

Abstract
Nitric oxide (NO•), an important signaling molecule and a component of inflammatory response, is involved in tumorigenesis. However, the quantity of NO• and the cellular micro-environment influences the role of NO• in tumor development. We used a genetic strategy to test the hypothesis that an inflammatory microenvironment with an enhanced level of NO• accelerates spontaneous tumor development. C. parvum–induced inflammation and increased NO• synthase-2 (NOS2) expression coincided with accelerated spontaneous tumor development, mostly lymphomas, in p53−/−NOS2+/+ C57BL6 mice when compared with the controls (P = 0.001). However, p53−/−NOS2−/− mice did not show any difference in tumor latency between C. parvum–treated and control groups. In C. parvum–treated p53−/−NOS2+/+ mice, tumor development was preceded by a higher expression of NOS2 and phosphorylated Akt-Ser473 (pAkt-Ser473) in spleen, increased cell proliferation measured by Ki-67 IHC in spleen and thymus, and a lower apoptotic index and CD95-L expression in spleen and thymus. C. parvum–treated p53−/−NOS2+/+ mice showed an increase in the number of Foxp3(+) T-reg cells, dendritic cells (DC), as well as increased CD80+, CD86+, CD40+, and CD83+ on DC in the spleen. Regulatory T-cells (T-reg) and the maturation of DC may modulate tumorigenesis. An increase in the FoxP3(+)T-reg cells in C. parvum–treated p53−/−NOS2+/+ mice indicates a role of NO• in the regulation of T-reg cells that may contribute to a protumor shift of the immune environment favoring an accelerated tumor development. These data provide genetic and mechanistic evidence that an inflammatory microenvironment and an increased level of NO• can accelerate tumor development.
Introduction
The two facets of nitric oxide (NO•) action, one as an important regulator of cellular function and the other as its toxic response, after inflammation toward invading pathogens, are well-established (1, 2). However, recent experimental findings are continuously defining and establishing the role of NO• in tumorigenesis (3, 4). NO• can both inhibit or enhance tumor development depending on the cellular microenvironment including the quantity of NO•, redox status, cell type, and cellular adaptation (3, 5, 6). The quantity of NO• that is produced in the cellular microenvironment is associated with the type of NO• synthase (NOS), a family of enzymes that catalyzes the conversion of L-arginine to Citruline and produces NO•. NOS1 and NOS3 are constitutively present and produce a small (nmol/L range), transient increase in NO• depending on the surge in Ca+2 level. However, NOS2, which is a Ca+2-independent isoform, is predominantly regulated through gene expression during a variety of stress conditions including inflammation and generates a larger quantity of NO• (micromoles per liter range) for a sustained period (7, 8).
p53 transrepresses NOS2 gene expression (9). p53-deficient mice, an animal model of the cancer-prone human Li-Fraumeni syndrome, produces about a 2-fold higher basal level of NO• when compared with the p53 wild-type (WT) mice (10). The administration of heat-inactivated C. parvum induces NOS2 expression and NO• production in different organs of mice accompanied by stimulation of the reticuloendothelial system and an increase in the level of cytokines (1, 10, 11). C. parvum–treatment enhanced natural killer (NK) activity and can suppress tumorigenesis (12). However, the suppression of cell-mediated tumor immunity by C. parvum has also been reported (13). We have previously reported that the administration of a single dose of heat-inactivated C. parvum to p53-deficient mice enhanced NOS2 expression in the spleen and NO• production (>100 μmol/L range), measured as nitrate and nitrite in urine and serum, which stayed at a higher level for a prolonged period of time when compared with p53 WT mice (10). In the present study, we have used a genetic strategy to investigate the role of a higher NO• level within an inflammatory microenvironment, induced by C. parvum, in regulating tumorigenesis in the p53 knockout mice in the presence or absence of NOS2.
Materials and Methods
p53 and NOS2 double knockout mice and treatment of heat-inactivated C. parvum
We generated p53 and NOS2 double knockout mice in >99% C57BL6 genetic background, as described earlier (5). Mice, 7- to 8 wk-old, were treated with a single i.p. dose of 100 mg/Kg body weight, of heat-inactivated C. parvum (Van Kampen Gr., Inc.) as described earlier (10). Mice were maintained in a climate control facility with food and water ad libitum at NCI, Frederick, and were monitored on a daily basis for any sign of morbidity. The generation of double knockout mice, treatment of C. parvum, and all the end points were approved by animal care and use committee of the NCI.
Spontaneous tumor incidence
p53-deficient mice with wild-type (WT), heterozygous, or null NOS2 alleles were divided into either C. parvum–treated or control groups and were compared for the latency of tumor development. Both C. parvum–treated and control groups were done at the same time. Kaplan-Meier survival curves were generated for C. parvum–treated and control groups of mice, and the Wilcoxon test was used to determine the statistical difference between survival curves. The control groups have been previously analyzed and reported for difference in latency of spontaneous tumor development among different genotypic combinations of p53 and NOS2 (5). The analyses of the following end points were done 10 d after the treatment of C. parvum..
Apoptosis assay
To determine the percentage of apoptotic cells, 5-μm-thick sections of the spleen and thymus from C. parvum–treated and control groups of 8- to 9-wk-old p53−/−NOS2+/+ mice, before the development of any tumor, were stained for apoptotic cells by terminal deoxynucleotidyl-transferase–mediated dUTP nick-end labeling (TUNEL) assay using Apoptag kit (Chemicon) following the manufacturer’s instructions. NIH image analysis software, imageJ,8 was used to count the stained apoptotic and unstained normal cells in 10 randomly selected fields in coded slides at −40 magnification. The assay was repeated twice.
Immunohistochemistry
Paraffin sections, 5-μm thick, from the spleen and thymus of C. parvum–treated and control p53−/−NOS2+/+ mice were incubated with either normal serum (negative controls) or antibodies against monoclonal anti-mouse Ki-67 (Dako), monoclonal anti-mouse NOS2 (BD Biosciences), and polyclonal anti-mouse Fas-L (Oncogene Research). Signals were amplified using biotinylated IgG (Dako), followed by horseradish peroxidase–conjugated avidin-biotin complex (Vector Laboratories); diaminobenzidine tetrahydrochloride (Pierce) was the chromogen. Immunohistochemistry (IHC) was repeated twice with positive and negative controls. Coded slides were evaluated by two independent observers as previously described (14).
Immunoblotting (Western analysis)
Whole protein extracts were prepared from the spleen of either C. parvum–treated or control p53−/−NOS2+/+ mice as described earlier (15). Protein extracts were separated by SDS-PAGE, electrotransfered onto the polyvinylidene difluoride membranes (Immobilon-P; Millipore), and probed with antibodies specific for NOS2 (Cayman), phAkt-Ser 473, and Akt (Cell Signaling). Cells positive for pAkt-Ser473 (293 embryonic kidney cell line) and purified NOS2 (Cayman) were used as a positive control for immunoblotting.
Isolation and quantitation of splenocytes
Spleens were harvested and single-cell suspension was made by placing the spleen in a Filtra-Bag (Fisher Scientific) and gently smoothing the tissue over the mesh. Lymphocytes were counted using a Sysmex KX-21 (Roche) automated cell counter.
Flow cytometric analysis
Flow cytometric analysis was performed on splenocytes according to a previously described procedure (16). The monoclonal antibodies used were CD4 FITC (clone GK1.5), FoxP3 PE (FJK-16s), MHC Class II APC (clone M5/114.15.2), CD11c PE-Cy5.5 (clone N418), CD80 Fitc (clone 16-101A1), CD86 PE (clone GL1), CD40 Fitc (HM40-3), and CD83 PE (clone Michel-17; each from eBioscience). Intracellular FoxP3 staining was performed according to the manufacturer’s suggested protocol. The number of splenic dendritic cells was determined by multiplying the number of splenocytes by the percentage of cells that stain for MHC Class II and CD11c. The number of splenic T-reg cells was determined by multiplying the number of splenocytes by the percentage of cells that stained for FoxP3.
Serum cytokine quantitation by Cytometric Bead Array
Mice were euthanized and blood was obtained by cardiac puncture. Serum was collected from the blood by centrifuging blood in a BD Microtainer SST Tube with Serum Separator (Pulmo Lab) at 10,000 g for 90 s. Serum (50 μL) was analyzed for cytokines using the mouse Cytometric Bead Array (CBA) inflammation kit (BD Biosciences) on a FACScan flow cytometer, affixed with a 488-nm laser (Becton Dickinson Immunocytometry Systems), according to the manufacturer’s suggested protocol.
Results
C. parvum–treatment induced NOS2 expression and accelerated tumorigenesis in p53-deficient mice with intact NOS2
p53−/−NOS2+/+ mice developed tumors rapidly after treatment with C. parvum when compared with controls (P = 0.001; Fig. 1). However, the treatment of p53−/−NOS2−/− mice with C. parvum did not affect the tumor latency compared with the controls (P = 0.41). Also, the C. parvum–treated p53−/−NOS2+/− mice did not show any significant difference in tumor latency, when compared with control mice (P = 0.20). Furthermore, there was no statistically significant difference in tumor latency between C. parvum–treated p53−/−NOS2+/+ and p53−/−NOS2−/− mice. The majority of the tumors were lymphomas with a dominance of T-cell lymphoma irrespective of the treated or control mice. Furthermore, 8- to 10-week-old, C. parvum–treated p53−/−NOS2+/+ mice showed an enhanced expression of NOS2 in the spleen when compared with the controls (P < 0.001; Fig. 2). However, p53−/−NOS2−/− mice did not have any detectable level of NOS1, NOS2, or NOS3 in either C. parvum–treated or control groups (Supplementary Fig. S1). These data indicate a role of NO• in the acceleration of tumorigenesis in C. parvum–treated p53−/−NOS2+/+ mice. Therefore, we further investigated the mechanistic role of NO• on tumorigenesis in C. parvum–treated and control p53−/−NOS2+/+ mice.
Figure 1.

Comparison of survival probability among C. parvum–treated and control p53-deficient mice with either WT, heterozygous, and null NOS2 status, using the Kaplan Meier survival curve. The Wilcoxon test was used to estimate the differences between survival curves of C. parvum–treated and control groups. •, censored animals (moribund mice that were sacrificed but did not show any tumor).
Figure 2.
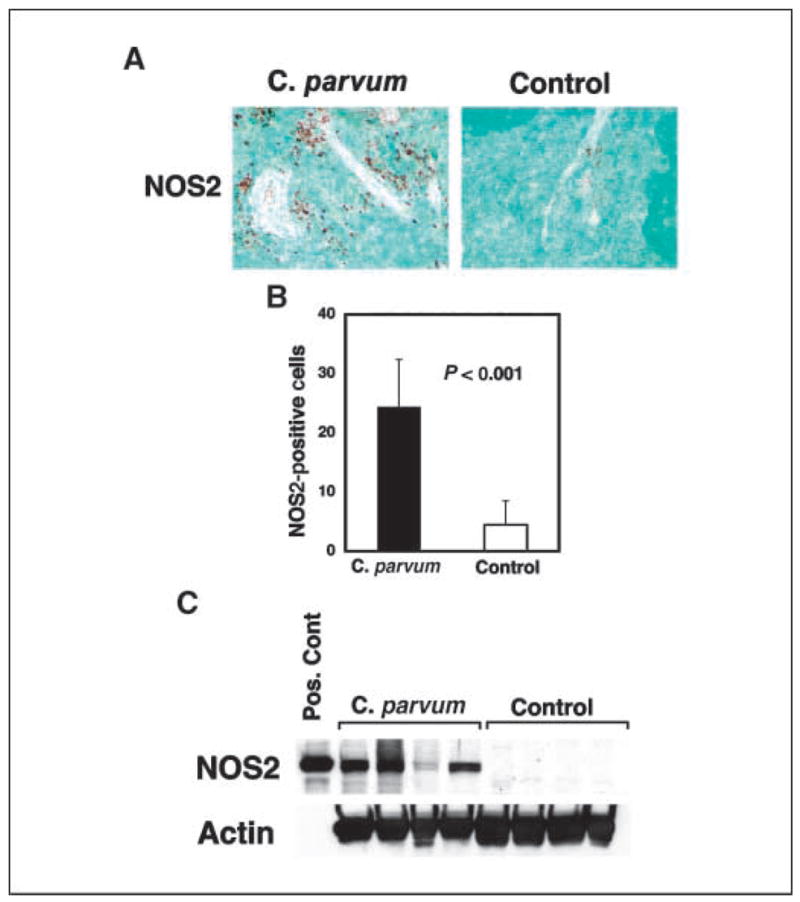
Increased NOS2 expression in C. parvum–treated p53-deficient mice 10 d after treatment. A and B, NOS2 expression in the spleen of C. parvum–treated and control p53−/−NOS2+/+ mice as determined by IHC. C, Western blot analysis of NOS2 in the spleen of C. parvum–treated and control p53−/−NOS2+/+ mice. Pos cont, positive control.
Apoptosis is decreased in C. parvum–treated p53−/−NOS2+/+ mice
To determine the apoptotic index, the spleen and thymus of 8- to 9-week-old, C. parvum–treated and control p53−/−NOS2+/+ mice were analyzed for apoptotic cells by TUNEL assay. The spleen and thymus of C. parvum–treated p53−/−NOS2+/+ mice showed a significantly lower number of apoptotic cells compared with control mice (P < 0.001; Fig. 3A).
Figure 3.

TUNEL assay and immunohistochemistry showing an increased number of apoptotic cells, as determined by staining with antidigoxigenin peroxidase (A), and increased expression of Ki-67 (B) and CD95-L (C) in the spleen and thymus of C. parvum–treated p53-deficient mice with WT NOS2, 10 d after the treatment.
Decreased expression of CD95-L in C. parvum–treated p53−/−NOS2+/+ mice
To study a possible mechanism of decreased apoptosis in C. parvum–treated p53−/−NOS2+/+ mice, we analyzed the expression of CD95-L in the spleen and thymus of 8- to 9-week-old, tumor-free, C. parvum–treated and control mice. C. parvum–treated p53−/−NOS2+/+ mice, showed a lower expression of CD95-L in the spleen (P < 0.001) and thymus (P = 0.13) when compared with the control mice (Fig. 3C).
Increased expression of Ki-67 in C. parvum–treated p53−/−NOS2+/+ mice
To measure the degree of proliferation, we analyzed the expression of Ki-67, a proliferation marker antigen, in the spleen and thymus of 8- to 9-week-old, tumor-free, C. parvum–treated and control p53−/−NOS2+/+ mice. The spleen and thymus of C. parvum–treated p53−/−NOS2+/+ mice showed a higher frequency of Ki-67–immunopositive cells when compared with the control mice (P < 0.01; Fig. 3B). However, C. parvum–treated or control groups of p53−/−NOS2−/− mice did not show any difference in the frequency of Ki-67–immunopositive cells (Supplementary Fig. S2).
Increased expression of phosphorylated Akt-Ser-473 in C. parvum–treated p53−/−NOS2+/+ mice
To determine the NO•-associated increase in the level of pAkt-Ser473, we determined the expression level of both pAkt-Ser473 as well as total Akt in the spleen of C. parvum–treated and control p53−/−NOS2+/+ mice. C. parvum–treated p53−/−NOS2+/+ mice showed an increased expression of pAkt-Ser473 when compared with control mice (Fig. 4). However, both C. parvum–treated and control p53−/−NOS2−/− mice showed very weak expression of ph-Akt ser473 (Supplementary Fig. S1).
Figure 4.

Western blot analysis showing an increased expression of pAkt-Ser473 in the spleen of C. parvum–treated p53−/−NOS2+/+ mice, 10 d after the treatment.
Increased level of IFNγ, tumor necrosis factor α, and interleukin 6 in C. parvum–treated p53−/−NOS2+/+ and p53−/−NOS2−/− mice
To study the immune modulation due to C. parvum treatment and increased NO• production, we analyzed the cytokine levels in serum or in the cultured splenocytes and thymocytes of 8- to 9-week-old, C. parvum–treated and control p53−/−NOS2+/+ and p53−/−NOS2−/− mice. IFNγ, tumor necrosis factor (TNF)α, and interleukin (IL)-6 were significantly higher in C. parvum–treated p53−/−NOS2+/+ and p53−/−NOS2−/− mice compared with the control mice (Fig. 5). However, serum IL-12p70, IL-5, and IL-10 level did not show any difference between C. parvum–treated and control groups (data not shown).
Figure 5.

The quantitation of cytokines in serum and in supernatant from cultured splenocytes and thymocytes of C. parvum–treated and control p53−/−NOS2+/+ and p53−/−NOS2−/− mice using the CBA on a FACScan flow cytometer, 10 d after the treatment. A single-cell suspension of the spleen and thymus was cultured for 48 h before the supernatant was used for analysis.
Increased number of splenocytes and FoxP3(+)T-regulatory cells in C. parvum–treated p53−/−NOS2+/+ mice
To investigate the role of C. parvum and NO• in the alteration of immune profile that can provide a conducive microenvironment for tumorigenesis, we analyzed the splenocytes of 8- to 9-week-old, C. parvum–treated and control p53−/−NOS2+/+ and p53−/−NOS2−/− mice. C. parvum–treated p53−/−NOS2+/+ mice showed an increase in the total number of splenocytes and an increased population of FoxP3(+)T-regulatory cells, which can be immunosuppressive (Fig. 6). p53−/−NOS2−/− mice did not show any difference in these end points among C. parvum–treated and control mice.
Figure 6.

Total number of splenocytes and spleen FoxP3(+) T-reg cells in C. parvum–treated and control p53−/−NOS2+/+ and p53−/−NOS2−/− mice by flow cytometric analysis, 10 d after the treatment, as described in Materials and Methods.
Increased number and maturation of dendritic cells in the spleen of p53−/−NOS2+/+ mice
To further investigate the possible alteration in the immune profile after C. parvum treatment or C. parvum–induced NOS2 expression, we analyzed the total number and maturation of dendritic cells in the spleen and lymph nodes of C. parvum–treated and control p53−/−NOS2+/+ mice. C. parvum–treated p53−/−NOS2+/+ mice showed a significant increase in the total number as well as CD80+, CD86+, CD40+, and CD83+ DC population in the spleen when compared with the control mice (Supplementary Fig. S3). However, there was no difference in the above-mentioned dendritic cell population in the lymph nodes.
Discussion
The role of NO• in cancer is complex and not clearly defined. Interestingly, both protumorigenic and antitumorigenic effects of NO• have been documented (3, 17, 18). A large amount of NO• is generated during inflammation, which then interacts with other reactive oxygen species to generate reactive nitrogen oxide species (19, 20). NO• mediates several signaling pathways and modulates crucial physiologic processes that are necessary for maintaining normal tissue homeostasis, for example, apoptosis, DNA repair, and cell-cycle (3). Prolonged exposure to NO•, as in chronic inflammatory diseases, can induce DNA damage and cellular proliferation, and can alter DNA repair and apoptosis generating a protumorigenic state. Posttranslational modification of key proteins such as p53, RB, or cyclooxygenase 2 is one of the several events that mediate the action of NO• (13–15, 21).
We have shown earlier that a modest increase in the basal NOS2 expression and NO• production can delay the development of cancer in p53−/−NOS2+/+ mice when compared with p53−/−NOS2−/− mice, indicating that basal level of NO• can be protective against spontaneous tumorigenesis in p53-deficient mice (5). In the present study, we tested the hypothesis that a proinflammatory state would increase tumorigenesis. We found an accelerated development of lymphomas and increased expression of NOS2 in C. parvum–induced inflammation in p53−/−NOS2+/+ mice compared with the control p53−/−NOS2+/+ mice. This observation is consistent with the antagonistic, proneoplastic role of NO•, depending on the quantity, concentration, and the microenvironment in tumorigenesis (3, 6).
We then investigated the possible mechanisms of NO•-mediated acceleration of tumorigenesis in p53−/−NOS2+/+ mice after C. parvum–induced inflammation. The process of apoptosis is a major determinant of tumorigenesis (22, 23). NO• possesses both proapoptotic and antiapoptotic functions (24). We observed a decrease in the apoptotic index in C. parvum–treated p53−/−NOS2+/+ mice when compared with the control p53−/−NOS2+/+ mice. NO•-mediated alteration in apoptosis may involve the inactivation of caspase proteases, a key mediator of cell death, by nitrosylation and alterations in ligands of the extrinsic apoptotic pathway (25). NO• has been found to block apoptosis by inhibiting the activation of CD95-L in a dose-dependent manner and is mediated through the alteration of AP1 binding to the CD95-L promoter (26). Consistent with these in vitro findings, we found a decrease in the expression of CD95-L in the spleen and thymus of C. parvum–treated p53−/−NOS2+/+ mice compared with the control p53−/−NOS2+/+ mice. Furthermore, treatment with the proinflammatory cytokine, IFNγ, decreased the expression of CD95-L in microglial cells (27). Also, C. parvum–treated p53−/−NOS2+/+ mice showed a higher frequency of Ki-67–immunopositive cells, an indicator of cellular proliferation, when compared with the control p53−/−NOS2+/+ mice.
The Akt family of serine/threonine kinases mediates signaling pathways that influences various cellular processes including proliferation and survival, and its hyperactivation is associated with human cancers (28, 29). However, recent studies have indicated an antimetastatic role of Akt (30). The activation of Akt is predominantly mediated by its phosphorylation at serine-473 and threonine-308, by phospho-inositide 3 (PI3)-kinase. Increased NO• production leads to the phosphorylation/activation of Akt through PI3-kinase pathway (31). An increased expression of NOS2 is associated with increased expression of phosphorylated Akt in breast cancer (29). In the present study, an increased expression of pAkt Ser-473 in the C. parvum–treated p53−/−NOS2+/+ mice could contribute to the decreased apoptosis and rapid tumor development.
The inflammatory response is mediated by well-orchestrated functions of inflammatory cells and a variety of mediators including cytokines, chemokines, and reactive oxygen and nitrogen species. Unresolved and prolonged inflammation can contribute to the development of cancer (3, 32). Cytokines are crucial mediators of inflammation and can both enhance and inhibit tumor development (33). Proinflammatory cytokines such as IFNγ, TNFα, IL-1β, and IL-6 transcriptionally transactivate NOS2, leading to enhanced NO• production (34). However, in turn, NO• can differentially modify the production/secretion of several different cytokines including TNFα, IL-6, and IL-8 and can modulate the inflammatory microenvironment (35–37). NO• up-regulates TNFα and down-regulates IL-6 in lipopolysaccharide- and IFNγ-treated murine macrophage cell line (35). Furthermore, NO•-mediated modulation in cytokine production is an important event in the process of wound healing (36). Recent studies have implicated IL-6 in both epithelial and nonepithelial tumor development (38, 39). Tumorigenic conversion of mammary stem cells is mediated by IL-6 through the up-regulation of Notch-3 ligand Jagged-1 leading to an increase in the hypoxic response protein carbonic anhydrase IX, aiding the survival of cells in hypoxic condition (40). Furthermore, evidence of an epidermal growth factor receptor/IL-6/signal transducers and activators of transcription 3 signaling cascade in the development of lung adenocarcinoma has been recently reported (41). C. parvum–treatment enhanced the expression of IFNγ, TNFα, and IL-6 in p53−/−NOS2+/+ as well as p53−/−NOS2−/− mice; however, the acceleration of tumor development was found only in the C. parvum–treated p53−/−NOS2+/+ mice. This could be, in part, due to the cytokine-mediated increase in NOS2 and elevated level of NO• in p53−/−NOS2+/+ mice.
T-reg cells are involved in the maintenance of immunologic tolerance by suppressing various immune responses, thus, providing protection from autoimmune diseases (42). However, an increased production of T-reg cells can inhibit the generation of effector T cells and result in the development of tolerance against the tumor (43). An increase in the number of T-reg cells is common in several cancers (43). NO• converts CD4+CD25− cells into CD4+CD25+ T-reg cells, which can counter-balance NO•-mediated Th1 amplification and a potential for autoimmune disease (44, 45). An increase in the FoxP3(+)T-reg cells in C. parvum–treated p53−/−NOS2+/+ mice indicates a role of NO• in the regulation of T-reg cells that may contribute to a protumor shift of the immune environment favoring an accelerated tumor development in these mice.
Dendritic cells are strong antigen-presenting cells that are involved in the initiation of T-cell–dependent immune response and serve as a link between innate and adaptive immune responses (46). The activation of dendritic cell is associated with an anti-tumor response and, therefore, can be used as vaccines in immunotherapy of cancer (47). NO• regulates the maturation of dendritic cell and increases their ability to activate T lymphocytes (48). However, NO• generated by mouse dendritic cell in response to IFNγ and LPS induced apoptosis and inhibited T-cell proliferation (49). Therefore, dendritic cell can have dual role as an inducer or inhibitor of T-cell responses after exposure to NO•. Furthermore, dendritic cell can also expand antigen-specific CD4+CD25+Foxp3+ T-reg cells that can inhibit an immune response (50). Interestingly, we observed an increase in the total number and maturation of dendritic cell population in the spleen of C. parvum–treated p53−/−NOS2+/+ mice with accelerated tumor development. However, the exact role of dendritic cell in tumorigenicity in the present model system is not clear. One hypothesis, consistent with our data, is the dendritic cell–mediated increase in T-reg cells in C. parvum–treated p53−/−NOS2+/+ mice contributes to rapid tumor development.
Because the C. parvum–induced inflammation did not change the latency of tumor development in p53−/−NOS2−/− mice, the rapid tumor development in C. parvum–treated p53−/−NOS2+/+ mice is genetic evidence of a role of NO•. We propose a dose-dependent model of NO•-mediated modulation of tumorigenesis in p53-deficient mice, in which an increase in NO• production under inflammatory conditions can inhibit apoptosis, increase proliferation, and modulate the immune profile, giving rise to an internal milieu that is conducive to the tumor growth.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
We thank Dorothea Dudek-Creaven for editorial assistance and Mohammad A. Khan for the technical advice in immunohistochemical analysis.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Rees DD, Cunha FQ, Assreuy J, Herman AG, Moncada S. Sequential induction of nitric oxide synthase by Corynebacterium parvum in different organs of the mouse. Br J Pharmacol. 1995;114:689–93. doi: 10.1111/j.1476-5381.1995.tb17193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murad F. Shattuck Lecture, Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355:2003–11. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 3.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 4.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–34. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 5.Hussain SP, Trivers GE, Hofseth LJ, et al. Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res. 2004;64:6849–53. doi: 10.1158/0008-5472.CAN-04-2201. [DOI] [PubMed] [Google Scholar]
- 6.Lancaster JR, Jr, Xie K. Tumors face NO problems? Cancer Res. 2006;66:6459–62. doi: 10.1158/0008-5472.CAN-05-2900. [DOI] [PubMed] [Google Scholar]
- 7.Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. 2000;6:347–73. [PMC free article] [PubMed] [Google Scholar]
- 8.Geller DA, Billiar TR. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998;17:7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- 9.orrester K, Ambs S, Lupold SE, et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci U S A. 1996;93:2442–7. doi: 10.1073/pnas.93.6.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambs S, Ogunfusika MO, Merriam WG, Bennett WP, Billiar TR, Harris CC. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc Natl Acad Sci U S A. 1998;95:8823–8. doi: 10.1073/pnas.95.15.8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geller DA, Nussler AK, Di Silvio M, et al. Cytokines, endotoxin, and glucocorticoids regulate the expression of inducible nitric oxide synthase in hepatocytes. Proc Natl Acad Sci U S A. 1993;90:522–6. doi: 10.1073/pnas.90.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiltrout RH, Mathieson BJ, Talmadge JE, et al. Augmentation of organ-associated natural killer activity by biological response modifiers. Isolation and characterization of large granular lymphocytes from the liver. J Exp Med. 1984;160:1431–49. doi: 10.1084/jem.160.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cycloox-ygenase-2. Science. 2005;310:1966–70. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 14.Ying L, Marino J, Hussain SP, et al. Chronic inflammation promotes retinoblastoma protein hyper-phosphorylation and E2F1 activation. Cancer Res. 2005;65:9132–6. doi: 10.1158/0008-5472.CAN-05-1358. [DOI] [PubMed] [Google Scholar]
- 15.Hofseth LJ, Saito S, Hussain SP, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A. 2003;100:143–8. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fogler WE, Volker K, Watanabe M, et al. Recruitment of hepatic NK cells by IL-12 is dependent on IFN-γ nd VCAM-1 and is rapidly down-regulated by a mechanism involving T cells and expression of Fas. J Immunol. 1998;161:6014–21. [PubMed] [Google Scholar]
- 17.Xie K, Huang S. Contribution of nitric oxide-mediated apoptosis to cancer metastasis inefficiency. Free Radic Biol Med. 2003;34:969–86. doi: 10.1016/s0891-5849(02)01364-3. [DOI] [PubMed] [Google Scholar]
- 18.Ridnour LA, Thomas DD, Donzelli S, et al. The biphasic nature of nitric oxide responses in tumor biology. Antioxid Redox Signal. 2006;8:1329–37. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 19.Li CQ, Trudel LJ, Wogan GN. Genotoxicity, mito-chondrial damage, and apoptosis in human lymphoblastoid cells exposed to peroxynitrite generated from SIN-1. Chem Res Toxicol. 2002;15:527–35. doi: 10.1021/tx010171x. [DOI] [PubMed] [Google Scholar]
- 20.Tretyakova NY, Burney S, Pamir B, et al. Peroxynitrite-induced DNA damage in the supF gene: correlation with the mutational spectrum. Mutat Res. 2000;447:287–303. doi: 10.1016/s0027-5107(99)00221-3. [DOI] [PubMed] [Google Scholar]
- 21.Marnett LJ, Wright TL, Crews BC, Tannenbaum SR, Morrow JD. Regulation of prostaglandin biosynthesis by nitric oxide is revealed by targeted deletion of inducible nitric-oxide synthase. J Biol Chem. 2000;275:13427–30. doi: 10.1074/jbc.275.18.13427. [DOI] [PubMed] [Google Scholar]
- 22.Wright KM, Deshmukh M. Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle. 2006;5:1616–20. doi: 10.4161/cc.5.15.3129. [DOI] [PubMed] [Google Scholar]
- 23.Li CQ, Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 24.Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–9. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 25.Kim PK, Zamora R, Petrosko P, Billiar TR. The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol. 2001;1:1421–41. doi: 10.1016/s1567-5769(01)00088-1. [DOI] [PubMed] [Google Scholar]
- 26.Melino G, Bernassola F, Catani MV, et al. Nitric oxide inhibits apoptosis via AP-1-dependent CD95L trans-activation. Cancer Res. 2000;60:2377–83. [PubMed] [Google Scholar]
- 27.Frigerio S, Silei V, Ciusani E, Massa G, Lauro GM, Salmaggi A. Modulation of fasligand (Fas-L) on human microglial cells: an in vitro study. J Neuroimmunol. 2000;105:109–14. doi: 10.1016/s0165-5728(99)00227-1. [DOI] [PubMed] [Google Scholar]
- 28.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 29.Prueitt RL, Boersma BJ, Howe TM, et al. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer. 2007;120:796–805. doi: 10.1002/ijc.22336. [DOI] [PubMed] [Google Scholar]
- 30.Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandau KB, Faus HG, Brune B. Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem Biophys Res Commun. 2000;278:263–7. doi: 10.1006/bbrc.2000.3789. [DOI] [PubMed] [Google Scholar]
- 32.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 34.de Vera ME, Shapiro RA, Nussler AK, et al. Transcriptional regulation of human inducible nitric oxide synthase (NOS2) gene by cytokines: initial analysis of the human NOS2 promoter. Proc Natl Acad Sci U S A. 1996;93:1054–9. doi: 10.1073/pnas.93.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deakin AM, Payne AN, Whittle BJ, Moncada S. The modulation of IL-6 and TNF-α release by nitric oxide following stimulation of J774 cells with LPS and IFN-γ. Cytokine. 1995;7:408–16. doi: 10.1006/cyto.1995.0056. [DOI] [PubMed] [Google Scholar]
- 36.Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and wound repair: role of cytokines? Nitric Oxide. 2002;7:1–10. doi: 10.1016/s1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- 37.Kim YM, Talanian RV, Li J, Billiar TR. Nitric oxide prevents IL-1β and IFN-γ-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1β-converting enzyme) J Immunol. 1998;161:4122–8. [PubMed] [Google Scholar]
- 38.Schafer ZT, Brugge JS. IL-6 involvement in epithelial cancers. J Clin Invest. 2007;117:3660–3. doi: 10.1172/JCI34237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 40.Sansone P, Storci G, Tavolari S, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao SP, Mark KG, Leslie K, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–56. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–7. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 43.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–11. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 44.Niedbala W, Cai B, Liew FY. Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis. 2006;65(Suppl 3):iii37–40. doi: 10.1136/ard.2006.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niedbala W, Wei XQ, Campbell C, Thomson D, Komai-Koma M, Liew FY. Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor β 2 expression via cGMP. Proc Natl Acad Sci U S A. 2002;99:16186–91. doi: 10.1073/pnas.252464599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 47.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 48.Paolucci C, Burastero SE, Rovere-Querini P, et al. Synergism of nitric oxide and maturation signals on human dendritic cells occurs through a cyclic GMP-dependent pathway. J Leukoc Biol. 2003;73:253–62. doi: 10.1189/jlb.0902447. [DOI] [PubMed] [Google Scholar]
- 49.Lu L, Bonham CA, Chambers FG, et al. Induction of nitric oxide synthase in mouse dendritic cells by IFN-γ, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol. 1996;157:3577–86. [PubMed] [Google Scholar]
- 50.Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev. 2006;212:314–29. doi: 10.1111/j.0105-2896.2006.00422.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).