

Abstract
The mobilities of transmembrane adhesion proteins are key underlying physical factors that contribute to neutrophil adhesion and arrest during inflammation. Here we present a novel (to our knowledge) fluorescence recovery after photobleaching system and a complementary analytical model to measure the mobility of the four key receptors involved in the adhesion cascade: L-selectin, PSGL-1, Mac-1, and LFA-1 for resting, spherical, and human neutrophils. In general, we find that β2 integrins (Mac-1, LFA-1) have mobilities 3–7 times faster than rolling associated molecules (L-selectin; PSGL-1), but that the mobilities within each of these groups are indistinguishable. Increasing temperature (room temperature versus 37°C) results in increased mobility, in all cases, and the use of a bivalent antibody label (mAb versus Fab) decreases mobility, except in the case of rolling associated molecules at room temperature. Disrupting the actin cytoskeleton increased mobility except that the highest mobilities measured for integrins (D = 1.2 × 10−9 cm2/s; 37°C, Fab) are not affected by actin poisons and approach the expected value for free diffusion. Although evidence of cytoskeletal hindrance of integrin mobility has been found in other systems, our data suggest such hindrance does not limit bulk integrin diffusion in resting neutrophils over distances and times important for adhesive plaque formation.
INTRODUCTION
Resting neutrophils passively circulate in the bloodstream or roll on endothelial walls. Rolling involves the binding of the specific adhesion molecules L-selectin and PSGL-1 (rolling associated adhesion molecules, or RAAMs) to their endothelial counter-receptors (1,2). At sites of inflammation, many rolling neutrophils will arrest on the endothelial wall due to a sudden firm adhesion mediated by the binding of neutrophil β2 integrins, LFA-1 (αLβ2) and Mac-1 (αMβ2), to endothelial ICAM-1. Firm arrest is a prerequisite for transmigration into extravascular space where neutrophils police tissue for the presence of infectious agents. Although much is known about the biochemical cues and signaling pathways responsible for the transition from rolling to firm adhesion, many of the physical parameters that ultimately determine how firm adhesion occurs remain poorly characterized.
The process of rolling is partly facilitated by the strategic positioning of key adhesion molecules on the resting neutrophil's surface. This surface can be described as a topologically complex ruffled membrane (3) containing numerous peaks and valleys. Electron microscopy (EM) studies on fixed cells have shown that RAAMs are concentrated on membrane peaks (4–8) where they can readily access the endothelial surface during rolling. By contrast, EM studies of β2 integrins suggest that these receptors are sequestered in membrane folds where their interaction with the endothelium should be minimal (5,9). These static images suggest that rolling is optimized by the position of RAAMs at membrane peaks and that firm arrest requires membrane rearrangement to expose β2 integrins to the endothelial surface.
There are several dynamic mechanisms that might allow β2 integrins to engage the endothelium and create firm adhesions. First, neutrophils deform as they roll along the endothelial surface (10,11). Such deformation might be sufficient to flatten the neutrophil surface topography and bring integrins into close contact with endothelium. Similarly, shape changes occurring upon neutrophil activation could flatten the surface and create the opportunity for integrin-based adhesion. Finally, it is possible that the tethers confining β2 integrins to valleys are dynamic so that a small number of receptors diffuse to regions of close contact and form bonds with the endothelium. Regardless of the mechanism of initial bond formation, the size and strength of adhesion are thought to grow through the recruitment of β2 integrins to the contact zone (12), and so the mobility of β2 integrins in the plasma membrane is key to determining the kinetics of firm adhesion. Because the conversion of β2 integrins from low to high affinity states is also essential for firm adhesion (13), it is likely that a combination of high integrin number and high ligand affinity gives rise to neutrophil firm adhesion.
Investigators have measured the mobility of β2 integrins by single particle tracking (SPT) and fluorescence recovery after photobleaching (FRAP). In SPT, the diffusive motions of 40–1000 nm beads coupled to integrins through antibody linkages are followed with high-resolution video tracking (14–18). Depending on the size of the tracer bead and the number of Fab and IgG molecules bound, SPT measurements may be tracking more than one integrin. Because of this, the measurements made using SPT can be exquisitely sensitive to transmembrane anchoring of integrins to cytoskeleton. Indeed, increases in integrin mobility have been measured by SPT after cytoskeletal disruption and cellular activation (14,18). However, because only a single cytoskeletal tether is sufficient to slow the motion of a diffusing particle, SPT does not provide a reliable estimate of the most mobile integrins on the cell. Because particle movements in SPT are assumed to be in a plane, investigators have typically studied lateral mobility in cell membranes of flat cells. To address this issue with spherical leukocytes, researchers have used poly-L-lysine-coated coverslips to adhere and flatten the cells before particle tracking (14–16). This approach is problematic for neutrophils, which undergo activation and initiate crawling on adhesive surfaces. Thus, although SPT offers a quantitative and qualitative measure of lateral mobility, its use on resting neutrophils is not without difficulty.
Another technique for measuring lateral mobility is FRAP. In this technique, the entire cell surface is typically labeled with a fluorescent antibody to a specific target molecule (19,20). Fluorescence is locally bleached using high intensity excitation, and the evolution of fluorescence is monitored to infer lateral mobility. Because of the limits of focused laser dimensions, FRAP experiments measure an effective diffusion coefficient over micron length scales. This is particularly useful for the study of neutrophil adhesion where the contact zone between a neutrophil and endothelium is also on the order of microns. To our knowledge, the only published example of a FRAP-based measurement of integrin mobility on human neutrophils is by Mukherjee et al. (21), who report a diffusion coefficient for Mac-1 but no other molecules. There have also been several other studies that have investigated the lateral mobility of integrins on neutrophil-like cells (14,16,22); however, no studies have investigated L-selectin or PSGL-1 lateral mobility.
Although FRAP may be well suited to measuring lateral mobility over micron length scales, its typical use is in measuring lateral mobility in planar surfaces. Adaptation of existing methods of analysis is needed for application to neutrophils. The first challenge in performing FRAP on a resting neutrophil is accounting for the spherical geometry. Typical FRAP systems pass a high intensity laser through the objective of an inverted microscope to create a circular photobleached spot. For cells spread over a coverslip surface, fluorescence recovery can be reasonably interpreted using models of diffusion in a plane (20); however, for curved surfaces, the analysis is considerably more complex (23). For a spherical cell, the bottom surface of the cell is most accessible (21) but may be deformed or adherent to the supporting coverslip. Additionally, the laser beam likely extends through the cell to photobleach molecules on the top surface. If these additional bleached molecules are not accounted for, the analysis will overestimate the immobile fraction. With these challenges to study molecules on a spherical surface, it is no surprise that like SPT studies, most FRAP measurements of receptor mobility have been made on artificially flattened cells (14–16,18).
Here we overcome the technical challenges with a new, to our knowledge, experimental and analytical approach to FRAP measurements on a spherical surface. We measure the lateral mobility of key adhesion molecules, LFA-1, Mac-1, L-selectin, and PSGL-1, under various experimental conditions. Specifically, we explore temperature, label valency, and cytoskeletal disruption. Our results suggest that under all conditions, RAAMs are significantly slower to diffuse than β2 integrins. Additionally, β2 integrins, when labeled with Fab fragments and imaged at 37°C, diffuse (D = 1.2 × 10−9 cm2/s) at rates just a factor of three less than freely diffusive proteins in membrane blebs.
METHODS
Neutrophil preparation and labeling
Two microliters of whole blood obtained via finger stick was diluted into 80 uL Hanks' balanced salt solution without Ca2+ or Mg2+ plus 10 mM HEPES. To visualize adhesion molecule localization and dynamics, cells were labeled with 3uL (1ug) fluorescent monoclonal antibodies (mAbs). All antibodies were labeled with the AlexaFluor 488 or 546 (AF488 and AF546) antibody labeling kits (Molecular Probes, Eugene, OR). All neutrophils adhesion molecules were visualized with the following mouse monoclonal antibodies: LFA-1, anti-CD11a (HI111, eBioscience, San Diego, CA); Mac-1, anti-CD11b (ICRF44, eBioscience, San Diego, CA); L-selectin, anti-CD62L (DREG-56, eBioscience); and PSGL-1, anti-CD162 (PL1/3E2-25-5, Ancell, Bayport, MN). Monovalent Fab fragments were made from HI111 and PL1/3E2-25-5 using a Pierce Mouse IgG1 Fab and F(ab′)2 Preparation Kit (Product No. 44880, Pierce, Rockford, IL). Cells were labeled at room temperature for 15 min and washed three times for 5 min at 170 g. Cells were handled according to protocols used for more than 20 years in the Waugh laboratory for studies on passive neutrophils. With these procedures, for example, we've established that LFA-1 is primarily in the inactive state and exhibits minimal adhesion to ICAM-1 (24).
Some cells were also treated with varying concentrations of cytochalasin D (100 nM, 250 nM, 500 nM, 1 μM, and 2 μM) in dimethylsulfoxide. Dimethylsulfoxide concentrations were kept below 0.1% for all treatments, and carrier control studies showed no significant difference in mobility measurements from untreated cells (n = 14). Cells were treated for a minimum of 15 min with cytochalasin D at room temperature after antibody labeling. We found short exposures of higher concentrations (1 or 2 μM) produced similar effects to 250 nM, but chose lower concentrations at longer times because it was more reliable and reproducible. Cytochalasin D and other routine reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Once labeled with antibody, cells were imaged within 60 min and then discarded. The total time from finger prick to final measurement per experiment was kept to <2 h. We also performed experiments at 37°C using a heated stage and heated oil objective. The temperature was tuned on the stage and objective to produce a near uniform 37°C temperature measurement on the coverslip using multiple thermocouples submerged in media. In experiments conducted at 37°C, the cells were allowed 20 min to adjust from room temperature before measurements were taken. Since experimentation was performed on whole blood samples to minimize neutrophil activation and processing, neutrophils were identified by eye with differential interference contrast (DIC) based on a segmented polymorphonuclear appearance. Wright's stain confirmed that ∼95% of white cells identified with this approach were in fact neutrophils in control experiments.
Antibody control experiments
Because surface proteins can be internalized by membrane trafficking, there was some concern that fluorescent antibodies might be associated with internal stores just below the membrane surface. To guard against this possibility, we labeled surface Mac-1 with anti-CD11b (ICRF44) tagged to AF488. After three washes, we labeled with a noncompetitive anti-CD11b (Bear1, Abcam, Cambridge, MA) tagged to AF546 and washed again. Identical green and red fluorescent images were obtained after the final wash, suggesting the Mac-1 molecules remained on the cell surface throughout the labeling procedure. We also performed a complementary set of competitive antibody tests with ICRF-AF488 labeled cells exposed to ICRF-AF546. More than 95% of the cells fluoresced green with no red signal, indicating antibody saturation as well as no upregulation of Mac-1 during labeling procedures. Reversing the sequence of antibody labels gave similar results. These controls confirm that labeled receptors are associated with the plasma membrane and not internal stores.
We performed several control experiments that attest to the specificity of fluorescence labeling with mAbs. In addition to general nonspecific binding, we were aware of the potential for mAbs to fluorescently label neutrophil Fc receptors (25). In one study, we quantified the fluorescence obtained using our standard labeling procedures to the fluorescence obtained when these procedures were preceded by incubation of cells with an unlabeled, nonspecific human IgG antibody to block Fc receptors. We performed these experiments for both CD11a and CD62L and noted no significant changes in average fluorescence or the fluorescence distribution when the blocking step was included. We also sought to directly label Fc sites using a fluorescein isothiocyanate (FITC)-tagged nonspecific mouse IgG antibody (Clone 679.1Mc7, IOTest, Marseille, France). When cells were labeled with both a FITC-tagged nonspecific mouse mAb and anti-CD11a/ AF546, there was a regular CD11a fluorescent signal and surface distribution in the AF546 channel and no signal in the FITC channel. Together these studies suggest that signals from Fc receptor labeling and any other forms of nonspecific labeling are insignificant.
FRAP
After antibody labeling, cells were transferred to HBSS supplemented with 1% (v/v) heat-inactivated fetal bovine serum (Hyclone, Logan, UT) and placed in custom cell observation chambers. We found that 1% fetal bovine serum prevented glass activation of neutrophils by the coverslip floors of observation chambers, but allowed a small degree of adhesion that prevented cells from rolling during measurements. Observation chambers were placed on the stage of an inverted Zeiss Axiovert 200 m with a 1.45 NA 100× objective (Zeiss, Thornwood, NY). All preparation steps and measurements were done at room temperature. Before photobleaching, a cell equator was brought into focus using epi-illumination. Epi-illumination utilized a 100 W mercury arc lamp attenuated 75% with a neutral density filter. Cells were photobleached using a 5 mW laser beam from a solid state Coherent Sapphire 488 Laser (Coherent, Santa Clara, CA) reshaped by a cylindrical lens so that a rectangular band ∼2.5 μm × 10 μm was focused at the objective focal plane. A motorized mirror was used to switch between epi-illumination and laser light paths, which were both directed into the epi-illumination optical path of the microscope. Photobleaching duration was typically 2 s. Interlaced fluorescence and DIC time-lapse images were collected before and after the photobleaching burst using a Cooke Sensicam EM (Romulus, MI). Photobleaching recovery was followed over different timescales depending on the rate of recovery (varying from Δt = 1 s to Δt = 10 s for a minimum of 10 frames after photobleaching). The total fluorescence from each image was compared to subsequent frames, and any inadvertent photobleaching resulting from observation of the cells was corrected for by a linear interpolation. In practice, the signal decreased by much less than 10% over an image set. Small cell movements were corrected for by registering the center of the photobleach band in digitized fluorescence profiles (see Analysis). Any cells seen rolling, rotating, or activating in the DIC images were discarded. All microscope control, image acquisition, and processing were performed with custom MATLAB (The MathWorks, Natick, MA) algorithms.
Cell volume reconstruction
Twenty-five epi-fluorescence slices, spaced 500 nm apart, were used to construct a three-dimensional image of L-selectin on the neutrophil surface. Image acquisition was performed in <7 s using a motorized z axis. In MATLAB, an isosurface was used to connect voxels of similar fluorescence intensity in three dimensions. Due to the voxel size of 500 nm tall × 128 nm × 128 nm, there is less information in the vertical direction leading to reconstructive artifact at the top and bottom of the sphere, where the slope of the surface is smallest. However, the reconstruction before and after photobleaching illustrates the clean and rectangular cross section of the band (Fig. 1 A).
FIGURE 1.

Photobleaching a spherical resting neutrophil. (A) Three-dimensional reconstruction of a neutrophil fluorescently labeled for L-selectin before and after photobleaching shows a nearly uniform rectangular bleach cross section in each focal plane. For purposes of analysis, the optical axis is designated as y and the focal plane as x-z. (B) Rotated orientation illustrates the coordinate system for analysis with symmetry about the z axis. φ is the only direction in which fluorescence gradients exist. (C) MC simulations model diffusion of fluorescent “molecules” along φ that step forward or backward during each time step. (D) Width and area of simulation elements in φ is a function of the sine of φ, making it more likely that a molecule will diffuse toward the equator than toward the pole (see text). (E) Postphotobleach image with the coordinate system overlaid in the same orientation as B.
ANALYSIS
Photobleaching experiments and data acquisition
To perform photobleaching recovery experiments on the surface of the resting neturophil, we developed what to our knowledge are new experimental and analytic frameworks suited to the spherical geometry of these cells. Experimentally, we sought to photobleach an equatorial band around the neutrophil. This geometry exploits spherical symmetry so that recovery occurs equally from the two poles along lines of longitude and reduces the mathematical description of recovery to one dimension. We achieved an equatorial bleach band by inserting a cylindrical lens in the laser path to reshape the laser beam into a rectangular band that measures 2.5 um × 10 um at its focus. Three-dimensional surface reconstruction of a photobleached cell confirmed that focusing the laser at the neutrophil's mid plane resulted in an equatorial bleached band of approximately uniform width (Fig. 1 A).
Fluorescence recovery was monitored at the neutrophil's midplane. This imaging plane intersects the neutrophil at its widest cross section along the microscope's optical axis (Fig. 1) and it minimizes out-of-plane fluorescence contributions in our nonconfocal imaging system. More importantly, this imaging plane defines a meridional plane that intersects the equatorial bleach band perpendicularly, so that the angular coordinate in our images becomes the primary direction of fluorescence recovery from pole to equator. Thus, our experiments naturally align with a spherical coordinate system for analysis in which the polar angle φ lies between the axis of symmetry (z axis) for the bleached cell and a vector identifying positions on the membrane surface (Fig. 1 E).
We digitized membrane fluorescence using a custom MATLAB algorithm that created 100 equally sized boxes around the cell periphery (Fig. 2). The boxes were centered on the plasma membrane and the average intensity in each box was calculated to create a fluorescence profile at every time point. Note that because the bleach band bisects the cell, there are two bleached membrane spans per image (Fig. 2). Thus, each circular fluorescence profile was divided into two linear profiles representing a polar coordinate range between φ = 0 and φ = 180° and with a bleached span roughly centered in the profile (Fig. 2 C). Fluorescence minima in the profiles were aligned in the time sequence to eliminate slight shifts in cell position. Cells that exhibited obvious rolling or activation were not included in our analysis.
FIGURE 2.

Fluorescence intensity analysis after photobleaching. (A) Characteristic epi-fluorescence images labeled with mAb for LFA-1 at 23°C on a resting neutrophil following photobleaching are shown. (B) Perimeter of the cell at the midplane is traced in MATLAB to determine fluorescence signal at each position of φ. An algorithm calculates average curvature from neighboring points and redraws 100 boxes of 0.5 μm thickness (fewer are shown in the figure for clarity). (C) Fluorescence signal profiles are shown for the above images. The initial fluorescence intensity values at each position are used to populate a MC simulation to model diffusion. Scale bar is 5 μm.
Analytical model
We developed numerical and analytic models to interpret fluorescence recovery dynamics. Analytically, we solved the ideal diffusion problem implied by our experimental system. The mathematical statement of this problem is given by the equation governing diffusion on the surface of a sphere:
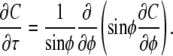 |
(1) |
Here φ is the usual spherical polar angle, and τ is a dimensionless time, defined by τ = D t/R2, where D is the surface diffusivity, t is the experimental time and R is the radius of the sphere. The initial fluorescence profile is defined as
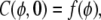 |
(2) |
where
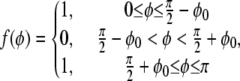 |
(3) |
where φo is the half-width of the photobleached band centered at φ = π/2. In the Appendix, we solve this initial value problem using separation of variables and obtain a series solution involving Legendre polynomials.
A major difficulty with the analytic solution is its use of an idealized initial profile that is not realized in practice. Because the laser photobleach profile is not a “notch” function and because some receptor diffusion occurs during the 2 s bleaching interval, the initial fluorescence condition is more complex than assumed by our analytical solution. For these reasons we developed a one-dimensional Monte-Carlo (MC) model that allowed us to simulate diffusion starting from the initial measured fluorescence profile.
Monte Carlo simulation
Following the example of Dunn et al. (26), we developed a MC simulation of diffusion by assuming that each gray scale value corresponded to a diffusible fluorescent “molecule”. A 12-bit camera was used to measure the intensity of fluorescence around the equatorial cell membrane. Peak intensities were typically between 2000 and 3000, and so simulations were densely populated with diffusible units. The starting frame of the simulation was populated with interpolated intensities from the earliest postbleach profile. To account for the variable area of the nonrectangular grid elements along φ (Fig. 1), the image fluorescence was first transformed by multiplying by sine φ. Fluorescent units diffused in φ over a 200 point spatial grid spanning a hemisphere (φ = 0 to φ = π). Each fluorescent unit was made to step a distance of one grid point with each iteration of the simulation. To account for the narrowing of the elements toward the poles, the probabilities of stepping toward the equator, Pe, or pole Pp are unequal and defined locally by
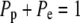 |
(5) |
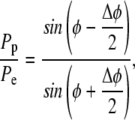 |
(6) |
where Δφ is the interval between grid points (π/200). The likelihood of moving to a neighboring element is dependent on the relative width of the boundaries. It is more likely for a “molecule” to cross the larger boundary than the smaller boundary during a time step. As shown in the schematic (Fig. 1 C), a “molecule” in A3 is more likely to move to A4 than A2 because the boundary (B34) between A3 and A4 is larger. Since there is symmetry about the z axis and no θ gradients exist, there is no net movement of “molecules” in θ. (Note: if the likelihood of moving forward or backward in φ were equal, the number of “molecules” in every element at equilibrium would be the same, yet we would obtain the nonphysical result that the surface concentrations would be unequal.)
The diffusion coefficient is calculated by determining the number of simulation steps needed to mimic the fluorescence profiles of the recorded data in a time-lapse series. Beginning with the initial fluorescence profile, simulations were carried out for ∼1000 steps. The time interval represented by a simulation step was systematically varied until a minimum error between the experimental and simulated fluorescence profiles was found. The diffusion coefficient is related to the optimum time interval Δtopt by
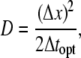 |
(7) |
where Δx is the linear spacing between grid points. The value of Δx varies with the size of a cell perimeter, but was typically ∼130 nm.
RESULTS
Validation of Monte Carlo analysis
The MC simulation method was validated using several approaches. First, we confirmed that model surfaces with uniform fluorescence distributions remained uniform throughout a simulation. In this check, we populated each element as the inverse of sine φ (Fig. 1) to create a uniform density. We then ran the simulation for thousands of iterations and observed no deviation from a constant density across the surface. This check ensures against implementation errors that might create or destroy “molecules” as a simulation progresses.
Next, we compared numerical MC simulations against analytical solutions with an idealized initial fluorescence profile (Eqs. 1–3). In this case, we created an initial condition with a cell circumference of 25 μm and a bleach band width of 3 μm. Within the simulated photobleach region, all molecules were bleached at the start of the simulation. We modeled recovery with an iteration time and step size corresponding to a diffusion coefficient of 1 × 10−10 cm2/s. We compared the MC simulation to the analytical solution using identical parameters at 60, 120, and 240 s. There was excellent agreement at all times with only slight deviations at the edges of the simulation area (Fig. 3 A). Deviations at the boundaries are due to the small element size at the poles of the sphere in the MC simulation and the resulting small number of ‘molecules’ in those elements. Statistical variations due to small numbers at these extreme regions led to some variation in the simulated fluorescence signal. To avoid artifacts from these fluctuations, we confined our calculation of recovery rates to the center bleached region where these fluctuations were minimal. Next, the MC simulated recovery was matched to the analytical solution at 60, 120, and 240 s by a least-square regression scheme (Fig. 3 B). As described above, the regression routine determined the best fit iteration time. Additionally, we compared MC simulations with immobile fractions. Consistent with the analytical model, the best fit occurred for no immobile fraction. We then replotted the curves as a function of diffusion coefficient, illustrating that the best fit was 1.0 × 10−10 cm2/s, matching the analytical input (Fig. 3 C). Additionally, MC simulations of the analytical solution with varying immobile fractions converged with the predefined diffusion coefficient and appropriate immobile fraction (data not shown).
FIGURE 3.

Fluorescence recovery on a spherical surface is modeled with a MC simulation. (A) Fluorescence recovery profiles of an analytical solution and a numerical MC model have strong agreement. To validate our numerical method, we compared the recovery profiles of a MC simulation to an analytical solution of a model cell (see Appendix). A model cell with a 25 μm circumference is bleached by a rectangular 3 μm wide band using the geometry defined in Fig. 1. Using the analytical solution, recovery is drawn at 60, 120. and 240 ss for a diffusion coefficient of 1 × 10−10 cm2/s (solid line). A MC simulation was run using the same initial conditions and shows strong agreement at the same time points (dashed line). (B) Least-squares fit determines the best iteration time for the MC simulation of the model cell. In this case, the MC simulation was run for 1000 steps to create an iteration library. Assuming the time step varies from 0.1 to 4 s, the simulation profiles are compared to the iteration library and a least-squares fit of the normalized error is used to select the best iteration time and immobile fraction. In the case of the model cell, MC analysis showed a best-fit iteration time of 0.87 s with no immobile fraction as expected. (C) Best-fit curves were redrawn as a function of diffusion coefficient. Consistent with the analytical model, the best fit occurred at D = 1.0 × 10−10 cm2/s.
Application to experiments
Analytic solutions to FRAP experiments require use of an idealized initial profile that is not realized in practice. To overcome this and other analysis hurdles, we used our MC model to simulate and measure the diffusion coefficient of adhesion molecules on the neutrophil surface. After confirming that our MC simulation matched analytical models, we applied this model to interpret FRAP experiments on human neutrophils. The MC simulation is run and sampled at higher spatial resolution than we can obtain with optical images. To minimize high frequency noise in the MC simulation and to give similar spatial resolution to the fluorescent images, we ran both sets of data through a fifth-order low-pass Butterworth filter with a 500nm−1 cutoff. This minimized least-squares regression artifacts due to signal noise.
For each of the four receptors under study, we compared the recovery profiles to simulated data with varying immobile fractions from 0 to 40%. For each sample, we varied the percentage of the simulated “molecules” that were fixed in their original postphotobleach position. The remaining “molecules” randomly diffused in the MC simulation. We applied least-squares routines to identify the simulation iteration time and immobile fraction that most closely matched the experimental results. For almost all cases, the assumption of no immobile fraction produced the best fit (Fig. 4, A–D). For this reason, we present all data in the following sections assuming a fully mobile recovery. Note that this assumption does not imply that the molecules under study diffuse through the plasma membrane without hindrance. If the molecules interact transiently with the cytoskeleton or other “immobile” elements as is traditionally assumed, the appearance of a single mobility will follow if these interactions are short-lived compared to the timescale of experiments. A single phase of recovery would also result if the receptors were permanently bound to larger elements that are diffusing. To show the strong agreement between the fluorescence data and the best-fit MC simulation, we present the recovery kymographs of a typical data-simulation pair (Fig. 4, E and F).
FIGURE 4.

Least-squares fit determines MC iteration time and immobile fraction. Characteristic least-squares error analysis is shown for (A) CD11a, (B) CD11a treated with cytochalasin D, (C) CD62L, and (D) CD62L treated with cytochalasin D. In all cases, the best fit of the data was found without an immobile fraction, validating our use of this assumption. (E and F) Kymograph comparison of data and best-fit MC simulation. Time-lapse fluorescence recovery profiles of one band on a cell labeled with mAb for L-selectin were compiled and shown in a pseudo-color surface kymograph. The MC simulation profiles that produced the best-fit iteration time were compiled into a kymograph for comparison.
β2 integrins are more mobile than RAAMs on the resting neutrophil
In our first experiments, we set out to investigate the differences in lateral mobility between the two classes of adhesion molecules on the resting neutrophil. The RAAMs, L-selectin, and PSGL-1 are predominately responsible for mediating neutrophil rolling, whereas the β2 integrins, LFA-1, and Mac-1 help mediate firm adhesion. When referring to the \measurements of the antibody-labeled molecules, we will refer to each by the CD designation of the antibody target: CD11a (αL subunit of LFA-1), CD11b (αM subunit of Mac-1), CD62L (L-selectin), and CD162 (PSGL-1). We first measured the lateral mobility of these four molecules at room temperature using fluorescent mAbs. Using mAb labels, we found that the β2 integrins, (anti-CD11a and anti-CD11b) have mobilities three times greater than the RAAMs (anti-CD62L and anti-CD162); Table 1, Fig. 5 A.
TABLE 1.
Effects of antibody valency and temperature on mobility
| Temp (°C) | Label | n | D (10−10 cm2/s) | Mean ± SE (10−10 cm2/s) | |
|---|---|---|---|---|---|
| CD11a | 23 | mAb | 16 | 1.3 | 0.2 |
| Fab | 15 | 3.7 | 0.4 | ||
| 37 | mAb | 13 | 5.0 | 0.7 | |
| Fab | 11 | 12.1 | 2.2 | ||
| CD11b | 23 | mAb | 15 | 1.1 | 0.2 |
| CD62L | 23 | mAb | 15 | 0.5 | 0.1 |
| Fab | 10 | 0.3 | 0.1 | ||
| 37 | mAb | 19 | 1.1 | 0.3 | |
| Fab | 22 | 1.8 | 0.2 | ||
| CD162 | 23 | mAb | 20 | 0.3 | 0.1 |
FIGURE 5.

β2 integrins have significantly greater lateral mobility than RAAMs. (A) Lateral mobility of LFA-1 (CD11a), Mac-1 (CD11b), L-selectin (CD62L), and PSGL-1 using fluorescently tagged mAb at 23°C. There was no significant difference between CD11a and CD11b and no difference between CD62L and CD162. Grouped as classes of molecules, β2 integrins are statistically more mobile than RAAMs. There are also significant differences between each of the β2 integrins and either of the RAAMs (p < 0.01). (B) Effect of antibody valency and temperature on CD11a mobility. The measured lateral mobility of CD11a increases significantly when the label is changed from divalent mAb to monovalent Fab. There was also a significant increase in lateral mobility when increasing the temperature from 23°C to 37°C for both mAb and Fab labels. (C) Effect of antibody valency and temperature on CD62L mobility. Although there is no statistical difference between mAb and Fab labels for CD62L at 23°C, there is significant difference at 37°C. Like CD11a, Fab mobility is significantly greater at 37°C compared to 23°C.
Effects of temperature and label valency
We next set out to determine the effect of antibody valency and experimental temperature on measured diffusion coefficients. Compared to SPT beads, which can have several antibodies on their surfaces, fluorescently tagged mAbs are less likely to induce clustering. However, even with mAbs, it is possible to cross-link two target molecules to create artificially large structures that may diffuse differently than native receptors. Thus, to test for effects of label valency, we used Fab fragments that can only bind one target molecule. These Fab fragments were covalently conjugated with the same Alexa Fluor dye as mAbs, but because the Fabs are smaller molecules, fewer fluorophores were bound to each molecule and the fluorescence signal was less bright. This created a technical barrier to testing the diffusion of CD11b and CD162 because the signal intensity for these molecules was already low when using the mAb label. However, the Fab label brightness for CD11a and CD62L was sufficient to perform reliable measurements.
For CD11a, Fab labeling resulted in a >2 times increase in lateral mobility at both room temperature and at 37°C compared to mAb at the same temperatures (Table 1, Fig. 5 B). For CD62L, there was no significant change in mobility with Fab labels at room temperature, but there was a 1.6 times increase in mobility at 37°C (Table 1, Fig. 5 C). Similar diffusion coefficient differences have been previously reported between mAb and Fab labeling (27). Regarding experimental temperature control, increasing the temperature from 23°C to 37°C resulted in an ∼3 times increase in diffusion coefficients under each condition, for both CD11a and CD62L. A similar difference (>2 times) between diffusion coefficients at room temperature and 37°C was reported for protein in C3H/10T1/2 cells (28). Our results confirm the significance of both experimental temperature and label valency on measured lateral mobility.
Effects of cytochalasin D
Both RAAM's and β2 integrins are thought to be physically linked to the actin cytoskeleton in resting neutrophils (7,29,30). For this reason, we tested the effects of disrupting the actin cytoskeleton on the mobility of CD11a and CD62L at 37°C. First, we tested a range of concentrations of the actin disrupting agent, cytochalasin D. At 100 nM cytochalasin D, both cell morphology and CD11a and CD62L lateral mobility were not obviously perturbed. At 250 nM cytochalasin D, we began to see slight morphological changes in some cells (Fig. 6), but the majority of cells appeared normal. This level of treatment resulted in at least a two times increase in lateral diffusion of CD62L labeled with either mAb or Fab at 37°C (Fig. 6 A, Table 2). At 250 nM cytochalasin D, CD11a labeled with mAb also increased mobility by a factor of two, but there was no significant increase for cells labeled with anti-CD11a Fab (Fig. 6 B, Table 2). Cells treated with 500 nM and higher concentrations of cytochalasin D showed a more dramatic morphological change in almost all cases. At 500 nM concentration, diffusion coefficients did not further increase and in most cases retreated back to 100 nM or control levels. At 2.0 μM, dramatic morphological changes were seen and the distribution of fluorescence became highly clustered and difficult to model with our MC analysis. This dose response suggests that a low concentration of cytochalasin D (250 nM) disrupts the cytoskeleton enough to release some restraints on lateral diffusion. Higher concentrations result in severe disruption of the actin cytoskeleton leading to the formation of surface aggregates and decreasing mobility of surface molecules.
FIGURE 6.

Dose-dependent response of cytochalasin D on lateral mobility at 37°C. (A) Effects of cytochalasin D on CD62L mobility. Cells labeled for CD62L with mAb display maximal mobility at 250 nM cytochalasin D. Lateral diffusion coefficients for CD62L labeled with Fab showed a similar trend with the maximum lateral mobility at 250 nM cytochalasin D. Characteristic images of cells labeled for CD62L with Fab show that increasing concentrations of cytochalasin D causes morphological changes. At concentrations >500 nM, cells became highly irregular in shape and label-internalized. (B) Effects of cytochalasin D on CD11a mobility. Cells labeled for CD11a with mAb showed minimal change in lateral mobility at 100 nM cytochalasin D, but a significant increase in lateral mobility with 250 nM cytochalasin D (p < 0.05). Mobility decreased at higher concentrations of cytochalasin D. Lateral diffusion coefficients for CD11a labeled with Fab were the highest measured and did not increase further with cytochalasin D. Characteristic images of cells labeled for CD11a with Fab show that increasing concentrations of cytochalasin D causes morphological changes. In all experiments, cells were pretreated with varying concentrations of cytochalasin D for 15 min and then imaged at 37°C. Scale bar is 5 μm.
TABLE 2.
Effects of cytochalasin D on mobility
| Label | Cyto D (nM) | n | D (10−10 cm2/s) | Mean ± SE (10−10 cm2/s) | |
|---|---|---|---|---|---|
| CD11a | mAb | 0 | 13 | 5.0 | 0.7 |
| 100 | 12 | 5.0 | 1.2 | ||
| 250 | 14 | 9.8 | 2.3 | ||
| 500 | 9 | 4.2 | 0.6 | ||
| Fab | 0 | 11 | 12.1 | 2.2 | |
| 100 | 9 | 15.2 | 2.7 | ||
| 250 | 9 | 14.3 | 2.1 | ||
| 500 | 7 | 16.4 | 3.7 | ||
| CD62L | mAb | 0 | 19 | 1.1 | 0.3 |
| 100 | 12 | 1.2 | 0.1 | ||
| 250 | 11 | 2.8 | 0.5 | ||
| 500 | 14 | 2.2 | 0.5 | ||
| Fab | 0 | 22 | 1.8 | 0.2 | |
| 100 | 10 | 2.1 | 0.3 | ||
| 250 | 10 | 4.3 | 1.7 | ||
| 500 | 12 | 2.7 | 0.4 |
Because the mobility of Fab-labeled CD11a at 37°C was the highest value we had measured and did not increase with cytochalasin D treatment, we conducted further FRAP experiments with lipid labels to ensure that the CD11a Fab values were within the dynamic range of our system. We made four FRAP measurements of the lipid label probe octadecyl rhodamine B (ORB). Rapid recovery of this lipid label produced a diffusion coefficient (D = 4 × 10−8 cm2/s) more than an order of magnitude greater than anti-CD11a Fab at 37°C. These results confirmed that all our FRAP measurements on adhesion molecules were well within the detection limits of our system.
DISCUSSION
During the early stages of the inflammation cascade, neutrophils roll on, and then firmly adhere to endothelium. Although the adhesion molecules and several signaling pathways involved in the transition from rolling to firm adhesion are known, the underlying physical mechanisms remain unclear. A common view is that RAAMs facilitate rolling via their localization to the tips of neutrophil microvilli (1,2), whereas integrins are prevented from interacting with the endothelial surface because they exist in low affinity conformations and are bound to cytoskeletal linkages that sequester them away from microvilli tips (5,9,13). Upon cell activation, integrins switch to a high-affinity conformation (31) and become mobile (32,33) in the neutrophil membrane. In this way, the strength of firm adhesions grows through the continuous recruitment of high affinity integrin receptors to the site of endothelial contact. Despite the prevalence of this mechanistic view of neutrophil firm arrest, many elements of the paradigm need to be tested more rigorously.
As part of an effort to build a thorough physical picture of neutrophil arrest and transmigration, we have measured the mobility of key adhesion molecules on human neutrophils in their naturally spherical resting state. We believe our work distinguishes itself from prior literature in which neutrophils or lymphocytes were artificially flattened (14–16,18), or for which receptor mobility was measured on spherical neutrophils in membranes within molecular reach of cover slip surfaces (21). While such experimental configurations allowed investigators to use standard photobleaching setups or SPT, the configurations have the potential to alter the mobility of the receptors being measured. Instead we designed a photobleaching system to measure receptor mobility at the midplane of spherical neutrophils, far from coverslip surfaces. Our approach required the codevelopment of new mathematical models for interpretation of measurements. We developed both an analytical solution to the problem of recovery of an equatorial bleach band and a Monte Carlo solution that allowed us to begin with a nonideal initial bleach profile. Our work is also distinguished from prior work because it is the only existing attempt, to our knowledge, to measure the mobility of the four principal neutrophil receptors involved with the transition to firm arrest (14,16,21,22). It is also notable that the neutrophils examined here were labeled and viewed in dilutions of whole blood. Although our cell preparations did involve gentle centrifugation to remove fluorescent label from the solution, they did not create high concentrations of isolated neutrophils (red cells outnumber neutrophils 1000:1 throughout our preps). We chose this approach because of a concern that standard neutrophil isolation procedures can cause homotypic interactions and lead to artificial arrangements of receptors (13).
Mobility differences between β2 integrins and RAAMs
The primary objective of this research was the measurement of RAAM and β2 integrin mobilities on the plasma membrane of resting neutrophils. Our photobleaching data indicate that LFA-1, a β2 integrin, is 3–7 times more mobile than L-selectin under nearly all of the conditions tested. Interestingly, at room temperature with mAb labeling, the β2 integrins (LFA-1 and Mac-1) have similar mobilities to each other as do the two RAAMs (L-selectin and PSGL-1). Given the topological segregation of RAAMs and β2 integrins, one plausible explanation for the different mobilities is differences in the dynamics of the linkages between these receptors and the underlying cytoarchitecture. According to studies in lymphocytes, the bridging proteins for both L-selectin and PSGL-1 in microvilli and ruffles appear to be members of the ezrin and moesin protein family (34,35). β2 integrin subunits, on the other hand, can bind to the cytoskeleton through the actin binding proteins talin, filamin, and α-actinin, each of which is found in neutrophils (29,31,36).
Fab and IgG measurements suggest cytoskeletal interaction
We report a 40–60% decrease in the diffusion coefficient when using fluorescently tagged mAb against CD11a and CD62L compared to fluorescently tagged Fab at 37°C. A similar difference was also reported in a study of MHC Class I diffusion on HeLa cells (27). This difference in measurement could be due to the size of the diffusing antigen-antibody complex: mAb has two antigen binding sites whereas Fab has only one. Recent work by Gambin et al. has revisited the original theories of protein diffusion in lipid membranes and developed a new model (37). They show protein diffusion is strongly dependent on physical dimension and predict lateral diffusion is proportional to the inverse of protein radius. This finding is consistent with the differences in the diffusion coefficient we report between a potential mAb-receptor dimer and a Fab-receptor monomer.
Another explanation for the differences between mAb and Fab measurements is the existence of membrane corrals or other cytoskeletal interactions. Reviews by Kusumi and others (17,38), explore evidence of membrane corrals or compartments that limit the free diffusion proteins with a cytoplasmic tail. In these models, proteins move through the plasma membrane in a restricted manner that involves diffusion within individual compartments, a wait time, and then a hop to an adjacent compartment. This process is repeated over and over again, but is only observable on very short timescales (much less than 33 ms video rate). The cytoplasmic tail of proteins is corralled by cytoskeletal fences just beneath the plasma membrane, potentially forming protein pickets within these membrane compartments. Kusumi and others argue that cytoskeletal fences or picket lines dramatically reduce the likelihood of a molecular complex traversing a fence compared to a single protein because all members of the complex must traverse simultaneously (17). This idea can be extended to our experimental results of mAb and Fab diffusion. Since mAbs can bind two transmembrane proteins, they are much more likely than Fabs to be hindered, even just momentarily, by membrane corrals. Because the timescale of our data is tens of seconds and shows a single, fully recoverable population, interactions that hinder mobility must be much less than 10 s. Thus our results are consistent with corral or compartment residency times of milliseconds (17).
Comparisons to prior studies
It is informative to compare our integrin mobility measurements to those measured previously by SPT. Values for β2 integrin diffusion coefficients measured in early experiments using SPT on untreated transformed B-cells were 50 times smaller than values reported here (14). This is not surprising because SPT measures the mobility of the slowest receptor bound to the tracking bead, whereas FRAP measures the aggregate diffusion of the whole receptor population. Recent work by Cairo and others (18) on Jurkat cells provided insight into this disparity by dividing SPT data into millisecond (Dmicro) and second (Dmacro) timescales. Dmacro was further broken down into Dmobile and Dimmobile fractions. Reported values for Dimmobile, which were likely tracking beads representative of the slowest integrin targets, were typically 10 times smaller than Dmobile and comparable to early SPT data on integrins (14). Interestingly, measurements by Cairo and others of the fast mobile fraction (Dmobile) over 1.5 s yielded values on E6.1 and lymphocyte cells (18) that bracket our measurements of CD11a labeled with monovalent Fab at room temperature.
There is only one previous report that studied the lateral mobility of either RAAMs or β2 integrins on human neutrophils. In this report, Mukherjee and others investigated CD11b and reported a diffusion coefficient of 3.1 × 10−10 cm2/s, with a 62% immobile fraction using FRAP at room temperature (21). If we were to force such an immobile fraction to our data, our measured mobility of CD11b labeled with mAb at room temperature would be similar, although the quality of the data fit would be significantly worse.
When comparing our measurements to previous reports, it is important to note the significant effects of temperature as well as label valency on the diffusion coefficient. We report an ∼3 times increase in lateral mobility when switching from a divalent label (mAb) to a monovalent label (Fab). In SPT, it is difficult to ensure that only one target molecule is tethered for each bead, particularly when beads become much larger than the antibody-target complex (18). Temperature is also a source of variation; we as well as others note a 2–3 times increase in lateral mobility when increasing the temperature from 23°C to 37°C (28). Taken together, in our own data we see an order of magnitude difference between CD11a measurements with mAb at room temperature compared to Fab at 37°C. These experimental variations as well as differences in cell types may explain (at least in part) the inconsistencies in integrin lateral mobilities from one report to another.
Effects of cytoskeletal disruption
Results obtained with the actin poison cytochalasin D directly relate to the role of cytoskeletal linkages in limiting diffusion rates. Cells treated with cytochalasin D typically show increased lateral mobility of tethered proteins in the plasma membrane. Researchers studying β2 integrins have seen this effect in several cases in different cell types (14,18). In a review by Kusumi et al., the authors were careful to point out that cytoskeletal treatments can produce confusing results (38). A disruption of the cytoskeletal network may lead to increased fragments near the cytoplasmic side of the plasma membrane. Transmembrane proteins influenced by membrane corrals may experience minimal release by cytochalasin D or even a hindered effect as cytoskeletal debris leads to additional diffusion hurdles.
In our work we conducted dose dependent responses to cytochalasin D to find an ideal concentration for restraint release. In the case of L-selectin, lateral mobility increased significantly with low concentrations of cytochalasin D. We found that 250 nM cytochalasin D produced a significant increase in effective lateral mobility when receptors were tagged with either mAb or Fab at 37°C. Higher concentrations produced lower lateral mobility and even higher dosages resulted in poor cell morphologies that precluded analysis. For LFA-1, 250 nM cytochalasin D also increased lateral mobility when tagged with mAb. Interestingly, when tagged with Fab, LFA-1 did not show significant increases in lateral mobility under any tested concentration. This implies that LFA-1 is only lightly associated with the cytoskeleton in a resting neutrophil and/or has a highly dynamic cytoskeletal linkage. Our LFA-1 data, however, are still consistent with the idea of membrane corrals: mAb-labeled LFA-1 as a potential dimer is more likely to be influenced by the cytoskeletal fences than monovalent Fab. Thus the mAb-labeled receptors are expected to have a more significant mobility increase upon light cytoskeletal disruption with cytochalasin D.
A good model of free protein diffusion in cell membranes is the membrane “bleb”, a cell protrusion devoid of cytoskeleton (39). Protein diffusion measurements in blebs can be more than 10 times greater in these regions compared to the main cell body (40). Early work by Tank and others reported diffusion coefficients in cytoskeleton-free blebs to be 3–4 × 10−9 cm2/s for several different proteins at 22°C (40). In red cells lacking the principal components of the cytoskeleton, Koppel and others report the diffusion coefficient of proteins to be 2.5 × 10−9 cm2/s at room temperature (41). To compare these measurements against our measurements of LFA-1 mobility using Fab labeling, we must account for the complexity of the ruffled, resting spherical neutrophil surface compared to a smooth bleb. The ruffled neutrophils surface is reported to be 2.1 times larger than an equivalent simple sphere (42). Using theory developed by King (43) to account for diffusion over a ruffled surface, we calculate that our measured diffusion coefficients may be as much as 65% smaller than the actual lateral diffusion in the plane of the membrane. Taking into account these correction factors, we estimate in-plane diffusion coefficients on human neutrophils to be as high as 1.1 × 10−9 cm2/s (23°C) and 3.4 × 10−9 cm2/s (37°C) for LFA-1, and 1.3 × 10−10 cm2/s (23°C) and 5.1 × 10−10 cm2/s (37°C) for L-selectin. At room temperature, protein diffusion in cytoskeleton-free blebs is just three times faster than our corrected value for LFA-1 on smooth a surface. This further suggests that LFA-1 is not tightly associated with the cytoskeleton in resting neutrophils.
CONCLUSION
The most significant implication of our findings relates to the mechanism of neutrophil adhesion. Specifically, the current paradigm, which holds that firm arrest is mediated by the diffusion of individual integrins after the proteolysis of cytoskeletal tethers (32,33), must be revisited. Unlike SPT experiments, our analysis here measures the mobility of integrins over the length scales and timescales that are relevant to adhesion strengthening at endothelial contact zones. Our data suggest that integrins may diffuse to these zones at rates that are minimally affected by cytoskeletal anchors or membrane corrals. For LFA-1 labeled with monovalent Fab, we report an effective diffusion coefficient of 1.2 × 10−9 cm2/s, which is ∼1 order faster than L-selectin (1.8 × 10−10 cm2/s). In fact, LFA-1 lateral mobility of 1.2 × 10−9 cm2/s is just a factor of three less than free diffusion of proteins in membrane blebs when accounting for the ruffled neutrophil surface (42,43).
APPENDIX
Solution for fluorescence recovery after photobleaching on a sphere
We outline here the solution of the diffusion problem specified by Eqs. 1–3. We look for separable solutions of the form C(φ, τ) = F(τ)G(φ), and we find
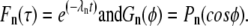 |
(A1) |
Here λn = n(n + 1) and Pn is the nth Legendre polynomial, with n = 0, 1, 2… The solution to the diffusion problem is obtained by superposing the separated solutions:
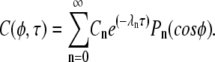 |
(A2) |
The coefficients Cn are determined from the initial condition f(φ) (given by Eq. 3) and from the orthogonality of the Legendre polynomials:
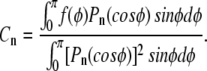 |
(A3) |
The result is
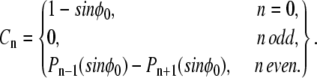 |
(A4) |
Editor: Michael Edidin.
References
- 1.Norman, K. E., K. L. Moore, R. P. McEver, and K. Ley. 1995. Leukocyte rolling in vivo is mediated by P-selectin glycoprotein ligand-1. Blood. 86:4417–4421. [PubMed] [Google Scholar]
- 2.von Andrian, U. H., J. D. Chambers, L. M. McEvoy, R. F. Bargatze, K. E. Arfors, and E. C. Butcher. 1991. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc. Natl. Acad. Sci. USA. 88:7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majstoravich, S., J. Zhang, S. Nicholson-Dykstra, S. Linder, W. Friedrich, K. A. Siminovitch, and H. N. Higgs. 2004. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich syndrome protein (WASp) for their morphology. Blood. 104:1396–1403. [DOI] [PubMed] [Google Scholar]
- 4.Bruehl, R. E., T. A. Springer, and D. F. Bainton. 1996. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J. Histochem. Cytochem. 44:835–844. [DOI] [PubMed] [Google Scholar]
- 5.Erlandsen, S. L., S. R. Hasslen, and R. D. Nelson. 1993. Detection and spatial distribution of the beta 2 integrin (Mac-1) and L-selectin (LECAM-1) adherence receptors on human neutrophils by high-resolution field emission SEM. J. Histochem. Cytochem. 41:327–333. [DOI] [PubMed] [Google Scholar]
- 6.Ivetic, A., O. Florey, J. Deka, D. O. Haskard, A. Ager, and A. J. Ridley. 2004. Mutagenesis of the ezrin-radixin-moesin binding domain of L-selectin tail affects shedding, microvillar positioning, and leukocyte tethering. J. Biol. Chem. 279:33263–33272. [DOI] [PubMed] [Google Scholar]
- 7.Pavalko, F. M., D. M. Walker, L. Graham, M. Goheen, C. M. Doerschuk, and G. S. Kansas. 1995. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via alpha-actinin: receptor positioning in microvilli does not require interaction with alpha-actinin. J. Cell Biol. 129:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore, K. L., K. D. Patel, R. E. Bruehl, F. Li, D. A. Johnson, H. S. Lichenstein, R. D. Cummings, D. F. Bainton, and R. P. McEver. 1995. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 128:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tohya, K., and M. Kimura. 1998. Ultrastructural evidence of distinctive behavior of L-selectin and LFA-1 (alphaLbeta2 integrin) on lymphocytes adhering to the endothelial surface of high endothelial venules in peripheral lymph nodes. Histochem. Cell Biol. 110:407–416. [DOI] [PubMed] [Google Scholar]
- 10.Lei, X., M. B. Lawrence, and C. Dong. 1999. Influence of cell deformation on leukocyte rolling adhesion in shear flow. J. Biomech. Eng. 121:636–643. [DOI] [PubMed] [Google Scholar]
- 11.Jadhav, S., C. D. Eggleton, and K. Konstantopoulos. 2005. A 3-D computational model predicts that cell deformation affects selectin-mediated leukocyte rolling. Biophys. J. 88:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simon, S. I., and C. E. Green. 2005. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu. Rev. Biomed. Eng. 7:151–185. [DOI] [PubMed] [Google Scholar]
- 13.Carman, C. V., and T. A. Springer. 2003. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15:547–556. [DOI] [PubMed] [Google Scholar]
- 14.Kucik, D. F., M. L. Dustin, J. M. Miller, and E. J. Brown. 1996. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest. 97:2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin, T., and J. Li. 2002. Dynamitin controls beta 2 integrin avidity by modulating cytoskeletal constraint on integrin molecules. J. Biol. Chem. 277:32963–32969. [DOI] [PubMed] [Google Scholar]
- 16.Zhou, X., J. Li, and D. F. Kucik. 2001. The microtubule cytoskeleton participates in control of beta2 integrin avidity. J. Biol. Chem. 276:44762–44769. [DOI] [PubMed] [Google Scholar]
- 17.Kusumi, A., H. Ike, C. Nakada, K. Murase, and T. Fujiwara. 2005. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin. Immunol. 17:3–21. [DOI] [PubMed] [Google Scholar]
- 18.Cairo, C. W., R. Mirchev, and D. E. Golan. 2006. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 25:297–308. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson, K., Z. Derzko, E. S. Wu, Y. Hou, and G. Poste. 1976. Measurement of the lateral mobility of cell surface components in single, living cells by fluorescence recovery after photobleaching. J. Supramol. Struct. 5:565(417)–576(428). [DOI] [PubMed] [Google Scholar]
- 20.Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee, G., B. Rasmusson, J. G. Linner, M. T. Quinn, C. A. Parkos, K. E. Magnusson, and A. J. Jesaitis. 1998. Organization and mobility of CD11b/CD18 and targeting of superoxide on the surface of degranulated human neutrophils. Arch. Biochem. Biophys. 357:164–172. [DOI] [PubMed] [Google Scholar]
- 22.Kim, M., C. V. Carman, W. Yang, A. Salas, and T. A. Springer. 2004. The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. J. Cell Biol. 167:1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koppel, D. E., M. P. Sheetz, and M. Schindler. 1980. Lateral diffusion in biological membranes. A normal-mode analysis of diffusion on a spherical surface. Biophys. J. 30:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lomakina, E. B., and R. E. Waugh. 2004. Micromechanical tests of adhesion dynamics between neutrophils and immobilized ICAM-1. Biophys. J. 86:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleit, H. B., S. D. Wright, and J. C. Unkeless. 1982. Human neutrophil Fc gamma receptor distribution and structure. Proc. Natl. Acad. Sci. USA. 79:3275–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zicha, D., I. M. Dobbie, M. R. Holt, J. Monypenny, D. Y. Soong, C. Gray, and G. A. Dunn. 2003. Rapid actin transport during cell protrusion. Science. 300:142–145. [DOI] [PubMed] [Google Scholar]
- 27.Georgiou, G., S. S. Bahra, A. R. Mackie, C. A. Wolfe, P. O'Shea, S. Ladha, N. Fernandez, and R. J. Cherry. 2002. Measurement of the lateral diffusion of human MHC class I molecules on HeLa cells by fluorescence recovery after photobleaching using a phycoerythrin probe. Biophys. J. 82:1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson, K., D. O'Dell, and J. T. August. 1984. Lateral diffusion of an 80,000-dalton glycoprotein in the plasma membrane of murine fibroblasts: relationships to cell structure and function. J. Cell Biol. 99:1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sampath, R., P. J. Gallagher, and F. M. Pavalko. 1998. Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J. Biol. Chem. 273:33588–33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calderwood, D. A. 2004. Talin controls integrin activation. Biochem. Soc. Trans. 32:434–437. [DOI] [PubMed] [Google Scholar]
- 31.Calderwood, D. A. 2004. Integrin activation. J. Cell Sci. 117:657–666. [DOI] [PubMed] [Google Scholar]
- 32.Ni, N., C. G. Kevil, D. C. Bullard, and D. F. Kucik. 2003. Avidity modulation activates adhesion under flow and requires cooperativity among adhesion receptors. Biophys. J. 85:4122–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Kooyk, Y., and C. G. Figdor. 2000. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 12:542–547. [DOI] [PubMed] [Google Scholar]
- 34.Alonso-Lebrero, J. L., J. M. Serrador, C. Dominguez-Jimenez, O. Barreiro, A. Luque, M. A. del Pozo, K. Snapp, G. Kansas, R. Schwartz-Albiez, H. Furthmayr, F. Lozano, and F. Sanchez-Madrid. 2000. Polarization and interaction of adhesion molecules P-selectin glycoprotein ligand 1 and intercellular adhesion molecule 3 with moesin and ezrin in myeloid cells. Blood. 95:2413–2419. [PubMed] [Google Scholar]
- 35.Ivetic, A., J. Deka, A. Ridley, and A. Ager. 2002. The cytoplasmic tail of L-selectin interacts with members of the Ezrin-Radixin-Moesin (ERM) family of proteins: cell activation-dependent binding of Moesin but not Ezrin. J. Biol. Chem. 277:2321–2329. [DOI] [PubMed] [Google Scholar]
- 36.Watts, R. G., and T. H. Howard. 1994. Role of tropomyosin, alpha-actinin, and actin binding protein 280 in stabilizing Triton insoluble F-actin in basal and chemotactic factor activated neutrophils. Cell Motil. Cytoskeleton. 28:155–164. [DOI] [PubMed] [Google Scholar]
- 37.Gambin, Y., R. Lopez-Esparza, M. Reffay, E. Sierecki, N. S. Gov, M. Genest, R. S. Hodges, and W. Urbach. 2006. Lateral mobility of proteins in liquid membranes revisited. Proc. Natl. Acad. Sci. USA. 103:2098–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kusumi, A., C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo, and T. Fujiwara. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34:351–378. [DOI] [PubMed] [Google Scholar]
- 39.Scott, R. E., R. G. Perkins, M. A. Zschunke, B. J. Hoerl, and P. B. Maercklein. 1979. Plasma membrane vesiculation in 3T3 and SV3T3 cells. I. Morphological and biochemical characterization. J. Cell Sci. 35:229–243. [DOI] [PubMed] [Google Scholar]
- 40.Tank, D. W., E. S. Wu, and W. W. Webb. 1982. Enhanced molecular diffusibility in muscle membrane blebs: release of lateral constraints. J. Cell Biol. 92:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koppel, D. E., M. P. Sheetz, and M. Schindler. 1981. Matrix control of protein diffusion in biological membranes. Proc. Natl. Acad. Sci. USA. 78:3576–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans, E., and A. Yeung. 1989. Apparent viscosity and cortical tension of blood granulocytes determined by micropipet aspiration. Biophys. J. 56:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King, M. R. 2004. Apparent 2-D diffusivity in a ruffled cell membrane. J. Theor. Biol. 227:323–326. [DOI] [PubMed] [Google Scholar]