

Abstract
To understand the pathogenesis of chronic inflammatory disease, we analyzed an experimental mouse model of a chronic lung disease that resembles asthma and chronic obstructive pulmonary disease (COPD) in humans. In this model, chronic lung disease develops after infection with a common type of respiratory virus is cleared to trace levels of noninfectious virus. Unexpectedly, the chronic inflammatory disease arises independently of an adaptive immune response and is driven by IL-13 produced by macrophages stimulated by CD1d-dependent TCR-invariant NKT cells. This innate immune axis is also activated in the lungs of humans with chronic airway disease due to asthma or COPD. These findings provide new insight into the pathogenesis of chronic inflammatory disease with the discovery that the transition from respiratory viral infection into chronic lung disease requires persistent activation of a novel NKT cell-macrophage innate immune axis.
It has been widely speculated that infections are linked to the development of chronic inflammatory diseases. Although the connection between infection and chronic disease is uncertain, it likely depends on an aberrant immune response. In particular, it is believed that the innate immune system mediates the acute response to an infectious agent 1, while an atypical adaptive immune response may cause chronic inflammation 2. Furthermore, infection-induced alterations in the adaptive immune response that produce T cell or antibody-mediated damage to host tissues may underlie the pathogenesis of a variety of common diseases, ranging from asthma to diabetes 3–6. For example, it has been suggested that common respiratory viruses, such as respiratory syncytial virus (RSV) or human metapneumovirus (hMPV) cause chronic inflammation that leads to asthma 7,8. Despite these speculations, we do not know how a chronic disease develops long after apparent clearance of the inciting infection.
To investigate the possible link between acute viral infection and chronic inflammation, we developed a mouse model of chronic inflammatory lung disease. The infectious agent used in this model is Sendai virus (SeV), which is a mouse parainfluenza virus that is similar to other paramyxoviruses (e.g., RSV, hMPV, and human parainfuenza virus) that more commonly infect humans. However, mice are relatively resistant to infection with human pathogens such as RSV and hMPV. By contrast, SeV replicates at high efficiency in the mouse lung, and SeV infection causes injury and inflammation of the small airways (i.e., bronchiolitis) that is indistinguishable from the comparable condition in humans. This acute response is followed by a delayed but permanent switch to chronic airway disease that is characterized by mucus production (mucous cell metaplasia) and increased airway reactivity to inhaled methacholine (airway hyperreactivity) 9–11. The features of chronic disease manifest in this experimental mouse model are characteristic of human chronic airway diseases such as asthma and chronic obstructive pulmonary disease (COPD). To obtain insight into the pathogenesis of chronic inflammatory disease, we further characterized this distinct mouse model. We identified a new type of innate immune response that drives the development of chronic airway disease. This disease mechanism is therefore distinct from the dogma that the adaptive immune response is solely responsible for chronic inflammatory disease. This unusual immune activity develops after infectious virus is cleared and remaining virus in the lung is detectable only by an ultrasensitive PCR-based assay. This new immune axis is of special interest because it provides the first indication of how respiratory viral infection is linked to the development of chronic inflammatory disease. Moreover, we demonstrate that the immune alterations identified using this mouse model are also present in humans with chronic inflammatory airway disease due to asthma or COPD.
Results
Persistent disease and IL-13-producing macrophages
After C57BL/6 mice are infected with SeV, chronic disease is manifest by mucous cell metaplasia (with increased lung levels of Muc5ac mRNA and Muc5ac immunostaining) and airway hyperreactivity (with increased pulmonary resistance in response to inhaled methacholine). Evidence of chronic airway disease is first detected on Day 21 post-inoculation (PI), but disease does not become maximal until Day 49 PI (Supplementary Fig. 1). Similar to the time course for appearance of airway disease, IL-13 production is also first detected on Day 21 PI and reaches maximal levels on Day 49 PI. Furthermore, mice treated with an IL-13 decoy receptor (sIL-13Rα2-Fc) 12,13 or gene targeted to be IL-13-deficient (Il13−/−), no longer developed either mucous cell metaplasia or airway hyperreactivity at any time after viral infection (Fig. 1a, Supplementary Fig. 1, and Supplementary Results). By viral plaque-forming assay, infectious virus is no longer detectable in the lung by Day 12 PI 9,11,14, and by real-time PCR assay, SeV-specific RNA is decreased to trace levels by Day 49 PI (Supplementary Fig. 1).
Figure 1.
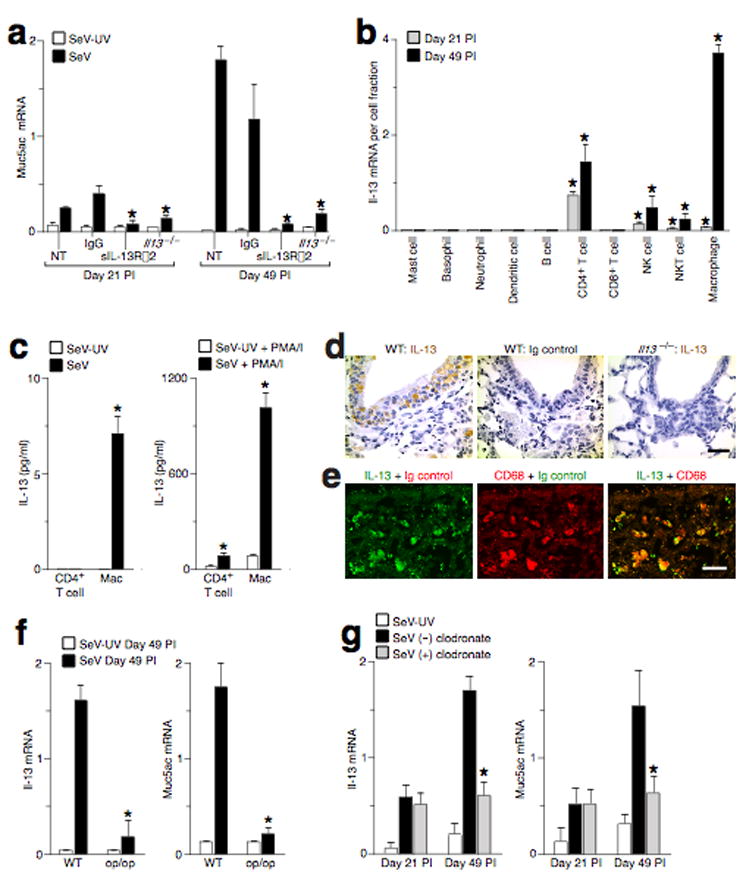
IL-13-producing macrophages drive chronic airway disease after viral infection. (a) Wild-type (WT) and Il13−/− mice were inoculated with SeV or UV-inactivated SeV (SeV-UV), and WT mice were untreated (NT) or treated with control IgG or sIL-13Rα2-Fc from Day 12 to 49 PI. Lung levels of Muc5ac mRNA were determined at Day 21 and 49 PI. (*) indicates a significant decrease from corresponding untreated control. (b) Lung cell subsets were isolated at Day 21 and 49 PI and were analyzed for Il-13 mRNA levels. Values represent Il-13 mRNA per cell fraction. (c) Lung CD4+ T cells and macrophages from Day 49 PI were cultured without or with PMA (20 ng/ml) and ionomycin (500 ng/ml) for 24 h at 37 °C, and IL-13 was determined in cell culture media. (*) indicates a significant difference from corresponding SeV-UV control. (d) Airway sections from WT or Il13−/− mice at Day 49 PI were immunostained for IL-13. (e) Airway sections from WT mice at Day 49 PI were immunostained for IL-13 and CD68. Bars = 20 μm. (f) Op/op mice at Day 49 PI were assessed for lung Il-13 and Muc5ac mRNA levels. (*) indicates a significant decrease from corresponding SeV-UV control value. (g) WT mice were assessed after being treated with clodronate (+) or empty control (−) liposomes from Day 12 to 49 PI. (*) indicates a significant decrease from corresponding value for empty liposome treatment.
We next investigated the cellular sources of IL-13 in the lung after clearance of infectious virus. At PI Day 21, CD4+ T cells (CD3+CD4+) contributed the highest amount of Il-13 mRNA among immune cells in the lung, but macrophages (Mac1+CD68+) became the predominant cellular source of Il-13 mRNA by Day 49 PI (Fig. 1b, Supplementary Fig. 2, and Supplementary Results). We also found that lung macrophages cultured from SeV (but not SeV-UV) Day 49 PI expressed Il-13 mRNA and produced IL-13 protein (Fig. 1c and Supplementary Fig. 2). IL-13 protein release was further increased by treatment with PMA-ionomycin. With and without PMA-ionomycin treatment, IL-13 protein production was increased in lung macrophages compared to CD4+ T cells and in cells isolated from SeV-inoculated mice compared to SeV-UV-inoculated control mice. The observed pattern for IL-13 protein production was consistent with the analysis of Il-13 mRNA levels. Furthermore, cells with typical macrophage morphology and CD68 expression were positive for IL-13 immunostaining in alveolar, interstitial, and epithelial locations in wild-type but not in Il13−/− mice (Fig. 1d,e and data not shown). Together, these findings provide evidence that lung macrophages are a significant cell source of chronic IL-13 production after viral infection. Analysis of macrophage-deficient or macrophage-depleted mice indicated that IL-13 and Muc5ac production after SeV infection depended on the presence of macrophages (Fig. 1f,g and Supplementary Results).
NKT cell requirement for macrophage activation
Analysis of MHC Class II-deficient H2-Ab1−/−, CD4−/−, and CD8a−/− T-cell deficient mice; mice that were antibody-depleted of CD4+ T cells, CD8+ T cells, or both CD4+ and CD8+ T cells showed the usual development of mucous cell metaplasia and IL-13-producing macrophages after viral infection (Supplementary Fig. 3 and Supplementary Results). The unexpected findings with T cell blockade led us to investigate whether a new type of cellular mechanism could mediate a chronic immune response.
In addition to T cells and macrophages, we found that an increased number of NKT cells were recruited into the lung at Day 49 PI (Fig. 1b, Supplementary Fig. 2, and Supplementary Results). Further analysis of the lung NKT cell population indicated that CD4− as well as CD4+ NKT cells were a source of Il-13 mRNA in the lung (Fig. 2a–c). Chronic NKT cell activation after viral infection was marked by a relatively selective increase in Il-13 mRNA production (Supplementary Fig. 4). This pattern of NKT cell activation is distinct from the other inflammatory conditions, especially the response to inhaled allergen 15. Nonetheless, the persistent production of Il-13 mRNA and the accumulation of NKT cells in the lung suggested that this cell population might act in a regulatory role to drive the chronic production of IL-13 by macrophages in the lung. The majority of mouse NKT cells express the semi-invariant Vα14-Jα18 TCR chain that recognizes glycolipid antigen presented by the non-polymorphic CD1d, an MHC Class I-like protein 16,17. Consistent with the involvement of NKT cells in this chronic lung disease, mice that were deficient in NKT cells due to deletion of the Cd1d gene (Cd1d1−/−) or Ja18 gene segment (Traj18−/−) 1,18 had decreases in the levels mucous cell metaplasia and airway hyperreactivity as well as IL-13-producing macrophages at PI Day 49 (Fig. 2d–f, Supplementary Fig. 4, and Supplementary Results).
Figure 2.
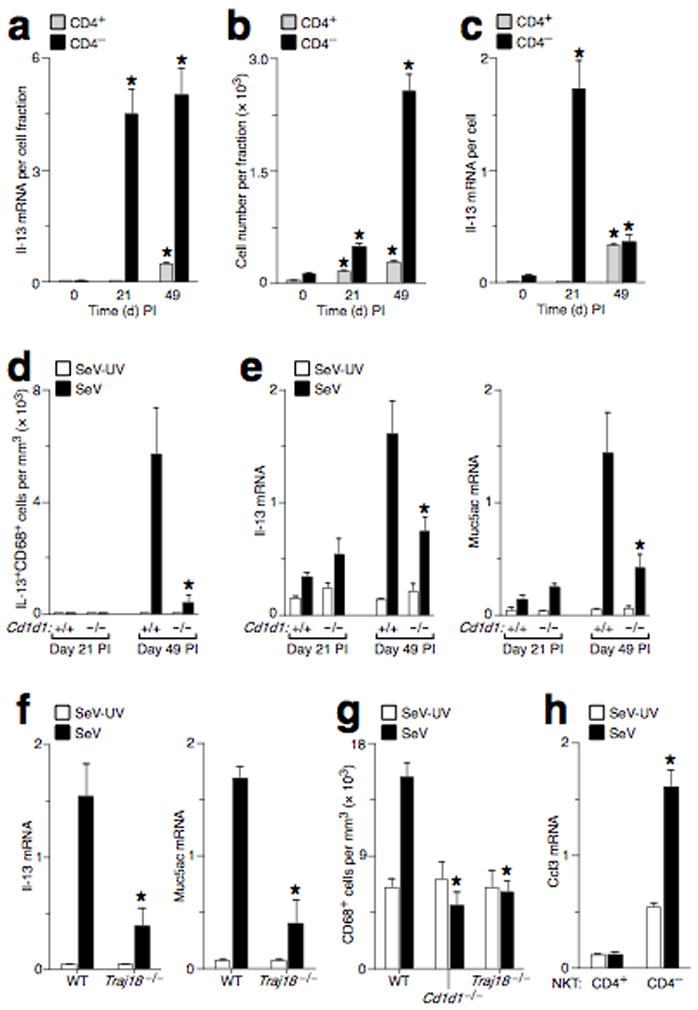
Activated NKT cells are required for chronic IL-13-producing macrophages in the lung after viral infection. (a) CD4+ and CD4− NKT cells were purified from mouse lungs at Day 0, 21, and 49 PI and were analyzed for Il-13 mRNA levels. Values represent units of Il-13 mRNA per cell fraction. (b) Corresponding number of cells in each NKT cell fraction. (c) Corresponding values for units of Il-13 mRNA per cell. For (a–c), (*) indicates a significant difference from Day 0 PI. (d) WT and Cd1d1−/− mice were inoculated with SeV or SeV-UV, and lung sections were immunostained for IL-13+CD68+ cells. (e) Using conditions in (d), mouse lungs were analyzed for Il-13 and Muc5ac mRNA levels. (f) WT and Traj18−/− mice were inoculated with SeV or SeV-UV, and lungs at Day 49 PI were analyzed for Il-13 and Muc5ac mRNA levels. (g). WT and Cd1d1−/− mice were inoculated with SeV or SeV-UV, and lung sections at Day 49 PI were immunostained for CD68+ cells. For (d–g), (*) indicates a significant decrease from corresponding value for WT mice. (h) CD4+ and CD4− NKT cells were purified from mouse lungs at PI Day 49 and were analyzed for Ccl3 mRNA by real-time PCR. (*) indicates a significant increase from SeV-UV.
Direct NKT cell-macrophage interaction
Based on the findings in NKT cell-deficient mice, we postulated that NKT cells might directly influence the population of IL-13-producing macrophages in the lung after viral infection. In support of a role for NKT cell-dependent recruitment of macrophages, we found that lung macrophage levels were decreased in NKT cell-deficient mice and that purified lung NKT cells produced increased levels of macrophage chemokines (especially Ccl3) after viral infection (Fig. 2g,h, Supplementary Fig. 5, and Supplementary Results).
To determine whether NKT cells might directly stimulate macrophage production of IL-13, we established a system for co-culture of purified NKT cells and macrophages. In this system, we found that lung NKT cells induced macrophage production of Il-13 mRNA and protein (Fig. 3a and Supplementary Results). The NKT cell effect on macrophage production of IL-13 was not found for CD4+ T cells and was inhibited by treatment with anti-CD1d mAb, indicating that this NKT cell-macrophage immune axis required direct contact of Jα18-TCR on the NKT cell with CD1d on the macrophage. Furthermore, NKT cell-dependent activation of lung macrophages was increased by 10-fold in the presence of TCR-CD1d ligand α-GalCer, achieving IL-13 production levels similar to PMA-ionomycin stimulation of macrophages (Fig. 3b). NKT cell-derived IL-13 was also necessary for NKT cell-dependent activation of macrophage IL-13 production, since activation was lost if co-culture was performed with NKT cells from Il13−/−mice. To determine whether CD4 expression also influenced NKT cell-macrophage interaction, we studied liver NKT cells to provide a more abundant source of CD4+ and CD4− NKT cells. Pure preparations of either CD4+ or CD4− liver NKT cells were both capable of activating lung macrophages when CD1d glycolipid ligand is provided (Fig. 3c). Thus, while CD4+ NKT cells have been the focus for studies of the response to allergen in the lung 15, it appears that CD4 expression is not necessary for NKT cell capacity to drive macrophage production of IL-13. Instead, NKT cell-dependent macrophage activation appears to depend primarily on invariant TCR-CD1d and IL-13–IL-13 receptor (IL-13R) interactions.
Figure 3.
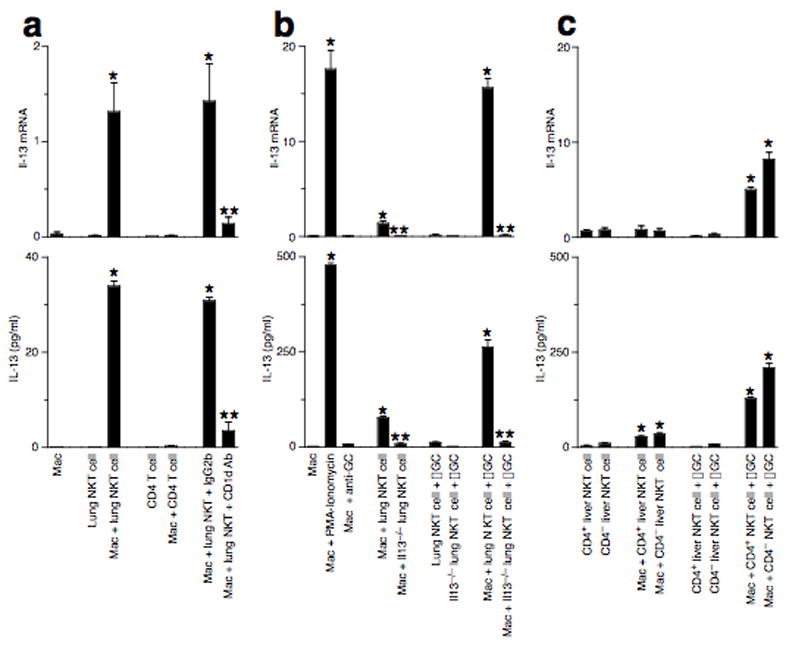
Direct lung NKT cell-macrophage interaction generates IL-13-producing macrophages. (a) Lung macrophages were cultured alone or with lung NKT cells or CD4+ T cells for 24 h at 37 °C. Lung macrophages were also treated with anti-CD1d mAb or control IgG2b. (b) Culture conditions are the same as (a), but also include stimulation using PMA-ionomycin or α-GalCer (αGC). (c) Conditions are the same as (b), but macrophages were cultured with CD4+ and CD4− liver NKT cells. For (a–c), adherent macrophages were analyzed for Il-13 mRNA, and cell media was analyzed for corresponding IL-13 protein levels. Note 10–20-fold increase in y-axis for (b,c) compared to (a). (*) indicates a significant increase from macrophage and NKT cell alone, and (**) indicates a significant decrease from corresponding value for lung NTK cell-macrophage co-culture.
IL-13R signalling and alternatively activated macrophages
We next examined the downstream events in the NKT cell-macrophage-IL-13 immune axis. Analysis of gene expression in the lung indicated that the only significant postviral change among 148 cytokines or cytokine receptors was induction of Il-13 receptor alpha chain (Il13ra1) gene expression (Supplementary Fig. 6). Induction of IL-13Rα1 expression was driven by NKT cells and was restricted mainly to lung macrophages (Fig. 4a–c, Supplementary Fig. 7, and Supplementary Results). Moreover, IL-13-dependent activation of the IL-13 receptor (IL-13R) was necessary for the development of IL-13-producing macrophages (Fig. 4d,e).
Figure 4.
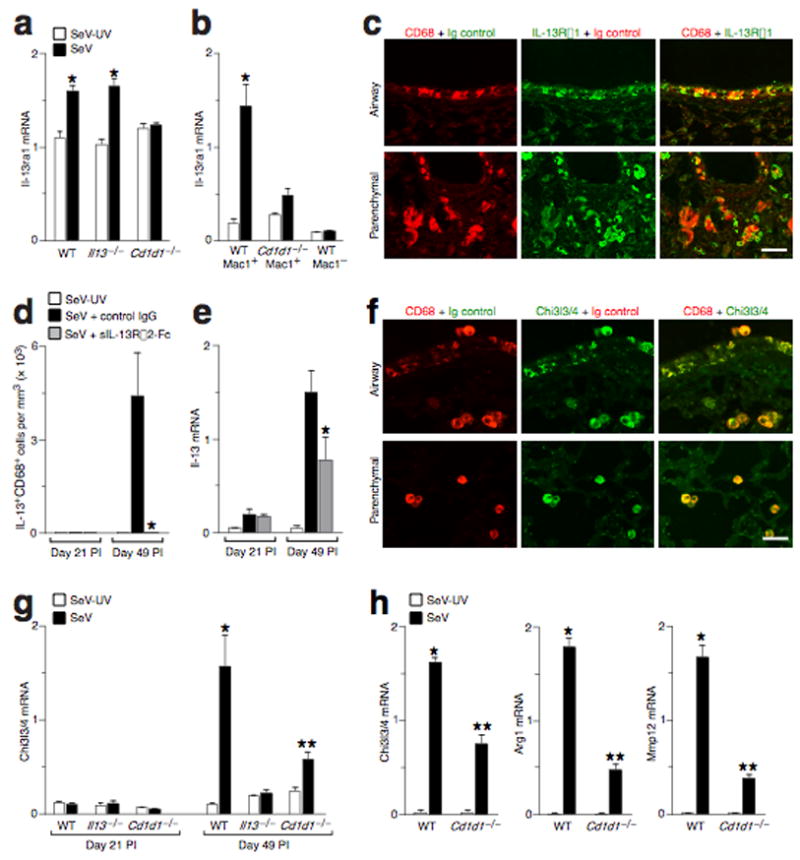
NKT cells drive IL-13R expression to upregulate IL-13 production and alternative activation of macrophages after viral infection. (a) Lung levels of Il-13ra1 mRNA in WT, Cd1d1−/−, and Il13−/− mice at Day 49 PI. (b) Levels of Il-13ra1 mRNA in macrophages (Mac1+ and CD3− NK1.1−GR1−B220−CD11c−) and non-macrophages (Mac1−) purified from WT and Cd1d1−/− mice at Day 49 PI. Similar results were obtained for Mac1+CD68+ versus Mac1+CD68− cell subsets (data not shown). For (a,b), (*) indicates a significant increase from SeV-UV. (c) Representative photomicrographs of lungs sections immunostained for CD68 and Il-13rα1 at Day 49 PI. Bar = 50 μm. (d) Quantitative analysis of IL-13+CD68+ macrophages in mice inoculated with SeV-UV or SeV and treated with either control IgG or sIL-13Rα2-Fc from Day 12 to 49 PI. (e) Lung levels of Il-13 mRNA determined for conditions in (d). For (d,e), (*) indicates significant decrease from control IgG. (f) Representative photomicrographs of lungs sections immunostained for CD68 and Chi3l3/4 at Day 49 PI. Bar = 50 μm. (g) Lung levels of Chi3l3/4 mRNA in WT, Il13−/−, and Cd1d1−/−mice at Days 21 and 49 PI. (h) Levels of Chi3l3/4, Arg1, and Mmp12 mRNA in lung macrophages purified from WT and Cd1d1−/− mice at Day 49 PI. For (g,h), (*) indicates a significant increase from corresponding SeV-UV value, and (**) indicates a significant decrease from corresponding WT control value.
Further examination of microarray data followed by real-time PCR and immunostaining revealed that macrophage production of IL-13 was associated with increased macrophage expression of Chi3l4, Fizz1, Arg1, Mmp12, and Alox12e genes (Fig. 4f and Supplementary Fig. 7). This pattern of gene expression is characteristic of an alternative pathway for activation of macrophages 19,20. Like IL-13 production, this type of macrophage activation also depended on NKT cells and IL-13–IL-R interaction, since it was decreased in Cd1d1−/− and Il13−/− mice after viral infection (Fig. 4f–h, Supplementary Fig. 7, and Supplementary Results). Together, the findings imply that increased levels of macrophage IL-13R could respond to IL-13 produced by NKT cells or macrophages to amplify macrophage production of IL-13 and consequent macrophage activation via the alternative pathway.
NKT cell-macrophage pathway in human chronic airway disease
We next determined if there was evidence of an activated NKT cell-macrophage immune axis in humans with chronic airway disease. Based on the established association of acute respiratory viral infection and the subsequent development of chronic asthma, we first investigated whether the NKT cell-macrophage-IL-13 pathway was activated in subjects with asthma. We detected an increased number of macrophages that immunostained positive for IL-13 in bronchoalveolar lavage (BAL) samples obtained from subjects with severe asthma relative to samples from healthy control subjects (Supplementary Fig. 7).
To obtain suitable samples of lung tissue for more detailed analysis, we analyzed lung tissue obtained from lung transplant recipients with severe COPD and lung donors that did not have COPD. The lung tissue obtained from COPD subjects exhibited significant mucous cell metaplasia as evidenced by an increased number of MUC5AC+ mucous cells and higher levels of MUC5AC mRNA in the lung (Fig. 5a,b). Consistent with our findings in the mouse model of chronic lung disease, we also detected an increased level of IL-13 mRNA in COPD lungs with chronic mucous cell metaplasia (Fig. 5b). The increase in IL-13 mRNA levels was associated with an increase in the number of cells that immunostained for IL-13 protein in COPD lung tissue (Fig. 5c). These IL-13+ cells were identified as lung macrophages based on typical morphology and positive immunostaining for CD68. Furthermore, these IL-13+CD68+ macrophages were found at increased numbers in lungs from COPD subjects compared to non-COPD controls (Fig. 5d). In addition, we were able to detect NKT cells in lung tissue based on immunostaining for the invariant Vα24 T cell receptor chain (Fig. 5e). In concert with increased levels of mucous cells, IL-13 production, and IL-13+ macrophages, we also found that Vα24+ NKT cells were present in increased numbers in the lungs of subjects with COPD compared to non-COPD controls (Fig. 5f). Together, the findings suggest that an innate NKT cell-macrophage-IL-13 immune axis may be activated in human disease conditions that are similar to the virus-induced mouse model of chronic airway disease.
Figure 5.
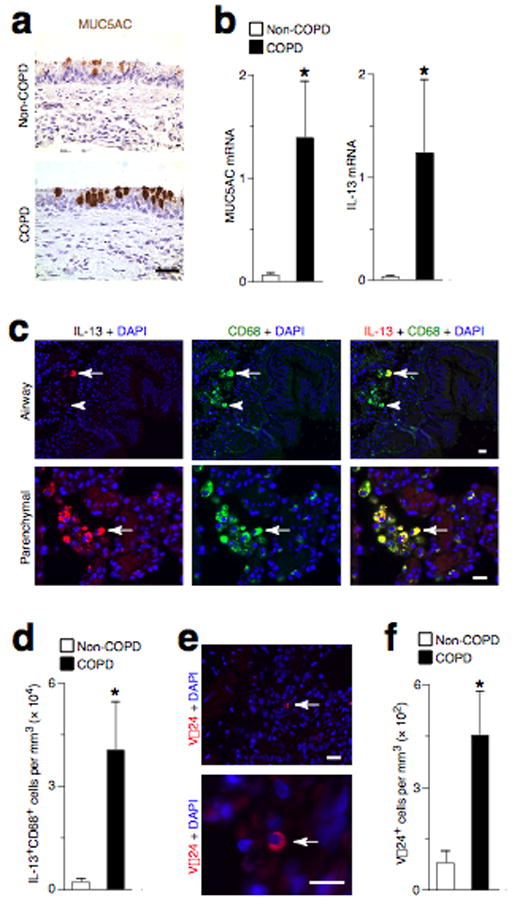
Increased IL-13-producing macrophages and Vα24-expressing NKT cells in humans with COPD and mucous cell metaplasia. (a) Representative photomicrographs of lung sections from a COPD subject undergoing lung transplantation and a non-COPD lung transplant donor were immunostained for MUC5AC. (b) Levels of MUC5AC and IL-13 mRNA in lungs from COPD subjects and non-COPD controls. (c) Representative photomicrographs of lung sections from COPD and non-COPD subjects that were immunostained for IL-13 and CD68. Arrows indicate an IL-13+CD68+ macrophage; arrowheads indicate an IL-13−CD68+ macrophage. (d) Quantitative analysis of data from (c) in lungs from COPD subjects and non-COPD controls. (e) Representative photomicrographs of lung sections from COPD subjects that were immunostained for Vα24. Arrows indicate Vα24+ NKT cells. (f) Quantitative analysis of data from (e) in COPD subjects and non-COPD lung transplant donors. All bars = 50 μm. For (b,d,f), n = 5 per group, and (*) indicates a significant increase from non-COPD control value.
DISCUSSION
This report defines a new immune pathway for the development of chronic inflammatory lung disease (diagrammed in Fig. 6). To identify this pathway, we analyzed an experimental model of chronic airway disease that develops after viral infection. In this model, the inciting virus remains detectable in the lung at trace levels, and an invariant CD4− NKT cell population is activated in the lung. This cell population appears to be specially programmed to recruit and activate lung macrophages. Activation of macrophages requires NKT cell production of IL-13 and direct contact between the invariant Vα14Jα18-TCR on lung NKT cells and CD1d on macrophages. The activated lung macrophages produce IL-13 and overexpress the IL-13 receptor, and this combination of events establishes a positive feedback loop that promotes the persistent expression of IL-13 as well as other IL-13-induced genes that are characteristic of an alternative pathway for macrophage activation. The persistent production of IL-13 causes chronic mucous cell metaplasia and airway hyperreactivity that are characteristic of chronic inflammatory airway diseases such as asthma and COPD. This analysis provides the first evidence that IL-13-producing macrophages are present in increased numbers in the lungs of patients with asthma and COPD. We also show that an orthologous population of invariant Vα24Jα18-TCR NKT cells is found at increased levels in the lung tissue of patients with COPD. Together, these findings in this experimental model identify an innate NKT cell-macrophage immune axis that enables a viral infection to initiate a chronic inflammatory lung disease. Analysis of human lung samples obtained from patients with asthma and COPD indicate that components of this pathway may also contribute to chronic airway diseases that are common in the human population.
Figure 6.
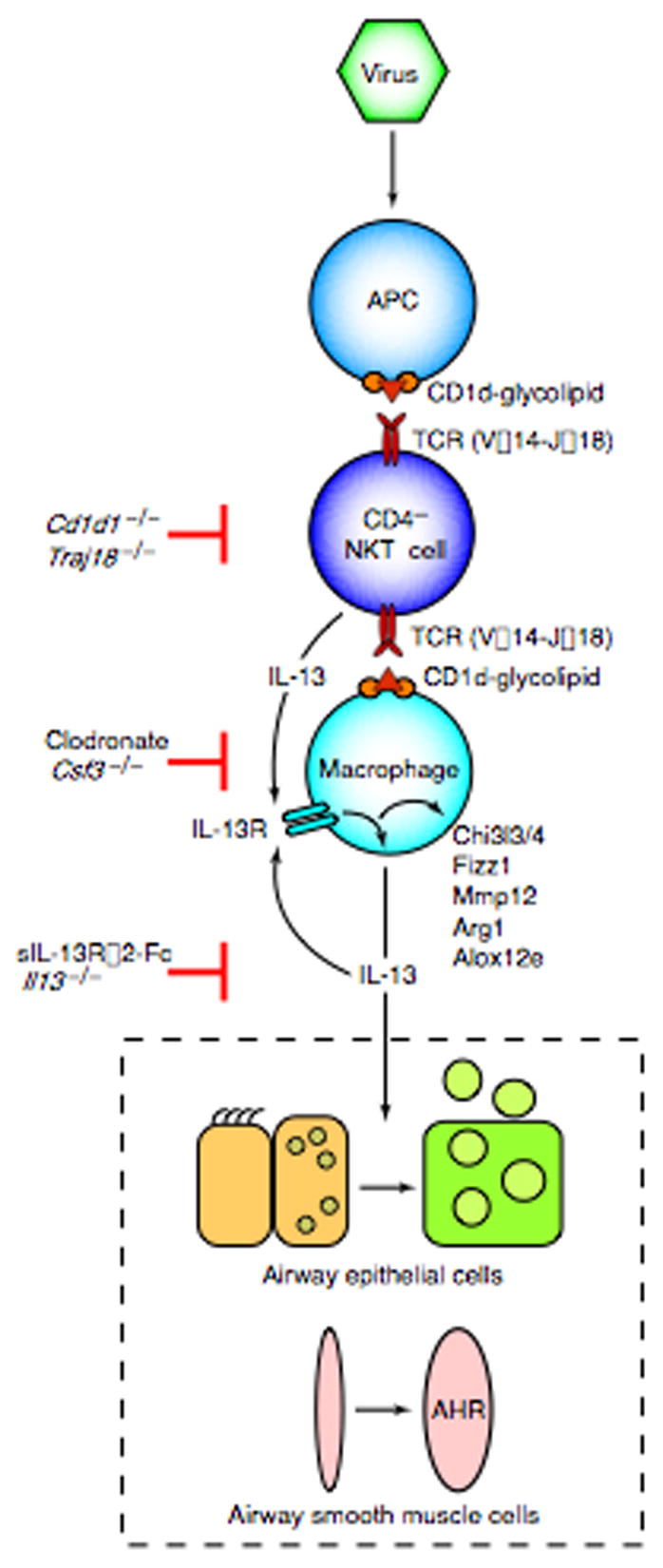
Scheme for an NKT cell-macrophage immune axis leading to chronic airway disease after viral infection. Virus may directly or indirectly facilitate CD1d-dependent antigen presentation and consequent activation of invariant CD4− NKT cells. NKT cells then interact directly with lung macrophages via contact between invariant Vα14 TCR and glycolipid-loaded CD1d. This interaction leads to increased expression of IL-13R and production of IL-13 that drives a positive feedback loop to amplify IL-13 production and alternative activation of macrophages, including Chi3l3/4, Fizz1, Mmp12, Arg1, and Alox12e gene expression. Persistent IL-13 production also drives differentiation of airway epithelial cell precursors towards mucous cells (mucous cell metaplasia) and airway smooth muscle cells to become more reactive to contractile agonists (airway hyperreactivity or AHR).
Our findings for NKT cell-macrophage function are distinct from previous reports of immune abnormalities in chronic inflammatory disease in general, and lung disease in particular. For example, invariant NKT cells were necessary for airway hyperreactivity in a mouse model of allergen-induced asthma and were found in increased numbers in allergic asthma patients 15,21–23. However, subsequent reports indicated that NKT cells were not necessary for airway inflammation in the mouse model and that the numbers of invariant NKT cells in BAL fluid or endobronchial biopsies were no different from normal in either asthma or COPD patients 24,25. The present findings highlight the need for a more complete assessment of the innate immune response. Thus, respiratory viral infection provided a more effective stimulus than acute allergen challenge for chronic activation of the immune system in an experimental mouse model. In addition, a proper assessment of a small subset of lymphocytes was better accomplished with the relatively larger amounts of sample tissue that can be obtained from lung resection or transplantation in patients with airway disease. Finally, a synchronized analysis of the mouse model in combination with human patients was essential to establish a previously unrecognized role for NKT cells and IL-13-producing macrophages in the inflammatory process that drives chronic disease.
The presence of a persistent innate immune response suggests that there must be ongoing immune stimulation. In that regard, we did not detect any evidence of infectious virus by 2 weeks after inoculation. However, using highly sensitive PCR probes and virus-clean isolation rooms, we were able to detect low levels of virus-specific RNA in lung tissue until at least PI Day 49. Whether viral persistence is necessary for a chronic immune response is still being defined. For example, conventional DCs (cDCs) are sites of virus uptake, are activated acutely and perhaps chronically after SeV infection, and are capable of activating NKT cells at low levels of antigen 26–28. Our results (like that of others) indicate that invariant NKT cells may also react to CD1d-expressing macrophages by a mechanism that does not require but can be enhanced by high-affinity agonists such as αGalCer 29. Additional work must therefore be directed at the cause of chronic NKT cell-macrophage activation after viral infection, and the possibility that viral remnants contained in cDCs, macrophages, or other cell types might drive this process.
Our results provide a model for persistent activation of an innate immune axis and present a new paradigm for the pathogenesis of chronic inflammatory lung disease. Previously, immune pathways promoting IL-13 production were viewed primarily as protection against parasitic infection 19,30. Infection by extracellular parasites is known to stimulate a T helper type 2 (Th2) cell response with production of IL-4 and IL-13 that can in turn activate macrophages by an alternative pathway 19. A similar type of response is found after allergen challenge in mice and in allergic asthma in humans 31,32. By contrast, intracellular bacteria and viruses characteristically activate a Th1 response with production of IFN-γ and consequent activation of macrophages via the classical pathway. Development of this IFN-dependent pathway is proposed to downregulate the allergic response and protect against allergic airway disease 33. However, these general patterns were defined largely defined within the context of an acute immune response 34. The present observations indicate that viruses can also trigger long-term activation of NKT cells and achieve chronic production of IL-13 by macrophages themselves. In combination with IL-13 receptor signaling, this innate mechanism can establish a state of persistent macrophage activation that is typical of the alternative pathway. Presumably this mechanism evolved to mount a long-term innate immune response independently of CD4+ or CD8+ T cells and thereby enable a response to low-level endemic pathogens that are poorly presented by MHC but adequately presented by CD1d. However, in at least some genetic backgrounds, we show that this type of innate immune activation can also lead to the development of chronic inflammatory airway disease in an experimental model and that similar immune activation occurs in patients with asthma and COPD. Indeed, there is already a well-established clinical relationship between acute viral infection and the subsequent development of chronic asthma later in childhood and perhaps adulthood 7. Other clinical studies will need to be done to test whether patients with COPD may share this same link to earlier infection. The present results provide new markers of innate immune activation to follow how infection leads to chronic inflammatory disease in this group of patients and perhaps others.
METHODS
For details, see Supplementary Methods online.
Mice generation and treatment
We received breeding pairs of Il13−/− mice from Tom Wynn (NIAID, Bethesda, MD), Cd1d1−/− mice from Albert Bendelac (Univ. Chicago, Chicago, IL), and Traj18−/− mice from Masaru Taguchi (RIKEN, Yokohama, Japan). We obtained Il4−/−, Op/op, and wild-type C57BL/6J mice from Jackson Laboratory and H2-Ab1−/− mice from Taconic Farms. All mice strains were fully backcrossed onto the C57BL/6 background. For IL-13 blockade, we obtained sIL-13Rα2-Fc from Sandy Goldman (Wyeth Pharmaceuticals) and administered this agent at 200 μg/mouse by subcutaneous injection beginning on PI Day 12 and then at 3-day intervals. For macrophage depletion, we treated mice with clodronate-containing or control liposomes prepared as described previously 35. We administered liposomes by intraperitoneal or retro-orbital intravenous injection using 2 mg at Day 14 after inoculation and then 0.5 mg at 3-d intervals thereafter until d 21 or 49 after inoculation, and macrophage depletion was monitored as described previously 35. We used Sendai virus (Sendai/52, Fushimi Strain) to inoculate mice intranasally with virus or UV-inactivated virus as described previously 9,10,35,36. We delivered SeV at 1 × 105 pfu per mouse but reduced the inoculum to 0.5 × 105 pfu for immunocompromised mice (Op/op and H2-Ab1−/−mice) to ensure survival. The Animal Studies Committee of Washington University School of Medicine approved all experimental protocols.
Analysis of mRNA and SeV-specific RNA
We purified RNA from homogenized lung tissue or isolated cells using the RNeasy mini kit (Qiagen), and we made cDNA from RNA with the High Capacity cDNA Archive kit (Applied Biosystems). We quantified target mRNA and viral RNA levels with real-time PCR using specific fluorogenic probe-primer combinations and Fast Universal PCR Master Mix systems (Applied Biosystems). We performed oligonucleotide microarray analysis as described previously 11,35.
Cell isolation and culture
We purified immune cell subsets from lung cell suspensions by FACS using rat anti-mouse mAb and a Mo-Flo high-speed flow cytometer (Dako Cytomation). For macrophage-NKT cell co-culture, we purified macrophages from lung cell suspensions using OptiPrep (Sigma-Aldrich) followed by adherence to 8-well Lab-Tek II chambers (Nunc). In some experiments, we blocked macrophages with anti-mouse CD16/CD32 (1 μg per 1 × 106 cells) for 15 min and then incubated them with anti-CD1d mAb or rat IgG2b control (50 μg/ml) for 30 min at 4 °C. We purified lung and liver NKT cells by FACS after initial enrichment with the MACS Separator (Miltenyi-Biotec) using FITC-labeled anti-CD3e mAb plus APC-labeled α-GalCer-analogue (PBS57)-loaded CD1d tetramer (supplied by the NIH Tetramer Facility) and anti-APC microbeads (Miltenyi-Biotec). We cultured macrophages and NKT cells (both >99% purity) at 5 × 104 and 5 × 103 cells per well, respectively, for 24 h at 37 °C in RPMI with 10% FBS. We collected cell media for ELISA, and then washed off NKT cells and lysed adherent macrophages in TRIzol for RNA isolation and real-time PCR assay.
Immunohistochemistry
We performed immunostaining with brightfield microscopy as described previously 9,10,36. For immunofluorescence, we used paraffinized sections that were hydrated and incubated in Antigen Unmasking Solution (Vector Labs) for antigen retrieval. We visualized staining with tyramide signal amplification with Alexa Fluor 555 or 594 (Invitrogen). We performed photomicrography and quantification of reporter by cell counting (cells per mm of basement membrane or mm3 of lung tissue) using Image J and NIH Image software as described previously 10,36,37.
Human subject samples
We obtained samples from COPD patients undergoing lung transplantation and non-COPD lung donors as described previously 10. We performed immunofluorescence microscopy using biotinylated goat anti-human IL-13 Ab (R&D Systems, 4 μg/ml), biotinylated mouse anti-human CD68 mAb (clone KP1, DakoCytomation, 5 μg/ml), and mouse anti-Vα24 mAb (Beckman Coulter, 2 μg/ml) as described above for mouse lung tissue sections. Sections were blocked with 5% goat serum or mouse IgG1 (eBioscience), stained with primary Ab and then with HRP-conjugated anti-mouse IgG secondary Ab, or avidin-HRP (Vector ABC Elite System). The Washington University Human Studies Committee approved all human studies, and informed consent was obtained from all human subjects.
Statistical analysis
We analyzed values for target mRNA, mucous cell, and airway reactivity levels using a one-way analysis of variance (ANOVA) for a factorial experimental design. If significance was achieved by one-way analysis, we performed post-ANOVA comparison of means using Scheffe’s F test. We compared PCR data by unpaired Student’s t-test. If variances were unequal, we applied Welch’s correction. Significance level for all analyses was 0.05. All values represent mean ± SEM.
Microarray data was deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under series accession number GSE10964.
Supplementary Material
Note: Supplementary information is available on the Nature Medicine website.
Acknowledgments
We thank W. Eades, C. Holley, and J. Hughes in the Siteman Cancer Center High Speed Cell Sorter Core Facility and J. Jung and K. Dolim at Roche Palo Alto for technical assistance. This work was supported by grants from the National Institutes of Health (Heart, Lung, and Blood Institute, Cancer Institute, and Institute of Allergy and Infectious Diseases), Barnes-Jewish Hospital Foundation, Martin Schaeffer Fund, and Alan A. and Edith L. Wolff Charitable Trust.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
E.Y.K. conducted mouse model experiments; J.T.B. conducted human sample experiments; A.C.P. conducted bioinformatics analysis; Y.Y. conducted co-culture experiments; E.A. generated viral preparations and viral monitoring reagents; M.H.G. assisted in flow cytometry experiments; L.A.B. assisted in NKT cell experiments; D.E.B. assisted in flow cytometry experiments; Y.A. established PCR assays; J. T. performed PCR assays; S. S. performed airway reactivity measurements; R. T. performed viral titer experiments; J.W.T. and J.D.M. assisted in mouse model experiments; M.C. provided asthma patient samples; D.P. assisted in human sample experiments; G.A.P. provided COPD patient samples; R.A.S. provided liposome preparations; J.D.A. assisted in bioinformatic analysis; G.P. conducted microarray experiments; and M.J.H. provided overall design of the project and construction of the article.
References
- 1.Mattner J, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 2.Seino K, Taniguchi M. Functional roles of NKT cells in the immune system. Frontiers Biosci. 2006;9:2577–2587. doi: 10.2741/1418. [DOI] [PubMed] [Google Scholar]
- 3.Busse WW, Lemanske RFJ. Asthma. New Engl J Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 4.Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol. 2003;3:1–8. doi: 10.1038/nri1084. [DOI] [PubMed] [Google Scholar]
- 5.Jones EY, Fugger L, Strominger JL, Siebold C. MHC Class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6:271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 6.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 7.Sigurs N, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 8.Hamelin ME, Prince GA, Gomez AM, Kinkead R, Boivin G. Human metapneumovirus infection induces long-term pulmonary inflammation associated with airway obstruction and hyperresponsiveness in mice. J Infect Dis. 2006;193:1634–1642. doi: 10.1086/504262. [DOI] [PubMed] [Google Scholar]
- 9.Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest. 2002;110:165–175. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyner JW, et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. 2006;116:309–321. doi: 10.1172/JCI25167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel AC, et al. Genetic segregation of airway disease traits despite redundancy of chloride channel calcium-activated (CLCA) family members. Physiol Genomics. 2006;25:502–513. doi: 10.1152/physiolgenomics.00321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grunig G, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wills-Karp M, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 14.Zhong W, Marshall D, Coleclough C, Woodland DL. CD4+ T cell priming accelerates the clearance of Sendai virus in mice, but has a negative effect on CD8+ T cell memeory. J Immunol. 2000;164:3274–3282. doi: 10.4049/jimmunol.164.6.3274. [DOI] [PubMed] [Google Scholar]
- 15.Akbari O, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyper-reactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 16.Bendelac A, et al. CD1 recognition by mouse NK1+ T lymphocytes. Science. 1995;268:863–865. doi: 10.1126/science.7538697. [DOI] [PubMed] [Google Scholar]
- 17.Kowano T, et al. CD1d-restricted and TCR-mediated activation of Va14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 18.Cui J, et al. Requirement for Va14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–162. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 19.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 20.Pouladi MA, et al. Interleukin-13-dependent expression of matrix metalloproteinase-12 is required for the development of airway eosinophilia in mice. Am J Respir Cell Mol Biol. 2004;30:84–90. doi: 10.1165/rcmb.2003-0051OC. [DOI] [PubMed] [Google Scholar]
- 21.Lisbonne M, et al. Cutting edge: invariant Va14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J Immunol. 2003;171:1637–1641. doi: 10.4049/jimmunol.171.4.1637. [DOI] [PubMed] [Google Scholar]
- 22.Akbari O, et al. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. New Engl J Med. 2006;354:1117–1129. doi: 10.1056/NEJMoa053614. [DOI] [PubMed] [Google Scholar]
- 23.Sen Y, et al. Va24-invariant NKT cells from patients with allergic asthma express CCR9 at high frequency and induce Th2 bias of CD3+ T cells upon CD226 engagement. J Immunol. 2005;175:4914–4926. doi: 10.4049/jimmunol.175.8.4914. [DOI] [PubMed] [Google Scholar]
- 24.Das J, et al. Natural killer T cells and CD8+ T cells are dispensable for T cell-dependent allergic airway inflammation. Nat Med. 2006;12:1345–1346. doi: 10.1038/nm1206-1345. [DOI] [PubMed] [Google Scholar]
- 25.Vijayanand P, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. New Engl J Med. 2007;356:1410–1422. doi: 10.1056/NEJMoa064691. [DOI] [PubMed] [Google Scholar]
- 26.Grayson MH, et al. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol. 2007;179:1438–1448. doi: 10.4049/jimmunol.179.3.1438. [DOI] [PubMed] [Google Scholar]
- 27.Grayson MH, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. 2007;204:2759–2769. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng L, et al. Efficient activation of Va14 invariant NKT cells by foreign lipid antigen is associated with concurrent dendritic cell-specific self recognition. J Immunol. 2007;178:2755–2762. doi: 10.4049/jimmunol.178.5.2755. [DOI] [PubMed] [Google Scholar]
- 29.Hegde S, et al. NKT cells direct monocytes into a DC differentiation pathway. J Leukoc Biol. 2007;81:1224–1235. doi: 10.1189/jlb.1206718. [DOI] [PubMed] [Google Scholar]
- 30.Skold M, Behar SM. Role of CD1d-restricted NKT cells in microbial immunity. Infect Immun. 2003;71:5447–5455. doi: 10.1128/IAI.71.10.5447-5455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb DC, McKenzie ANJ, Foster PS. Expression of the Ym2 lectin-binding protein is dependent on interleukin (IL)-4 and IL-13 signal transduction. J Immunol. 2001;276:41969–41976. doi: 10.1074/jbc.M106223200. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Z, et al. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 33.von Mutius E. Allergies, infections, and the hygiene hypothesis–the epidemiological evidence. Immunobiol. 2007;212:433–439. doi: 10.1016/j.imbio.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Holtzman MJ, Sampath D, Castro M, Look DC, Jayaraman S. The one-two of T helper cells: does interferon-γ knockout the Th2 hypothesis for asthma? Am J Respir Cell Mol Biol. 1996;14:316–318. doi: 10.1165/ajrcmb.14.4.8600934. [DOI] [PubMed] [Google Scholar]
- 35.Tyner JW, et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–1187. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter MJ, Kajiwara N, Karanja P, Castro M, Holtzman MJ. IL-12 p40 production by barrier epithelial cells during airway inflammation. J Exp Med. 2001;193:339–352. doi: 10.1084/jem.193.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampath D, Castro M, Look DC, Holtzman MJ. Constitutive activation of an epithelial signal transducer and activator of transcription (Stat1) pathway in asthma. J Clin Invest. 1999;103:1353–1361. doi: 10.1172/JCI6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Medicine website.