

Abstract
The brain’s resistance to ischemic injury can be transiently augmented by prior exposure to a sub-lethal stress stimulus, i.e. preconditioning. It has been reported that Toll-like receptors (TLRs) are involved in the preconditioning-induced protective effect against ischemic brain injury. In this study, we investigated the effect of preconditioning with a TLR2 specific ligand, Pam3CSK4, on focal cerebral ischemia/reperfusion (I/R) injury in mice. Pam3CSK4 was administered systemically 24 hrs before the mice were subjected to focal cerebral ischemia (1 hr) followed by reperfusion. Cerebral infarct size was determined, blood brain barrier (BBB) permeability was evaluated, and expression of tight-junction proteins were examined after focal cerebral I/R. Results showed that pre-treatment with Pam3CSK significantly reduced brain infarct size (1.9 ± 0.5% vs 9.4 ± 2.2%) compared with the untreated I/R group. Pam3CSK4 pre-treatment also significantly reduced acute mortality (4.3% vs 24.2%), preserved neurological function (8.22 ± 0.64 vs 3.91 ± 0.57), and attenuated brain edema (84.61 ± 0.08% vs 85.29 ± 0.09%) after cerebral I/R. In addition, Pam3CSK4 pre-treatment preserved BBB function as evidenced by decreased leakage of serum albumin (0.528 ± 0.026 vs 0.771 ± 0.059) and Evans Blue (9.23 ± 0.72 μg/mg vs 12.56 ± 0.65μg/mg) into brain tissue. Pam3CSK4 pre-treatment also attenuated the loss of the tight junction protein occludin in response to brain I/R injury. These results suggest that TLR2 is a new target of ischemic preconditioning in the brain and preconditioning with a TLR2 specific ligand will protect the brain from I/R injury.
Keywords: Toll-like receptor 2, Pam3CSK4, Cerebral ischemia, Blood-brain barrier, Mouse
1. Introduction
The brain’s resistance to ischemic injury can be transiently augmented by preconditioning, which is defined as a brief exposure to a harmful stimulus at a dose below the threshold for tissue injury that provides robust protection against, or tolerance to, the injurious effects of a subsequent more severe insult (Hertzog et al. 2003). Tolerance to experimental brain ischemia can be induced by a variety of stimuli that elicit a stress response, such as transient cerebral ischemia (Barone et al. 1998), cytokines (Beltrami et al. 2003) and endotoxin (Jiang et al. 2007). Recent evidence indicates that Toll-like receptors (TLRs) are involved in the preconditioning-induced tolerance to ischemia (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997;Stevens et al. 2008). Toll-like receptors (TLRs) are a family of signal transduction molecules that play a critical role in the induction of innate and adaptive immunity (Medzhitov et al. 1997). Preconditioning with TLR4 (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997) and TLR9 (Stevens et al. 2008) ligands has been reported to induce neuroprotection against ischemic injury. By way of example, the TLR4 ligand, lipopolysaccharide (LPS), can induce protection against ischemia-induced brain injury (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997). However, whether preconditioning induced by other TLR ligands can induce neuroprotection in ischemic injury remains uninvestigated. Pam3CysSerLys4 (Pam3CSK4) is a specific ligand for TLR2 (Aliprantis et al. 1999). We hypothesized that Pam3CSK4 may induce preconditioning that might protect the brain from I/R injury. To test this hypothesis, C57BL/10ScSn (wild type) mice were pretreated with Pam3CSK4 twenty four hours before focal cerebral ischemia/reperfusion (I/R) induced by middle cerebral artery occlusion (MCAO). We observed that pretreatment of mice with Pam3CSK4, a TLR2 specific ligand, decreased mortality after I/R, attenuated ischemia-induced neurological deficits, reduced infarct size and brain edema. In addition, Pam3CSK4 preconditioning also preserved blood–brain barrier (BBB) function after cerebral I/R by modulating the expression of tight junction (TJ) proteins, zonula occluden-1 (ZO-1) and occludin in the brain vasculature. Although the mechanisms remain to be elucidated, the present study demonstrates, for the first time, that preconditioning with Pam3CSK4 prevents focal cerebral I/R injury, which may involve modulation of vascular tight junction proteins and preservation of BBB integrity.
2. Materials and Methods
2.1. Animals
Male mice (C57BL/10ScSn, 25~30g) were obtained from Jackson Laboratory and maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU). The experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996). The animal care and experimental protocols were approved by the ETSU Committee on Animal Care.
2.2. Focal cerebral ischemia/reperfusion
Focal cerebral ischemia/reperfusion (I/R) was induced by occlusion of the middle cerebral artery (MCAO) on the left side according to previously published methods (Yang et al. 1999) with modifications. Briefly, mice were subjected to anesthesia by 5.0% Isoflurane and maintained by inhalation of 1.5% to 2% Isoflurane driven by 100% oxygen flow. Mice were ventilated (110 breaths/min with volume 0.5ml) and body temperature was regulated at 37.0°C by surface water heating. Following the skin incision, the left common carotid artery (CCA), the external carotid artery (ECA) and the internal carotid artery (ICA) were carefully exposed. Microvascular aneurysm clips were applied to the left CCA and the ICA. A coated 6-0 filament (6023PK, Doccol Corp. CA, USA) was introduced into an arteriotomy hole, fed distally into the ICA and advanced 11 mm from the carotid bifurcation. The ICA clamp was removed and focal cerebral ischemia started. After ischemia for 60 minutes, the filament and the CCA clamp were gently removed (reperfusion starts). The collar suture at the base of the ECA stump was tightened. The skin was closed, anesthesia discontinued, and the animal allowed to recover in pre-warmed cages. Control mice underwent a neck dissection and coagulation of the external carotid artery, but no occlusion of middle cerebral artery.
2.3. Experimental design
Mice were assigned to four groups: sham control (S), focal cerebral ischemia/reperfusion (I/R), Sham + Pam3CSK4 (S-Pam) and I/R + Pam3CSK4 (I/R-Pam). To examine the permeability of the BBB, brains were harvested from the mice subjected to one hr of ischemia followed by 6 hrs of reperfusion. Mortality and neurological score were evaluated in mice which were subjected to one hour of ischemia followed by reperfusion for 24 hrs. To evaluate infarct size, brains were harvested and stained with thiphenyltetrazolium chloride (TTC). To examine the levels of tight junction proteins, brain tissues were harvested and cellular proteins isolated (Hua et al. 2007b). For immunohistochemistry staining, mice were anesthetized with Ketamine and transcardially perfused with normal saline followed by 30 ml of 4% buffered paraformaldehyde, pH 7.4. The brain was removed, postfixed, embedded in paraffin and cut into sections (7μm) as described previously (Hua et al. 2007b).
2.4. Pam3CSK4 pre-treatment
Pam3CSK4 (Catalog number: tlrl-pms, InvivoGen, San Diego, CA) was dissolved in sterile endotoxin-free water and injected intraperitoneally (i.p., 2mg/Kg Body weight) 24 hours prior to MCAO or sham surgical operation. This optimal dose was selected from a series of dose-ranging studies.
2.5. Evaluation of neurological score
All mice were scored by a blinded investigator using a neurological evaluation instrument described previously (Garcia et al. 1995) with modification. Briefly, the scoring system included five principle tasks: spontaneous activity over a 3-minute period (0–3), symmetry of movement (0–3), open-field path linearity (0–3), beam walking on a 3cm × 1 cm beam (0–3), and response to vibrissae touch (1–3) (Shimamura et al. 2006b). The scoring system ranged from 0 to 15, in which 15 is a perfect score and 0 is death due to cerebral I/R injury.
2.6. Assessment of cerebral infarct size
The infarct size was determined as described previously (Reglodi et al. 2002). Twenty-four hrs after I/R, mice were sacrificed and perfused with ice cold phosphate buffered saline (PBS) via the ascending aorta. Brains were removed and sectioned coronally into 2-mm-thick slices. The slices were stained with 2% TTC solution at 37°C for 15 min followed by fixation with 10% formalin neutral buffer solution (pH 7.4). The infarct areas were traced and quantified with an image-analysis system. Unstained areas (pale color) were defined as ischemic lesions. The areas of infarction and the areas of both hemispheres were calculated for each brain slice. An edema index was calculated by dividing the total volume of the left hemisphere by the total volume of the right hemisphere. The actual infarct volume adjusted for edema was calculated by dividing the infarct volume by the edema index. Infarct volumes are expressed as percentage of the total brain volume ± S.E.M.
2.7. Assessment of brain edema (brain water content)
Brain water content was measured as described previously (Shimamura et al. 2006a). The harvested brains were divided into left and right hemisphere. The brain samples were weighed immediately after dissection (wet weight) and then dried at 105°C for 24 hours (dry weight). The percentage of water content was calculated with the formula [(wet weight − dry weight)/wet weight] × 100%.
2.8. Western Blots
Cellular proteins were prepared from ischemic cerebral hemispheres (Hua et al. 2007b), electrophoresed with SDS-polyacrylamide gel and transferred onto Hybond ECL membranes (Amersham Pharmacia, Piscataway, NJ) (Hua et al. 2007a; Hua et al. 2005;Hua et al. 2007b). The ECL membranes were incubated with the appropriate primary antibody followed by incubation with peroxidase-conjugated secondary antibodies (Cell Signaling Technology, Inc.). The signals were detected with the ECL system (Amersham Pharmacia). The same membranes were probed with anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase, Biodesign, Saco, Maine) after being washed with stripping buffer. The signals were quantified by scanning densitometry and computer-assisted image analysis. The primary antibodies used in the present study were goat anti-serum albumin (ab19194, Abcam, Cambridge, MA), rabbit anti-Occludin (N-term) (40–6100, Invitrogen, South San Francisco, CA) and rabbit anti-zonula occludens-1 (ZO-1) (61–7300, Invitrogen, South San Francisco, CA).
2.9. Immunohistochemistry (IHC)
The IHC staining for tight junction protein ZO-1 were performed as described previously (Hua et al. 2005;Hua et al. 2007b). The primary antibody employed was rabbit anti-ZO-1 (40–6100, Invitrogen, South San Francisco, CA). The biotinylated secondary antibody and antibody–biotin–avidin–peroxidase complexes (ABC reagent, SC-2018) for ZO-1 IHC staining were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Slides processed without primary antibodies served as negative controls.
2.10. Assessment of blood-brain barrier permeability (BBBP)
Six hrs after reperfusion following MCAO for 60 minutes, mice were anesthesized and perfused with Evan’s Blue dye (4%; 2.5 ml/kg) as described previously (Shimamura et al. 2006a). Two hrs after infusion, the mice were euthanized and perfused with ice-cold PBS via the ascending aorta until the perfusion buffer was clear from the right atrium. The brains were removed. The left hemispheres were homogenized with 2.0 ml PBS and centrifuged at 1,000 x g for 30 min. Supernatants were mixed with equal amounts of 100% trichloroacetic acid sufficient to precipitate proteins. The supernatants were measured for the absorbance of Evans Blue dye color at 610 nm with a spectrophotometer. Evans Blue content was expressed as μg/g brain tissue.
2.11. Statistical analysis
Comparison for mortality was accomplished with the Chi-square test. Continuous scale measurements were expressed by the mean and standard error of the mean (S.E.M) for each group. Group comparisons for neurological function (neurological score) were accomplished with the t-test (applied to score values) and by the chi-square test. ANOVA and the least significant difference procedure were used to assess the effectiveness of intervention and group differences for infarct size, brain edema, levels of albumin, Occludin, ZO-1 and content of Evan’s Blue dye. Probability levels of 0.05 or smaller were used for reporting statistical significance.
3. Results
3.1 Pam3CSK4 preconditioning significantly reduced cerebral infarct size following I/R
We have previously reported that TLR2 modulation with an immunomodulator protected the myocardium from ischemic injury (Li et al. 2003). To investigate whether modulation of TLR2 would induce protection against focal cerebral I/R injury, we administered Pam3CSK4, a TLR2 specific ligand, to mice 24 hrs before cerebral I/R. As shown in Figure 1, focal cerebral I/R significantly induced cerebral infarction. Modulation of TLR2 by Pam3CSK4 significantly reduced cerebral infarct volumes by 79.8% compared with the untreated I/R group (1.9 ± 0.5% vs 9.4 ± 2.2%, p<0.05).
Figure 1. Pre-treatment with Pam3CSK4 reduced infarct size following cerebral I/R injury.
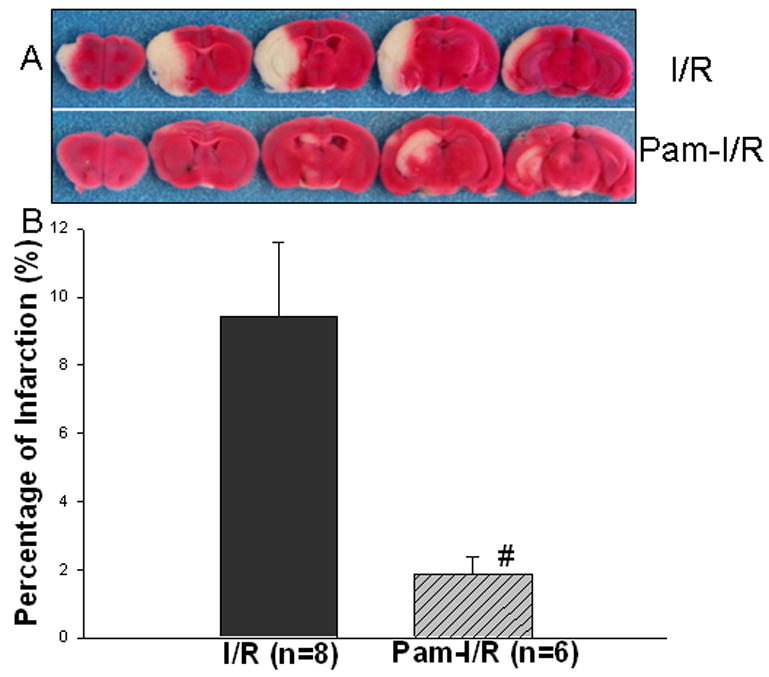
Pam3CSK4 was administered to mice (n=6) 24 hrs before cerebral ischemia (1 hr)/reperfusion (24 hrs) was induced. Untreated mice were also subjected to cerebral I/R. Infarct size was determined by TTC staining and expressed as the percentage of actual infarct volume in the total cerebral volume. The data are expressed as mean ± standard error (s.e.). Representative brain sections stained with TTC are shown above the graph. #compared with I/R, p<0.05.
3.2. Pam3CSK4 preconditioning attenuated the neurological deficits, brain edema, and mortality associated with focal cerebral I/R
Neurological score evaluation is an index for the degree of neurological deficits associated with stroke. Figure 2A shows that the neurological score was significantly decreased in the mice that were subjected to focal cerebral ischemia (1 hour) followed by reperfusion for 24 hrs. In Pam3CSK4 treated mice (Pam-I/R), the neurological score was significantly higher than that in the untreated I/R mice (8.22 ± 0.65 vs 3.91 ± 0.57, p<0.05), indicating that TLR2 modulation attenuated the neurological deficits associated with stroke. No neurological deficits were observed in the two sham control groups.
Figure 2. Pre-treatment with Pam3CSK4 preserved neurological function (A), reduced brain edema (B) and decreased mortality (C) following cerebral I/R.

Mice that were treated with (Pam-I/R) and without (I/R) Pam3CSK4 were subjected to ischemia (1 hr)/reperfusion (24 hrs). Respective sham surgical operated mice served as sham (S) and Pam-sham (Pam-S) controls. (A) Neurological function was evaluated using a scoring system ranging 0 to 15, with 15 being a perfect score and 0 being death due to cerebral I/R. (B) Brain water content was measured in cerebral tissues from the left hemisphere. (C) Mortality was monitored during reperfusion (within 24 hrs). The data are expressed as mean ± standard error (s.e.). *compared with sham; #compared with I/R, p<0.05.
Prevention of brain edema is critical for preservation of neurological function and survival following focal cerebral I/R. We examined the brain water content in cerebral tissue from the left hemisphere 24 hrs after cerebral I/R. As shown in Figure 2B, brain water content was significantly increased in the mice subjected to focal cerebral I/R compared with sham control (85.29 ± 0.09% vs 81.60 ± 0.17%). Focal cerebral I/R also resulted in significantly increased brain water content in Pam3CSK4-treated mice compared with Pam-sham control (84.61 ± 0.08% vs 81.57 ± 0.22%). However, brain water content in Pam3CSK4 treated I/R mice was significantly less compared with untreated I/R mice (84.61 ± 0.08% vs 85.29 ±0.09%, p<0.05). There was no significant difference in the brain water content between the sham control and Pam-sham groups.
We monitored the mortality in mice treated with and without Pam3CSK4 for 24 hrs after cerebral I/R. As shown in Figure 2C, eight of thirty-three mice (24%) in the untreated I/R group died within 24 hrs after reperfusion. In contrast, only one death (4.4%) was observed in the Pam3CSK4 pre-treated I/R group within the same time period. As expected, there were no deaths in sham and Pam-treated sham control groups.
3.3. Pam3CSK4 preconditioning attenuated blood-brain barrier permeability following cerebral I/R
A functional blood brain barrier plays a critical role in preventing brain damage from I/R. We examined whether modulation of TLR2 by Pam3SCK4 would preserve BBB function following cerebral I/R. Leakage of serum albumin into brain tissue is an established indicator for BBB function. Therefore, we examined the levels of serum albumin in brain tissue 24 hours after cerebral I/R. Figure 3A shows that cerebral I/R resulted in significantly increased levels of serum albumin in brain tissue compared with sham control (0.77 ± 0.06 vs 0.22 ± 0.067, p < 0.05). In contrast, the levels of serum albumin in brains from the Pam-I/R mice were significantly less when compared with control I/R mice (0.53 ± 0.03 vs 0.77 ± 0.06, p < 0.05). However, we did note that the levels of serum albumin in the brain tissue of Pam-treated mice was not maintained at control levels (0.53 ± 0.03 vs 0.22 ± 0.01, p < 0.05). As expected, the levels of serum albumin in brain tissue were low in both sham control groups.
Figure 3. Pre-treatment with Pam3CSK4 preserved Blood Brain Barrier (BBB) function following cerebral I/R.
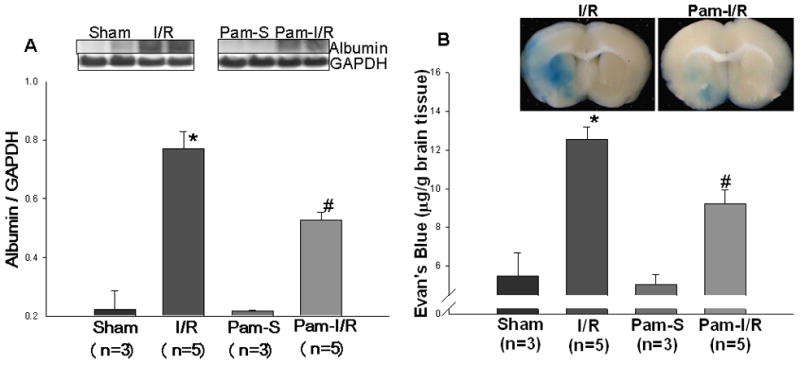
Mice that were treated with and without Pam3CSK4 were subjected to ischemia (1 hr) followed by reperfusion (24 hrs). Respective sham surgical operated mice served as sham (S) and Pam3CSK4 treated sham (Pam-S) controls. (A) Serum albumin in cerebral tissue was examined by Western blot. (B) Blood-brain barrier permeability was determined using the Evans Blue dye exudation method. Representative Evans Blue dye in brains is shown above the graph. The data are expressed as mean ± standard error (s.e.). *compared with sham, p<0.01; #compared with I/R, p<0.05.
The Evans Blue dye exudation technique is another method for evaluating BBB permeability. We measured the content of Evans Blue in the brain tissue 6 hrs after cerebral I/R. Figure 3B shows that cerebral I/R significantly increased the content of Evans Blue in the brain tissue of both I/R (12.56 ± 0.65 μg/mg tissue) and Pam3CSK4-treated I/R (Pam-I/R) (9.23 ± 0.72 μg/mg tissue) mice. However, the content of Evans Blue in the brain tissue of Pam-I/R mice was significantly lower than that in untreated I/R mice (9.23 ± 0.72 μg/mg vs 12.56 ± 0.65 μg/mg cerebral tissue). As expected, the content of Evans Blue in the brain was low in both sham and Pam-treated sham control mice.
3.4. Pam3CSK4 preconditioning increased ZO-1 levels in the brain tissues in the presence and absence of I/R injury
ZO-1 is an important protein in tight junctions (TJ) of the BBB. To investigate whether modulation of TLR2 will affect the expression of ZO-1 protein in the brains tissue, we analyzed tissue sections by immunohistochemical staining with specific antibody against ZO-1. Figure 4 shows that the staining of ZO-1 protein in cerebral vessels was weak in both sham control (A) and I/R mice (B). However, intense staining of ZO-1 in brain vessels was observed in both Pam-treated sham control and Pam-I/R mice (Fig. 4C and D). Western blot data indicate that modulation of TLR2 by Pam3CSK4 significantly increased the expression of ZO-1 protein in the presence or absence of cerebral I/R injury (Figure 4E).
Figure 4. Pre-treatment with Pam3CSK4 increased the expression of zonula occluden-1 (ZO-1) in cerebral vasculature in the presence and absence of I/R.
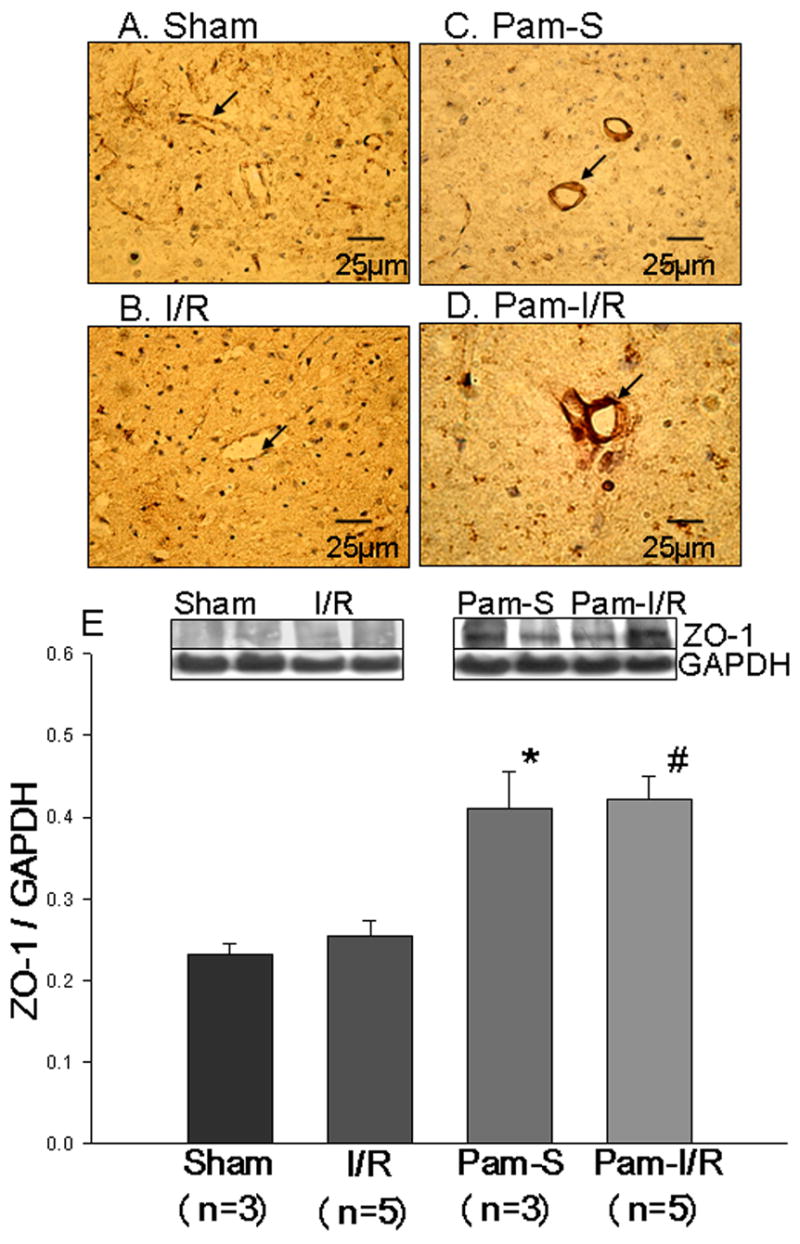
Immunohistochemical staining using specific antibody showed that there was weak expression of ZO-1 in cerebral vessels in sham (A) and I/R (B). Intense staining for ZO-1 was observed in Pam-S (C) and Pam-I/R (D). Western Blots (E) showed that treatment with Pam3CSK4 significantly increased the expression of ZO-1 compared with sham and I/R (untreated controls). The data are expressed as mean ± standard error (s.e.). *compared with sham, p<0.05; #compared with I/R, p<0.05.
3.5. Pam3CSK4 preconditioning attenuated the loss of Occludin in brain tissue following I/R injury
Occludin is an integral plasma membrane protein located specifically at tight junctions. We examined the expression of Occludin protein in brain tissue by Western blot. Figure 5 shows that the levels of Occludin were significantly reduced following cerebral I/R (0.35 ± 0.02 vs 0.72 ± 0.08, p<0.05) compared with the sham control. However, the decreased levels of Occludin in the brain tissue of I/R mice (0.35 ± 0.02) were significantly attenuated in Pam3CSK4 treated I/R mice (0.47 ± 0.04). There was no significant difference between sham control and Pam-treated sham control groups.
Figure 5. Pre-treatment with Pam3CSK4 prevented the decrease in Occludin following cerebral I/R.
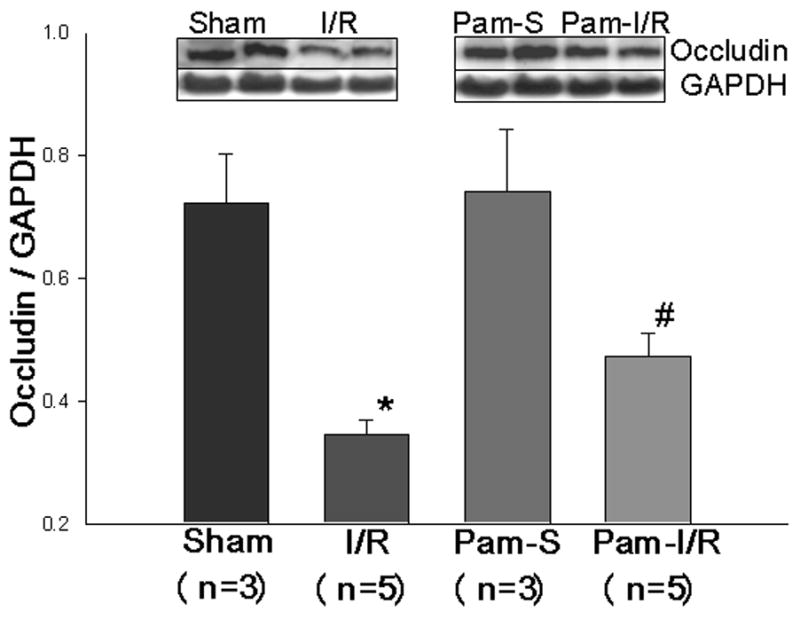
Cellular proteins were isolated from brain tissues of sham (S), cerebral I/R (I/R), Pam3SCK4 treated sham control (Pam-S), and Pam3SCK4 treated I/R (Pam-I/R) mice, respectively and subjected to Western blot with specific antibody against Occludin. The data are expressed as mean ± standard error (s.e.). *compared with sham, p<0.05; #compared with I/R, p<0.05.
4. Discussion
The most significant finding that emerged from the present study was that preconditioning with a TLR2 specific ligand significantly reduced cerebral infarct size and the mortality associated with focal cerebral I/R injury. TLR2 preconditioning also improved BBB function after cerebral I/R suggesting that preservation of BBB function could be one of the mechanisms by which a TLR2 specific ligand induced protection against cerebral I/R injury.
Toll-like receptors (TLRs) are a family of signal transduction molecules that play a critical role in the induction of innate and adaptive immunity (Medzhitov et al. 1997). TLRs have been reported to be involved in preconditioning induced tolerance to ischemia (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997;Stevens et al. 2008). A widely recognized agent for preconditioning is bacterial LPS and its derivatives (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997). LPS, a TLR4 specific ligand (Jerala 2007), is a glycolipid that constitutes the major portion of the outermost membrane of Gram-negative bacteria (Trent et al. 2006). Systemic administration of low dose LPS provides protection against a variety of subsequent insults (Bordet et al. 2000;Puisieux et al. 2000;Dawson et al. 1999;Tasaki et al. 1997). Preconditioning with a TLR9 ligand can induce neuroprotection against ischemic injury via mechanisms that share common elements with LPS preconditioning via TLR4 (Stevens et al. 2008). Since bacterial lipoproteins are a family of proinflammatory cell wall components found in both Gram positive and Gram negative bacteria and the stimulatory activity of bacterial lipoproteins resides in their acylated amino terminus, we hypothesized that pretreatment with a TLR2 specific ligand, Pam3CysSerLys4 (Pam3CSK4), a synthetic tripalmitoylated lipopeptide that mimics the acylated amino terminus of bacterial lipoproteins, might induce a protective effect against brain ischemic injury. Our results demonstrate that Pam3CSK4 preconditioning significantly decreased cerebral infarct size, reduced mortality related to cerebral I/R and preserved neurologic function in the first 24 hours after I/R (Fig. 1 and 2). To best of our knowledge, these are the first data to demonstrate that preconditioning with a TLR2 specific ligand, Pam3CSK4, protects the brain from ischemic injury.
The exact mechanisms involved in preconditioning-induced ischemic tolerance in the brain are not well understood. Previous studies indicate that inflammatory cytokines (Tasaki et al. 1997;Nawashiro et al. 1997), endogenous brain superoxide (Bordet et al. 2000), bcl-2-protein, mitochondrial membranes (Brambrink et al. 2004), endothelium-derived nitric oxide (NO) and heat shock proteins (HSP) (Puisieux et al. 2000;Valentim et al. 2001) may be involved in the protective mechanisms of preconditioning. In addition, preconditioning has been reported to induce a vasculoprotective effect (Dawson et al. 1999) and to reduce BBB disruption and brain edema (Masada et al. 2002). The development of cerebral infarction is accompanied by the formation of severe brain edema, which increases intracranial pressure, leading to a compression of the microvasculature and further cerebro-circulatory disorders followed by secondary expansion of the infarct volume, all of which increases mortality (Klatzo et al. 1986). An interesting finding in the present study was that Pam3CSK4 preconditioning attenuated brain edema induced by focal cerebral I/R injury (Fig. 2). Attenuation of brain edema may not only may result in smaller infarct size and improved neurological function but may also reduce mortality due to I/R. When taken together, these effects may be responsible for the protective effect of Pam3CSK4 preconditioning in the mice subjected to cerebral I/R.
Previous studies indicate that cerebral I/R causes disruption of the brain-blood barrier (BBB), which accelerates the development of abnormal vascular permeability and exacerbation of post-ischemic edema (Cole et al. 1991;Yang and Betz 1994). Protection of the BBB has become an important target of stroke intervention (Veltkamp et al. 2005). The BBB is the interface between the peripheral circulation and the central nervous system (CNS) which serves to limit the free passage of hormones, drugs, and other neuroactive and neurotoxic substances into the central nervous system. Our study showed that Pam3CSK4 preconditioning attenuated the increased BBB permeability induced by cerebral I/R, which was evaluated by brain albumin levels and Evans Blue (EB) exudation. Leakage of EB into cerebral tissue was clearly related to infarct area, which was observed predominantly in the caudoputamen, thalamus and the cerebral neocortex ipsilateral to the MCAO. However, the EB dye content in the ischemic cerebral hemisphere of brains from the Pam3CSK4 preconditioned group (Pam-I/R) was significantly less than that in the I/R group. These results provided evidence that Pam3CSK4 preconditioning attenuated damage to the BBB, with decreased BBB leakage following focal cerebral I/R injury. Although the possibility that brain parenchymal cells may participate in the development of tolerance after Pam3CSK4 preconditioning can not be ruled out, our data strongly indicates that the Pam3CSK4 preconditioning induced a protective effect, in part, by preserving BBB integrity after cerebral ischemia.
It is well known that cerebral I/R results in a marked decrease of cerebral blood flow, leading to disruption of the BBB (Nagel et al. 2008;Nagaraja et al. 2007). The mechanisms postulated to be involved include the acute destruction of the endothelium (Westergaard et al. 1976), oxygen free radicals causing peroxidation of lipids in cell membranes, failure of membrane ATPase and changes in ion homeostasis (Nelson et al. 1992). However, the mechanisms by which Pam3CSK4 preconditioning preserves BBB integrity after I/R is not known and worthy of further investigation.
The anatomical substrate of the BBB is the cerebral microvascular endothelium, which, together with astrocytes, pericytes, neurons, and the extracellular matrix, constitute a “neurovascular unit” that is essential for the health and function of the central nerve system (CNS) (Hawkins and Davis 2005). The continuous layer of cerebrovascular endothelial cells is connected by tight junctions (TJ), which are an intricate complex of tight junction proteins, such as Occludin and zonula occludens-1 (ZO-1) (Hawkins and Davis 2005;Hawkins and Davis 2005). Disruption of the tight junction by disease or drugs can lead to impaired BBB function and thus may compromise the CNS. Therefore, maintaining tight junction proteins at certain levels may be critical for preservation of BBB function during cerebral I/R. The expression and subcellular localization of TJ proteins are modulated by several intrinsic signaling pathways. Recent reports indicate that TLR2 regulated epithelial barrier function by enhancing the tight junction (TJ) protein zonula occludens-1 (ZO-1) (Cario et al. 2004). TLR2-mediated TJ regulation critically determines susceptibility to intestinal injury and inflammation (Cario et al. 2007). The TLR2 specific ligand, Pam3CSK4, has been shown to suppress mucosal inflammation and apoptosis by restoring TJ-associated integrity of the intestinal epithelium in vivo (Cario et al. 2004;Cario et al. 2007). TLR2 has been reported to be expressed in vascular endothelial cells and response to TLR2 ligands (Chen et al. 2007). In addition, weak TLR2 expression has also been observed in cerebral vascular endothelial cells in the ischemic brain (Ziegler et al. 2007). In the present study, we investigated whether Pam3CSK4 preconditioning modulated the expression of TJ proteins. We found that Pam3CSK4 preconditioning significantly increased the expression of ZO-1 protein in the cerebral vasculature in the absence and presence of cerebral I/R. Interestingly, Pam3CSK4 preconditioning significantly attenuated the loss of Occludin protein induced by cerebral I/R. Our data suggest that over expression of ZO-1 protein and preventing in the loss of Occludin protein could be another mechanism by which Pam3CSK4 precondition attenuates BBB dysfunction following focal cerebral I/R.
In conclusion, we present evidence that preconditioning with a TLR2 specific ligand, Pam3CSK4, will reduce infarct size and mortality associated with focal cerebral I/R injury. Our findings suggest that maintaining the integrity of the blood brain barrier may be one mechanism by which preconditioning with Pam3CSK4 protects the brain from I/R injury. While the protective mechanisms have not been fully elucidated, the present data demonstrate for the first time that TLR2 is a new target of ischemic preconditioning in the brain and that preconditioning with TLR2 ligand may be an effective approach to prevent the debilitating and life threatening consequences of ischemic stroke.
Acknowledgments
This work was supported by AHA Scientist Development Grant 0830481N and AHA postdoctoral fellowship 0625348B to FH; ETSU RDC Grant to RLK; NIH GM53552 to DLW and HL071837 to CL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, Feuerstein GZ. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–1950. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Bordet R, Deplanque D, Maboudou P, Puisieux F, Pu Q, Robin E, Martin A, Bastide M, Leys D, Lhermitte M, Dupuis B. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab. 2000;20:1190–1196. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- Brambrink AM, Noga H, Astheimer A, Heimann A, Kempski O. Pharmacological preconditioning in glocal cerebral ischemia. Acta Neurochir Suppl. 2004;89:63–66. doi: 10.1007/978-3-7091-0603-7_8. [DOI] [PubMed] [Google Scholar]
- Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology. 2004;127:224–238. doi: 10.1053/j.gastro.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- Chen S, Wong MH, Schulte DJ, Arditi M, Michelsen KS. Defferential expression of Toll-like receptor-2 (TLR2) and responses to TLR2 ligands between human and murine vascular endothelial cells. J Endotoxin Res. 2007;13:281–296. doi: 10.1177/0968051907085096. [DOI] [PubMed] [Google Scholar]
- Cole DJ, Matsumura JS, Drummond JC, Schultz RL, Wong MH. Time- and pressure-dependent changes in blood-brain barrier permeability after temporary middle cerebral artery occlusion in rats. Acta Neuropathol. 1991;82:266–273. doi: 10.1007/BF00308811. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Furuya K, Gotoh J, Nakao Y, Hallenbeck JM. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. J Cereb Blood Flow Metab. 1999;19:616–623. doi: 10.1097/00004647-199906000-00004. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hertzog PJ, O’Neill LA, Hamilton JA. The interferon in TLR signaling: more than just antiviral. Trends Immunol. 2003;24:539. doi: 10.1016/j.it.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Comm. 2005;338:1118–1125. doi: 10.1016/j.bbrc.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C. Protection against Myocardial Ischemia/Reperfusion Injury in TLR4 Deficient Mice is Mediated through a Phosphoinositide 3-Kinase Dependent Mechanism. J Immunol. 2007a;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007b;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerala R. Structural biology of the LPS recognition. International Journal of Medical Microbiology. 2007;297:353–363. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Jiang J, Wang W, Sun YJ, Hu M, Li F, Zhu DY. Neuroprotective effect or curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur J Pharmacol. 2007;561:54–62. doi: 10.1016/j.ejphar.2006.12.028. [DOI] [PubMed] [Google Scholar]
- Klatzo I, Seida M, Wagner HG, Ting P. Features of ischemic brain edema. In: Krieglstein J, editor. Pharmacology of Cerebral Ischemia. Elsevier; Amsterdam: 1986. pp. 23–30. [Google Scholar]
- Li C, Ha T, Kelley J, Gao X, Qiu Y, Kao RL, Browder W, Williams DL. Modulating Toll-like receptor mediated signaling by (1-->3)-β-D-glucan rapidly induces cardioprotection. Cardiovascular Research. 2003;61:538–547. doi: 10.1016/j.cardiores.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Masada T, Hua Y, Xi G, Ennis SR, Keep RF. Effect of ischemic preconditioning on edema formation and cerebrovascular injury following focal cerebral ischemia. Acta Neurochir Suppl. 2002:2002–265. doi: 10.1007/978-3-7091-6738-0_68. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Nagaraja TN, Keenan KA, Brown SL, Fenstermacher JD, Knight RA. Relative distribution of plasma flow markers and red blood cells across BBB openings in acute cerebral ischemia. Neurology Research. 2007;29:78–80. doi: 10.1179/174313206X153815. [DOI] [PubMed] [Google Scholar]
- Nagel S, Su Y, Horstmann S, Heiland S, Gardner H, Koziol J, Martinez-Torres FJ, Wagner S. Minocycline and hypothermia for reperfusion injury after focal cerebral ischemia in the rat: effects on BBB breakdown and MMP expression in the acute and subacute phase. Brain Research. 2008;1188:206. doi: 10.1016/j.brainres.2007.10.052. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- Nelson CW, Wei EP, Povlishock JT, Kontos HA, Moskowitz MA. Oxygen radicals in cerebral ischemia. Am J Physiol. 1992;263:H1356–H1362. doi: 10.1152/ajpheart.1992.263.5.H1356. [DOI] [PubMed] [Google Scholar]
- Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R. Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischemic tolerance. Eur J Pharmacol. 2000;389:71–78. doi: 10.1016/s0014-2999(99)00893-6. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Somogyvari-Vigh A, Szanto Z, Kertes E, Lenard L, Arimura A, Lengvari I. Effects of pretreatment with PACAP on the infarct size and functional outcome of rat permanent focal cerebral ischemia. Peptides. 2002;23:2227–2234. doi: 10.1016/s0196-9781(02)00262-0. [DOI] [PubMed] [Google Scholar]
- Shimamura N, Matchett G, Solaroglu I, Tsubokawa T, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 reduces blood-brain barrier breakdown in focal ischemia in rats. J Neurosci Res. 2006a;84:1837–1847. doi: 10.1002/jnr.21073. [DOI] [PubMed] [Google Scholar]
- Shimamura N, Matchett G, Yatsushige H, Calvert JW, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 ameliorates focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Stroke. 2006b;37:1902–1909. doi: 10.1161/01.STR.0000226991.27540.f2. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, Lessov NS, Simon RP, Stenzel-Poore MP. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008 doi: 10.1038/sj.jcbfm.9600606. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- Trent MS, Stead CM, Tran AX, Hankins AX. Diversity of endotoxin and its impact on pathogenesis. Journal of Endotoxin Research. 2006;12:205–223. doi: 10.1179/096805106X118825. [DOI] [PubMed] [Google Scholar]
- Valentim LM, Geyer AB, Tavares A, Cimarosti H, Worm PV, Rodnight R, Netto CA, Salbego CG. Effects of global cerebral ischemia and preconditioning on heat shock protein 27 immunocontent and phosphorylation in rat hippocampus. Neuroscience. 2001;107:43–49. doi: 10.1016/s0306-4522(01)00325-6. [DOI] [PubMed] [Google Scholar]
- Veltkamp R, Siebing DA, Sun L, Heiland S, Bieber K, Marti HH, Nagel S, Schwab S, Schwaninger M. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke. 2005;36:1679–1783. doi: 10.1161/01.STR.0000173408.94728.79. [DOI] [PubMed] [Google Scholar]
- Westergaard E, Go KG, Klatzo I, Spatz M. Increased permeability of cerebral vessels to horseradish peroxidase induced by ischemia in Mongolian gerbils. Acta Neuropathol (Berl) 1976;35:307–325. [PubMed] [Google Scholar]
- Yang GY, Betz AL. Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke. 1994;25:1658–1665. doi: 10.1161/01.str.25.8.1658. [DOI] [PubMed] [Google Scholar]
- Yang GY, Gong C, Qin Z, Liu XH, Lorris BA. Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res. 1999;69:135–143. doi: 10.1016/s0169-328x(99)00007-8. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Röhr C, Prinz V, König J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]