

Abstract
Nitroalkene derivatives of linoleic acid (nitrolinoleic acid; LNO2) and nitro-oleic acid (OA-NO2) are endogenous lipid products with potent anti-inflammatory properties. The present study was undertaken to evaluate the therapeutic potential of OA-NO2 in a mouse model of renal ischemia-reperfusion (I/R) injury. B6129SF2/J mice were subjected to bilateral renal ischemia for 30 min, followed by 24 h of reperfusion. Fifty minutes after ischemia, mice received intraperitoneal (ip) injections of OA-NO2 (500 μg/kg; I/R OA-NO2), vehicle for OA-NO2 (i.e., 0.8 ml/kg ethanol; I/R veh), or oleic acid (500 μg/kg; I/R OA) every 6 h during the 24-h recovery period. A sham-operated group was not subjected to ischemia and received 0.8 ml/kg ethanol ip every 6 h during the 24-h recovery period (sham veh). While plasma urea and creatinine were elevated (P < 0.05) in I/R veh vs. sham veh mice, the severity was less (P < 0.05) in I/R OA-NO2 animals. Indices of histological damage, polymorphonucleocyte infiltration, together with expression of intracellular adhesion molecule-1, interleukin-1β, and tumor necrosis factor-α, p47phox, and gp91phox were greater in I/R veh vs. sham veh mice, but were attenuated (P < 0.05) in I/R OA-NO2 animals. Because indices of renal dysfunction were similar between I/R veh and I/R OA mice (P > 0.05), but less (P < 0.05) in I/R OA-NO2 animals compared with both groups, protection from bilateral renal ischemia is afforded by the nitrated but not free form of oleic acid. Together, delayed administration of nitrated fatty acid OA-NO2 attenuates renal I/R injury in the mouse likely via inhibition of the inflammatory response.
Keywords: nitrated fatty acids, acute renal failure, inflammation
renal ischemia and reperfusion (I/R) injury is a common cause of acute renal failure (ARF) in patients (1, 7). In ischemic ARF, there is reduced renal blood flow, an impaired ultrafiltration coefficient, and tubular obstruction (16, 43). These events result in tissue hypoxia and subsequently trigger pathophysiological processes that include polymorphonucleocyte (PMN) infiltration into the kidney, together with activation of proinflammatory cytokines, reactive nitrogen species, reactive oxygen species (ROS), and the coagulation system (2, 42). If the intensity and/or duration of ischemia is sufficient, these processes can lead to cell death and compromise renal function (6, 39, 44). Although mechanisms responsible for renal I/R injury in experimental animal models have been investigated extensively, pharmacological interventions designed to target a specific factor in this process have not been efficacious in the clinical setting (1). Despite the advances in critical care medicine, the high mortality and morbidity rate from ARF has remained unchanged over the past 50 years (5, 9, 41). Thus a need exists to identify new intervention strategies.
Recently, nitrated free fatty acids (NO2-FA), notably nitroalkene derivatives of linoleic acid (nitrolinoleic acid; LNO2) and nitro-oleic acid (OA-NO2), are found to be endogenous molecules with several attractive signaling properties (4, 38). These derivatives are formed via nitric oxide-dependent oxidative reactions (31, 36, 37). Plasma concentrations are ∼0.5 μM for LNO2 (4) and ∼0.6 μM for OA-NO2 (4) in healthy human blood, and the combined blood levels of these two fatty acid derivatives exceed 1 μM, indicating their capability to act in physiological concentration ranges. Nitroalkenes display potent anti-inflammatory properties as they inhibit human neutrophil superoxide generation, degranulation, and integrin expression (8), and they also attenuate lipopolysaccharide-induced secretion of proinflammatory cytokines in cultured macrophages (10). Additionally, nitroalkenes are found to be a robust endogenous ligand for peroxisome proliferator-activated receptor-γ (PPARγ) although they also activate PPARα and PPARδ at increasing concentrations (4, 38). PPARs have emerged as a novel therapeutic target for the treatment of metabolic syndrome as well as other inflammatory diseases such as ischemic ARF (17). Finally, nitroalkenes exhibit other signaling properties, such as releasing nitric oxide (28). However, to date there are no prior studies to examine their biological function in vivo. Herein, the present study examines the therapeutic potential of OA-NO2 in a mouse model of renal I/R injury generated by 30 min of bilateral ischemia followed by 24-h reperfusion.
METHODS
Materials.
9-Nitro-oleic acid and 10-nitro-oleic acid are two regioisomers of nitro-oleic acid (OA-NO2), which are formed by nitration of oleic acid (OA) in approximately equal proportions in vivo (4). The two compounds (9-nitro-oleic acid: catalog no. 10008042; 10-nitro-oleic acid: catalog no. 10008043) were purchased from Cayman Chemical (Ann Arbor, MI), dissolved in ethanol, and used as a 1:1 mixture of the isomers. OA was purchased from the same company (catalog no. 90260) and dissolved in the same solvent. All other reagents were purchased from Sigma-Aldrich unless otherwise specified.
Animals and treatment groups.
All protocols were approved by the Institutional Animal Care and Use Committee of the University of Utah. Three-month-old male B6129SF2/J mice were maintained in a temperature-controlled barrier facility with a 12:12-h light-dark cycle and were given free access to standard laboratory chow and tap water. Animals were anesthetized initially with 2–5% isoflurane, placed on a heated surgery table, and maintained on 2% isoflurane while breathing spontaneously. After a 1-cm incision in the abdomen was made, the left and right renal arteries were located, isolated, and occluded for 30 min using microaneurysm clips to produce ischemia. After removal of the clips, verification of reperfusion, and closing of the incision in two layers, anesthesia was terminated and 1 ml of 37°C saline was injected into the abdomen to supplement fluid loss. One hour after the commencement of ischemia, the animals were randomly divided to receive intraperitoneal (ip) injections of OA-NO2 (500 μg/kg), vehicle for OA-NO2 (i.e., 0.8 ml/kg ethanol), or oleic acid (500 μg/kg) every 6 h. A sham-operated group underwent identical surgical procedures except that microaneurysm clamps were not applied; these mice received 0.8 ml/kg ethanol ip every 6 h. In a separate experiment, the I/R animals received a single injection of OA-NO2 (2.0 mg/kg) or ethanol (0.8 ml/kg). After 24 h, mice were anesthetized using 2% isoflurane, 500–600 μl of blood was obtained from the inferior vena cava, and both kidneys were excised.
Measurement of MPO.
MPO was measured in one kidney as an indicator of PMN infiltration. Immediately upon excision, the kidney was weighed, homogenized in 10 mmol/l potassium phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide, centrifuged for 15 min at 1,500 g at 4°C, and the supernatant was used to assess MPO activity (ng/100 mg wet wt) using spectrophotometric techniques according to the manufacturer's guidelines (Hycult Biotechnology, Uden, The Netherlands).
Measurement of circulating TNF-α.
Circulating. TNF-α was measured by using a commercially available enzyme immunoassay kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Measurement of biochemical parameters.
Blood samples from anesthetized mice were obtained after renal ischemia-reperfusion for 24 h. Plasma levels of urea and creatinine were measured using standard urease assay/conductivity and picric acid reactions, respectively.
Morphological studies.
Kidneys were hemisectioned and fixed by direct immersion in 3% paraformaldehyde for 16 h. Following embedding in paraffin, 4-μm sections were prepared and stained with hematoxylin and eosin and analyzed with light microscope. Renal pathology scores were generated by an observer who was blind as to the identity of the specimen. The following parameters were scored on a 1–3 scale as described elsewhere (40): 0, normal histology; 1, tubular cell swelling, brush-border loss, with up to of tubular profile showing nuclear loss; 2, as for score 1, but greater than and less than of tubular profile shows nuclear loss; and 3, greater than of tubular profile shows nuclear loss. The histological score for each kidney was calculated by addition of all 100 scores with a maximum score of 300.
Real-time RT-PCR.
Under isoflurane anesthesia, kidneys were harvested and preserved in RNAlater solution (Ambion, Austin, TX). The tissue samples in the RNAlater solution were kept on ice after sampling, stored overnight at 4°C, and kept frozen at −20°C until RNA extraction. Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA), and cDNA was synthesized using SuperScript (Invitrogen). The sequence of oligonucleotides used for real-time PCR (RT-PCR) is listed as follows: ICAM-1 sense:5′-GGTCGAAGGT-GGTTCTTCTG-3′ and ICAM1 antisense:5′-CCAAGCGTCCGTCTCGT-3′; p47phox sense: 5′-GTCGTGGAGAAGAGCGAGAG-3′ and p47phox antisense: 5′-CGCTTTGATGGTTACATACGG-3′; gp91phox sense: 5′-CCGTATTGTGGGAGACTGGA-3′ and gp91phox antisense: 5′-CTTGAGAATGGAGGCAAAGG-3′; and GAPDH sense: 5′-GTCTTCACTACCATGGAGAAGG-3′ and GAPDH antisense: 5′- TCATGGATGACCTTGGCCAG-3′. Real-time RT-PCR was performed using Sybergreen and the ABI Prism 7900 Sequence Detection System. The amplification was carried out for 40 cycles with conditions of 15-s denaturation at 95°C.
Immunoblotting.
The lysates of the kidney were stored at −80°C until assayed. Protein concentrations were determined using Coomassie reagent. An equal amount of the whole tissue protein (60 μg) was denatured at 100°C for 10 min, separated by SDS-PAGE, and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS), followed by incubation for 1 h with rabbit polyclonal antibody against IL-1β (catalog no. sc-7884, Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal antibody against TNF-α (catalog no. sc57469, Santa Cruz Biotechnology), or mouse monoclonal antibody against α-tubulin (catalog no. T5168, Sigma, St. Louis, MO). The blots were washed with TBS followed by incubation with goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody. Immune complexes were detected using ECL methods. The immunoreactive bands were quantified using the Gel and Graph Digitizing System (Silk Scientific, Tustin, CA).
Measurement of thiobarbituric acid-reactive substances.
The measurement of thiobarbituric acid-reactive substances (TBARS) in the mouse kidney was based on the formation of malondialdehyde (MDA) by using a commercially available TBARS Assay kit (catalog no. 10009055, Cayman Chemical) according to the manufacturer's instructions.
Statistical analysis.
Values shown represent means ± SE. Data were analyzed using one-way ANOVA followed by a Bonferroni posttest. A P value <0.05 was considered significant.
RESULTS
Renal injury.
After 24 h of reperfusion, plasma urea and creatinine were 5.6- and 9.3-fold greater, respectively, in I/R vehicle-treated vs. sham vehicle-treated mice [plasma blood urea nitrogen (BUN): 198.7 ± 22.8 vs. 35.6 ± 2.7 mg/dl, P < 0.01; plasma creatinine: 2.6 ± 0.47 vs. 0.26 ± 0.05 mg/dl, P < 0.01] (Fig. 1). While the increases in plasma urea and creatinine were 3.9- and 5.7-fold were greater, respectively in I/R OA-NO2 vs. sham vehicle-treated mice, it was less severe compared with I/R vehicle-treated animals (plasma BUN: 139.2 ± 5.9 mg/dl; plasma creatinine: 1.62 ± 0.31 mg/dl in the I/R OA-NO2 group) (Fig. 1). Compared with sham vehicle-treated group, I/R vehicle-treated mice exhibited marked renal pathologies such as tubular necrosis, dilation of renal tubules, and luminal casts. Remarkably, these histological changes were attenuated in I/R OA-NO2 mice (Fig. 2A). These mice had a significantly reduced semiquantitative histological damage score in the renal cortex compared with I/R vehicle-treated mice (Fig. 2B). These data indicate that functional and histological indices of renal I/R injury were attenuated by OA-NO2 treatment during the 24-h reperfusion period.
Fig. 1.
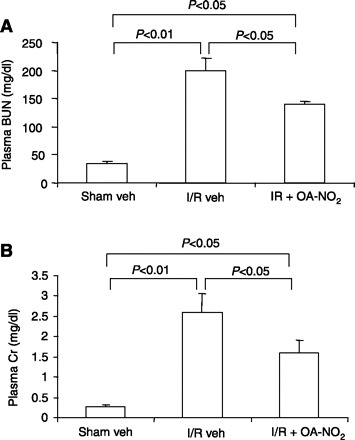
Effect of nitro-oleic acid (OA-NO2) on acute renal failure induced by ischemia-reperfusion (I/R). Male 3-mo-old B6129SF2/J mice were subjected to bilateral renal ischemia for 30 min by clamping of the renal artery, followed by reperfusion. One hour after the ischemia, vehicle (I/R veh) or OA-NO2 (I/R OA-NO2) was administered at 500 μg/kg every 6 h via intraperitoneal injection. Twenty-four hours after I/R, the animals were killed for evaluation of renal injury. Sham-operated mice served as a control (Sham veh). Values are means ± SE. A: plasma blood urea nitrogen (BUN). B: plasma creatinine (Cr). Sham veh: n = 5; I/R veh: n = 6; I/R OA-NO2: n = 6.
Fig. 2.
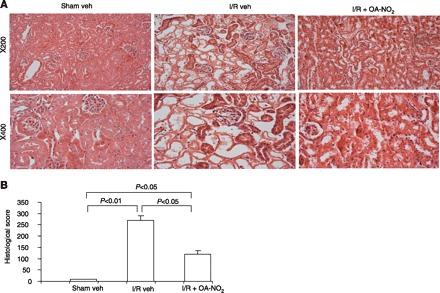
A: representative photomicrographs (hematoxylin and eosin staining, magnification ×200 and ×400) of renal cortex of the kidneys of sham veh, I/R veh, and I/R OA-NO2 mice. B: semiquantitative analysis of histological appearance. Values are means ± SE. Sham veh: n = 5; I/R veh: n = 6; I/R OA-NO2: n = 6.
Renal MPO levels.
MPO is a predominant enzyme in monocytes and in the azurophilic granules of PMN and is released upon neutrophil activation. This enzyme has been shown to play a detrimental role in the induction of renal I/R injury (30). Therefore, we determined tissue MPO levels using ELISA. Renal MPO levels were 4.6-fold greater in I/R vehicle-treated vs. sham vehicle-treated mice (Fig. 3). Strikingly, the increase in renal MPO was almost completely abolished in I/R OA-NO2 mice, demonstrating the ability of OA-NO2 treatment during reperfusion to attenuate I/R-induced PMN infiltration (Fig. 3).
Fig. 3.
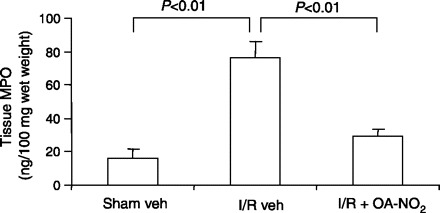
ELISA analysis of kidney MPO in sham veh, I/R veh, and I/R OA-NO2 mice. The values were normalized by wet kidney weight. Values are means ± SE. Sham veh: n = 5; I/R veh: n = 6; I/R OA-NO2: n = 6.
Expression of proinflammatory cytokines and adhesion molecules.
By real-time RT-PCR, renal expression of both IL-1β and ICAM1 exhibited a >10-fold increase in I/R vehicle-treated vs. sham vehicle-treated mice. Compared with I/R vehicle-treated mice, this increase was significantly attenuated in I/R OA-NO2 mice (Fig. 4). Similar results were obtained concerning circulating TNF-α as assessed by enzyme immunoassay (Fig. 4), and kidney protein abundance of TNF-α and IL-1β by immunoblotting (Fig. 5). Taken together, OA-NO2 ameliorates the release of proinflammatory cytokines and adhesion molecules in response to renal I/R.
Fig. 4.
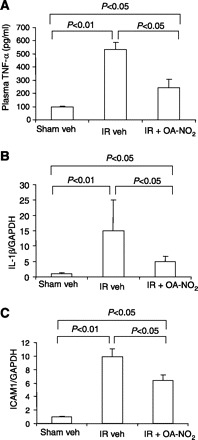
Levels of proinflammatory cytokines and adhesion molecules in sham veh, I/R veh, and I/R OA-NO2 mice. A: ELISA analysis of circulating TNF-α. B: real-time RT-PCR of renal IL-1β expression. C: real-time RT-PCR of renal ICAM1 expression. The expression was normalized by GAPDH. Values are means ± SE. Sham veh: n = 5; I/R veh: n = 6; I/R OA-NO2: n = 6.
Fig. 5.
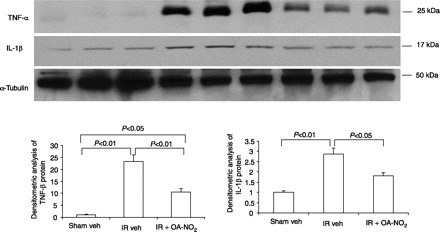
Immunoblotting analysis of TNF-α and IL-1β in the kidneys of sham veh, I/R veh, and I/R OA-NO2 mice. A: representative blots for TNF-α and IL-1β. α-Tubulin served as a loading control. B: densitometric analysis of TNF-α protein. C: densitometric analysis of IL-1β protein. Values are means ± SE; n = 3/group. Densitometric data were generated from 1 representative of 2–3 separate experiments.
Renal oxidative stress.
Renal p47phox and gp91phox were 24.5- and 2.0-fold greater, respectively, in I/R vehicle-treated vs. sham vehicle-treated mice (Fig. 6). While the increase in p47phox (15.5-fold) and gp91phox (1.2-fold) was greater in I/R OA-NO2 vs. sham vehicle-treated mice, it was less severe compared with I/R vehicle-treated animals (Fig. 6). The levels of kidney TBARS, an index of lipid peroxidation, were significantly elevated in I/R vehicle-treated vs. sham vehicle-treated mice and were almost completely normalized in I/R OA-NO2 mice (Fig. 6). These data suggest that OA-NO2 lessened NADPH oxidase subunit activation and ROS generation in response to I/R.
Fig. 6.
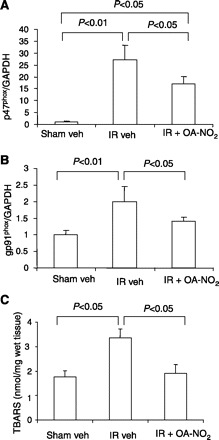
Real-time RT-PCR analysis of the expression of p47phox (A) and gp91phox (B) and measurements of thiobarbituric acid-reactive substances (TBARS) in the kidneys of sham veh, I/R veh, and I/R OA-NO2 mice. Values are means ± SE. Sham veh: n = 5; I/R veh: n = 6; I/R OA-NO2: n = 6.
Effect of a single injection of OA-NO2.
Of note, the above-discussed data were all obtained with multiple ip injections of OA-NO2 (500 μg/kg every 6 h). We also examined the effect of a single ip injection of OA-NO2 (2 mg/kg) on I/R-induced renal dysfunction. Compared with I/R vehicle-treated mice, the I/R OA-NO2 (single injection) mice had a modest attenuation of the increase in plasma BUN and creatinine, but this did not reach statistical significance (plasma BUN: 202.8 ± 3.9 vs. 163.8 ± 15.8 mg/dl, P > 0.05; plasma creatinine: 2.8 ± 0.1 vs. 2.0 ± 0.5 mg/dl, n = 4, P > 0.05). These findings suggest that multiple injections are likely required to produce the full beneficial effect of OA-NO2 at least in the mouse model of renal I/R injury.
Effect of OA on I/R-induced renal injury.
These experiments determined whether the protective effect of OA in response to renal I/R is specific to the nitrated (i.e., OA-NO2) or native form (i.e., OA). Functional (Fig. 7) and histological (Fig. 8) indices of renal dysfunction were similar in kidneys from I/R vehicle-treated and I/R OA-treated mice, documenting the lack of renoprotection with OA.
Fig. 7.
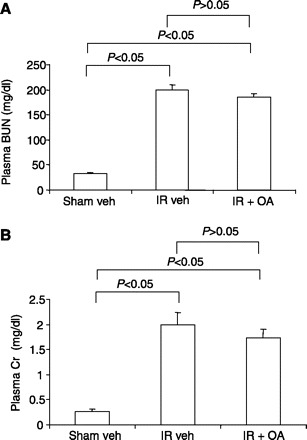
Effects of oleic acid (OA) on renal dysfunction in mice subjected to I/R. Shown are plasma BUN (A) and Cr (B) in sham veh, I/R veh, and I/R OA mice. Male 3-mo-old B6129SF2/J mice were subjected to bilateral renal ischemia for 30 min by clamping of the renal artery, followed by reperfusion. One hour after the ischemia, vehicle (I/R veh) or OA (I/R OA) was administered at 500 μg/kg every 6 h via intraperitoneal injection. Of note, the sham veh and I/R veh mice in these experiments were separate from those described in Fig. 1. Twenty-four hours after I/R, the animals were killed for evaluation of renal injury. Values are means ± SE; n = 5/group.
Fig. 8.
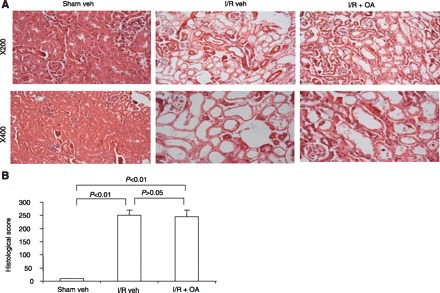
A: representative photomicrographs (hematoxylin and eosin staining, magnification ×200 and ×400) of renal cortex of the kidneys of sham veh, I/R veh, and I/R OA mice. B: semiquantitative analysis of histological appearance. Values are means ± SE; n = 5/group.
DISCUSSION
NO2-FA, notably nitroalkene derivatives of LNO2 and OA-NO2, are recently identified as endogenous signaling molecules; they are detected at micromolar concentrations in healthy human plasma (4). In vitro evidence suggests that these nitrated lipids possess potent anti-inflammatory properties and also function as endogenous PPARγ ligands (4, 38), suggesting their therapeutic potential for an array of human diseases, such as inflammatory diseases. In the present study, we tested the hypothesis that OA-NO2 attenuates injury in a mouse model with 30 min of bilateral renal ischemia and 24 h of reperfusion. Functional and histological indices of glomerular dysfunction, renal PMN infiltration, proinflammatory cytokines, and indices of oxidative stress were less severe in mice treated with OA-NO2 compared with those wherein vehicle or OA was administered during reperfusion. Of note, this observation was made with delayed administration of OA-NO2, which is of clinical importance since, in a clinical setting, ARF is usually diagnosed after the insult has already occurred. To our knowledge, these data provide the first evidence that OA-NO2 protects from renal I/R injury in the mouse.
Bilateral renal ischemia for 30 min followed by 24-h reperfusion produced significant renal injury in the present study. First, I/R-induced glomerular dysfunction, as reflected by elevated plasma urea and creatinine concentrations, was present in vehicle-treated vs. sham-operated mice. Furthermore, histological indices of tubular injury in kidneys from mice that completed I/R support the aforementioned functional evidence. Second, renal I/R produced marked increases in MPO activity in vehicle-treated animals, indicating that PMN infiltration into kidney tissue was more pronounced compared with sham-operated animals. Third, proinflammatory mediators measured in renal tissue (i.e., ICAM-1, IL-1β) and plasma (i.e., TNF-α) were activated to a greater degree in I/R vs. sham-operated mice. Finally, indices of oxidative stress, including the expression of p47phox and gp91phox, the key subunits of NADPH oxidase, and the levels of TBARS, were elevated in response to I/R. Collectively, these data indicate that our I/R protocol evoked significant renal injury. To test whether OA-NO2 ameliorates indices of renal I/R injury, we administered this compound at 1 h of ischemia, and every 6 h for 24 h thereafter. Plasma urea and creatinine, renal tubular damage, MPO activity, proinflammatory cytokines, and p47phox and gp91phox gene expression were less in I/R OA-NO2 vs. I/R vehicle-treated mice. As such, functional and histological damage, PMN infiltration, inflammation, and indices of oxidant stress, respectively, were attenuated by the nitrated fatty acid OA-NO2.
Inflammation has been recognized as a critical factor in the development and progression of I/R injury. Specifically, inflammation is an important mechanism to initiate and maintain renal cell injury as it potentates both necrosis (26, 27) and apoptosis (11–13). Early in the 1980s, Romson and coworkers (34) observed that neutrophil depletion reduced myocardial injury resulting from temporary artery occlusion. Similarly, prevention of PMN adherence using a monoclonal antibody against the β-chain of the CD18 complex significantly attenuated I/R-induced microvascular permeability in the intestine (19). Neutrophil invasion of damaged tissues constitutes the local inflammatory response in part by releasing their lysosomal constitutes including MPO. MPO, a 140-kDa protein present in the cytoplasm of neutrophils, represents one of the most abundant enzymes released on neutrophil activation (21). Renal I/R injury is associated with neutrophil infiltration (18) and increased tissue MPO activity (29, 32). A definitive role of MPO in renal I/R injury is recently demonstrated using MPO knockout mice (MPO −/−) (30). These mice exhibited improved renal function after I/R compared with their wild-type controls. Interestingly, MPO release by activated neutrophils is important for the activation and adherence of other neutrophils (20, 22). In line with this notion, neutrophil accumulation was significantly reduced in Mpo −/− mice (30). As expected, we found that I/R vehicle-treated mice had a marked increase in renal MPO levels compared with sham controls. Strikingly, the rise of MPO after I/R was almost completely blocked by delayed administration of OA-NO2, suggesting that inhibition of neutrophil activation may represent a primary mechanism for the renoprotective action of the nitrated lipid. Indeed, there is in vitro evidence that LNO2 inhibited phorbol 12-myristate 13-acetate- or N-formyl-Met-Leu-Phe-evoked neutrophil activation, as assessed by measurements of superoxide generation, Ca2+ influx, elastase release, and CD11b expression (8).
A major superoxide-generating system in activated neutrophils is NADPH oxidase, which is composed of membrane-associated subunits, p22phox, gp91phox, and cytosolic subunits, p47phox, p67phox, and p40phox. Among these subunits, gp91phox and p47phox are of particular importance as the former contains the catalytic domain and the latter is necessary for cytosolic subunit translocation and for initiation of NADPH oxidase assembly (3, 14, 15). We found that OA-NO2 was effective in reducing I/R-induced expression of both gp91phox and p47phox. Considering that the activated neutrophils may constitute a major source of ROS production during I/R, it seems reasonable to speculate that the reduction of NADPH oxidase expression by OA-NO2 might be the result of reduced neutrophil infiltration. It is also possible that OA-NO2 may exert a direct inhibitory effect on gene expression of NADPH oxidase. Of note, OA-NO2 treatment produced a nearly complete blockade of I/R-induced increases in renal TBARS, which cannot be fully explained by the partial effect on NADPH oxidase expression. Therefore, it seems reasonable to speculate that other sources than NADPH oxidase may also contribute to I/R-induced ROS generation and may serve as an additional target of OA-NO2.
In the present study, the anti-inflammatory properties of OA-NO2 in the mouse model of renal I/R injury was also evidenced by amelioration of the production of proinflammatory cytokines (i.e., TNF-α and IL-1β) and adhesion molecules (i.e., ICAM1), which may involve not only neutrophils but also other inflammatory cells such as macrophages and lymphocytes. We found that OA-NO2 significantly reduced I/R-induced increases in renal IL-1β, and ICAM1 expression and circulating TNF-α. In general, these findings agree with the observation that LNO2 and OA-NO2 exerted a direct inhibitory effect on secretion of various proinflammatory cytokines as well as the NF-κB activation in RAW264.7 macrophages exposed to lipopolysaccharide (10).
A significant body of evidence demonstrates renoprotective actions of all three PPAR subtypes, α, δ, and γ, in renal I/R injury. Pharmacological activation of each one of the PPAR subtypes was able to ameliorate I/R-induced renal dysfunction and histological damage (17, 33, 35). Conversely, genetic deficiency of ether PPARα or PPARδ in mice accelerated renal I/R injury (23, 33). Although all three PPAR subtypes shared anti-inflammatory and antioxidant properties, they may protect against renal I/R injury via different mechanisms. PPARα provided protection likely via activation of fatty acid β-oxidation (33), a mechanism that also appeared to protect against cisplatin-induced nephrotoxicity (24, 25), while PPARδ may act via activation of the PKB/Akt pathway, leading to the increased spread of renal tubular epithelial cells (23). Through a poorly characterized mechanism, PPARγ agonists exerted protective effects against not only I/R but also various kidney injuries including diabetic nephropathy, cisplatin-induced nephrotoxicity, experimental glomerulonephritis, cyclosporine-induced renal injury, and hypertensive nephropathy. OA-NO2 is reported to activate all three PPAR subtypes, with significant activation of PPARγ at 100 nM and PPARα and PPARδ at ∼300 nM (4), suggesting possible involvement of PPARs in mediating the renoprotective action of OA-NO2. Our study is limited in the lack of measurement of tissue concentrations of OA-NO2. Therefore, it is unclear whether the tissue concentrations of OA-NO2 achieved with our experimental protocol will be in a range for selective activation of PPARγ or activation of all PPAR subtypes. It is also possible that OA-NO2 may act independently of PPARγ or other PPAR subtypes. In line with the latter possibility, the anti-inflammatory properties of OA-NO2 in cultured macrophages were shown to be independent of PPARγ (10).
Renal I/R is the most common cause of ARF. An urgent need exists to define mechanisms responsible for the onset and severity of ARF and to identify new therapeutic targets and intervention strategies. Herein, we show that the endogenous nitrated lipid OA-NO2 ameliorates injury in response to bilateral renal I/R in the mouse. The attenuation of functional and histological indices of injury likely resulted from the ability of OA-NO2 to reduce inflammation, PMN infiltration, and ROS generation. Existing treatments of renal injury in response to I/R generally have not been efficacious. The present findings provide important insights into the use of endogenous nitrated fatty acids in this regard. Future studies are warranted to understand their mechanism (s) of action more precisely.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant RO-1 HL-079453 and a Merit Review from the Department of Veterans Affairs (to T. Yang). H. Liu was an exchange graduate student and was partially supported by Shandong University.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med 357: 797–805, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Akbulut G, Dilek ON, Kahraman A, Koken T, Serteser M. The correlation between renal tissue oxidative stress parameters and TNF-alpha levels in an experimental model of ischemia-reperfusion injury in mice. Ulus Travma Acil Cerrahi Derg 11: 11–16, 2005 [PubMed] [Google Scholar]
- 3.Babior BM. Activation of the respiratory burst oxidase. Environ Health Perspect 102, Suppl 10: 53–56, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem 280: 42464–42475, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bove T, Calabro MG, Landoni G, Aletti G, Marino G, Crescenzi G, Rosica C, Zangrillo A. The incidence and risk of acute renal failure after cardiac surgery. J Cardiothorac Vasc Anesth 18: 442–445, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Burns AT, Davies DR, McLaren AJ, Cerundolo L, Morris PJ, Fuggle SV. Apoptosis in ischemia/reperfusion injury of human renal allografts. Transplantation 66: 872–876, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee PK. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: a comprehensive review. Naunyn Schmiedebergs Arch Pharmacol 376: 1–43, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O'Donnell VB. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circ Res 91: 375–381, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Conlon PJ, Stafford-Smith M, White WD, Newman MF, King S, Winn MP, Landolfo K. Acute renal failure following cardiac surgery. Nephrol Dial Transplant 14: 1158–1162, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem 281: 20450–20463, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daemen MA, de Vries B, Buurman WA. Apoptosis and inflammation in renal reperfusion injury. Transplantation 73: 1693–1700, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Daemen MA, de Vries B, van't Veer C, Wolfs TG, Buurman WA. Apoptosis and chemokine induction after renal ischemia-reperfusion. Transplantation 71: 1007–1011, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Daemen MA, van 't Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104: 541–549, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallin JI, Leto TL, Rotrosen D, Kwong CH, Malech HL. Delineation of the phagocyte NADPH oxidase through studies of chronic granulomatous diseases of childhood. Curr Opin Immunol 4: 53–56, 1992 [DOI] [PubMed] [Google Scholar]
- 15.Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem 279: 51715–51718, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Gilbert RE, Kelly DJ, Atkins RC. Novel approaches to the treatment of progressive renal disease. Curr Opin Pharmacol 1: 183–189, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Guan Y. Peroxisome proliferator-activated receptor family and its relationship to renal complications of the metabolic syndrome. J Am Soc Nephrol 15: 2801–2815, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis 34: 384–399, 1999 [DOI] [PubMed] [Google Scholar]
- 19.Hernandez LA, Grisham MB, Twohig B, Arfors KE, Harlan JM, Granger DN. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol Heart Circ Physiol 253: H699–H703, 1987 [DOI] [PubMed] [Google Scholar]
- 20.Johansson MW, Patarroyo M, Oberg F, Siegbahn A, Nilsson K. Myeloperoxidase mediates cell adhesion via the alpha M beta 2 integrin (Mac-1, CD11b/CD18). J Cell Sci 110: 1133–1139, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol 77: 598–625, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, Meinertz T, Baldus S. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA 102: 431–436, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letavernier E, Perez J, Joye E, Bellocq A, Fouqueray B, Haymann JP, Heudes D, Wahli W, Desvergne B, Baud L. Peroxisome proliferator-activated receptor beta/delta exerts a strong protection from ischemic acute renal failure. J Am Soc Nephrol 16: 2395–2402, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol 289: F469–F480, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Li S, Wu P, Yarlagadda P, Vadjunec NM, Proia AD, Harris RA, Portilla D. PPARα ligand protects during cisplatin-induced acute renal failure by preventing inhibition of renal FAO and PDC activity. Am J Physiol Renal Physiol 286: F572–F580, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Lieberthal W, Koh JS, Levine JS. Necrosis and apoptosis in acute renal failure. Semin Nephrol 18: 505–518, 1998 [PubMed] [Google Scholar]
- 27.Lieberthal W, Nigam SK. Acute renal failure. II. Experimental models of acute renal failure: imperfect but indispensable. Am J Physiol Renal Physiol 278: F1–F12, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O'Donnell VB, Eiserich JP, Freeman BA. Nitrolinoleate, a nitric oxide-derived mediator of cell function: synthesis, characterization, and vasomotor activity. Proc Natl Acad Sci USA 99: 15941–15946, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Neblina F, Toledo-Pereyra LH, Mirmiran R, Paez-Rollys AJ. Time dependence of Na-nitroprusside administration in the prevention of neutrophil infiltration in the rat ischemic kidney. Transplantation 61: 179–183, 1996 [DOI] [PubMed] [Google Scholar]
- 30.Matthijsen RA, Huugen D, Hoebers NT, de Vries B, Peutz-Kootstra CJ, Aratani Y, Daha MR, Tervaert JW, Buurman WA, Heeringa P. Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am J Pathol 171: 1743–1752, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol 12: 83–92, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Patel NS, Sharples EJ, Cuzzocrea S, Chatterjee PK, Britti D, Yaqoob MM, Thiemermann C. Pretreatment with EPO reduces the injury and dysfunction caused by ischemia/reperfusion in the mouse kidney in vivo. Kidney Int 66: 983–989, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARα-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol 278: F667–F675, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 67: 1016–1023, 1983 [DOI] [PubMed] [Google Scholar]
- 35.Ruan X, Zheng F, Guan Y. PPARs and the kidney in metabolic syndrome. Am J Physiol Renal Physiol 294: F1032–F1047, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Rubbo H, Parthasarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch Biochem Biophys 324: 15–25, 1995 [DOI] [PubMed] [Google Scholar]
- 37.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem 269: 26066–26075, 1994 [PubMed] [Google Scholar]
- 38.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci USA 102: 2340–2345, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheridan AM, Bonventre JV. Pathophysiology of ischemic acute renal failure. Contrib Nephrol: 7–21, 2001 [DOI] [PubMed]
- 40.Sivarajah A, Chatterjee PK, Patel NS, Todorovic Z, Hattori Y, Brown PA, Stewart KN, Mota-Filipe H, Cuzzocrea S, Thiemermann C. Agonists of peroxisome-proliferator activated receptor-gamma reduce renal ischemia/reperfusion injury. Am J Nephrol 23: 267–276, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Star RA. Treatment of acute renal failure. Kidney Int 54: 1817–1831, 1998 [DOI] [PubMed] [Google Scholar]
- 42.Stenvinkel P. Interactions between inflammation, oxidative stress, and endothelial dysfunction in end-stage renal disease. J Ren Nutr 13: 144–148, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996 [DOI] [PubMed] [Google Scholar]
- 44.Venkatachalam MA, Bernard DB, Donohoe JF, Levinsky NG. Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int 14: 31–49, 1978 [DOI] [PubMed] [Google Scholar]