [PubMed] [Google Scholar]
- 139.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 173: 3589–3593, 2004. [DOI] [PubMed] [Google Scholar]
- 140.Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, Di Cesare S, Bonetto V, Casoni F, Carrozzo R, Federici G, Piemonte F. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. J Biol Chem 278: 42588–42595, 2003. [DOI] [PubMed] [Google Scholar]
- 141.Perez VI, Lew CM, Cortez LA, Webb CR, Rodriguez M, Liu Y, Qi W, Li Y, Chaudhuri A, Van Remmen H, Richardson A, Ikeno Y. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radic Biol Med 44: 882–892, 2008. [DOI] [PubMed] [Google Scholar]
- 142.Pompella A, Corti A, Paolicchi A, Giommarelli C, Zunino F. Gamma-glutamyltransferase, redox regulation and cancer drug resistance. Curr Opin Pharmacol 7: 360–366, 2007. [DOI] [PubMed] [Google Scholar]
- 143.Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem 282: 18427–18436, 2007. [DOI] [PubMed] [Google Scholar]
- 144.Raguso CA, Regan MM, Young VR. Cysteine kinetics and oxidation at different intakes of methionine and cystine in young adults. Am J Clin Nutr 71: 491–499, 2000. [DOI] [PubMed] [Google Scholar]
- 145.Ramirez A, Ramadan B, Ritzenthaler JD, Rivera HN, Jones DP, Roman J. Extracellular cysteine/cystine redox potential controls lung fibroblast proliferation and matrix expression through upregulation of transforming growth factor-β. Am J Physiol Lung Cell Mol Physiol 293: L972–L981, 2007. [DOI] [PubMed] [Google Scholar]
- 146.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 7: 619–626, 2005. [DOI] [PubMed] [Google Scholar]
- 147.Richie JP, Leutzinger Y, Parthasarathy S, Malloy V, Orentreich N, and Zimmerman JA. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J 8: 1302–1307, 1994. [DOI] [PubMed] [Google Scholar]
- 148.Sagher D, Brunell D, Hejtmancik JF, Kantorow M, Brot N, Weissbach H. Thionein can serve as a reducing agent for the methionine sulfoxide reductases. Proc Natl Acad Sci USA 103: 8656–8661, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17: 2596–2606, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423: 769–773, 2003. [DOI] [PubMed] [Google Scholar]
- 151.Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P Jr, Reed RL, and Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free Radic Biol Med 24: 699–704, 1998. [DOI] [PubMed] [Google Scholar]
- 152.Sanz A, Caro P, Ayala V, Portero-Otin M, Pamplona R, Barja G. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J 20: 1064–1073, 2006. [DOI] [PubMed] [Google Scholar]
- 153.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30: 1191–1212, 2001. [DOI] [PubMed] [Google Scholar]
- 154.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909–1911, 2005. [DOI] [PubMed] [Google Scholar]
- 155.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6: 280–293, 2007. [DOI] [PubMed] [Google Scholar]
- 156.Schwartz DR, Sack MN. Targeting the mitochondria to augment myocardial protection. Curr Opin Pharmacol 8: 160–165, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shan X, Jones DP, Hashmi M, Anders MW. Selective depletion of mitochondrial glutathione concentrations by (R,S)-3-hydroxy-4-pentenoate potentiates oxidative cell death. Chem Res Toxicol 6: 75–81, 1993. [DOI] [PubMed] [Google Scholar]
- 158.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal 7: 348–366, 2005. [DOI] [PubMed] [Google Scholar]
- 159.Shioji K, Kishimoto C, Nakamura H, Masutani H, Yuan Z, Oka S, Yodoi J. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation 106: 1403–1409, 2002. [DOI] [PubMed] [Google Scholar]
- 160.Shioji K, Nakamura H, Masutani H, Yodoi J. Redox regulation by thioredoxin in cardiovascular diseases. Antioxid Redox Signal 5: 795–802, 2003. [DOI] [PubMed] [Google Scholar]
- 161.Sies H Nicotinamide nucleotide compartmentation. In: Metabolic Compartmentation, edited by Sies H. London: Academic, 1982, p. 205–231.
- 162.Sies H, Chance B. The steady state level of catalase compound I in isolated hemoglobin-free perfused rat liver. FEBS Lett 11: 172–176, 1970. [DOI] [PubMed] [Google Scholar]
- 163.Sies H, Jones DP. Oxidative stress. In: Encyclopedia of Stress (2nd ed.), edited by Fink G: Elsevier, 2007, p. 45–48.
- 164.Sies H, Summer KH. Hydroperoxide-metabolizing systems in rat liver. Eur J Biochem 57: 503–512, 1975. [DOI] [PubMed] [Google Scholar]
- 165.Snary D, Allen A, Pain RH. Structural studies on gastric mucoproteins: lowering of molecular weight after reduction with 2-mercaptoethanol. Biochem Biophys Res Commun 40: 844–851, 1970. [DOI] [PubMed] [Google Scholar]
- 166.Snyder GH, Cennerazzo MJ, Karalis AJ, Field D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry 20: 6509–6519, 1981. [DOI] [PubMed] [Google Scholar]
- 167.Spina MB, Cohen G. Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc Natl Acad Sci USA 86: 1398–1400, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Staab CA, Alander J, Brandt M, Lengqvist J, Morgenstern R, Grafstrom RC, Hoog JO. Reduction of S-nitrosoglutathione by alcohol dehydrogenase 3 is facilitated by substrate alcohols via direct cofactor recycling and results in GSH-controlled formation of glutathione transferase inhibitors. Biochem J 413: 493–504, 2008. [DOI] [PubMed] [Google Scholar]
- 169.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25: 207–218, 2003. [DOI] [PubMed] [Google Scholar]
- 170.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta 1703: 135–140, 2005. [DOI] [PubMed] [Google Scholar]
- 171.Starke DW, Chock PB, Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem 278: 14607–14613, 2003. [DOI] [PubMed] [Google Scholar]
- 172.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature 401: 79–82, 1999. [DOI] [PubMed] [Google Scholar]
- 173.Sykes MC, Mowbray AL, Jo H. Reversible glutathiolation of caspase-3 by glutaredoxin as a novel redox signaling mechanism in tumor necrosis factor-alpha-induced cell death. Circ Res 100: 152–154, 2007. [DOI] [PubMed] [Google Scholar]
- 174.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 51: 794–798, 1991. [PubMed] [Google Scholar]
- 175.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J 21: 1695–1703, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tapia PC Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med Hypotheses 66: 832–843, 2006. [DOI] [PubMed] [Google Scholar]
- 177.Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res 89: 224–236, 2001. [DOI] [PubMed] [Google Scholar]
- 178.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem 278: 19603–19610, 2003. [DOI] [PubMed] [Google Scholar]
- 179.Thomas DD, Liu X, Kantrow SP, Lancaster JR Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci USA 98: 355–360, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem 281: 25984–25993, 2006. [DOI] [PubMed] [Google Scholar]
- 181.Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys 319: 1–9, 1995. [DOI] [PubMed] [Google Scholar]
- 182.Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci USA 88: 4328–4332, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tribble DL, Jones DP. Oxygen dependence of oxidative stress. Rate of NADPH supply for maintaining the GSH pool during hypoxia. Biochem Pharmacol 39: 729–736, 1990. [DOI] [PubMed] [Google Scholar]
- 184.Ueno M, Masutani H, Arai RJ, Yamauchi A, Hirota K, Sakai T, Inamoto T, Yamaoka Y, Yodoi J, Nikaido T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem 274: 35809–35815, 1999. [DOI] [PubMed] [Google Scholar]
- 185.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423: 773–777, 2003. [DOI] [PubMed] [Google Scholar]
- 186.Wallace DC Mitochondrial diseases in man and mouse. Science 283: 1482–1488, 1999. [DOI] [PubMed] [Google Scholar]
- 187.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem 276: 47763–47766, 2001. [DOI] [PubMed] [Google Scholar]
- 188.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett 543: 144–147, 2003. [DOI] [PubMed] [Google Scholar]
- 189.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem 278: 33408–33415, 2003. [DOI] [PubMed] [Google Scholar]
- 190.Webb CL Arsenicals. In: Enzyme and Metabolic Inhibitors, edited by Webb CL. New York: Academic, 1966, p. 337–365.
- 191.Webb CL Comparison of SH reagents. In: Enzyme and Metabolic Inhibitors, edited by Webb CL. New York: Academic, 1966, p. 595–819.
- 192.Webb CL Mercurials. In: Enzyme and Metabolic Inhibitors, edited by Webb CL. New York: Academic, 1965, p. 729–985.
- 193.Webb CLN Ethylmaleimide. In: Enzyme and Metabolic Inhibitors, edited by Webb CL. New York: Academic, 1966, p. 1–283.
- 194.Webb CL Sulfhydryl reagents. In: Enzyme and Metabolic Inhibitors, edited by Webb CL. New York: Academic, 1965, p. 635–653.
- 195.Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res 60: 6688–6695, 2000. [PubMed] [Google Scholar]
- 196.Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther WT, Matthews B, St John G, Nathan C, Brot N. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch Biochem Biophys 397: 172–178, 2002. [DOI] [PubMed] [Google Scholar]
- 197.Winterbourn CC, Metodiewa D. The reaction of superoxide with reduced glutathione. Arch Biochem Biophys 314: 284–290, 1994. [DOI] [PubMed] [Google Scholar]
- 198.Woo JR, Kim SJ, Jeong W, Cho YH, Lee SC, Chung YJ, Rhee SG, Ryu SE. Structural basis of cellular redox regulation by human TRP14. J Biol Chem 279: 48120–48125, 2004. [DOI] [PubMed] [Google Scholar]
- 199.Wullner U, Loschmann PA, Schulz JB, Schmid A, Dringen R, Eblen F, Turski L, Klockgether T. Glutathione depletion potentiates MPTP and MPP+ toxicity in nigral dopaminergic neurones. Neuroreport 7: 921–923, 1996. [DOI] [PubMed] [Google Scholar]
- 200.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J 11: 653–665, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xia YM, Hill KE, Burk RF. Effect of selenium deficiency on hydroperoxide-induced glutathione release from the isolated perfused rat heart. J Nutr 115: 733–742, 1985. [DOI] [PubMed] [Google Scholar]
- 202.Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34: 496–502, 2003. [DOI] [PubMed] [Google Scholar]
- 203.Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med 176: 270–276, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yong VW, Perry TL, Krisman AA. Depletion of glutathione in brainstem of mice caused by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine is prevented by antioxidant pretreatment. Neurosci Lett 63: 56–60, 1986. [DOI] [PubMed] [Google Scholar]
- 205.Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys 465: 119–126, 2007. [DOI] [PubMed] [Google Scholar]
- 206.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res 94: 1483–1491, 2004. [DOI] [PubMed] [Google Scholar]
- 207.Zhao T, Heberlein K, Jonas C, Jones DP, Hu X. New double quantum coherence filter for localized detection of glutathione in vivo. Magn Reson Med 55: 676–680, 2006. [DOI] [PubMed] [Google Scholar]
- 208.Ziegler DM Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu Rev Biochem 54: 305–329, 1985. [DOI] [PubMed] [Google Scholar]