

Abstract
A basic question for theories of learning and memory is whether neuronal plasticity suffices to guide proper memory recall. Alternatively, information processing that is additional to readout of stored memories might occur during recall. We formulate a “lock-and-key” hypothesis regarding cerebellum-dependent motor memory in which successful learning shapes neural activity to match a temporal filter that prevents expression of stored but inappropriate motor responses. Thus, neuronal plasticity by itself is necessary but not sufficient to modify motor behavior. We explored this idea through computational studies of two cerebellar behaviors and examined whether deep cerebellar and vestibular nuclei neurons can filter signals from Purkinje cells that would otherwise drive inappropriate motor responses. In eyeblink conditioning, reflex acquisition requires the conditioned stimulus (CS) to precede the unconditioned stimulus (US) by >100 ms. In our biophysical models of cerebellar nuclei neurons this requirement arises through the phenomenon of postinhibitory rebound depolarization and matches longstanding behavioral data on conditioned reflex timing and reliability. Although CS–US intervals <100 ms may induce Purkinje cell plasticity, cerebellar nuclei neurons drive conditioned responses only if the CS–US training interval was >100 ms. This bound reflects the minimum time for deinactivation of rebound currents such as T-type Ca2+. In vestibulo-ocular reflex adaptation, hyperpolarization-activated currents in vestibular nuclei neurons may underlie analogous dependence of adaptation magnitude on the timing of visual and vestibular stimuli. Thus, the proposed lock-and-key mechanisms link channel kinetics to recall performance and yield specific predictions of how perturbations to rebound depolarization affect motor expression.
INTRODUCTION
Research to date on the biological mechanisms of long-term memory has focused primarily on candidate mechanisms for memory formation, such as neuronal plasticity. But to what degree are the phenomenological properties of memory determined by biological mechanisms of memory recall? Studies on recall mechanisms have concerned reconsolidation processes that accompany retrieval (Debiec et al. 2006; Doyere et al. 2007), network attractor theories of associative memory (Hopfield 1982; Wills et al. 2005), and expression of learned reflexes (du Lac et al. 1995; Mauk and Donegan 1997; Medina and Mauk 2000; Medina et al. 2000). However, the electrophysiological dynamics that occur during recall might have an important role in shaping qualities such as memory reliability and generalization. Thus, a basic question is whether these dynamics function primarily as a readout mechanism for retrieving stored memories or also perform additional processing of the stored information. Pattern completion is one aspect of associative memory recall for which candidate biological mechanisms have been identified (Nakazawa et al. 2002). Nonetheless, the existing literature on recall has generally assumed that the electrophysiological dynamics of recall should facilitate effective readout, i.e., retrieving the appropriate memory in response to a stimulus. The possibility that some constraints on memory expression might also be enacted at recall has not been widely considered.
Recent work on cerebellar memory systems indicates there are multiple loci of neuronal plasticity and at least two different brain areas of memory storage with distinct induction kinetics (Boyden et al. 2004; De Zeeuw and Yeo 2005; Hansel et al. 2001; Lang et al. 1999; Ohyama and Mauk 2001; Ohyama et al. 2003a). According to two-stage models of cerebellar learning, the numerous synapses in cerebellar cortex support flexible and rapid acquisition of new associations, whereas subsequent plasticity in the deep cerebellar or vestibular nuclei allows long-lasting memory storage (Boyden et al. 2004; du Lac et al. 1995; Mauk 1997; Mauk and Donegan 1997; Miles and Lisberger 1981). Purkinje cells in the cerebellar cortex receive inputs from approximately 105 parallel fibers and project outputs to the deep cerebellar and vestibular nuclei in a highly convergent manner, with each nuclear cell influenced indirectly by 107–108 parallel fibers (Mauk 1997; Napper and Harvey 1988). Given the vast number of potential network states in the cerebellar cortex, a rich set of training experiences might lead to network states that encode undesirable or inappropriate movements. The plausibility of this occurring is indicated by behavioral and computational studies that suggest the distribution of synaptic plasticity levels might evolve in a complex manner throughout learning experience, rather than purely reversing course during extinction or relearning (Kimpo et al. 2005; Mauk and Ohyama 2004). An example of an undesirable motor response is one executed in response to sensory cues that are reliably associated with rewarding or aversive stimuli but that arrive too late to be predictive of an appropriate motor action. Are there memory recall mechanisms that selectively prevent the expression of inappropriate motor responses, despite significant induction of synaptic plasticity? Or does plasticity induction always suffice to modify cerebellar-mediated motor behavior? At least for some forms of associative motor learning mediated by noncerebellar memory systems, it has been shown that associative memory storage by itself can be insufficient to modify behavior (Barnet et al. 1997).
To explore these issues, we formulated a “lock-and-key” hypothesis stating that the induction of plasticity is necessary but not sufficient to modify motor behavior. There is the additional requirement that plasticity must shape the dynamics of neural activity (the “key”) to match a temporal filter (the “lock”) that selectively precludes inappropriate motor responses to sensory stimuli. We examined this hypothesis in the context of two cerebellum-dependent behaviors, classical eyeblink conditioning (Christian and Thompson 2003) and adaptation of the vestibulo-ocular reflex (VOR) (Ito 1982; Miles and Lisberger 1981), for which there exist longstanding, rich behavioral data sets (Gormezano et al. 1962; Raymond and Lisberger 1996). If our hypothesis is true, what biological mechanisms might serve as the lock for these two behaviors?
This paper focuses on rebound currents in the deep cerebellar nuclei (DCN) and medial vestibular nuclei (MVN) neurons as candidate lock mechanisms, because it is well established that these currents perform significant temporal transformations of hyperpolarizing inputs, such as those from cerebellar Purkinje cells believed to trigger learned movements. Rebound channels, such as low voltage-activated (T-type) and hyperpolarization-activated cation (h) channels, are expressed at sufficient density to generate robust postinhibitory rebound depolarizations in DCN and MVN neurons, the output neurons of cerebellar circuits (Aizenman and Linden 1999; Aizenman et al. 1998; Jahnsen 1986a; Llinás and Muhlethaler 1988; Sekirnjak and du Lac 2002). For both behaviors studied, rebound channel kinetics emerge as crucial determinants of the minimum allowable duration between a sensory cue and a trained motor response. If the delay between the cue and a well-timed response is less than the time needed to activate rebound channels fully from the neuronal resting potential, the magnitude of the learned response declines or vanishes, thereby enacting the lock mechanism. This proposal represents a direct link from channel kinetics to learning performance and yields specific predictions of how learning performance is affected by perturbations to the rebound process.
In eyeblink conditioning, key aspects of the behavior that remain poorly understood concern stimulus timing. Training of a reliable reflex requires the conditioned stimulus (CS), such as a tone, to start at least ∼100 ms prior to the unconditioned stimulus (US), such as an air puff to the eye (Fig. 1A) (Gormezano et al. 1962; Ohyama et al. 2003b). Even after averaging data over multiple subjects, there remains a steep dependence of reflex acquisition on the CS–US training interval (Fig. 1B), with the expression of conditioned blinks falling sharply for intervals <100 ms (Ohyama et al. 2003b; Salafia et al. 1980; Schneiderman and Gormezano 1964; Smith 1968; Smith et al. 1969). What is the mechanistic basis for this effect? Analogous, unexplained dependencies on stimulus timing have been reported for VOR adaptation, in which the magnitude of learned eye movements depends on the timing between pulsed visual and vestibular training stimuli (Raymond and Lisberger 1996).
FIG. 1.

Neural pathways and stimulus timing requirements for eyeblink conditioning. A: neural pathways involved in delay eyeblink conditioning. Cerebellar climbing fibers (CFs) originate in the inferior olive (IO) and convey activity driven by the unconditioned stimulus (US). Mossy fibers (MFs) originate in the pons and convey activity driven by the conditioned stimulus (CS). The Golgi (Go) and granule (Gr) cell network processes the CS-driven signals. Purkinje (Pkj) cells receive synaptic inputs from parallel fiber (PF) axons of Gr cells. Pkj cells send GABAergic projections to neurons in the deep cerebellar nuclei (DCN) that drive conditioned motor responses via the red nucleus (RN). B: the reliability of conditioned responses to a CS in trained rabbits, as a function of the CS–US interstimulus interval (ISI) used in training. Data were collected from classic studies of Smith et al. (1969; solid black line and black squares), Salafia et al. (1980; dotted blue line and blue diamonds), Smith (1968; solid red line and red triangles), and Schneiderman and Gormezano (1964; dotted green line and green circles).
According to current thinking in the field an important mechanism of memory formation is the long-term depression (LTD) of cerebellar parallel fiber (PF) to Purkinje cell synapses induced by synchronous activation of PF and climbing fiber (CF) inputs to Purkinje cells (Albus 1971; Ito 1989; Ito and Kano 1982; Marr 1969). In eyeblink conditioning, it is thought that PF and CF inputs respectively convey signals regarding the CS and the US (Hesslow et al. 1999; Mauk et al. 1986; McCormick et al. 1985; Steinmetz et al. 1989; Steinmetz et al. 1986), and that LTD resulting from repeated CS–US pairings leads to a conditioned reflex to the CS alone. This is proposed to occur since LTD should diminish the efficacy of CS-driven input to Purkinje cells, allowing disinhibition of deep cerebellar nuclei (DCN) neurons that receive GABAergic Purkinje cell inputs and drive conditioned reflexes (Albus 1971). In VOR adaptation, CFs and PFs respectively convey visual and vestibular information, and LTD is proposed to allow adaptive increases in VOR amplitude by reducing the strength of PF inputs signaling ipsiversive head rotation (Ito 1989). Although other cerebellar plasticity mechanisms exist (Boyden et al. 2004; De Zeeuw and Yeo 2005; Hansel et al. 2001), multiple strains of mice with disrupted LTD show deficits in eyeblink conditioning and VOR adaptation (Feil et al. 2003; Kishimoto et al. 2001; Koekkoek et al. 2003, 2005; Miyata et al. 2001; Shibuki et al. 1996). Nonetheless, accounts of cerebellar-mediated learning based solely on LTD do not easily explain the full range of behavioral data (Boyden and Raymond 2003; Boyden et al. 2006; Kimpo et al. 2005; Medina and Mauk 1999; Ohyama and Mauk 2001; Ohyama et al. 2003a).
One issue concerns whether Purkinje cells purely inhibit motor responses. Purkinje cells might be partly excitatory in their net effect, due to postinhibitory depolarization in their target DCN and MVN neurons (Aizenman et al. 1998; Jahnsen 1986a,b; Llinás and Muhlethaler 1988; Sekirnjak and du Lac 2002). Another issue concerns the possible role in learning of long-term potentiation (LTP) at the PF–Purkinje cell synapse. LTP and LTD induction at this synapse are spike-timing dependent (Abbott and Nelson 2000), with LTP induced by unpaired PF or asynchronous PF–CF input (Coesmans et al. 2004; Wang et al. 2000). Maximal LTD induction seems to occur for PF activity that slightly precedes CF activity by 50–100 ms, which likely reflects the kinetics of postsynaptic Ca2+ dynamics (Doi et al. 2005). LTD induction can occur with either PF or CF activity occurring first, but delays of ≳200 ms are ineffective with either ordering (Wang et al. 2000). It has been suggested that disinhibition of cerebellar nuclei neurons and spike-timing dependent plasticity suffice to explain the requirement in eyeblink conditioning for the CS–US interval to be >100 ms (Wang et al. 2000). However, this has never been demonstrated explicitly using either computational modeling or experimental manipulation of behavior. A main difficulty is that the empirically determined rules for LTD induction suggest LTD should occur at short CS–US intervals that do not lead to acquisition of conditioned reflexes in behavioral experiments (Wang et al. 2000). Furthermore, the dependence of learning performance on the CS–US interval appears much steeper than that of spike-timing dependent plasticity at the PF–Purkinje cell synapse (Salafia et al. 1980; Schneiderman and Gormezano 1964; Smith 1968; Smith et al. 1969; Wang et al. 2000). Thus, the degree to which conditioned reflex acquisition is shaped by physiological mechanisms other than spike-timing dependent plasticity remains an important issue for experimental investigation.
Here, we consider the novel possibility that significant shaping of learned motor expression might occur through the electrophysiological mechanisms of memory recall. In our work LTP and LTD emerge as complementary processes, both of which are important for memory formation as well as for memory clearance. This contrasts with the common view of LTD and LTP as opposing processes, one allowing memory storage and the other clearance (Boyden and Raymond 2003; Coesmans et al. 2004; Lev-Ram et al. 2003). Because plasticity induction is spike-timing dependent, we begin by considering the timing of sensory driven activity in the PF axons of cerebellar granule cells. Using a series of electrical compartmental models of increasing complexity, we simulate responses of DCN and MVN cells to learned sensory cues. This allows us to validate quantitatively the data from our DCN cell simulations against the classic behavioral data on eyeblink conditioning (Salafia et al. 1980; Schneiderman and Gormezano 1964; Smith 1968; Smith et al. 1969), by comparing the percentage of trials with successful responses as found experimentally to data generated by our models.
Comparison of the VOR adaptation magnitude in our modeling to that in behavioral studies suggests postinhibitory rebounds might play a role in multiple cerebellum-dependent behaviors. Based on the results of our biophysical models we provide an algorithmic description of the “lock-and-key” mechanism as a temporal filter. Learning experience that successfully modifies motor behavior shapes neural activity to match this temporal filter. Unsuccessful training can yield comparable magnitudes of synaptic plasticity, but the resulting patterns of Purkinje cell activity do not trigger learned motor responses. We have organized the following sections so that readers who wish to omit the computational details may skip the following methods section without loss of logical continuity.
METHODS
General simulation procedures
We created compartmental models of DCN and MVN cells in the NEURON (Hines and Carnevale 1997) and MATLAB software environments and set model parameters using empirically determined values whenever possible. Fortunately, much is known about DCN cells from in vitro studies. We found that values determined from measurements in DCN and MVN cells, rather than other cell types, facilitated consistency with behavioral data. The current balance equation describing the balance of capacitive and ionic currents, Cm(dV/dt) = −∑ Iionic, was integrated over time using the MATLAB function ode45 for deterministic one-compartment simulations, an Euler method for one-compartment simulations with stochastic synaptic inputs, or NEURON′s implicit Euler method for two-compartment simulations. In all simulations, timesteps were less than or equal to 0.1 ms and the membrane capacitance Cm was 1 μF/cm2.
Voltage-dependent currents obeyed equations of the form I = ḡxψy(V − Vrev), where ḡ is the maximum conductance and Vrev is the reversal potential. Activation variables, x, followed first-order kinetics defined by dx/dt = φx[αx(V)(1 − x) − βx(V)(x)], where αx and βx are forward and backward rates and φx = Q10ΔT/10°C is a temperature factor. Q10 was 1.4 for T-type current and 2.3 for all other conductances. ΔT is the difference between the physiological temperature of 37°C used for all simulations and the temperature at which experimental measurements of channel kinetics were made. Inactivation variables, y, obeyed analogous expressions. Steady-state and relaxation time constants are given in terms of αx and βx: x∞ = αx/(αx + βx) and τx = 1/[φx(αx + βx)].
The firing rates of Purkinje cells were modeled to be from a cerebellar network after behavioral training. Electrophysiological data from in vivo recordings were used to constrain background firing rates, rPkj,b = 40 Hz (Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006), and the modulation of Purkinje cell firing rates due to learning-related cerebellar plasticity; low and high Purkinje cell spike rates following depression and potentiation of parallel fiber inputs were rPkj,d = 20 Hz and rPkj,p = 100 Hz, respectively (Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006).
Given these basic constraints the average Purkinje cell spike rate, RPkj(t), was determined by first convolving a smooth plasticity function, S(Δt), whose argument is the relative delay between activity in parallel fibers and climbing fibers (Fig. 2A), with a boxcar function, U(t), representing a US of 10 ms duration. The result of this convolution was multiplied by a smooth function, C(t), representing a CS of duration no less than a minimum interval, tCSmin = 50 ms
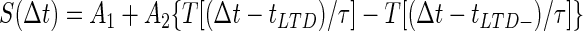 |
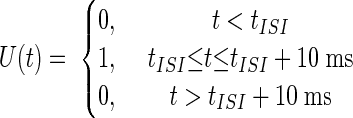 |
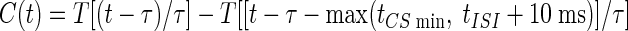 |
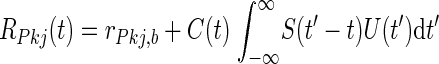 |
where tISI is the interstimulus interval (ISI) between the CS and US onset times. tLTD− = −10 ms and tLTD = 75 ms, respectively, set the minimum and maximum allowable delay between CS-driven parallel fiber and US-driven climbing fiber activity for induction of LTD, and τ = 10 ms is a characteristic transition time describing the smooth temporal evolution of neural dynamics. We used the smoothing function T[x], to ensure gradual changes in spike rates
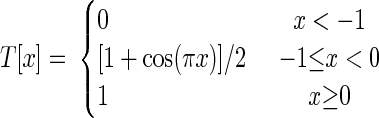 |
The constants A1 and A2 were set such that the minimum and maximum Purkinje cell firing rates for a long ISI were rPkj,d = 20 Hz and rPkj,p = 100 Hz, respectively (Table 1) (Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006). These plasticity rules led to gradual transitions between distinct average firing rate values over intervals of about 20 ms, approximating the observed intervals over which Purkinje cells modulate their spiking rates during expression of motor learning (Berthier and Moore 1986; King et al. 2001).
FIG. 2.

Cerebellar memory formation based on temporally sparse granule cell coding and bidirectional plasticity at the PF–Purkinje cell synapse. A: the relative timing of PF and CF activation sets the propensity toward long-term depression (LTD) or long-term potentiation (LTP). Maximal LTD induction arises when PF activity precedes CF activity by up to a time, tLTD, of about 75 ms, but LTD can also occur when CF activity slightly precedes PF activity (Coesmans et al. 2004; Wang et al. 2000). B: in classical eyeblink conditioning, individual PFs are assumed to exhibit elevated activity during only a brief portion of the CS. By the plasticity rule in A, some PF inputs will be strengthened and others depressed, depending on the relative timing of PF and US-driven CF activity. DCN cells receive input from populations of Purkinje cells whose activity reflects aggregate input from CS-activated PFs. C: repeated CS–US training (top) leads to biphasic CS-driven Purkinje cell spiking due to the bidirectional plasticity shown in B. In subjects that received forward training (bottom left), spiking rises and then falls relative to baseline (red curve). In subjects that received backward training (bottom right), spiking falls and then rises (blue curve). The red arrows (bottom left) correspond to tLTD.
TABLE 1.
Parameters for compartmental simulations of DCN neurons
| Parameter | Description | Value | Model(s) | Reference(s) |
|---|---|---|---|---|
| Cm | Membrane capacitance | 1 μF/cm2 | 1,2,3 | |
| Vrest | Resting membrane potential | −58 mV | 1,2,3 | Aizenman and Linden 1999; Llinás and Muhlethaler 1988 |
| VCa | Ca2+ reversal potential | 140 mV | 1,2,3 | Maincn and Scjnowski 1996 |
| Vsyn,Pkj | GABAergic reversal potential, determined by Cl− gradient | −75 mV | 1,2,3 | Jahnsen 1986b; Llinás and Muhlethaler 1988 |
| Vsyn,MF | Glutamatergic reversal potential | 0 mV | 1,2,3 | Anchisi et al. 2001 |
| VNa | Na+ reversal potential | 50 mV | 3 | Mainen and Sejnowski 1996 |
| VK | K+ reversal potential | −90 mV | 3 | Jahnsen 1986b; Mainen and Sejnowski 1996 |
| rPkj,b | Background Purkinje cell spike rate | 40 Hz | 1,2,3 | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rPkj,d | Reduced Purkinje cell spike rate due to LTD of parallel fiber inputs | 20 Hz | 1,2,3 | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rPkj,p | Elevated Purkinje cell spike rate due to LTP of parallel fiber inputs | 100 Hz | 1,2,3 | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rMF,b | Background mossy fiber spike rate | 10 Hz | 1,2,3 | Freeman Jr and Nicholson 1999; Nicholson and Freeman Jr 2002 |
| rMF,CS | CS-driven mossy fiber spike rate | 50 Hz | 1,2,3 | Freeman Jr and Nicholson 1999; Nicholson and Freeman Jr 2002 |
| ḡT | Maximum T-type Ca2+ conductance in Models 1 and 2 | 0.5 mS/cm2 | 1,2 | |
| ḡT | Maximum T-type Ca2+ conductance in phase plane modeling | 0.3 mS/cm2 | 1,2 | |
| ḡT | Maximum T-type Ca2+ conductance in Model 3, somatic compartment | 5 μS/cm2 | 3 | |
| ḡT | Maximum T-type Ca2+ conductance in Model 3, dendritic compartment | 0.5 mS/cm2 | 3 | |
| Wsyn,Pkj | Maximum total conductance of Purkinje cell synapses | 0.2 mS/cm2 | 1,2,3 | |
| Wsyn,MF | Maximum total conductance of mossy fiber synapses | 4 μS/cm2 | 1,2,3 | |
| ḡHVA | Maximum high-voltage-activated Ca2+ conductance in Model 2 | 0.15 mS/cm2 | 2 | |
| ḡHVA | Maximum high-voltage-activated Ca2+ conductance in Model 2 phase plane | 0.09 mS/cm2 | 2 | |
| ḡHVA | Maximum high-voltage-activated Ca2+ conductance in Model 3, somatic compartment | 0.3 mS/cm2 | 3 | |
| ḡHVA | Maximum high-voltage-activated Ca2+ conductance in Model 3, dendritic compartment | 0.15 mS/cm2 | 3 | |
| ḡSK | Maximum Ca2+-dependent K+ conductance | 32 μS/cm2 | 3 | |
| ḡNa | Maximum Hodgkin-Huxley type fast Na+ conductance | 144 mS/cm2 | 3 | |
| ḡKv | Maximum Hodgkin-Huxley type K+ conductance | 56 mS/cm2 | 3 | |
| τm | Membrane time constant | 12 ms | 1,2,3 | Jahnsen 1986b; Llinás and Muhlethaler 1988 |
| τsyn,Pkj | GABAergic synaptic time constant | 14 ms | 1,2,3 | Anchisi et al. 2001 |
| τsyn,MF | Glutamatergic synaptic time constant | 23 ms | 1,2,3 | Anchisi et al. 2001 |
| Nsyn,Pkj | Number of Purkinje cell inputs | 50 | 2,3 | |
| Nsyn,MF | Number of mossy fiber inputs | 10 | 2,3 | |
| R | Ratio of Purkinje to mossy fiber inputs | 5 | 2,3 | Chan-Palay 1973 |
| gc | Intercompartmental coupling | 0.53 μS/cm2 | 3 | Mainen and Sejnowski 1996; Pinsky and Rinzel 1994 |
| ρ | Percentage of membrane surface area occupied by somatic compartment | 5% | 3 |
The symbol, description, value, and literature citations are given for each parameter in Models 1–3 of DCN cells.
CS-driven modulation of the rate of mossy fiber spiking was also constrained by data from in vivo electrophysiological recordings and was expressed as
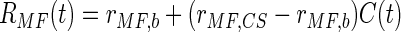 |
where rMF,b = 10 Hz is the background firing rate for mossy fibers and rMF,CS = 50 Hz is the spiking rate of mossy fibers during presentation of the conditioning stimulus (Freeman Jr and Nicholson 1999; Nicholson and Freeman Jr 2002).
Model 1: a single-compartment model of DCN neurons
We modeled a DCN cell with a single electrical compartment that included leak (IL) and T-type Ca2+ (IT) currents, as well as synaptic currents due to inputs from Purkinje cells (Isyn,Pkj) and mossy fibers (Isyn,MF). Membrane voltage dynamics were determined by time integration of the current balance equation: Cm(dV/dt) = −IT − IL − Isyn,Pkj − Isyn,MF. The passive current, IL = gL(V − VL), was an admixture of two components: a tonic mixed-cation current that is characteristic of DCN cells and has a −30 mV reversal potential (Raman et al. 2000) and a standard leak current with −75 mV reversal potential (Jahnsen 1986b; Llinás and Muhlethaler 1988). Total leak conductance, gL, and leak reversal potential, VL, were determined by the DCN cell's resting potential of Vrest = −58 mV (Aizenman and Linden 1999; Llinás and Muhlethaler 1988), and the observed membrane time constant of about 12 ms (Jahnsen 1986a; Llinás and Muhlethaler 1988)
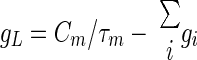 |
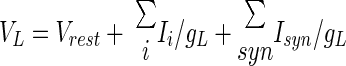 |
where gi and Ii are, respectively, the steady-state conductance and current density at Vrest for each of the active conductances, and Isyn represents the steady-state synaptic current densities determined by Vrest and the background rate of spiking of each type of synaptic input.
Parameter values for Purkinje and mossy fiber synaptic inputs were constrained by physiological measurements: Vsyn,Pkj = −75 mV (Jahnsen 1986b; Llinás and Muhlethaler 1988), τsyn,Pkj = 14 ms (Anchisi et al. 2001), and Vsyn,MF = 0 mV (Anchisi et al. 2001) (Table 1). Glutamatergic synapses in the DCN have significant α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA) components (Anchisi et al. 2001). As a simplification, τsyn,MF for mossy fibers was chosen to be 23 ms by weighting the AMPA and NMDA decay time constants at −60 mV by the measured relative amplitudes of AMPA and NMDA glutamatergic input (Anchisi et al. 2001). T-current was the sole voltage-dependent current: IT = ḡTnl(V − VCa), with VCa = 140 mV (Mainen and Sejnowski 1996). T-type kinetics were adapted from a model of the α1G T-type channel (McRory et al. 2001), which is highly expressed in the DCN (Talley et al. 1999). Steady-state values of the gating variables, n∞ and l∞, were modified to fit measurements of T-type currents in DCN cells (Gauck et al. 2001). Measurements of T-type currents in DCN neurons were made at room temperature, so we used a Q10 of 1.4 for temperature adjustment to produce rebound depolarizations at 37°C (Jahnsen 1986b). After temperature adjustment these expressions were
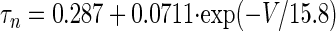 |
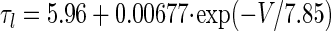 |
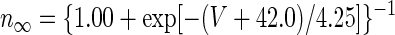 |
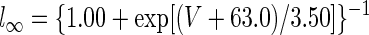 |
where in this and all subsequent expressions numerical parameters with dimensions of time and voltage are expressed in units of milliseconds and millivolts, respectively.
Unfortunately, several DCN cellular parameters could not be tightly constrained by biophysical data. T-type channels appear to be most dense in DCN cell distal dendrites (Gauck et al. 2001). Thus, estimation of total T-type conductance from somatic recordings is difficult. However, in initial simulations we identified a broad range of T-type conductance values over which rebound depolarizations occurred, indicating that the occurrence of postinhibitory rebound is not highly sensitive to the value of the T-type conductance density. In all subsequent single-compartment simulations (Models 1 and 2) we chose ḡT = 0.5 mS/cm2, within the middle of this identified range.
The weight of Purkinje cell synaptic input, Wsyn,Pkj = 0.2 mS/cm2, was chosen to be near the middle of a range of values capable of inducing physiological 10 to 15 mV changes in membrane voltage when input firing rates were modulated. The mossy fiber synaptic conductance weight, Wsyn,MF = 4 μS/cm2, was chosen such that mossy fiber input alone was insufficient to drive the cell to rebound. Without this stipulation there would be little dependence of learned responses on Purkinje cell input, contrary to experimental findings. In Model 1 all synaptic inputs were simulated in a deterministic fashion. Each synaptic conductance density, gsyn(t), was determined by convolving the input spike rate with an exponential function of time constant τsyn and amplitude Wsyn, which represented the conductance response (Table 1).
Model 2: a single-compartment model of dendritic Ca2+ spiking
Model 2 was the same as Model 1 but with two modifications. First, we added a high-voltage-activated (HVA) Ca2+ current, so we could study how graded T-current-mediated rebound depolarizations led to the initiation of HVA Ca2+ spikes. The conductance model for IHVA was identical to that used by Mainen and Sejnowski (1996). Second, membrane voltage dynamics were no longer deterministic. Instead, the time-varying synaptic conductance density was determined by the stochastic arrival of action potentials at times selected independently, as governed by Poisson statistics and the instantaneous spike rate. The number of Purkinje cell (Nsyn,Pkj = 50) and mossy fiber (Nsyn,MF = 10) inputs roughly matched the ratio, R, of GABAergic to glutamatergic synapses found in the DCN (Chan-Palay 1973) (Table 1). The occurrence of a presynaptic spike on any of these independent individual inputs led to an instantaneous jump in synaptic conductance of amplitude Wsyn,Pkj/Nsyn,Pkj or Wsyn,MF/Nsyn,MF, which then declined exponentially with time constant τsyn,Pkj or τsyn,MF, respectively (Table 1).
Model 3: a two-compartment model of DCN neurons
A DCN cell model with dendritic and somatic compartments was used to test the effect of rebound conductances on Na+ spiking output. The potentials of dendritic (Vd) and somatic (Vs) membranes were determined by the currents flowing in each compartment (Pinsky and Rinzel 1994)
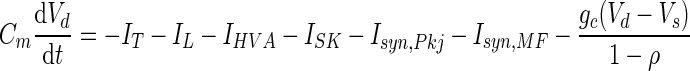 |
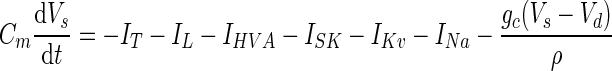 |
where the coupling between the two compartments was specified by the conductance between compartments, gc, and the ratio of somatic membrane surface area to total cell surface area, ρ. The passive current, IL, was determined independently in both compartments from Vrest = −58 mV and the membrane time constant τm = 12 ms. The somatic compartment exhibited spontaneous Na+ spiking, so we determined IL by setting the INa gating variables to m∞(Vrest) and h∞(Vrest), and the IKv gating variable to n∞(Vrest). Stochastically arriving synaptic currents entered the dendritic compartment. For computational ease, we modeled the spike rate of Purkinje cells using a simple formula that closely approximates the rate function used in Models 1 and 2
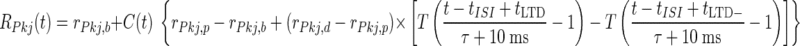 |
Active somatic currents were T-type Ca2+, high-voltage-activated Ca2+ (IHVA; Gauck et al. 2001), Ca2+-activated K+ (ISK; Raman et al. 2000), fast Na+ (INa), and delayed rectifying K+ (IKv). The simulation used the total Ca2+ current, ICa, to determine the internal Ca2+ concentration, which controlled the gating of ISK. In addition to IL, the dendrite had T-type currents (Gauck et al. 2001), IHVA (at half the density as in the soma; see Gauck et al. 2001), ISK, and synaptic input. Conductance models for IHVA, ISK, IKv, and ICa were identical to those used by Mainen and Sejnowski (1996), including parameter values. INa was based on the model of Mainen and Sejnowski (1996) (see also Hines and Carnevale 2001; Schaefer et al. 2007). For Na+ channel activation, ψ = 3 with kinetics determined by
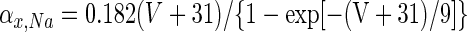 |
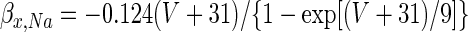 |
For Na+ channel inactivation, ψ = 1 and
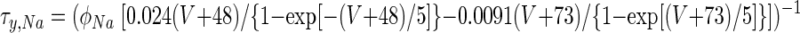 |
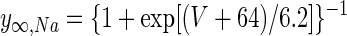 |
Conductance densities and the voltage dependence of INa gating were chosen to reproduce the observed tonic DCN cell firing rate of about 25 Hz (Aksenov et al. 2005; Jahnsen 1986a; Raman et al. 2000) and spike width of about 1.5 ms (Aizenman and Linden 1999; Llinás and Muhlethaler 1988). Reversal voltages were VNa = 50 mV, VK = −90 mV, and VCa = 140 mV (Jahnsen 1986b; Mainen and Sejnowski 1996).
Coupling parameters, gc = 0.53 μS/cm2 and ρ = 0.05, were chosen to ensure each compartment had relatively independent dynamics while still permitting dendritic voltage deflections to affect somatic spiking. Synaptic weights were set by the same criteria as for the one-compartment models, with Wsyn,Pkj = 0.2 mS/cm2 and Wsyn,MF = 4 μS/cm2. As in Models 1 and 2, in the dendrite ḡT = 0.5 mS/cm2 was set near the middle of a broad range of values that allowed rebound depolarization. In the soma ḡT = 5 μS/cm2, reflecting the lower density of low-voltage-activated Ca2+ channels in this compartment (Gauck et al. 2001). The densities of Ca2+-activated K+ conductance, ḡSK = 32 μS/cm2, and of high-voltage-activated Ca2+ conductance in the soma, ḡHVA = 0.3 mS/cm2, and dendrite, ḡHVA = 0.15 mS/cm2, had scarcely any effect on the probability of HVA Ca2+ spiking in response to synaptic inputs across a broad range of conductance densities and were chosen to reduce the duration of the Ca2+ spike to physiologically realistic values (Jahnsen 1986b; Llinás and Muhlethaler 1988). Hodgkin-Huxley conductances, ḡNa = 144 mS/cm2 and ḡKv = 56 mS/cm2, were chosen to reproduce the experimental observation of spontaneous spiking in the soma at Vrest = −58 mV.
Phase plane analysis of rebound depolarizations
To study whether a memory recall mechanism based on rebound depolarization would be robust, we reduced Model 1 to a system of two dynamical degrees of freedom amenable to graphical phase plane analysis. This involved an approximation in which the T-type activation variable was set equal to its asymptotic value, n = n∞(V), reducing the dynamical variables to only the T-type inactivation variable (l) and the membrane voltage (V). Because this approximation increased the membrane excitability, resulting in larger magnitude rebounds, we decreased the density of T-type Ca2+ channels to ḡT = 0.3 mS/cm2 as a compensatory measure. The system's dynamical trajectories within the (V, l) phase plane could then be fruitfully studied by determination of the two nullclines, on which the time derivatives vanish
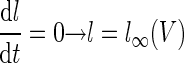 |
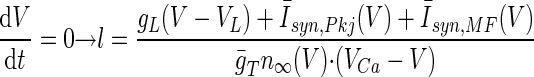 |
where the Purkinje cell and mossy fiber synaptic input currents Īsyn(V) = gsyn(V − Vsyn) represent the mean synaptic currents as determined from the synaptic weights and background firing rates. Both time derivatives vanish at the intersection point of the two nullclines, so this is a fixed-point of the dynamics. Fixed-points during the neuronal resting state (stage 1), the CS–US interstimulus interval up until tLTD before the expected US onset (stage 2), and the remaining portion of the CS (stage 3), were found using the MATLAB function fzero to solve for the intersection of the nullclines. Linear stability analysis within a neighborhood of the resting (stage 1) fixed-point at V = −58 mV revealed that this fixed-point is stable for ḡT < 1.28 mS/cm2. The dynamical trajectories near this fixed-point exhibit damped oscillations for ḡT ≥ 0.20 mS/cm2. The density of T-type channels used in our studies (Table 1) results in a stable spiral fixed-point.
A map of rebound magnitudes in the phase plane (Fig. 6C) was built by numerically integrating the equations of motion using MATLAB's Runge–Kutta initial-value differential equation solver, ode45. A series of evenly spaced initial points was chosen along the boundary lines of the phase plane, defined by V = −72 mV and l = 0, and trajectories were integrated forward in time using the current balance equation and the constant synaptic input values of stage 3. Integration proceeded until the trajectories reached the stage 3 fixed-point. Each trajectory formed a contour (level curve) on the phase plane map with the contour amplitude given by the maximum depolarization achieved on that trajectory.
To determine the voltage threshold curve for firing all-or-none Ca2+ spikes (Fig. 6D), we added a high-voltage-activated (HVA) Ca2+ conductance (Mainen and Sejnowski 1996) to the phase plane treatment of Model 1 in which n relaxes instantaneously to its asymptotic value, n = n∞(V) (Fig. 6, A–C). This yielded a deterministic version of Model 2 that produced virtually the same trajectories as those of Model 1 over the voltage range, V < −35 mV, in which the HVA channels are largely closed (Fig. 6D). As before, the instantaneous activation of T-type currents led to increased membrane excitability, for which we compensated by decreasing the density of T-type Ca2+ channels to ḡT = 0.3 mS/cm2 and the density of HVA Ca2+ channels to ḡHVA = 0.09 mS/cm2. We solved for the dynamical trajectories by integrating the equations of motion forward in time starting at a series of initial points distributed along two boundary lines of the phase plane, defined by V = −72 mV and l = 0. The trajectories fell into two classes depending on whether the T-current-mediated rebound was of sufficient magnitude to cross the threshold for generation of a HVA Ca2+ spike.
Phase plane movies showing model trajectories
Movies of deterministic (Movies S1 and S2 using Model 1) and stochastic (Movie S3 using Model 2) voltage trajectories were created in MATLAB.1 As in the phase plane analysis of Fig. 6, Movies S1 and S2 relied on the mathematical approximation of instantaneous relaxation of the T-channel activation variable to its asymptotic value. The motion of the V nullcline was determined by solving the equation dV/dt = 0 for l, using the steady-state values of the synaptic conductances, gsyn, that would be attained given constant Purkinje cell and mossy fiber spiking at rates equal to their instantaneous values. Numerical integration of the current balance equation used a maximum timestep of 0.1 ms. In Movie S3 the synaptic conductances were modulated by the independent but stochastic arrivals of spikes from 50 Purkinje cells and 10 mossy fibers.
Single-compartment model of MVN neurons
We created a simple one-compartment model of MVN cells (Table 2) obeying the current balance equation, Cm(dV/dt) = −IL − Ih − Isyn,Pkj − Isyn,MF, in which the reversal potential and conductance values for the leak current, IL = gL(V − VL), were determined by the membrane time constant (12 ms), the resting potential (−58 mV), and Ih = ḡhq(V − Vh), where Vh = −20 mV is the mixed-cation reversal potential (Dickson et al. 2001; Pape 1996) and q is the activation variable. Synaptic inputs were modeled in a deterministic fashion, as in Model 1 above, with synaptic time constants τsyn,Pkj = 8.9 ms and τsyn,MF = 5.5 ms (Chun et al. 2003) and synaptic weights Wsyn,Pkj = 0.5 mS/cm2 and Wsyn,MF = 4 μS/cm2.
TABLE 2.
Parameters for compartmental simulations of MVN neurons
| Parameter | Description | Value | Reference(s) |
|---|---|---|---|
| Cm | Membrane capacitance | 1 μF/cm2 | |
| Vrest | Resting membrane potential | −58 mV | du Lac and Lisberger 1995b; Straka et al. 2005 |
| Vsyn,Pkj | GABAergic reversal potential, determined by Cl− gradient | −75 mV | Hille 2001 |
| Vsyn,MF | Glutamatergic reversal potential | 0 mV | Chun et al. 2003 |
| Vh | Ih mixed-cation reversal potential | −20 mV | Dickson et al. 2001; Pape 1996 |
| rPkj,b | Background Purkinje cell spike rate | 40 Hz | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rPkj,d | Reduced Purkinje cell spike rate due to LTD of parallel fiber inputs | 20 Hz | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rPkj,p | Elevated Purkinje cell spike rate due to LTP of parallel fiber inputs | 100 Hz | Berthier and Moore 1986; Jirenhed et al. 2007; Kotani et al. 2006 |
| rMF,b | Background mossy fiber spike rate | 10 Hz | Freeman Jr and Nicholson 1999; Nicholson and Freeman Jr 2002 |
| rMF,CS | Mossy fiber spike rate during head rotation | 50 Hz | Freeman Jr and Nicholson 1999; Nicholson and Freeman Jr 2002 |
| ḡh | Maximum h-type cation conductance | 3 mS/cm2 | |
| Wsyn,Pkj | Maximum total conductance of Purkinje cell synapses | 0.5 mS/cm2 | |
| Wsyn,MF | Maximum total conductance of mossy fiber synapses | 4 μS/cm2 | |
| τm | Membrane time constant | 12 ms | du Lac and Lisberger 1995a, b |
| τsyn,Pkj | GABAergic synaptic time constant | 8.9 ms | Chun et al. 2003 |
| τsyn,MF | Glutamatergic synaptic time constant | 5.5 ms | Chun et al. 2003 |
| τq | h-current activation time constant | 400 ms |
The symbol, description, value, and literature citations are given for each parameter used in simulations of MVN cells.
Measured h-current time constants vary broadly across cell types, but detailed measurements of h-current in MVN cells have not yet been made. The HCN2 isoform appears to be the predominant subtype of h-channel in the vestibular nuclei (Santoro et al. 2000), and the kinetics of this isoform are consistent with the activation time constant of hyperpolarization-activated rebound burst firing in the MVN, measured to be ∼620 ms at 31°C (Sekirnjak and du Lac 2002). Following Sekirnjak and du Lac (2002) we modeled Ih kinetics with a fixed time constant, τq. To determine the steady-state voltage dependence for HCN2 we fit measurements of total h-current obtained in a Xenopus oocyte expression system (see Fig. 9C in Santoro et al. 2000)
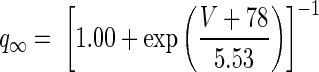 |
ḡh = 3 mS/cm2 was chosen to be within a range of values that produced rebound depolarization. We used τq = 400 ms for MVN simulations shown in Fig. 7, C and E, because this value approximated the time constant that generated the largest ratio of rebound amplitude between the long ISI condition and the zero ISI condition. Rebound amplitude in the zero ISI condition varied by only about 3 mV across a wide range of time constants, 50 ms < τq < 1,000 ms.
Linear–nonlinear (LN) model of lock-and-key mechanism
For our algorithmic description of memory retrieval we generated a set of “key” activity patterns, K(t), using the CS-driven waveforms for the instantaneous Purkinje cell spike rates arising for ISI values ranging from 0 to 200 ms. The Purkinje cell spiking rates were the same as those for biophysical Model 1. We created a linear filter
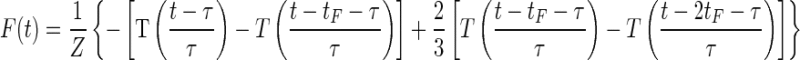 |
where Z is a normalization constant chosen to be the maximum absolute value of the linear response, T[t] is the smooth transition function (see General stimulation procedures), tF = 20 ms, and τ = 10 ms. The filtered key activity was determined by the convolution
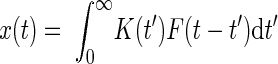 |
Finally, this signal was passed through an exponential nonlinearity, M(t) = G[x(t)] = exp[h * x(t)], where h = 12 is a gain factor. The response amplitude for a given ISI value (Fig. 8E) was determined by the peak value of M(t) normalized by the amplitude attained for a long ISI of 200 ms.
RESULTS
A theoretical framework for cerebellum-dependent learning and memory
Cerebellar granule cells number in the tens of billions but individually appear to be rarely active, producing only a few spikes at a time in response to mossy fiber input (Chadderton et al. 2004). Such transient activation implies that after behavioral training and plasticity induction at PF–Purkinje cell synapses, presentation of a learned sensory cue should drive a biphasic modulation of population Purkinje cell activity (Fig. 2). For example, in classical conditioning transient CS-driven granule cell activity that is concurrent with US-driven CF activity will lead to LTD at PF–Purkinje cell synapses (Fig. 2A). CS-driven granule cell activity that is asynchronous with CF activity will lead to LTP. During subsequent CS input, the net effect of LTD and LTP induction at distinct PF–Purkinje cell synapses will be biphasic modulation of the aggregate Purkinje cell activity received by a DCN neuron (Fig. 2, B and C). Similarly, overlapping pulses of vestibular and visual input in VOR adaptation will also lead to biphasic modulation of Purkinje cell activity. This general pattern of modulation does not hinge on the details of granule cell coding but is contingent on there being spike-timing-dependent bidirectional plasticity and subsets of granule cells in which sensory-driven activity lasts for only portions of the sensory cue duration (Buonomano 1994; Mauk and Donegan 1997; Medina et al. 2000).
We explored the conditions under which biphasic activation of Purkinje cells leads to reliable postinhibitory rebound depolarization of their target neurons that drive learned motor responses. In classical conditioning, whether Purkinje cell spiking first rises and then falls in response to a learned CS, or vice versa, depends on whether the CS and US were paired with a “forward” (CS–US) or “backward” (US–CS) ordering. The two patterns of aggregate Purkinje cell activity should be quite distinct in their propensity to induce DCN cell rebounds. A rise and then fall of Purkinje cell spiking appears well suited to induce rebounds by causing a hyperpolarization and then a depolarization in DCN target cells. The DCN cell resting potential is about −58 mV (Aizenman and Linden 1999), at which T-channels are largely inactivated (Fig. 3). The initial hyperpolarization allows T-channels to deinactivate and the ensuing depolarization allows them to activate. The opposite pattern of Purkinje cell spiking resulting from backward training should be a poor initiator of DCN cell rebounds because the initial depolarization will heighten T-channel inactivation and should largely preclude rebounds. To test these ideas, we performed compartmental modeling of DCN cells to explore whether such a disparity in rebound generation could account for the observed differences in behavioral responses following backward versus forward classical conditioning. For our modeling, we described the timing dependence of LTP and LTD induction on the interval between paired activation of PF and CF afferents as a smooth function that permits LTD for PF activity anticipating CF activity by up to a time tLTD ≃ 75 ms (Fig. 2A) (see methods). This timing dependence mimics that of the experimental data (Wang et al. 2000). The maximal levels of LTD and LTP induction in our models did not depend on the CS–US training interval. By comparison, the durations of each phase of the biphasic Purkinje cell activity did vary with the CS–US interval. This distinction allowed us to focus initially on the signal processing performed by the DCN cells rather than on effects that depend on plasticity amplitude. We subsequently explored how changes in plasticity amplitude, as quantified through the resulting changes in Purkinje cell spike rates, affect a rebound-based mechanism for memory recall in the DCN cells.
FIG. 3.

Compartmental modeling of T-type Ca2+ current rebounds in DCN cells. Compartmental simulations of a Purkinje target neuron in the DCN involved 3 models of increasing complexity. A: Model 1 has one electrical compartment, contains T (gT) and leak (gL) conductances, and receives glutamatergic mossy fiber and GABAergic Purkinje cell inputs. Membrane voltage follows a deterministic time course. B: Model 2 adds high-voltage-activated Ca2+ (gHVA) channels. Synaptic inputs arrive stochastically, leading to membrane potential fluctuations and nondeterministic dynamics. C: Model 3 has dendritic and somatic compartments, coupled by a conductance gc. Synaptic inputs are localized to the dendrite, approximating empirical findings. The soma has fast Na+ (gNa) and delayed rectifier K+ (gKv) conductances. Both compartments have leak, T, Ca2+-activated K+ (SK), and HVA Ca2+ conductances. Synaptic inputs arrive stochastically, leading to nondeterministic dynamics. D: voltage dependence of the activation (dashed red curve) and inactivation (solid blue curve) gating variables for the T-type conductance in DCN neurons. At the resting potential (about −58 mV, dashed vertical line), T-currents are largely inactivated. Hyperpolarization deinactivates T-currents, allowing activation during subsequent depolarization. E: voltage dependence of the T-channel activation (dashed red curve) and inactivation (solid blue curve) time constants. Parameter dependencies in D and E are based on Gauck et al. (2001) and McRory et al. (2001).
Memory recall in a one-compartment model DCN neuron
We studied whether forward and backward patterns of biphasic Purkinje cell spiking could lead to distinct patterns of rebound activity in DCN cells after presentation of a classically conditioned stimulus. We created a series of compartmental DCN cell models that received inputs from both Purkinje cells and mossy fibers and we interpreted the resulting rebounds as the initiators of conditioned motor responses. The simplest model (Model 1) had one electrical compartment, lacked fast-spiking capability, and had only leak, T-type, and synaptic conductances (Fig. 3A). This allowed us to focus initially on rebound generation, apart from issues studied later concerning membrane potential noise and downstream readout. Kinetic parameters for T-currents were obtained from in vitro measurements in DCN cells (Gauck et al. 2001; McRory et al. 2001). Deinactivation can occur within about 20–100 ms of hyperpolarization from the resting potential and activation can then occur within a few milliseconds during subsequent depolarization (Fig. 3, D and E). Conductance densities were set to reproduce the observed resting potential of −58 mV and membrane time constant of about 12 ms (Aizenman and Linden 1999; Jahnsen 1986a; Llinás and Muhlethaler 1988).
We compared the model's responses to forward and backward patterns of biphasic Purkinje cell input. In our initial studies, the forward CS–US interstimulus interval (ISI) was ≥200 ms, more than sufficient delay for reliable conditioning in rabbits (Fig. 1B) (Ohyama et al. 2003b). Mossy fiber excitation rose during the entire CS but was insufficient to drive a rebound during baseline or elevated Purkinje cell spiking. This is consistent with data supporting a key role for Purkinje cells in generating properly timed reflexes via the suppression of early, mossy fiber-driven responses to the CS, which can be unveiled by blocking Purkinje cell inputs to the DCN (Ohyama and Mauk 2001; Perrett et al. 1993). We found that biphasic Purkinje cell input shaped by forward training led to rebounds that initiated as Purkinje cell spiking transitioned from an elevated to a diminished rate, about tLTD prior to the expected US onset (Fig. 4A, red traces). Hence, rebounds could drive blinks that anticipate the US. We then tested the effect of varying the ISI value. With backward training there was insufficient deinactivation of T-currents to generate rebounds (Fig. 4A, blue traces). With positive ISI values <100 ms, rebounds occurred but with diminished amplitude, since there was insufficient time for T-channel deinactivation during the brief increase in Purkinje cell spiking (Fig. 4A, orange trace). Thus, rebound generation occurred selectively for sufficiently positive ISI values and anticipated US arrival.
FIG. 4.

DCN cell rebounds require a minimum CS–US ISI and sufficient expression of cerebellar LTP and LTD. A: the time course of CS-driven depolarization in Model 1 (Fig. 3A). If prior training involved a sufficiently positive ISI, the CS-driven rebound is of large amplitude and occurs at a time approximately tLTD before the expected US (red traces). If training involved an insufficient ISI value, CS-driven rebounds do not occur (blue traces). For short ISI values, rebounds are diminished in amplitude (orange trace). The color bar indicates the ISI values, which are also marked above the graph with the color corresponding arrowheads for each voltage trace. Rebounds occur prior to the expected US, indicating anticipatory responses. B: rebound amplitude varies with the degree to which the CS drives biphasic Purkinje cell activity. This, in turn, depends on having sufficient expression of both PF–Purkinje cell LTP and LTD (Fig. 2). Driving a large-amplitude rebound in the DCN cell requires that during the first phase of biphasic activity the Purkinje cell spiking rate rises well above the spontaneous frequency of 40 Hz. The 3 voltage traces (blue, cyan, red traces) in B1 occurred with the color corresponding, Purkinje cell peak spiking rates shown in B2. Lower peak spiking rates reflect lower expression levels of LTP. The arrowhead indicates the ISI value of 200 ms. C: driving a large-amplitude rebound in the DCN cell also requires that during the second phase of biphasic activity the Purkinje cell spiking frequency drops below the 40 Hz spontaneous rate. The 3 voltage traces in C1 (blue, cyan, red) were created using the color corresponding, Purkinje cell minimum spiking rates shown in C2. The higher rates reflect lesser degrees of LTD. The arrowhead indicates the ISI value of 200 ms.
We also explored the dependence of rebound generation on the graded magnitude of LTP and LTD at the PF–Purkinje cell synapse, as quantified through the resulting elevation and diminution in Purkinje cell spike rates, respectively (Fig. 4, B and C). Rebound generation in the DCN cell required biphasic Purkinje cell spiking, with both a sufficient elevation and subsequent decline in spiking needed for large-amplitude rebounds (∼50 mV). Ample levels of both LTP and LTD would thus be needed to induce sufficient biphasic variation in Purkinje cell spiking. These findings held across a broad range of T-channel densities, opening the possibility that DCN cell T-currents help shape the differences in conditioned reflex expression following backward and forward training (Ohyama et al. 2003b).
Readout mechanisms of rebound depolarization and correspondence to conditioned behavior
If rebounds induce learned motor action, how do DCN cells convey rebound magnitudes via the rate of Na+ spikes sent to premotor areas? The graded amplitude of pure T-current-mediated rebounds indicates these low-voltage-activated events are not stereotyped Ca2+ spikes. Real DCN neurons do exhibit Ca2+ spikes, mediated by high-voltage-activated Ca2+ channels and, as in other cell types, dendritic Ca2+ spikes may be good triggers of somatic Na+ spike bursts (Jahnsen 1986b; Llinás and Muhlethaler 1988). We reasoned that the amplitude of T-current-mediated rebounds should set the likelihood of crossing the voltage threshold for Ca2+ spike generation, with membrane potential fluctuations influencing the degree of variability. Smaller-amplitude rebounds that occur with shorter ISI values would be less likely to cross the Ca2+ spike threshold. Within this framework we interpret a Ca2+ spike as the initiator of signals sent downstream to drive a conditioned motor response.
To test whether this readout mechanism would be able to convert the amplitude of rebound depolarization into the probability of Ca2+ spike generation, we examined an enhanced one-compartment model that included high-voltage-activated Ca2+ channels (Model 2, Fig. 3B) and membrane potential fluctuations due to stochastic arrival of synaptic inputs (methods). This contrasts with Model 1, in which both synaptic inputs and membrane voltage followed deterministic time courses. In Model 2 a biphasic pattern of Purkinje cell input resulting from forward training with a long ISI value led reliably to a T-current-mediated rebound of sufficient magnitude to trigger a Ca2+ spike. These Ca2+ spikes were properly timed, prior to the expected US. Backward training led to small rebounds and virtually no Ca2+ spiking. Forward training with a short ISI value led to unreliable Ca2+ spiking, with the amplitude of the T-current-mediated rebound being sufficient to trigger a Ca2+ spike on some trials but not others (Fig. 5A). Thus, as the ISI value varied, the amplitude of T-current-driven rebounds set the probability of crossing the Ca2+ spike threshold (Fig. 5B, closed green triangles). Of prime interest, the shape of the curve describing this response probability as a function of the ISI closely resembles that obtained in rabbit eyeblink conditioning studies (Fig. 5B, open red squares, diamonds, and downward facing triangles), validating the plausibility of a rebound-based recall mechanism. The sum of tLTD and the T-channel inactivation time constant determine the temporal offset of the curve from the origin. It follows that experimental manipulations lengthening the time needed for T-channel deinactivation during the ISI are predicted by our theory to cause a rightward shift of the behavioral data curve (discussion).
FIG. 5.

Readout of rebounds via Ca2+ spikes leads to a dependence of the response reliability on the CS–US ISI. A: sample voltage traces during CS presentation in Model 2 (Fig. 3B) in the presence of membrane potential fluctuations from noisy synaptic inputs. At an intermediate ISI of 100 ms, a T-current-mediated rebound depolarization triggers a Ca2+ spike during one trial (dashed red line) but not another (solid blue line). B: the reliability of learned responses in Model 2 (closed green triangles) and Model 3 (closed blue circles), defined as the probability of generating a dendritic Ca2+ spike in response to a test CS, plotted as a function of the ISI. Classic data on the reliability of conditioned blinks in trained rabbits are replotted from Fig. 1 (open red symbols) (Salafia et al. 1980; Smith 1968; Smith et al. 1969), showing the similarity to the model data. A tLTD of 75 ms was used for the model data, consistent with empirical data indicating tLTD is in the range of about 50–200 ms (Wang et al. 2000). C and D: example voltage traces from the dendritic and somatic compartments of Model 3 (Fig. 3C) during CS presentation with an ISI of 200 ms. A T-mediated rebound depolarization leads to a high-voltage-activated dendritic Ca2+ spike (C) that drives a rise in the somatic Na+ spike rate (D). E: the corresponding time courses of the activation (n, solid red curve) and inactivation (l, dashed blue curve) gating variables during the Ca2+ spike.
We examined readout issues in greater depth using a two-compartment model DCN cell (Model 3, Fig. 3C) that included a dendrite and soma, as well as channels mediating dendritic Ca2+ and somatic Na+ spikes (see methods). The somatic and dendritic compartments were only weakly coupled, which was intended to mimic the electrotonic isolation between the cell body and the long distal dendrites of DCN cells where T-channels appear to be most dense, >100 μm from the cell body (Gauck et al. 2001). This is consistent with the observation that Purkinje cell input triggers DCN cell rebounds much more effectively than somatic hyperpolarization of comparable magnitude (Aizenman and Linden 1999). Synaptic inputs in Model 3 arrived stochastically, inducing membrane potential fluctuations. As in real DCN cells, a tonic cation current induced a basal rate of somatic spiking at about 25 Hz (Aksenov et al. 2005; Jahnsen 1986a; Raman et al. 2000). Simulations revealed that a dendritic rebound induces a Ca2+ spike, which in turn drives a corresponding increase in the rate of somatic Na+ spikes (Fig. 5, C–E). This increase represents a plausible signal from the DCN cell to downstream pathways for driving learned motor output (Fig. 1A). Forward training with an ISI >100 ms virtually always led to such a spike burst. Na+ spike bursts occurred with lower probability under the same conditions that failed to produce large-amplitude rebounds in Model 1, such as backward training or forward training with a short ISI. Across ISI values the probability of a Na+ spike burst closely matched the behavioral dependence of conditioned blinking on the ISI value as observed in rabbits (Fig. 5B, closed blue circles).
Phase plane analysis of rebound generation as a robust mechanism for recall
To further explore the basic dynamics and robustness of rebound mechanisms, we studied DCN neuronal dynamics using a phase plane analysis of Model 1. Prior applications of such analysis to other neuron types have provided considerable insight into Ca2+ spike generation, spike bursting, and transitions between “up” and “down” activity states (Fitzhugh 1960; Loewenstein et al. 2005; Rinzel and Ermentrout 2001; Rush and Rinzel 1994). As is common in phase plane analysis, we focused on the slow dynamic variables that set the relevant timescale. Here, these variables are membrane voltage (V) and the T-type channel inactivation variable (l). The latter has a voltage-dependent time constant of about 10–100 ms (Fig. 3E), close to the minimum ISI for reliable memory retrieval (Fig. 1B). By comparison, the time constant for T-current activation is about 1–10 ms, considerably faster than motor memory recall and rebound depolarization. Because of this separation of timescales we approximated T-type activation as occurring instantaneously and thus restricted to the (V, l) plane. Rebounds may then be viewed as trajectories in this two-dimensional (2-D) phase plane (Fig. 6) .
FIG. 6.

Phase plane analysis of CS-driven rebounds. A: membrane voltage time course (blue curve) in response to a CS that initiates at time t = 0 in Model 1, under the approximation of instantaneous relaxation of the T-channel activation variable to its asymptotic value. The rebound peaks at a time about 40 ms prior to the expected US at 200 ms after CS onset. Dashed vertical lines delineate 3 stages of the phase plane trajectory in B. B: the state trajectory (blue curve) in the 2-dimensional (2-D) phase plane defined by the voltage (V) and T-type inactivation variable (l), corresponding to the voltage trace in A. The open black circle marks the fixed-point in the resting state (stage 1). The open green triangle marks the fixed-point from CS onset until approximately tLTD prior to the expected US (stage 2). The open red square marks the fixed-point during the remainder of the CS (stage 3). According to longstanding convention, channels are completely inactivated when l = 0 (Hodgkin and Huxley 1952). C: a color plot conveying the amplitude of the rebound that occurs during stage 3 for the state trajectory passing through each point in the phase plane of B and converging toward the stage 3 fixed-point (open red square). Warmer hues indicate the larger rebounds (color bar) that initiate if during stage 2 the system has successfully entered the “memory reliability zone” near the stage 2 fixed-point (open green triangle). White curves are example state trajectories. D: the addition of high-voltage-activated (HVA) Ca2+ channels to the phase plane analysis of C reveals those stage 3 trajectories that lead to a Ca2+ spike (red trajectories) and those that do not (blue trajectories). All of the trajectories closely concur with those in Model 1 (C) in the voltage range V < −35 mV over which the HVA Ca2+ channels are largely closed. The red trajectories, which initiate within the reliability zone near the stage 2 fixed-point (green triangle), cross the Ca2+ spike threshold and allow successful readout of the rebound (Fig. 5, A and B). Horizontal dotted lines indicate the resting potential of −58 mV in A–D. Solid and dashed black curves in B, C, and D are nullclines during the resting state for the l and V variables, respectively, on which the time derivatives dl/dt and dV/dt, respectively, vanish during stage 1.
Phase plane analysis of Model 1 revealed the key ingredients for rebounds. The analysis can best be understood by breaking a CS presentation into three stages: the initial resting condition, the ISI, and the remainder of the CS following the ISI (Fig. 6A). During each stage, the system has a unique attractive fixed-point at the intersection of the V and l nullclines, the curves on which the time derivatives dV/dt and dl/dt, respectively, vanish (Fig. 6B). The three fixed-points and the ISI value are the chief determinants of the dynamics. At rest (stage 1), the system resides at a fixed-point location at which the T-current is mainly inactivated (open black circle in Fig. 6B and Movies S1 and S2). At CS onset and during the ISI (stage 2), mossy fiber and Purkinje cell input to the DCN cell shift the fixed-point location to a potential at which the T-channel deinactivates (open green triangle in Fig. 6B and Movies S1 and S2). The system approaches the stage 2 fixed-point during the ISI, starting from the resting position (Fig. 6B; Movies S1 and S2). The ISI value determines the duration and proximity of the system's approach. At about tLTD prior to the moment of the expected US (stage 3), Purkinje cell activity declines and the fixed-point shifts to a third location that is depolarized relative to rest (open red square in Fig. 6B and Movies S1 and S2). This initiates a rebound that is well timed for driving an anticipatory reflex. More precisely, there is a family of trajectories that undergo rebound depolarization during stage 3, with rebound amplitude a strict function of the (V, l) values attained by the end of stage 2. A 2-D color map of rebound amplitude as a function of (V, l) reveals the basis for the sharp dependence on the ISI value and the stage 2 and stage 3 fixed-point locations (Fig. 6C). In turn, these fixed-point locations depend critically on the degree of biphasic Purkinje cell spiking and thus on the levels of LTP and LTD attained during training.
Stage 3 rebound trajectories with the greatest depolarization initiate in a neighborhood of the (V,l) plane that may be viewed as a memory recall “reliability zone” from which a large rebound will occur without fail (Fig. 6C, red shaded region). The level of LTP and the peak Purkinje cell spiking rate are important because they determine the proximity of the stage 2 fixed-point to the reliability zone. However, even with sufficient LTP if the ISI is too brief the system does not have time to reach the reliability zone during stage 2, leading to a small or no rebound (Fig. 6C, blue shaded region; Movie S2). The rebound amplitude also hinges on the location of the stage 3 fixed-point, due to the dependence of T-channel activation on the reduction in Purkinje spike rate and the level of LTD.
To understand the implications of these observations for a readout mechanism based on Ca2+ spike generation (Fig. 5), using Model 2 we determined the set of stage 3 trajectories in the (V, l) plane that lead to a Ca2+ spike (see methods). Large-amplitude rebounds that initiated within the reliability zone passed furthest above the spiking threshold (Fig. 6D). Rebounds that initiated elsewhere either failed to reach or just crossed threshold. In the presence of membrane potential noise, this implies that if the system reaches the reliability zone the probability of Ca2+ spike generation is high. Much as in our two-compartment simulations, this probability falls dramatically as the ISI is shortened (Movie S3 and Fig. 5B). The Ca2+ spike voltage threshold does not vary much across a wide range of HVA Ca2+-channel density (data not shown), indicating Ca2+ spiking is a robust readout of whether the system has entered the recall reliability zone. Thus the phase plane analysis illuminates key features of a rebound-based memory recall mechanism, including conditions for reliable recall.
Role of postinhibitory rebounds in VOR gain adaptation
Because cerebellar circuitry is highly conserved, rebound depolarization might serve multiple forms of cerebellar memory recall. For example, floccular Purkinje cells involved in horizontal VOR adaptation project to target cells in the vestibular nuclei that also exhibit significant rebound depolarization in vitro mediated by hyperpolarization-activated currents (Sekirnjak et al. 2003; Serafin et al. 1991). These currents require further characterization and are expressed to varying degrees across MVN cell types, but as a group the MVN neurons receiving input from the floccular Purkinje cells exhibit exceptionally pronounced rebound burst spiking (Sekirnjak et al. 2003). The currents involved seem to include the h-type cation current and probably some amount of Na+ and T-type Ca2+ currents (Sekirnjak and du Lac 2002; Serafin et al. 1991; Smith et al. 2002). Regardless of the current identities, the empirically determined time constant (∼620 ms) describing the duration of hyperpolarization needed for maximal rebound burst firing is considerably longer than that for DCN cells (Fig. 2E) (Sekirnjak and du Lac 2002). Might rebound depolarization and the need for a long period of hyperpolarization underlie some of the temporal asymmetries seen in behavioral studies of VOR adaptation?
Well-known primate behavioral studies have shown that the amplitude of learned VOR responses depends on the relative timing of vestibular and visual stimuli in a manner resembling the dependence on CS–US timing in classical conditioning. Raymond and Lisberger (1996) repeatedly paired a vestibular stimulus, a 600 ms pulse of head rotation, with a brief visual stimulus consisting of moving dots. The visual motion stimulus was presented at one of three different ISI values: a zero ISI condition analogous to backward conditioning (Fig. 7C, left), a short forward ISI of 225 ms (Fig. 7C, middle), and a long forward ISI of 450 ms (Fig. 7C, right). A learned VOR response developed in all cases, but the response amplitude grew as the ISI lengthened. Such dependence on the ISI may be analogous to that seen in eyeblink conditioning. Could rebounds underlie this effect? The timescale of the behavioral effect is similar to that of h-current activation.
FIG. 7.

Vestibular nuclei cell rebounds lead to temporally asymmetric vestibulo-ocular reflex (VOR) adaptation. A: vestibulo-cerebellar pathways for VOR horizontal gain adaptation involve Purkinje cells (Pkj) that project to target neurons within the medial vestibular nucleus (MVN). Neurons in the MVN project to brain stem motor nuclei (MNs) that drive eye movement. Slip of the visual scene on the retina is conveyed to the cerebellum via climbing fibers (CFs). Information about head velocity arrives via mossy fibers (MFs) originating in the vestibular ganglia (VG), is processed within the Golgi (Go) and granule (Gr) cell network, and reaches Purkinje cells by way of parallel fibers (PFs). Conjunctive arrival of CF and PF signals is thought to induce synaptic plasticity at the PF–Pkj synapse that underlies gain adaptation. B: a one-compartment model of an MVN Purkinje target neuron that contains h- (gh) and leak (gL) conductances and receives glutamatergic mossy fiber and GABAergic Purkinje cell input. Membrane voltage follows a deterministic time course. C: primate behavioral data from well-known studies in which pulses of head rotation (top, black bars) were paired during training with moving dot visual stimuli (top, gray bars) at 3 distinct ISIs. During later testing with pulsed head rotations in the dark, the learned component of VOR expression increased markedly with greater ISI values (bottom, green, blue, and red curves) (Raymond and Lisberger 1996). D: relative rebound amplitude as a function of the h-current activation time constant τq. The plot shows the maximum depolarization from the resting potential following training with zero (green), short (blue), and long (red) ISIs, normalized for each value of τq by the maximum depolarization of the zero ISI trajectory (green). Dashed black line indicates τq of 400 ms used for simulations shown in E. E: voltage traces (top) and state trajectories (bottom) from the model MVN cell in response to a test pulse of head rotation following training with the 3 different ISI values shown in C. The 3 state trajectories (bottom) traverse the 2-D phase plane defined by the voltage (V) and the activation level of the h-current relative to that at rest (h). Horizontal dashed lines in the top panels indicate the resting potential of −58 mV. The solid and dashed black curves in the bottom panels are the nullclines during the resting state for V and h, respectively, on which their respective time derivatives vanish. Vertical dashed lines in C and E mark the period of head rotation.
To study the issue we created a simple, one-compartment model of an MVN cell in which h-currents mediated rebounds (Fig. 7B). The model is analogous to Model 1 of a DCN cell in that the model has only one compartment and lacks the channels responsible for the fast spontaneous spiking that MVN cells exhibit. The single compartment thus better mimics a dendrite than a soma. As before, we interpreted the rebounds as signals driving learned motor responses. MVN simulations used a fixed time constant, τq, for Ih activation, as in the MVN cell model of Sekirnjak and du Lac (2002).
We used a stimulus protocol based on the Raymond–Lisberger experiments and found the largest rebounds arise when the visual stimulus occurs during the latter portion of the vestibular impulse (Fig. 7E). Longer ISI values allow more time for h-currents to activate at hyperpolarized voltages, heightening rebound depolarization. Rebound amplitude also depends on biphasic Purkinje cell spiking and thus on the levels of LTP and LTD induced during training (data not shown). By varying τq over a range of values, we found that a value of ∼400 ms generated the largest ratio of rebound amplitudes between the long ISI condition and the zero ISI condition (Fig. 7, D and E). This value of ∼400 ms for τq at physiological temperature appears consistent with the empirical value of about 620 ms measured at 31–33°C by Sekirnjak and du Lac (2002). Thus, the amplitude dependence of learned eye movements on the ISI value might stem from variable levels of current flow through hyperpolarization-activated conductances such as h. However, the component of the learned response that is independent of the ISI value is unlikely to be driven by rebounds and is beyond the scope of our present model, which seeks to account only for the ISI-dependent component.
DISCUSSION
We have presented a lock-and-key hypothesis on how the expression of memory responses may undergo filtering via neurophysiological mechanisms active during memory retrieval. This hypothesis and our computational work exploring a candidate rebound-based lock-and-key mechanism were prompted by data suggesting that backward-ordered classical eyeblink conditioning as well as noncerebellar forms of aversion conditioning can lead to latent memory storage or changes in neural activity, despite a lack of conditioned responses (Barnet et al. 1997; Gould and Steinmetz 1996). The complex manner in which plasticity might evolve across a large network of synaptic connections throughout learning experience also suggests some constraints on motor memory expression might be implemented via neurophysiological mechanisms of recall (Mauk and Ohyama 2004).
We explored these ideas through computational studies of two cerebellar behaviors by examining whether DCN and MVN cells can filter signals from Purkinje cells to influence response timing and prevent certain motor responses. Biophysical models of these two cell types that incorporate rebound channels lead to consistent explanations for behavioral data on cerebellar motor learning. These models make direct links between ion channel kinetics and memory expression and, particularly for eyeblink conditioning, yield specific predictions of how learning performance varies as a function of the relative timing of paired training stimuli. Within our lock-and-key framework for these models, subjects undergo both cerebellar LTP and LTD regardless of whether the training stimuli were presented in forward or backward order. As a result, the learned sensory cue drives biphasic Purkinje cell activity. Yet, this biphasic activity triggers rebound depolarization in the DCN cells and drives well-timed classically conditioned reflexes only if the training ISI was sufficiently positive. In this way, inappropriate motor responses to conditioned stimuli that do not precede the US sufficiently are avoided. Phase plane analysis reveals the basic ingredients for reliable reflex expression, including ample levels of both LTD and LTP. In MVN cells, rebound currents may underlie the variation of VOR adaptation magnitude with the relative timing of visual and vestibular training stimuli. Such effects hinge on the observed capabilities of both DCN and MVN cells for rebound depolarization.
Electrophysiological properties of DCN neurons are consistent with the rebound theory
In vitro studies of DCN cells have found that rebounds occur in both cerebellar slice and isolated cerebellum–brain stem preparations (Aizenman and Linden 1999; Jahnsen 1986a,b; Llinás and Muhlethaler 1988). MVN neurons also undergo rebounds in vitro, but there is more uncertainty about the channels involved (Sekirnjak and du Lac 2002, 2006). There is also indirect physiological and pharmacological evidence DCN cells rebound in vivo (Aksenov et al. 2005; Hesslow 1994a), including for a class of neurons with blink-related activity (Chen and Evinger 2006). Input from a single Purkinje cell induces a large conductance change in the DCN cell (Pedroarena and Schwarz 2003), which is sufficient to allow a modest postinhibitory rebound and increase in Na+ spike rate (M. Molineux, personal communication). Multiple Purkinje cells might drive larger rebounds and spike bursts in concert, such as through coordinated Purkinje cell spiking (Heck et al. 2002; Thier et al. 2000). The anatomical convergence of many hundreds of Purkinje cells onto each DCN neuron implies that the aggregate activity of a population of Purkinje cells influences DCN cell activity.
The in vivo extracellular recordings performed to date of DCN neurons during classical conditioning do not provide strong evidence either for or against our rebound theory. Single-unit and multi-unit recordings both reveal an increase in DCN spiking rate that precedes motor output (Berthier and Moore 1990; Choi and Moore 2003; McCormick and Thompson 1984a,b; McCormick et al. 1982). By comparison, evidence for a pause in spiking during early portions of the ISI is limited. Berthier and Moore reported some cells with reduced spiking at the beginning of the CS, although this pattern is not apparent in all single-unit recordings (Berthier and Moore 1990; Choi and Moore 2003). Multi-unit recordings of DCN cell activity do not exhibit a pause, but these recordings may not provide sufficient sensitivity to reveal a partial reduction in spiking within a subpopulation of recorded neurons (McCormick and Thompson 1984a,b; McCormick et al. 1982). Irrespective of these results, the rebound model does not make a strong prediction concerning DCN firing during early portions of the CS. During the early portion of the ISI, DCN neuron spiking might remain virtually unchanged, despite increased Purkinje input, due to the dendritic location of most Purkinje synapses and T-type channels (Fig. 5). Technically difficult in vivo intracellular recordings would be required to determine how subthreshold responses in DCN cells develop during conditioning.
Experimental predictions for studies of cerebellum-dependent motor learning
The lock-and-key hypothesis leads to a clear prediction that is testable independent of whether rebound depolarization provides a lock mechanism: Classical conditioning with a short (<100 ms) or backward CS–US interval should lead to plasticity in the cerebellar cortex despite the lack of reflex acquisition. Several experimental tests of this prediction are possible. Second-order classical conditioning, which can induce expression of previously latent first-order conditioning, might be useful for explicitly demonstrating that a memory of backward conditioning is formed (Barnet et al. 1997). Alternatively, studies of backward eyeblink conditioning using human brain imaging techniques might be capable of revealing plasticity-related effects (Cheng et al. 2007). Electrophysiological recordings of Purkinje cell spiking during reflex conditioning in decerebrate ferrets and guinea pigs have revealed changes in spiking patterns in response to forward training (Jirenhed et al. 2007; Kotani et al. 2003), and thus might be used to examine the effects of backward or short-interval conditioning. Recordings from rabbit Purkinje cells, but not DCN cells, were reported to show changes in activity patterns following backward US–CS pairings (Gould and Steinmetz 1996). This lends support to the lock-and-key hypothesis, but more experimental data are still needed.
Several other testable predictions emerge from our modeling of rebound dynamics during memory recall. Removal or blockade of DCN cell rebound conductances should hinder expression of conditioned blinks. To test this idea, T-channel blockers might be used to prevent rebounds in trained subjects (McDonough and Bean 1998; Porcello et al. 2003). Failure to impair conditioned blinks would cast serious doubt on our proposed rebound mechanism. A related test might be performed in trained animals during recordings of DCN cells that drive conditioned responses. Transient depolarization of these cells during the ISI in trained animals should prevent or diminish blink-related spiking activity by thwarting T-channel deinactivation. Although perturbation of one cell seems unlikely to disrupt the blink itself, stimulation of many DCN cells might have such an effect.
A corollary to this logic concerns inhibition of olivary cells by projections from GABAergic DCN cells that also receive Purkinje cell inputs and exhibit rebounds (De Zeeuw and Berrebi 1995; Teune et al. 1998). These connections appear critical for extinction of conditioned reflexes following unpaired CS presentations (Medina et al. 2002). Thus, blockade of rebound channels should hinder both expression and extinction of conditioned reflexes. A caveat is that rebound channel blockers or electrical stimulation applied to the DCN might alter climbing fiber input to the cerebellum through nucleo-olivary inhibitory feedback (Bengtsson et al. 2004; Hesslow and Ivarsson 1996). To dissociate the role of rebounds in excitatory versus inhibitory DCN neurons might require genetic tools for cell-type-specific manipulation, such as optogenetic techniques (Zhang et al. 2007).
Another prediction of the rebound theory is that blink-related Purkinje cells should exhibit, in the aggregate, biphasic patterns of CS-driven activity in conditioned subjects. This contrasts with Albus's proposal that GABAergic Purkinje cells should drive DCN cells through disinhibition (Albus 1971). The recordings made to date of the spiking activity of individual Purkinje neurons in trained animals have revealed a diversity of spiking patterns in response to the CS (Berthier and Moore 1986; Gould and Steinmetz 1996; Green and Steinmetz 2005; Jirenhed et al. 2007; Kotani et al. 2003, 2006; Tracy and Steinmetz 1998). Although multiple regions of cerebellar cortex have been implicated in eyeblink conditioning, most studies of Purkinje cell activity in classical conditioning have focused on a subset of these areas.
Recordings of Purkinje cell activity in trained rabbits from the cerebellar anterior lobe (Green and Steinmetz 2005) and cerebellar lobule HVI (Berthier and Moore 1986; Gould and Steinmetz 1996), both areas implicated in eyeblink conditioning, have revealed a mixture of activity increases and decreases in response to the CS. Excitatory responses might occur earlier in the ISI, with activity decreases occurring later (Green and Steinmetz 2005). Recordings from lobule HVI in decerebrate guinea pigs and ferrets have also revealed a mixture of responses (Jirenhed et al. 2007; Kotani et al. 2006). One particularly careful study, in which Purkinje cells were first identified as being responsive to the US prior to training, reported mainly decreases in Purkinje cell spiking in response to the CS after training (Jirenhed et al. 2007). However, this study also mentions that after training some cells undergo an increase in firing during the first 50–100 ms of the CS, followed by an abrupt drop in spiking. Overall, Purkinje cell recordings in trained animals are mainly consistent with the rebound theory but do not provide sufficient evidence to validate the prediction that aggregate Purkinje cell activity should be biphasic. Given the central role in the theory of such biphasic aggregate responses, it seems important that future studies testing this prediction should record from multiple Purkinje cells concurrently using multielectrode techniques.
If rebounds are instrumental in driving classically conditioned responses, the timing of training stimuli needed for successful conditioning should depend on rebound channel kinetics. Because the minimum ISI that leads to rebounds is set by the sum of the inactivation time constant and tLTD, an increase in the inactivation time constant should increase the minimum ISI for successful training. Likewise, slowing the kinetics of hyperpolarization-activated currents in MVN cells should increase the interval required between vestibular and visual inputs needed to generate the greatest changes in VOR gain.
Further predictions of the rebound theory concern classically conditioned subjects in which cerebellar LTD or LTP is impaired. Elimination of both LTD and LTP would prevent biphasic patterns of Purkinje cell spiking, precluding rebounds. Our simulations also suggest that if either LTP or LTD is partially impaired, rebound amplitude decreases and sporadically triggers readout by Ca2+ or Na+ spikes (data not shown). However, rebound-driven spiking remains well timed when it occurs. Thus some animals partially deficient in LTD or LTP might exhibit conditioned responses sporadically or of diminished amplitude, but with proper timing. Greater disruptions of cerebellar plasticity would disrupt response timing due to insufficient biphasic modulation of Purkinje cell spiking.
To date, several groups have studied eyeblink conditioning in mice with disrupted cerebellar LTD. Four strains of such mice exhibited blinks that occurred sporadically or with altered amplitude, but with the same distribution of time courses as that of wild-type mice (Kishimoto et al. 2001; Koekkoek et al. 2005; Miyata et al. 2001; Shibuki et al. 1996). It was reported that mice expressing a protein kinase C (PKC) inhibitor that impaired LTD had ill-timed residual blinks (Koekkoek et al. 2003). However, questions have been raised about this study regarding the degree to which motor learning was cerebellar in origin (Christian et al. 2004; Jirenhed et al. 2007; Koekkoek et al. 2003). It is possible PKC inhibition disrupts more than just LTD. Although further study is needed, the former four strains seem consistent with the rebound theory and suggest normal LTD levels might be unnecessary for normally timed responses.
Comparison to other work on the role of rebounds in cerebellar function
Several other authors have considered a potential role for DCN cell rebounds in cerebellar network function. Inspired by the observation that Purkinje cell activity effectively triggers rebounds in vitro (Aizenman and Linden 1999), Kistler and de Zeeuw (2003) created computational models emphasizing a potential role for rebounds in reverberatory olivo-cerebellar network activity through which Purkinje cells can affect their own subsequent activity and timed motor responses. In experiments on classically conditioned decerebrate cats, Hesslow (1994a,b) noted that brief electrical stimulation of the cerebellar cortex leads to a delayed activation of muscle fibers as assessed by electromyography. This finding was explained as arising from postinhibitory rebound depolarization in the DCN and supports a basic assumption underlying our models—that rebound-driven DCN cell activity can trigger motor action.
Aizenman and Linden (1998) found that DCN cell rebound depolarization and the associated spike burst are important for determining the polarity of gain changes at the Purkinje cell to DCN cell synapse. Mauk and collaborators (Mauk and Donegan 1997; Medina and Mauk 1999) performed extensive network modeling studies of cerebellar learning and suggested that the rise in intracellular Ca2+ due to DCN cell rebounds might be a potent trigger for plasticity at the mossy fiber to DCN cell synapse. A recent experimental study has demonstrated this effect, confirming the importance of postinhibitory rebound current for potentiation of mossy fiber to DCN cell synapses (Pugh and Raman 2006). However, in the original simulations the inefficacy of brief ISI values for classical conditioning arises from a minimum time interval for synchronization of granule cell activity following the CS, rather than from a rebound-based mechanism (Mauk and Donegan 1997). For ISI values shorter than the minimum for granule cell synchronization, plasticity in the cerebellar cortex is precluded. By comparison, our lock-and-key hypothesis is based on the supposition of at least some plasticity occurring with brief forward or backward ISI values.
A non-synaptic form of plasticity capable of increasing the intrinsic excitability of DCN and MVN neurons appears suited to raise the propensity for rebounds over the course of training. Bursts of synaptic input can induce DCN and MVN cells to display long-lasting gains in excitability due to increases in the maximum rebound current (Aizenman and Linden 2000; Smith et al. 2002; Zhang et al. 2004). A biphasic burst-and-pause pattern of Purkinje cell activity may be optimally suited to induce this increase (Aizenman and Linden 2000; Medina and Mauk 1999; Pugh and Raman 2006). Thus, rebound generation might be facilitated during training, perhaps reducing the number of Purkinje inputs needed to drive motor action. Overall, multiple cerebellar plasticity mechanisms in addition to those invoked here at the PF–Purkinje synapse are likely to occur during motor training and to shape both cerebellar dynamics and motor learning in a cooperative fashion (Boyden et al. 2004; D'Angelo et al. 1999; De Zeeuw and Yeo 2005; Hansel et al. 2001; Mauk and Donegan 1997; Medina and Mauk 1999; Soler-Llavina and Sabatini 2006). Collectively, the existing experimental data not only are consistent with our computational models but also reach well beyond, suggesting that rebound currents have multiple functions additional to the temporal filtering role explored here.
A role for rebounds in temporal shaping of learned responses
A long-standing debate has concerned the issue of whether cerebellar-mediated memory storage occurs within the cerebellar cortex or within the deep cerebellar or vestibular nuclei (du Lac et al. 1995; Ito 1982; McCormick and Thompson 1984a; Miles and Lisberger 1981). Consistent with the identification of multiple plasticity mechanisms in vitro, recent in vivo studies indicate both the cortex and the nuclei have a role in memory storage, but with distinct kinetics for plasticity induction and with distinct roles in setting the timing of motor expression (Kassardjian et al. 2005; Medina and Mauk 1999; Ohyama and Mauk 2001; Perrett et al. 1993; Shutoh et al. 2006). In both eyeblink conditioning and ocular reflex adaptation, shortly after the start of training motor memory is susceptible to extinction and is impaired by lesion or pharmacological disconnection of the cerebellar cortex. After a few days, motor memories are more resistant to extinction and can persist without Purkinje cell involvement (Kassardjian et al. 2005; Ohyama and Mauk 2001; Perrett et al. 1993; Shutoh et al. 2006).
However, even long after training, Purkinje cells appear important for setting the proper timing of learned responses. In eyeblink conditioning, disconnection of Purkinje cells' projections to the DCN leads to ill-timed, short-latency blink responses to the CS (Ohyama and Mauk 2001; Perrett et al. 1993). These short-latency blinks probably arise because without inhibitory input from Purkinje cells, DCN cells may be driven strongly by mossy fiber inputs starting from the onset of the CS. For VOR adaptation, the relative contributions of the flocullar Purkinje cells and the MVN neurons in shaping the temporal character of the learned motor response remain less clear. Our interpretation of the available data are that for VOR adaptation, a baseline component of the learned motor response that is not as sensitive to the relative timing of vestibular and visual training stimuli arises from mossy fiber-driven activity in the MVN (Fig. 7C). However, there also appears to be another component that is sensitive to the relative timing of the training stimuli (Raymond and Lisberger 1996); it is this second component that we propose has the proper form and kinetics to be explained by a rebound-based mechanism using a slow, hyperpolarization-activated current (Fig. 7E), such as that identified by Sekirnjak and du Lac (2002). Unlike eyeblink conditioning, for which there is no learned response with short ISI values, there is VOR adaptation following short-interval training, but at reduced amplitude relative to adaptation at longer ISI values (Raymond and Lisberger 1996). Thus, rebound mechanisms cannot quantitatively account for the full magnitude of VOR adaptation.
Generalizations of the proposed rebound mechanism
Our simulations involved specific choices of cellular parameters, but the ideas presented here on rebounds may have explanatory power surpassing that of our detailed models. One aspect of generality concerns the channels that mediate rebounds. It was first thought mainly T-type channels drive DCN cell rebounds, but new evidence also suggests a role for Na+ currents (Sangrey and Jaeger 2005). Further, rebound amplitude or kinetics may vary between individual DCN neurons due to differential expression of T-channel isoforms (Molineux et al. 2006). Identities of the rebound channels in MVN cells are still in question, but in addition to the h-type current there is evidence Na+ and low-voltage-activated Ca2+ currents play a role (Sekirnjak and du Lac 2002; Serafin et al. 1991). Further characterization of rebound channels may be important for understanding the precise timing relations seen in behavioral studies. However, the basic idea that rebounds shape temporally asymmetric behavioral responses is general and may apply to other cerebellum-dependent learning paradigms. For example, learned aspects of smooth visual pursuit exhibit temporal dependencies on the approximately 200 ms scale that might reflect requirements for rebound generation in cells of the cerebellar caudal fastigial nucleus (Medina et al. 2005).
Role of synaptic plasticity in memory formation and recall
A prevalent view holds that LTD and LTP are opposing mechanisms, with one encoding memories and the other erasing (Boyden and Raymond 2003; Coesmans et al. 2004; Lev-Ram et al. 2003). We are proposing a different view, in which both cerebellar LTD and LTP are needed for reliable recall. The lock mechanism based on rebound depolarization enforces the requirement for forward training, but nevertheless both LTD and LTP are induced during forward and backward training.
In our lock-and-key framework, backward conditioning and forward training at short intervals induces synaptic plasticity at parallel fiber to Purkinje cell synapses, despite the absence of behavioral output. In this respect, our work follows several prior models of cerebellar-mediated learning in which neuronal plasticity can occur without any change in motor behavior. Models of learning with two or more stages have been proposed for eyeblink conditioning (Mauk 1997; Mauk and Donegan 1997), VOR adaptation (Boyden et al. 2004; du Lac et al. 1995), and other motor behaviors (Smith et al. 2006). These models involve an intermediate stage of learning during which plasticity has occurred but learning is not yet expressed. More generally, any model of learning with two or more serial stages of plasticity inherently implies the possible existence of plasticity without expression of learning. We also present a two-stage model, but here the second stage of processing is implemented as temporal filtering occurring at memory recall rather than as plasticity occurring during memory formation. As with prior two-stage models of plasticity, our model also predicts the possibility of plasticity without behavior modification.
The idea of rebound generation as a temporal filter pertains to the ongoing debate about mechanisms that shape the dependence of classically conditioned responses on the CS–US ISI (Fig. 1B). Ideas from recent studies on synaptic plasticity and animal behavior have suggested conflicting explanations. Some authors have suggested spike-timing-dependent plasticity can account for the variation in performance with the ISI value (Wang et al. 2000). A timing-dependent plasticity rule in which PF activity preceding CF activity is optimal for LTD induction does create a distinction between backward and forward training (Fig. 2). However, the recent and older experimental data show that LTD can occur with forward or backward timing protocols (Fig. 2A) (Chen and Thompson 1995; Ito and Kano 1982; Wang et al. 2000), so another mechanism auxiliary to LTD seems needed to create conditioned reflexes selectively following forward but not backward training. Our model incorporates spike-timing-dependent plasticity, and tLTD is a key factor that causes the conditioned response to precede the expected US. However, it is rebound generation that precludes learned responses following backward training. In a sense, a memory of backward training is formed in the resulting patterns of LTD and LTP, but this memory is not retrieved in response to the CS.
This statement of the lock-and-key hypothesis fits well with studies of classical aversion conditioning, which have shown backward training leads to an associative memory of the US–CS pairing (Gallistel and Gibbon 2000). CS presentation alone does not yield conditioned motor responses, but the associative memory can be demonstrated through second-order conditioning (Barnet et al. 1997). Thus, at least for this form of aversion conditioning, mediated outside the cerebellum, the lock-and-key hypothesis appears to be correct. What mechanism prevents the stored memory from yielding a response to the CS? For aversion conditioning the answer remains unknown, but our study points to rebound depolarization as a candidate mechanism by which such behavioral filtering might occur for cerebellum-dependent behaviors.
An algorithmic lock-and-key description of memory retrieval
In sensory neuroscience, filters have been fruitfully used to describe receptive fields and spiking behavior in an algorithmic manner, apart from physiological details. Our study suggests an algorithmic lock-and-key description of cerebellar memory recall (Fig. 8) . The “lock” resides in the DCN or MVN as a temporal filter that requires specific “key” input activity to drive rebound spiking and a motor response. The key is the biphasic pattern of Purkinje cell activity that is shaped by training and driven by a learned sensory cue. Synaptic plasticity does not lead to learned responses unless key activity is shaped to match the temporal filter of the lock (Fig. 8, A and B). Backward training induces plasticity, but the resulting key activity does not fit the lock, precluding conditioned responses.
FIG. 8.

A lock-and-key description of memory recall. A: schematic of memory formation within the lock-and-key description. Paired PF and CF activity induces synaptic plasticity in the cerebellar cortex. This shapes Purkinje cell “key” activity in response to subsequent presentations of the learned sensory input. B: schematic of memory recall, in which the lock resides in the DCN. Key activity driven by learned sensory input is sent to the DCN via Purkinje cell axons. Not all keys will be successful at driving DCN cell activity and learned motor responses. The lock prevents inappropriate motor responses by filtering out nonmatching key activity. Partially matching key activity leads to unreliable recall or responses of diminished amplitude. C: an implementation of a lock-and-key mechanism using a linear–nonlinear (L-N) filter model, similar to those used to describe sensory receptive fields. Key activity, K(t), undergoes linear filtering according to F(t), then the result is filtered by a nonlinear threshold function, G(x). D: the L-N model enables responses, equal to G[K(t) * F(t)], to be made selectively to only those keys, K(t), that are shaped by training with sufficiently positive ISI values. E: response of the L-N model as a function of ISI in eyeblink conditioning. The results mimic those of DCN cell simulations (Figs. 4–6) and the classic rabbit behavioral data (Figs. 1B and 5B).
We found that a simple linear–nonlinear (L-N) filter model, inspired by those used to describe visual receptive fields (Baccus and Meister 2002; Korenberg and Hunter 1986), can accurately predict the responses of our biophysical models and the behavioral dependence of learned responses on ISI (Fig. 8, C–E). Thus from an algorithmic standpoint memory retrieval may involve a temporal filter that excludes certain behaviors and allows others.
The lock-and-key model of recall might be adaptable to other nondeclarative forms of memory involving feedforward networks. Striatal memory for motor sequences shows promising similarities, since both Purkinje cells and striatal medium spiny neurons receive diverse sensory information, undergo bidirectional plasticity, and are GABAergic projection neurons that trigger motor sequences (Fino et al. 2005). The similarity between these two GABAergic cell types also extends to their respective targets, because there is one class of striatal target neuron in the globus pallidum that is spontaneously active, has a baseline membrane potential of about −60 mV, and reliably undergoes postinhibitory rebound depolarization in response to electrical stimulation of striatal input fibers (Nambu and Llinás 1994). By analogy, corticostriatal plasticity alone may not represent a memory, since select patterns of striatal input activity to the globus pallidum may be needed to unlock motor expression. These ideas challenge classical theories of memory storage that focus almost exclusively on neuronal plasticity but avoid mention of how retrieval dynamics may help shape memory expression.
Neuroscientists have long sought the location and substance of memories. This quest led to the notion of the engram, the physical unit of memory, and to the later idea that changes in synaptic strength constitute a candidate substrate for memory (Bliss and Lomo 1973; Lashley 1950). Cognitive scientists have countered that synaptic plasticity alone does not account for the complex phenomenology of memory recall (Gallistel 1993; Martin et al. 2000). In this opposing view, a memory is a physical dynamic that occurs exclusively at recall. Plasticity may help shape this dynamic, but is not by itself a memory. Our computational models provide concrete examples in which it is difficult to identify any physically localized engram. Would the engram include the synapses that have undergone plasticity, the key activity that triggers a memory, the activity unlocked at a rebound, or all of the above? By itself plasticity seems a poor candidate for a complete engram, since it is insufficient to allow recall. Questions regarding the physical substance of memory may be misleading, neglecting that recall occurs due to a sequence of events culminating in a specific form of neural dynamics. This caveat regarding the substance of memory in our models might also apply broadly to multiple memory systems.
GRANTS
This work was supported by National Science Foundation Fellowships to D. Z. Wetmore and E. A. Mukamel. The work of M. J. Schnitzer on cerebellum is supported by the National Institutes of Health and the Klingenstein, Sloan, and Packard Foundations.
Supplementary Material
Acknowledgments
We thank M. Mauk and J. Raymond for helpful suggestions in writing the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The online version of this article contains supplemental data.
REFERENCES
- Abbott and Nelson 2000.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci Suppl 3: 1178–1183, 2000. [DOI] [PubMed] [Google Scholar]
- Aizenman and Linden 1999.Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol 82: 1697–1709, 1999. [DOI] [PubMed] [Google Scholar]
- Aizenman and Linden 2000.Aizenman CD, Linden DJ. Rapid, synaptically driven increases in the intrinsic excitability of cerebellar deep nuclear neurons. Nat Neurosci 3: 109–111, 2000. [DOI] [PubMed] [Google Scholar]
- Aizenman et al. 1998.Aizenman CD, Manis PB, Linden DJ. Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron 21: 827–835, 1998. [DOI] [PubMed] [Google Scholar]
- Aksenov et al. 2005.Aksenov DP, Serdyukova NA, Bloedel JR, Bracha V. Glutamate neurotransmission in the cerebellar interposed nuclei: involvement in classically conditioned eyeblinks and neuronal activity. J Neurophysiol 93: 44–52, 2005. [DOI] [PubMed] [Google Scholar]
- Albus 1971.Albus J A theory of cerebellar function. Math Biosci 10: 25–61, 1971. [Google Scholar]
- Anchisi et al. 2001.Anchisi D, Scelfo B, Tempia F. Postsynaptic currents in deep cerebellar nuclei. J Neurophysiol 85: 323–331, 2001. [DOI] [PubMed] [Google Scholar]
- Baccus and Meister 2002.Baccus SA, Meister M. Fast and slow contrast adaptation in retinal circuitry. Neuron 36: 909–919, 2002. [DOI] [PubMed] [Google Scholar]
- Barnet et al. 1997.Barnet RC, Cole RP, Miller RR. Temporal integration in second-order conditioning and sensory preconditioning. Anim Learn Behav 25: 221–233, 1997. [Google Scholar]
- Bengtsson et al. 2004.Bengtsson F, Svensson P, Hesslow G. Feedback control of Purkinje cell activity by the cerebello-olivary pathway. Eur J Neurosci 20: 2999–3005, 2004. [DOI] [PubMed] [Google Scholar]
- Berthier and Moore 1986.Berthier NE, Moore JW. Cerebellar Purkinje cell activity related to the classically conditioned nictitating membrane response. Exp Brain Res 63: 341–350, 1986. [DOI] [PubMed] [Google Scholar]
- Berthier and Moore 1990.Berthier NE, Moore JW. Activity of deep cerebellar nuclear cells during classical conditioning of nictitating membrane extension in rabbits. Exp Brain Res 83: 44–54, 1990. [DOI] [PubMed] [Google Scholar]
- Bliss and Lomo 1973.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232: 331–356, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden et al. 2006.Boyden ES, Katoh A, Pyle JL, Chatila TA, Tsien RW, Raymond JL. Selective engagement of plasticity mechanisms for motor memory storage. Neuron 51: 823–834, 2006. [DOI] [PubMed] [Google Scholar]
- Boyden et al. 2004.Boyden ES, Katoh A, Raymond JL. Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci 27: 581–609, 2004. [DOI] [PubMed] [Google Scholar]
- Boyden and Raymond 2003.Boyden ES, Raymond JL. Active reversal of motor memories reveals rules governing memory encoding. Neuron 39: 1031–1042, 2003. [DOI] [PubMed] [Google Scholar]
- Buonomano and Mauk 1994.Buonomano DV, Mauk AD. Neural network model of the cerebellum: temporal discrimination and the timing of motor responses. Neural Comput 6: 38–55, 1994. [Google Scholar]
- Chadderton et al. 2004.Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature 428: 856–860, 2004. [DOI] [PubMed] [Google Scholar]
- Chan-Palay 1973.Chan-Palay V Axon terminals of the intrinsic neurons in the nucleus lateralis of the cerebellum. An electron microscope study. Z Anat Entwicklungsgesch 142: 187–206, 1973. [DOI] [PubMed] [Google Scholar]
- Chen and Thompson 1995.Chen C, Thompson RF. Temporal specificity of long-term depression in parallel fiber–Purkinje synapses in rat cerebellar slice. Learn Mem 2: 185–198, 1995. [DOI] [PubMed] [Google Scholar]
- Chen and Evinger 2006.Chen FP, Evinger C. Cerebellar modulation of trigeminal reflex blinks: interpositus neurons. J Neurosci 26: 10569–10576, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng et al. 2007.Cheng DT, Disterhoft JF, Power JM, Ellis DA, Desmond JE. The effects of aging on human delay and trace eyeblink conditioning. Program No. 204.13. 2007 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2007. Online.
- Choi and Moore 2003.Choi JS, Moore JW. Cerebellar neuronal activity expresses the complex topography of conditioned eyeblink responses. Behav Neurosci 117: 1211–1219, 2003. [DOI] [PubMed] [Google Scholar]
- Christian et al. 2004.Christian KM, Poulos AM, Lavond DG, Thompson RF. Comment on “Cerebellar LTD and learning-dependent timing of conditioned eyelid responses.” Science 304: 211; author reply 211, 2004. [DOI] [PubMed] [Google Scholar]
- Christian and Thompson 2003.Christian KM, Thompson RF. Neural substrates of eyeblink conditioning: acquisition and retention. Learn Mem 10: 427–455, 2003. [DOI] [PubMed] [Google Scholar]
- Chun et al. 2003.Chun SW, Choi JH, Kim MS, Park BR. Characterization of spontaneous synaptic transmission in rat medial vestibular nucleus. Neuroreport 14: 1485–1488, 2003. [DOI] [PubMed] [Google Scholar]
- Coesmans et al. 2004.Coesmans M, Weber JT, De Zeeuw CI, Hansel C. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44: 691–700, 2004. [DOI] [PubMed] [Google Scholar]
- D'Angelo et al. 1999.D'Angelo E, Rossi P, Armano S, Taglietti V. Evidence for NMDA and mGlu receptor-dependent long-term potentiation of mossy fiber-granule cell transmission in rat cerebellum. J Neurophysiol 81: 277–287, 1999. [DOI] [PubMed] [Google Scholar]
- Debiec et al. 2006.Debiec J, Doyere V, Nader K, Ledoux JE. Directly reactivated, but not indirectly reactivated, memories undergo reconsolidation in the amygdala. Proc Natl Acad Sci USA 103: 3428–3433, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw and Berrebi 1995.De Zeeuw CI, Berrebi AS. Postsynaptic targets of Purkinje cell terminals in the cerebellar and vestibular nuclei of the rat. Eur J Neurosci 7: 2322–2333, 1995. [DOI] [PubMed] [Google Scholar]
- De Zeeuw and Yeo 2005.De Zeeuw CI, Yeo CH. Time and tide in cerebellar memory formation. Curr Opin Neurobiol 15: 667–674, 2005. [DOI] [PubMed] [Google Scholar]
- Dickson et al. 2000.Dickson CT, Magistretti J, Shalinsky MH, Fransén E, Hasselmo ME, Alonso A. Properties and role of I(h) in the pacing of subthreshold oscillations in entorhinal cortex layer II neurons. J Neurophysiol 83: 2562–2579, 2000. [DOI] [PubMed] [Google Scholar]
- Doi et al. 2005.Doi T, Kuroda S, Michikawa T, Kawato M. Inositol 1,4,5-trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J Neurosci 25: 950–961, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyere et al. 2007.Doyere V, Debiec J, Monfils MH, Schafe GE, Ledoux JE. Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat Neurosci 10: 414–416, 2007. [DOI] [PubMed] [Google Scholar]
- du Lac and Lisberger 1995a.du Lac S, Lisberger SG. Cellular processing of temporal information in medial vestibular nucleus neurons. J Neurosci 15: 8000–8010, 1995a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Lac and Lisberger 1995b.du Lac S, Lisberger SG. Membrane and firing properties of avian medial vestibular nucleus neurons in vitro. J Comp Physiol A Sens Neural Behav Physiol 176: 641–651, 1995b. [DOI] [PubMed] [Google Scholar]
- du Lac et al. 1995.du Lac S, Raymond JL, Sejnowski TJ, Lisberger SG. Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci 18: 409–441, 1995. [DOI] [PubMed] [Google Scholar]
- Feil et al. 2003.Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M, Konnerth A, De Zeeuw CI, Hofmann F. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol 163: 295–302, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino et al. 2005.Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci 25: 11279–11287, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzhugh 1960.Fitzhugh R Thresholds and plateaus in the Hodgkin–Huxley nerve equations. J Gen Physiol 43: 867–896, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman and Nicholson 1999.Freeman JH Jr, Nicholson DA. Neuronal activity in the cerebellar interpositus and lateral pontine nuclei during inhibitory classical conditioning of the eyeblink response. Brain Res 833: 225–233, 1999. [DOI] [PubMed] [Google Scholar]
- Gallistel 1993.Gallistel CR The Organization of Learning. Cambridge, MA: MIT Press, 1993, p. 662.
- Gallistel and Gibbon 2000.Gallistel CR, Gibbon J. Time, rate, and conditioning. Psychol Rev 107: 289–344, 2000. [DOI] [PubMed] [Google Scholar]
- Gauck et al. 2001.Gauck V, Thomann M, Jaeger D, Borst A. Spatial distribution of low- and high-voltage-activated calcium currents in neurons of the deep cerebellar nuclei. J Neurosci 21: RC158, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormezano et al. 1962.Gormezano I, Schneiderman N, Deaux E, Fuentes I. Nictitating membrane: classical conditioning and extinction in the albino rabbit. Science 138: 33–34, 1962. [DOI] [PubMed] [Google Scholar]
- Gould and Steinmetz 1996.Gould TJ, Steinmetz JE. Changes in rabbit cerebellar cortical and interpositus nucleus activity during acquisition, extinction, and backward classical eyelid conditioning. Neurobiol Learn Mem 65: 17–34, 1996. [DOI] [PubMed] [Google Scholar]
- Green and Steinmetz 2005.Green JT, Steinmetz JE. Purkinje cell activity in the cerebellar anterior lobe after rabbit eyeblink conditioning. Learn Mem 12: 260–269, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel et al. 2001.Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4: 467–475, 2001. [DOI] [PubMed] [Google Scholar]
- Heck et al. 2002.Heck D, Kummell F, Thach WT, Aertsena A. Dynamic correlation of neuronal activity in rat cerebellar cortex modulated by behavior. Ann NY Acad Sci 978: 156–163, 2002. [DOI] [PubMed] [Google Scholar]
- Hesslow 1994a.Hesslow G Correspondence between climbing fibre input and motor output in eyeblink-related areas in cat cerebellar cortex. J Physiol 476: 229–244, 1994a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslow 1994b.Hesslow G Inhibition of classically conditioned eyeblink responses by stimulation of the cerebellar cortex in the decerebrate cat. J Physiol 476: 245–256, 1994b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslow and Ivarsson 1996.Hesslow G, Ivarsson M. Inhibition of the inferior olive during conditioned responses in the decerebrate ferret. Exp Brain Res 110: 36–46, 1996. [DOI] [PubMed] [Google Scholar]
- Hesslow et al. 1999.Hesslow G, Svensson P, Ivarsson M. Learned movements elicited by direct stimulation of cerebellar mossy fiber afferents. Neuron 24: 179–185, 1999. [DOI] [PubMed] [Google Scholar]
- Hille 2001.Hille B Ion Channels of Excitable Membranes (3rd ed.). Sunderland, MA: Sinauer, 2001, p. xviii, 814.
- Hines and Carnevale 1997.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput 9: 1179–1209, 1997. [DOI] [PubMed] [Google Scholar]
- Hines and Carnevale 2001.Hines ML, Carnevale NT. NEURON: a tool for neuroscientists. Neuroscientist 7: 123–135, 2001. [DOI] [PubMed] [Google Scholar]
- Hodgkin and Huxley 1952.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield 1982.Hopfield JJ Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci USA 79: 2554–2558, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito 1982.Ito M Cerebellar control of the vestibulo-ocular reflex—around the flocculus hypothesis. Annu Rev Neurosci 5: 275–296, 1982. [DOI] [PubMed] [Google Scholar]
- Ito 1989.Ito M Long-term depression. Annu Rev Neurosci 12: 85–102, 1989. [DOI] [PubMed] [Google Scholar]
- Ito and Kano 1982.Ito M, Kano M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett 33: 253–258, 1982. [DOI] [PubMed] [Google Scholar]
- Jahnsen 1986a.Jahnsen H Electrophysiological characteristics of neurones in the guinea-pig deep cerebellar nuclei in vitro. J Physiol 372: 129–147, 1986a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen 1986b.Jahnsen H Extracellular activation and membrane conductances of neurones in the guinea-pig deep cerebellar nuclei in vitro. J Physiol 372: 149–168, 1986b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirenhed et al. 2007.Jirenhed DA, Bengtsson F, Hesslow G. Acquisition, extinction, and reacquisition of a cerebellar cortical memory trace. J Neurosci 27: 2493–2502, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassardjian et al. 2005.Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM. The site of a motor memory shifts with consolidation. J Neurosci 25: 7979–7985, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpo et al. 2005.Kimpo RR, Boyden ES, Katoh A, Ke MC, Raymond JL. Distinct patterns of stimulus generalization of increases and decreases in VOR gain. J Neurophysiol 94: 3092–3100, 2005. [DOI] [PubMed] [Google Scholar]
- King et al. 2001.King DA, Krupa DJ, Foy MR, Thompson RF. Mechanisms of neuronal conditioning. Int Rev Neurobiol 45: 313–337, 2001. [DOI] [PubMed] [Google Scholar]
- Kishimoto et al. 2001.Kishimoto Y, Kawahara S, Suzuki M, Mori H, Mishina M, Kirino Y. Classical eyeblink conditioning in glutamate receptor subunit delta 2 mutant mice is impaired in the delay paradigm but not in the trace paradigm. Eur J Neurosci 13: 1249–1253, 2001. [DOI] [PubMed] [Google Scholar]
- Kistler et al. 2003.Kistler WM, De Zeeuw CI. Time windows and reverberating loops: a reverse-engineering approach to cerebellar function. Cerebellum 2: 44–54, 2003. [DOI] [PubMed] [Google Scholar]
- Koekkoek et al. 2003.Koekkoek SK, Hulscher HC, Dortland BR, Hensbroek RA, Elgersma Y, Ruigrok TJ, De Zeeuw CI. Cerebellar LTD and learning-dependent timing of conditioned eyelid responses. Science 301: 1736–1739, 2003. [DOI] [PubMed] [Google Scholar]
- Koekkoek et al. 2005.Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, De Zeeuw CI. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47: 339–352, 2005. [DOI] [PubMed] [Google Scholar]
- Korenberg and Hunter 1986.Korenberg MJ, Hunter IW. The identification of nonlinear biological systems: LNL cascade models. Biol Cybern 55: 125–134, 1986. [DOI] [PubMed] [Google Scholar]
- Kotani et al. 2003.Kotani S, Kawahara S, Kirino Y. Purkinje cell activity during learning a new timing in classical eyeblink conditioning. Brain Res 994: 193–202, 2003. [DOI] [PubMed] [Google Scholar]
- Kotani et al. 2006.Kotani S, Kawahara S, Kirino Y. Purkinje cell activity during classical eyeblink conditioning in decerebrate guinea pigs. Brain Res 1068: 70–81, 2006. [DOI] [PubMed] [Google Scholar]
- Lang et al. 1999.Lang EJ, Sugihara I, Welsh JP, Llinás R. Patterns of spontaneous Purkinje cell complex spike activity in the awake rat. J Neurosci 19: 2728–2739, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley 1950.Lashley KS In search of the engram. Symp Soc Exp Biol 4: 454–482, 1950. [Google Scholar]
- Lev-Ram et al. 2003.Lev-Ram V, Mehta SB, Kleinfeld D, Tsien RY. Reversing cerebellar long-term depression. Proc Natl Acad Sci USA 100: 15989–15993, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás and Muhlethaler 1988.Llinás R, Muhlethaler M. Electrophysiology of guinea-pig cerebellar nuclear cells in the in vitro brain stem-cerebellar preparation. J Physiol 404: 241–258, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein et al. 2005.Loewenstein Y, Mahon S, Chadderton P, Kitamura K, Sompolinsky H, Yarom Y, Hausser M. Bistability of cerebellar Purkinje cells modulated by sensory stimulation. Nat Neurosci 8: 202–211, 2005. [DOI] [PubMed] [Google Scholar]
- Mainen and Sejnowski 1996.Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature 382: 363–366, 1996. [DOI] [PubMed] [Google Scholar]
- Marr 1969.Marr D A theory of cerebellar cortex. J Physiol 202: 437–470, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin et al. 2000.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649–711, 2000. [DOI] [PubMed] [Google Scholar]
- Mauk 1997.Mauk MD Roles of cerebellar cortex and nuclei in motor learning: contradictions or clues? Neuron 18: 343–346, 1997. [DOI] [PubMed] [Google Scholar]
- Mauk and Donegan 1997.Mauk MD, Donegan NH. A model of Pavlovian eyelid conditioning based on the synaptic organization of the cerebellum. Learn Mem 4: 130–158, 1997. [DOI] [PubMed] [Google Scholar]
- Mauk and Ohyama 2004.Mauk MD, Ohyama T. Extinction as new learning versus unlearning: considerations from a computer simulation of the cerebellum. Learn Mem 11: 566–571, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauk et al. 1986.Mauk MD, Steinmetz JE, Thompson RF. Classical conditioning using stimulation of the inferior olive as the unconditioned stimulus. Proc Natl Acad Sci USA 83: 5349–5353, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick et al. 1982.McCormick DA, Clark GA, Lavond DG, Thompson RF. Initial localization of the memory trace for a basic form of learning. Proc Natl Acad Sci USA 79: 2731–2735, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick et al. 1985.McCormick DA, Steinmetz JE, Thompson RF. Lesions of the inferior olivary complex cause extinction of the classically conditioned eyeblink response. Brain Res 359: 120–130, 1985. [DOI] [PubMed] [Google Scholar]
- McCormick and Thompson 1984a.McCormick DA, Thompson RF. Cerebellum: essential involvement in the classically conditioned eyelid response. Science 223: 296–299, 1984a. [DOI] [PubMed] [Google Scholar]
- McCormick and Thompson 1984b.McCormick DA, Thompson RF. Neuronal responses of the rabbit cerebellum during acquisition and performance of a classically conditioned nictitating membrane-eyelid response. J Neurosci 4: 2811–2822, 1984b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough and Bean 1998.McDonough SI, Bean BP. Mibefradil inhibition of T-type calcium channels in cerebellar Purkinje neurons. Mol Pharmacol 54: 1080–1087, 1998. [DOI] [PubMed] [Google Scholar]
- McRory et al. 2001.McRory JE, Santi CM, Hamming KS, Mezeyova J, Sutton KG, Baillie DL, Stea A, Snutch TP. Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem 276: 3999–4011, 2001. [DOI] [PubMed] [Google Scholar]
- Medina et al. 2005.Medina JF, Carey MR, Lisberger SG. The representation of time for motor learning. Neuron 45: 157–167, 2005. [DOI] [PubMed] [Google Scholar]
- Medina et al. 2000.Medina JF, Garcia KS, Nores WL, Taylor NM, Mauk MD. Timing mechanisms in the cerebellum: testing predictions of a large-scale computer simulation. J Neurosci 20: 5516–5525, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina and Mauk 1999.Medina JF, Mauk MD. Simulations of cerebellar motor learning: computational analysis of plasticity at the mossy fiber to deep nucleus synapse. J Neurosci 19: 7140–7151, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina and Mauk 2000.Medina JF, Mauk MD. Computer simulation of cerebellar information processing. Nat Neurosci Suppl 3: 1205–1211, 2000. [DOI] [PubMed] [Google Scholar]
- Medina et al. 2002.Medina JF, Nores WL, Mauk MD. Inhibition of climbing fibres is a signal for the extinction of conditioned eyelid responses. Nature 416: 330–333, 2002. [DOI] [PubMed] [Google Scholar]
- Miles and Lisberger 1981.Miles FA, Lisberger SG. Plasticity in the vestibulo-ocular reflex: a new hypothesis. Annu Rev Neurosci 4: 273–299, 1981. [DOI] [PubMed] [Google Scholar]
- Miyata et al. 2001.Miyata M, Kim HT, Hashimoto K, Lee TK, Cho SY, Jiang H, Wu Y, Jun K, Wu D, Kano M, Shin HS. Deficient long-term synaptic depression in the rostral cerebellum correlated with impaired motor learning in phospholipase C beta4 mutant mice. Eur J Neurosci 13: 1945–1954, 2001. [DOI] [PubMed] [Google Scholar]
- Molineux et al. 2006.Molineux ML, McRory JE, McKay BE, Hamid J, Mehaffey WH, Rehak R, Snutch TP, Zamponi GW, Turner RW. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc Natl Acad Sci USA 103: 5555–5560, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa et al. 2002.Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, Kato A, Carr CA, Johnston D, Wilson MA, Tonegawa S. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science 297: 211–218, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu and Llinás 1994.Nambu A, Llinás R. Electrophysiology of globus pallidus neurons in vitro. J Neurophysiol 72: 1127–1139, 1994. [DOI] [PubMed] [Google Scholar]
- Napper and Harvey 1988.Napper RM, Harvey RJ. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol 274: 168–177, 1988. [DOI] [PubMed] [Google Scholar]
- Nicholson and Freeman 2002.Nicholson DA, Freeman JH Jr. Neuronal correlates of conditioned inhibition of the eyeblink response in the anterior interpositus nucleus. Behav Neurosci 116: 22–36, 2002. [PubMed] [Google Scholar]
- Ohyama and Mauk 2001.Ohyama T, Mauk M. Latent acquisition of timed responses in cerebellar cortex. J Neurosci 21: 682–690, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama et al. 2003a.Ohyama T, Nores WL, Mauk MD. Stimulus generalization of conditioned eyelid responses produced without cerebellar cortex: implications for plasticity in the cerebellar nuclei. Learn Mem 10: 346–354, 2003a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama et al. 2003b.Ohyama T, Nores WL, Murphy M, Mauk MD. What the cerebellum computes. Trends Neurosci 26: 222–227, 2003b. [DOI] [PubMed] [Google Scholar]
- Pape 1996.Pape HC Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 58: 299–327, 1996. [DOI] [PubMed] [Google Scholar]
- Pedroarena and Schwarz 2003.Pedroarena CM, Schwarz C. Efficacy and short-term plasticity at GABAergic synapses between Purkinje and cerebellar nuclei neurons. J Neurophysiol 89: 704–715, 2003. [DOI] [PubMed] [Google Scholar]
- Perrett et al. 1993.Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. J Neurosci 13: 1708–1718, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky and Rinzel 1994.Pinsky PF, Rinzel J. Intrinsic and network rhythmogenesis in a reduced Traub model for CA3 neurons. J Comput Neurosci 1: 39–60, 1994. [DOI] [PubMed] [Google Scholar]
- Porcello et al. 2003.Porcello DM, Smith SD, Huguenard JR. Actions of U-92032, a T-type Ca2+ channel antagonist, support a functional linkage between I(T) and slow intrathalamic rhythms. J Neurophysiol 89: 177–185, 2003. [DOI] [PubMed] [Google Scholar]
- Pugh and Raman 2006.Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron 51: 113–123, 2006. [DOI] [PubMed] [Google Scholar]
- Raman et al. 2000.Raman IM, Gustafson AE, Padgett D. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci 20: 9004–9016, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond and Lisberger 1996.Raymond JL, Lisberger SG. Behavioral analysis of signals that guide learned changes in the amplitude and dynamics of the vestibulo-ocular reflex. J Neurosci 16: 7791–7802, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinzel and Ermentrout 2001.Rinzel J, Ermentrout B. Analysis of neural excitability and oscillations. In: Methods in Neuronal Modeling, edited by Koch C, Segev I. Cambridge, MA: MIT Press, 2001, p. 671.
- Rush and Rinzel 1994.Rush ME, Rinzel J. Analysis of bursting in a thalamic neuron model. Biol Cybern 71: 281–291, 1994. [DOI] [PubMed] [Google Scholar]
- Salafia et al. 1980.Salafia WR, Lambert RW, Host KC, Chiala NL, Ramirez JJ. Rabbit nictitating membrane conditioning: lower limit of the effective interstimulus interval. Anim Learn Behav 8: 85–91, 1980. [Google Scholar]
- Sangrey and Jaeger 2005.Sangrey TD, Jaeger D. Currents underlying hyperpolarization-induced rebound spiking in deep cerebellar nuclei neurons. Program No. 179.10. 2005 Abstract Viewer and Itinerary Planner. Washington, DC: Society for Neuroscience, 2005. Online.
- Santoro et al. 2000.Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20: 5264–5275, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer et al. 2007.Schaefer AT, Helmstaedter M, Schmitt AC, Bar-Yehuda D, Almog M, Ben-Porat H, Sakmann B, Korngreen A. Dendritic voltage-gated K+ conductance gradient in pyramidal neurones of neocortical layer 5B from rats. J Physiol 579: 737–752, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiderman and Gormezano 1964.Schneiderman N, Gormezano I. Conditioning of the nictitating membrane of the rabbit as a function of CS–US interval. J Comp Physiol Psychol 57: 188–195, 1964. [DOI] [PubMed] [Google Scholar]
- Sekirnjak and du Lac 2002.Sekirnjak C, du Lac S. Intrinsic firing dynamics of vestibular nucleus neurons. J Neurosci 22: 2083–2095, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirnjak and du Lac 2006.Sekirnjak C, du Lac S. Physiological and anatomical properties of mouse medial vestibular nucleus neurons projecting to the oculomotor nucleus. J Neurophysiol 95: 3012–3023, 2006. [DOI] [PubMed] [Google Scholar]
- Sekirnjak et al. 2003.Sekirnjak C, Vissel B, Bollinger J, Faulstich M, du Lac S. Purkinje cell synapses target physiologically unique brainstem neurons. J Neurosci 23: 6392–6398, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafin et al. 1991.Serafin M, de Waele C, Khateb A, Vidal PP, Muhlethaler M. Medial vestibular nucleus in the guinea-pig. II. Ionic basis of the intrinsic membrane properties in brainstem slices. Exp Brain Res 84: 426–433, 1991. [DOI] [PubMed] [Google Scholar]
- Shibuki et al. 1996.Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron 16: 587–599, 1996. [DOI] [PubMed] [Google Scholar]
- Shutoh et al. 2006.Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S. Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience 139: 767–777, 2006. [DOI] [PubMed] [Google Scholar]
- Smith et al. 2006.Smith MA, Ghazizadeh A, Shadmehr R. Interacting adaptive processes with different timescales underlie short-term motor learning. PLoS Biol 4: e179, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith 1968.Smith MC CS–US interval and US intensity in classical conditioning of the rabbit's nictitating membrane response. J Comp Physiol Psychol 66: 679–687, 1968. [DOI] [PubMed] [Google Scholar]
- Smith et al. 1969.Smith MC, Coleman SR, Gormezano I. Classical conditioning of the rabbit's nictitating membrane response at backward, simultaneous, and forward CS–US intervals. J Comp Physiol Psychol 69: 226–231, 1969. [DOI] [PubMed] [Google Scholar]
- Smith et al. 2002.Smith MR, Nelson AB, Du Lac S. Regulation of firing response gain by calcium-dependent mechanisms in vestibular nucleus neurons. J Neurophysiol 87: 2031–2042, 2002. [DOI] [PubMed] [Google Scholar]
- Soler-Llavina and Sabatini 2006.Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat Neurosci 9: 798–806, 2006. [DOI] [PubMed] [Google Scholar]
- Steinmetz et al. 1989.Steinmetz JE, Lavond DG, Thompson RF. Classical conditioning in rabbits using pontine nucleus stimulation as a conditioned stimulus and inferior olive stimulation as an unconditioned stimulus. Synapse 3: 225–233, 1989. [DOI] [PubMed] [Google Scholar]
- Steinmetz et al. 1986.Steinmetz JE, Rosen DJ, Chapman PF, Lavond DG, Thompson RF. Classical conditioning of the rabbit eyelid response with a mossy-fiber stimulation CS: I. Pontine nuclei and middle cerebellar peduncle stimulation. Behav Neurosci 100: 878–887, 1986. [DOI] [PubMed] [Google Scholar]
- Straka et al. 2005.Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol 76: 349–392, 2005. [DOI] [PubMed] [Google Scholar]
- Talley et al. 1999.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19: 1895–1911, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teune et al. 1998.Teune TM, van der Burg J, de Zeeuw CI, Voogd J, Ruigrok TJ. Single Purkinje cell can innervate multiple classes of projection neurons in the cerebellar nuclei of the rat: a light microscopic and ultrastructural triple-tracer study in the rat. J Comp Neurol 392: 164–178, 1998. [DOI] [PubMed] [Google Scholar]
- Thier et al. 2000.Thier P, Dicke PW, Haas R, Barash S. Encoding of movement time by populations of cerebellar Purkinje cells. Nature 405: 72–76, 2000. [DOI] [PubMed] [Google Scholar]
- Tracy and Steinmetz 1998.Tracy JA, Steinmetz JE. Purkinje cell responses to pontine stimulation CS during rabbit eyeblink conditioning. Physiol Behav 65: 381–386, 1998. [DOI] [PubMed] [Google Scholar]
- Wang et al. 2000.Wang SS, Denk W, Hausser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3: 1266–1273, 2000. [DOI] [PubMed] [Google Scholar]
- Wills et al. 2005.Wills TJ, Lever C, Cacucci F, Burgess N, O'Keefe J. Attractor dynamics in the hippocampal representation of the local environment. Science 308: 873–876, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al. 2007.Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature 446: 633–639, 2007. [DOI] [PubMed] [Google Scholar]
- Zhang et al. 2004.Zhang W, Shin JH, Linden DJ. Persistent changes in the intrinsic excitability of rat deep cerebellar nuclear neurones induced by EPSP or IPSP bursts. J Physiol 561: 703–719, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.