

Abstract
Antimicrobial peptides (AMPs) induce cytotoxicity by altering membrane permeability. The electrical properties of membrane-associated AMPs as well as their cellular effects have been extensively documented; however their three-dimensional structure is poorly understood. Gaining insight into channel structures is important to the understanding of the protegrin-1 (PG-1) and other AMP cytolytic mechanisms, and to antibiotics design. We studied the β-sheet channels morphology using molecular dynamics simulations. We modeled PG-1 channels as intrinsic barrel-stave and toroidal membrane pores, and simulated them in zwitterionic and anionic lipid bilayers. PG-1 channels consist of eight β-hairpins in a consecutive NCCN (N and C represent the β-hairpin's N- and C-termini) packing organization yielding antiparallel and parallel β-sheet channels. Both channels preserve the toroidal, but not the barrel-stave pores. The two lipid leaflets of the bilayer bend toward each other at the channels' edges, producing a semitoroidal pore with the outward-pointing hydrophobic residues preventing the polar lipid headgroups from moving to the bilayer center. In all simulated lipid environments, PG-1 channels divide into four or five β-sheet subunits consisting of single or dimeric β-hairpins. The channel morphology with subunit organization is consistent with the four to five subunits observed by NMR in the POPE/POPG bilayer. Remarkably, a β-sheet subunit channel motif is in agreement with Alzheimer ion channels modeled using the universal U-shape β-strand-turn-β-strand structure, as well as with high resolution atomic force microscopy images of β-amyloid channels with four to six subunits. Consistent with the toxic β-amyloid channels that are ion-conducting, the PG-1 channels permeate anions.
INTRODUCTION
Antimicrobial peptides (AMPs) are generally abundant in charges that allow their straightforward diffusion into membranes of invading microorganisms (1,2). Their cytotoxic activities mainly involve disrupting membranes, leading to the death of target cells (3). The mechanism, however, is still not understood fully. Recent observations suggested that peptides translocated into the interior of target cells could interfere with cellular metabolism (4). However, it is generally believed that the AMPs induce unregulated ion leakage through formation of channels embedded in membrane pores, mediating cellular permeability (3,5–7). Several models were proposed to explain the cell membrane rupturing events (7).
Protegrin-1 (PG-1, ∼2 kDa) is a small β-hairpin peptide consisting of 18 amino acids with a high content of cysteine (Cys) and positively charged arginine (Arg) residues (8). The peptide displays an antimicrobial activity (9), and thus is a very potent antibiotic peptide (10). PG-1 forms ion channels in cellular membranes (5,11). Single channel conductance of protegrin channels were reported recently (5); PG-1 and PG-3 (R4G) induce weak anion-selective channels and K+ leakage from liposomes. PG-3 also induces moderate cationic selectivity in the presence of bacterial lipopolysaccharide in planar phospholipids bilayers. Neutron diffraction of crystallized peptide-membrane on quartz plates (6) indicates that both PG-1 and magainin 2 make stable membrane pores in fully hydrated fluid membranes.
Solid-state NMR spectroscopy using 19F and 1H spin diffusion (11,12) suggests that PG-1 peptides form a β-barrel in an anionic membrane, whereas they aggregate into β-sheets on the surface of a membrane with cholesterol. In the anionic membrane, the β-barrel induces a toroidal membrane pore in which lipids surrounding the β-barrel have significant disorder and acyl chain reorientations. The pore diameter of the β-barrel, estimated from size-dependent membrane crossing by polyethylene glycol (PEG), is 2.1 nm. Based on both intermolecular distances and pore diameter, the β-barrel has the outer diameter of ∼4.2 nm. The oligomeric structure of PG-1 suggests that the β-barrel is an assembly of four to five β-sheets, where each sheet consists of two β-hairpins in the NCCN organization (where N and C represent N- and C-termini, blue and red, respectively in Fig. 1 a). Dimer formation is important for further oligomerization in the membrane: as the primary unit for assembly into ordered aggregates, the dimer could insert into the interior of the bilayer, clustering to form a multimer, including a channel. The recent model for PG-1 dimerization suggested that at low peptide concentration the dimer is bound to the lipid bilayer in such an NCCN organization, whereas at high concentration, the dimers begin to aggregate to form ordered oligomers (13). Two-dimensional solid-state NMR (14,15) of PG-1 aggregates show that a parallel β-sheet in the NCCN packing mode is the sole repeat motif in the ordered aggregates in the lipid bilayer.
FIGURE 1.

Topological diagrams (a) for the PG-1 channels in the antiparallel (left) and parallel (right) β-sheet arrangements in an NCCN packing mode. Blue and red represent the N-terminal and C-terminal residues, respectively, and yellow represents the loop residues in the β-hairpin. (b) Cartoon representations of the barrel-stave (left) and toroidal (right) membrane pores. Hollow cylinders represent PG-1 channels. (c) Schematic diagrams of the lipid headgroups for the zwitterionic POPC (left) and anionic POPG (right) lipids.
Membrane disruption by cytolytic peptides is implicated by the interaction of the peptide with lipid headgroups (16–19). The charged side chains of the peptide play a central role in the mechanism of peptide insertion and membrane disruption (12,16). In an earlier study of the PG-1 monomer on the lipid bilayers (20), we have reported that the β-hairpin induced the thinning effect in the anionic bilayer containing palmitoyl-oleyl-phosphatidylcholine (POPC) and palmitoyl-oleyl-phosphatidylglycerol (POPG) with a mole ratio of 4:1, whereas no thinning effect was observed for the zwitterionic bilayer composed of POPC. Further investigations of the dimers on the surface of POPC bilayer (21) have shown that they organize into a β-sheet motif with antiparallel (turn-next-to-tail association) and parallel (turn-next-to-turn association) arrangements in the NCCN packing mode. Although the antiparallel β-sheets have a more stable dimeric interface, the parallel β-sheets are more active in membrane disruption. This observation indicates that the parallel PG-1 dimer is biologically more relevant for the penetration into the membrane (11,13–15). The strong interactions of the monomeric or dimeric PG-1 with the polar lipid headgroups suggest that the cationic peptide adapts well to the amphipathic characteristics (22–24). However, the monomer and dimer simulations only provided the results for the peptides in the S (surface-bound) state, one of two distinct states for cytolytic peptides in lipid bilayers (19,25). Although the peptides in the S state give rise to membrane thinning effects, membrane-disrupting effects by forming ion channels with selective transport across the membrane are directly mediated by packing of the peptides in the I (membrane-inserted) state.
PG-1 cytotoxicity is believed to be due to the formation of ion channel in the membrane, leading to ion leakage (3,5,11). The mechanism underlying PG-1 induced cellular toxicity is similar to the cytotoxic activity of β-amyloid (Aβ). Unlike the cytotoxic peptides used in host defense, Aβ is an autocytotoxic peptide (3). Recent atomic force microscopy (AFM) experiments (26,27) reported that Aβ oligomers form heterogeneous ion channels in the cell membrane, destabilizing cellular ionic homeostasis and ultimately leading to cell death (28–31). At higher resolution, AFM images showed that the channels are assemblies of several subunits. Subsequent computational modeling of the Aβ channels provided a consistent picture of the subunit organization and channel dimensions (32,33). The atomistic models of the Aβ channels suggested dominant β-sheet motifs of the channel morphology in the membrane. In contrast to the naturally occurring functional ion-gated channels that mostly consist of interacting α-helices, currently known β-sheet channels are nonphysiological and their formations in the membrane are closely related to the cellular pathophysiology (26–36).
In the membrane, PG-1, a β-hairpin peptide, forms a β-sheet channel indicating that the PG-1 β-sheet channel could share a common morphological motif with other β-sheet channels. However, channel-forming cytolytic β-sheet peptides have been less intensively studied due to the complexity of the β-sheet motifs when involved in membrane toxic activities (37,38), whereas there are abundant studies for the amphipathic α-helical peptides (17,18,39,40). Obtaining atomic-level structures of β-sheet channels em bedded in the membrane is crucial, because providing atomistic channel conformations can accelerate drug development. Although PG-1 is a promising new pharmaceutical agent against harmful microbes, the atomistic structure of the PG-1 channel in the membrane pore remains poorly understood. Computer simulations of the detailed atomic models can offer three-dimensional structures for the membrane-inserted state of PG-1 channels at the atomic-level and provide information complementary to experimental approaches. In this study, we carried out extensive molecular dynamics (MD) simulations of the PG-1 β-sheet channels with different β-sheet arrangements in different lipid bilayer environments.
MATERIALS AND METHODS
We simulated PG-1 channels in different lipid environments. The channels consist of eight identical β-hairpins of PG-1, initially arranged to form a perfect annular shape of a single layer β-sheet. We used the monomer conformation of the β-hairpin from the NMR spectroscopy (8). Because the NMR-based β-hairpin structure was obtained in water environment, we carried out several preliminary simulations of the β-hairpin monomer and β-sheet dimer in POPC bilayer for 20 ns. From these preliminary trajectories, we obtained β-hairpin structures that were fully prerelaxed in the lipid environment. The β-hairpin has two β-strands and six intramolecular backbone hydrogen bonds (H-bonds). The β-hairpin was translated into a circular rim and then rotated eight times with respect to the pore axis, which is parallel to the molecular axis of the β-hairpin. The octameric PG-1 channel was initially constructed as an annular shape. Depending on the β-hairpin arrangement, the PG-1 channel has two potential β-sheet motifs; antiparallel (turn-next-to-tail) and parallel (turn-next-to-turn) β-sheets in a multimeric NCCN packing mode (Fig. 1 a) (14). The channel is minimized with a rigid body motion for the peptides to enhance the formation of intermolecular backbone H-bonds between the β-strands. The minimized channel is next embedded in the lipid bilayer. Two different bilayer topologies, intrinsic barrel-stave and toroidal membrane pores (Fig. 1 b) were initially prepared for the β-sheet channels. For the two topological bilayer settings, pure lipid bilayer composed of POPC and mixed lipid bilayer composed of POPC/POPG (mole ratio 4:1) (Fig. 1 c) were constructed. Thus, considering the bilayer topology and composition, each β-sheet channel has four different lipid environments, yielding four independent simulations for antiparallel (Aba-pc, Ato-pc, Aba-pg, and Ato-pg channels; where A represents antiparallel, “ba” denotes barrel-stave, and “to” denotes toroidal. “pc” and “pg” represent the zwitterionic and anionic bilayers, respectively) and parallel (Pba-pc, Pto-pc, Pba-pg, and Pto-pg channels; where P represents parallel) β-sheet channels.
A unit cell containing a channel, lipids, salts, and waters with almost 90,000 to 135,000 atoms, depending on the bilayer setting, is constructed. Because our simulation method closely follows the previous method for the PG-1 monomer (20), dimer (21), Aβ channel (32,33) simulations with the zwitterionic lipid bilayers, and the fusion domain of influenza hemagglutinin (HA) simulations with the anionic lipid bilayer (41), in this study we only briefly describe key parameters used for the PG-1 channel simulations. For the lipid bilayer, 240 lipids (120 lipids on each side) constitute the lateral cell. However, in the toroidal membrane pore, additional 150 lipids were used as the pore lining lipids. TIP3P waters were added and relaxed through a series of minimization and dynamics. The system contains NaCl at a concentration of 100 mM to satisfy a physiological salt concentration. The CHARMM program (42) was used to construct the set of starting points and to relax the systems to a production-ready stage. Fig. 2 shows the starting points for antiparallel (Fig. 2 a) and parallel (Fig. 2 b) β-sheet channels that enclose the initial structures of the water pores in a surface representation created by the HOLE program (43). Simulations for the initial construction and the pre-equilibration were carried out on the NPAT (constant number of atoms, pressure, surface area, and temperature) ensemble. For production runs to 30 ns, the NAMD code (44) on a Biowulf cluster at the NIH was used for the starting point. In the production simulations, the dynamics were carried out on both the NPAT and NPγT (constant number of atoms, pressure, surface tension, and temperature) ensembles with γ = 0. However, no significant differences were found for the use of different ensembles. For each system, at least two independent simulations were carried out to check the consistency in key findings. Averages were taken after 10 ns discarding initial transient.
FIGURE 2.

Starting points of the channels in a cartoon representation for the (a) antiparallel and (b) parallel β-sheet channels of PG-1. In the peptides, hydrophobic residues are shown in white, one polar (Tyr7) and three Gly (Gly2, Gly3, and Gly17) residues are shown in green, and six positively charged Arg residues (Arg1, Arg4, Arg9, Arg10, Arg11, and Arg18) are shown in blue. In each channel, the initial water pore structure calculated by the HOLE program (43) is embedded. For the pore structures in the surface representation, red denotes pore diameter of d < 0.8 nm, green denotes pore diameter in the range, 0.8 nm ≤ d ≤ 1.2 nm, and blue denotes pore diameter of d > 1.2 nm.
RESULTS
Dimensions of PG-1 channels composed of β-sheet subunits in the lipid bilayers
The conformations of environmentally relaxed PG-1 channels in a fully hydrated lipid bilayer can be obtained after 30 ns simulations with all-atom representations (Supplementary Material, Fig. S1). Fig. 3 shows the averaged channel structures of PG-1 in a cartoon representation for antiparallel (Fig. 3 a) and parallel (Fig. 3 b) β-sheet channels. Channels are in the lateral view from the bilayer, and the averaged structures of the water pores in a surface representation (created by the HOLE program (43)), are embedded in the channels. Initially, the PG-1 channels have a perfect annular morphology of single layered β-sheet (Fig. 2), but the annular shape disappears in the relaxed channel structures (Fig. S1). The PG-1 channels slightly increase the outer dimension. In the starting conformations, the antiparallel and parallel β-sheet channels of PG-1 have the outer diameters of ∼3.20 and ∼3.32 nm, respectively. After the simulations, the outer diameters increase to ∼3.47, ∼3.63, ∼3.53, and ∼3.62 nm for the Aba-pc, Ato-pc, Aba-pg, and Ato-pg channels (Fig. 4 a), and to ∼3.50, ∼3.66, ∼3.50, and ∼3.66 nm for the Pba-pc, Pto-pc, Pba-pg, and Pto-pg channels (Fig. 4 b), respectively. The PG-1 channels in the toroidal membrane pore have slightly larger outer diameter than those with the intrinsic barrel-stave setting. In contrast to the outer dimensions, the PG-1 channels slightly decrease the diameter of the water pore or stay close to the starting values of ∼1.07 and ∼1.02 nm for the antiparallel and parallel β-sheet channels, respectively. After the simulations, the pore diameters change to ∼0.81, ∼0.89, ∼1.06, and ∼0.94 nm for the Aba-pc, Ato-pc, Aba-pg, and Ato-pg channels (Fig. 4 a), and to ∼0.88, ∼0.92, ∼0.98, and ∼1.05 nm for the Pba-pc, Pto-pc, Pba-pg, and Pto-pg channels (Fig. 4 b), respectively. PG-1 channels in the anionic lipid bilayer have slightly larger water pores than those in the zwitterionic lipid bilayer. It can be seen from the figures that the pores become jagged tubes in the relaxed channel conformations, whereas they were straight cylindrical tubes in the initial channels (Fig. 2). Especially, the pores have narrow necks at both channel ends due to large fluctuations of the long Arg side chains at both termini and the loop of each β-hairpin; some side chains stretch into the interior of the water pore and may block the channel entrances. The inhomogeneous shapes of the water pores suggest that the subunits are mobile in the channels within the lipid environments.
FIGURE 3.

Channel structures averaged over the simulation in a ribbon representation for the (a) antiparallel and (b) parallel β-sheet channels of PG-1 are shown in the lateral view from the lipid bilayer. Peptides are colored according to the subunit organization in the channels. In each channel, the averaged water pore structure calculated by the HOLE program (43) is embedded. For the pore structures in the surface representation, red denotes pore diameter of d < 0.8 nm, green denotes pore diameter in the range, 0.8 nm ≤ d ≤ 1.2 nm, and blue denotes pore diameter of d > 1.2 nm.
FIGURE 4.

Averaged outer channel diameter and pore diameters (solid lines) as a function of the distance along the pore center axis for the (a) antiparallel and (b) parallel β-sheet channels of PG-1. Dotted lines represent the initial channel dimension at the starting points.
The relaxed PG-1 channels exhibit heterogeneous channel shapes (Fig. S1). The perfect annular conformation disappears and the channels break into several localized β-sheets during the simulations. Fig. 5 shows the averaged fraction of backbone hydrogen bonds (H-bond), 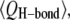 for the antiparallel (Fig. 5 a) and parallel (Fig. 5 b) β-sheet channels. We calculated the fraction using
for the antiparallel (Fig. 5 a) and parallel (Fig. 5 b) β-sheet channels. We calculated the fraction using 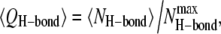 where 〈 〉 denotes the time average,
where 〈 〉 denotes the time average, 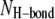 is the number of the intramolecular or intermolecular backbone H-bonds between the β-strands that are monitored during the simulations, and
is the number of the intramolecular or intermolecular backbone H-bonds between the β-strands that are monitored during the simulations, and 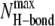 is the maximum possible number of the backbone H-bonds as described in the topological diagram of Fig. 1. Our results indicate that the monomeric N-C interfaces in each β-hairpin are stable, because the β-hairpin conformation is preserved through the formation of the intramolecular backbone H-bonds. However, the stability of the dimeric N-N and C-C interfaces between neighboring β-hairpins depend on the channel topology. In the aniparallel β-sheet channels, the C-C interfaces connect two PG-1 monomers through the formation of the intermolecular backbone H-bonds, whereas the N-N interfaces break the β-sheet network (Fig. 5 a). The four-leaf clover shapes in QH-bond indicate clearly that the channels divide into four dimeric β-sheet subunits with NCCN packing, and suggest that the aniparallel β-sheet channel is an (NCCN)k multimer, where k = 4. In contrast, in the parallel β-sheet channels, most N-N interfaces connect two β-hairpin monomers forming a β-sheet, whereas the C-C interfaces break the β-sheet network (Fig. 5 b). The channels divide into four to five subunits containing the β-hairpin monomers and the β-sheet dimers in (mostly) CNNC packing. The observation that parallel β-sheet channels favor the formation of CNNC dimer is inconsistent with the experimental result for the β-barrel with NCCN dimers (11). However, morphologically the PG-1 channels are composed of several subunits, similar to the β-barrel containing four to five parallel β-sheet subunits and to the Aβ17-42 channels consisting of three to six ordered subunits (32,33). From these observations we can conclude that in the membrane, β-sheet channels commonly break into subunits, i.e., membranes do not support intact β-sheet channels. This can also offer an explanation why functional ion-gated channels mainly consist of interacting α-helices rather than hydrogen-bonded β-strands.
is the maximum possible number of the backbone H-bonds as described in the topological diagram of Fig. 1. Our results indicate that the monomeric N-C interfaces in each β-hairpin are stable, because the β-hairpin conformation is preserved through the formation of the intramolecular backbone H-bonds. However, the stability of the dimeric N-N and C-C interfaces between neighboring β-hairpins depend on the channel topology. In the aniparallel β-sheet channels, the C-C interfaces connect two PG-1 monomers through the formation of the intermolecular backbone H-bonds, whereas the N-N interfaces break the β-sheet network (Fig. 5 a). The four-leaf clover shapes in QH-bond indicate clearly that the channels divide into four dimeric β-sheet subunits with NCCN packing, and suggest that the aniparallel β-sheet channel is an (NCCN)k multimer, where k = 4. In contrast, in the parallel β-sheet channels, most N-N interfaces connect two β-hairpin monomers forming a β-sheet, whereas the C-C interfaces break the β-sheet network (Fig. 5 b). The channels divide into four to five subunits containing the β-hairpin monomers and the β-sheet dimers in (mostly) CNNC packing. The observation that parallel β-sheet channels favor the formation of CNNC dimer is inconsistent with the experimental result for the β-barrel with NCCN dimers (11). However, morphologically the PG-1 channels are composed of several subunits, similar to the β-barrel containing four to five parallel β-sheet subunits and to the Aβ17-42 channels consisting of three to six ordered subunits (32,33). From these observations we can conclude that in the membrane, β-sheet channels commonly break into subunits, i.e., membranes do not support intact β-sheet channels. This can also offer an explanation why functional ion-gated channels mainly consist of interacting α-helices rather than hydrogen-bonded β-strands.
FIGURE 5.

Fractions of intermolecular and intramolecular backbone hydrogen bonds (H-bonds), QH-bond, for the (a) antiparallel and (b) parallel β-sheet channels of PG-1. Fractions of intramolecular H-bond in the N-C monomer interface are shown as white symbols under each peptide number, and the fractions of intermolecular H-bond in the N-N and C-C dimer interfaces are shown as gray symbols in between the peptide numbers. The peptides 2-3, 4-5, 6-7, and 8-1 contact with the N-N and dimer interface, whereas the peptides 1-2, 3-4, 5-6, and 7-8 contact with the C-C dimer interface.
Interactions of PG-1 channels with the lipid bilayers
Our simulations used various lipid environments for the PG-1 channels, which include different bilayer topologies (intrinsic barrel-stave versus toroidal membrane pores) and lipid compositions (zwitterionic versus anionic lipid bilayers). The various lipid settings in the simulations can help in pointing to a favorable environment for the PG-1 channels. To understand how the peptide-lipid interactions support the channel conformation in such complex lipid environments, we calculated the interaction energy of the PG-1 channels with the lipids. Fig. 6 a shows the averaged interaction energy of each PG-1 monomer in the channels with the lipids. The bar graphs with different heights clearly indicate that the peptide-lipid interactions are inhomogeneous; i.e., each PG-1 monomer interacts with lipids with different interaction energies. We speculate that the inhomogeneous interactions cause the shapes of the heterogeneous PG-1 channels. The total interaction energy of the PG-1 channel with the lipids can be calculated by the addition of the monomers interaction energies (Fig. 6 b). The total interaction energies of the antiparallel β-sheet channels with the lipids are −443.7 ± 77.3, −288.5 ± 93.7, −643.4 ± 52.9, and −489.6 ± 71.3 kcal/mol for the Aba-pc, Ato-pc, Aba-pg, and Ato-pg channels, respectively. The total interaction energies of the parallel β-sheet channels with the lipids are −469.7 ± 93.4, −262.4 ± 90.4, −566.3 ± 55.5, and −445.9 ± 66.4 kcal/mol for the Pba-pc, Pto-pc, Pba-pg, and Pto-pg channels, respectively. As expected, with the same bilayer topology, the PG-1 channels interact more strongly with lipids in the anionic lipid bilayer than those in the zwitterionic lipid bilayer. However, with the same lipid composition, the PG-1 channels interact more strongly with lipids in the semitoroidal membrane pore (the final membrane structure from the intrinsic barrel-stave pore setting; see below) than those in the toroidal membrane pore.
FIGURE 6.

(a) Interaction energies of each monomer in the channels with lipids for the antiparallel (upper) and parallel (lower) β-sheet channels of PG-1. (b) The total interaction energy of the PG-1 channels with lipid can be obtained by adding all monomers interaction energies.
Membrane pores induced by the PG-1 channels
PG-1 causes membrane permeation by forming an ion channel spanning a locally deformed membrane. There are three different membrane pore models for the AMP-induced membrane permeation: i), barrel-stave, ii), toroidal, and iii), carpet models (7). For a cylindrical bundle of peptides, the barrel-stave and toroidal models are biologically relevant to the channel-induced membrane pore. On the other hand, the carpet model is unsuitable for the channel-forming peptides, because peptides do not form a bundle but aggregate on the bilayer surface, covering the area around the dented membrane hole. Thus, our simulations used the barrel-stave and toroidal models of the membrane pore for the PG-1 channels. Fig. 7 a shows snapshots of the channel-bilayer systems at the end of the simulations of the channels in the anionic lipid bilayer. The channels satisfy the toroidal membrane pore, because the intrinsic pore is well preserved during the simulations. The three-dimensional density maps of the lipid headgroups (Fig. 7 b) clearly show that the two lipid leaflets bend toward each other and connect to form a continuous curvature. However, the channels do not satisfy the barrel-stave membrane pore, because the initially flattened and paralleled bilayer surfaces are not preserved (see Fig. 1 b). The inherent property of the barrel-stave membrane pore rapidly disappears during the initial transient <2 ns. Instead, the lipids lining the membrane pore have significant chain reorientations, producing local thinning of the bilayer near the channels. The height of the membrane-spanning channel is shorter than the bilayer thickness, allowing the lipids at both ends of the channel to pack in this curved way. The bending of the two lipid leaflets toward each other is similar to the formation of a toroidal membrane pore; however, unlike the toroidal pore, the two lipid leaflets are not in contact with each other. The hydrophobic residues located in the middle of the channels prevent the polar lipid headgroups from moving further into the bilayer center. Thus, we describe the membrane pores induced by the PG-1 channels as semitoroidal. The conformations of the channels are well preserved in the semitoroidal membrane pore.
FIGURE 7.

(a) Snapshots of the channel-bilayer systems at the end of simulations for the PG-1 channels in the anionic lipid bilayer. The first and third snapshots (from left) correspond to the bilayer with the intrinsic barrel-stave membrane pore, and second and fourth snapshots represent the bilayer with the intrinsic toroidal membrane pore for each β-sheet channel. In the surface representations of the channel structures, hydrophobic residues are shown in white, polar and Gly residues are shown in green, and positively charged residues are shown in blue. Phosphates in the lipid headgroups are shown as yellow beads and the lipid tails are shown as threads. Water molecules and ions are removed for clarity. (b) Three-dimensional density maps of the lipid headgroup for the anionic lipid bilayer. The intrinsic barrel-stave membrane pores (the first and third maps from left for the antiparallel and parallel β-sheet channels, respectively) become the semitoroidal membrane pores, whereas the intrinsic toroidal membrane pores (the second and fourth maps from the left for the antiparallel and parallel β-sheet channels, respectively) are well-preserved during the simulations. The density maps represent the angle view of the time averaged bilayer structure. The PG-1 channels in a ribbon representation in blue are embedded in the density maps.
Anionic conducting water pores of the PG-1 channels
The PG-1 channels preserve a water pore, and the size of the pore is wide enough for conducting water and ions. For each simulation the system contains Na+ and Cl− at a concentration of 100 mM. The PG-1 channels attract anions at the channel entrances with the positively charged Arg side chains, whereas the side chains avoid interacting with cations. Fig. 8 shows the potential of mean force (PMF) representing the relative free energy profile for water and ions across the bilayer. The PMF is calculated by using the equation of 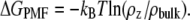 Here, kB is the Boltzmann constant, T is the simulation temperature, ρz is the ion density at the position z along the pore axis, and ρbulk is the ion density in the bulk region. An accurate equilibrium PMF relevant to ion permeation should be obtained from free energy calculations with the umbrella sampling method (45). Nevertheless, given the simulation trajectory without additional multiple equilibrium runs for the sampling, the ion-density-based PMF calculation is useful to estimate rough relative free energy changes for solvents, providing a general outline for solvent permeation through the pore (46,47). Thus, the absolute values in the free energy changes between the antiparallel and parallel β-sheet channels need to be compared carefully, because each system in the simulations has different numbers of salt, lipid, and water molecules. In addition, the lipid systems contain different bilayer compositions and topologies, indicating that the free energy differences for each system are relative to each other. As expected, the channels prevent Na+ crossing through the water pore with large free energy barrier. The cation interacts only with the phosphate headgroups at the lipid/water interface. In the toroidal membrane pore, the PG-1 channels yield free energy minima for Na+ at both edges of the channels, because the cation can slip into the bilayer through the lipid/water interface formed by the two merged lipid leaflets. However, this Na+ penetration occurs outside the channel, because the cation does not cross the water pore. In contrast to the cation, the PG-1 channels conduct anion and water through the water pore, because the PMF curves show low free energy profiles across the pore for Cl− and water. The PMF curves for water provide similar profiles for all channels, whereas the PMF curves for Cl− are different depending on the channel topology. For the antiparallel β-sheet channels, the PMF curves for Cl− show a small free energy barrier in the middle of the pore, whereas the barrier is relatively flat in the water pores of the parallel β-sheet channels. The flat barrier for the parallel β-sheet channels is due to four Arg4 side chains from every second monomer in the channel, because the side chains point into the interior of the water pore, luring more anions into the pore. We speculate that, without the free energy barrier, the water pores of the parallel β-sheet channels are more conductive for anions.
Here, kB is the Boltzmann constant, T is the simulation temperature, ρz is the ion density at the position z along the pore axis, and ρbulk is the ion density in the bulk region. An accurate equilibrium PMF relevant to ion permeation should be obtained from free energy calculations with the umbrella sampling method (45). Nevertheless, given the simulation trajectory without additional multiple equilibrium runs for the sampling, the ion-density-based PMF calculation is useful to estimate rough relative free energy changes for solvents, providing a general outline for solvent permeation through the pore (46,47). Thus, the absolute values in the free energy changes between the antiparallel and parallel β-sheet channels need to be compared carefully, because each system in the simulations has different numbers of salt, lipid, and water molecules. In addition, the lipid systems contain different bilayer compositions and topologies, indicating that the free energy differences for each system are relative to each other. As expected, the channels prevent Na+ crossing through the water pore with large free energy barrier. The cation interacts only with the phosphate headgroups at the lipid/water interface. In the toroidal membrane pore, the PG-1 channels yield free energy minima for Na+ at both edges of the channels, because the cation can slip into the bilayer through the lipid/water interface formed by the two merged lipid leaflets. However, this Na+ penetration occurs outside the channel, because the cation does not cross the water pore. In contrast to the cation, the PG-1 channels conduct anion and water through the water pore, because the PMF curves show low free energy profiles across the pore for Cl− and water. The PMF curves for water provide similar profiles for all channels, whereas the PMF curves for Cl− are different depending on the channel topology. For the antiparallel β-sheet channels, the PMF curves for Cl− show a small free energy barrier in the middle of the pore, whereas the barrier is relatively flat in the water pores of the parallel β-sheet channels. The flat barrier for the parallel β-sheet channels is due to four Arg4 side chains from every second monomer in the channel, because the side chains point into the interior of the water pore, luring more anions into the pore. We speculate that, without the free energy barrier, the water pores of the parallel β-sheet channels are more conductive for anions.
FIGURE 8.

PMF, ΔGPMF, representing the relative free energy profile for Na+, Cl−, and water as a function of the distance along the pore center axis for the antiparallel (left column) and parallel (right column) β-sheet channels of PG-1.
DISCUSSION AND CONCLUSIONS
We simulated the octameric PG-1 channels with different β-sheet arrangements in different lipid environments. To create two different β-sheet channel topologies, eight PG-1 β-hairpins were initially assembled into an annular β-sheet (Fig. 2) with both antiparallel (turn-next-to-tail) and parallel (turn-next-to-turn) β-sheet motifs in an NCCN (Fig. 1) packing mode (14). The annular β-sheet channels of PG-1 were next embedded in the lipid bilayers in both intrinsic barrel-stave and toroidal membrane pores (7). For each of the membrane pore types, both zwitterionic lipid bilayer composed of POPC and anionic lipid bilayer composed of POPC/POPG (mole ratio 4:1) were constructed. Thus, for each antiparallel and parallel β-sheet channel, four independent simulations were carried out with different lipid environments.
In our simulations, both the antiparallel and parallel β-sheet channels slightly increase the outer diameter from their starting values. In particular, the outer sizes of the channels in the toroidal membrane pore are slightly larger than those in the barrel-stave membrane pore. In the torus shape of the toroidal pore, the channels are directly in contact with the hydrophilic lipid headgroups, allowing the long Arg side chains at both ends of the cylindrical channels an easy stretch into the amphipathic interface. The outward movement of the Arg side chains causes a slight increase in the overall channel size. In contrast to the outer dimension, the PG-1 channels slightly decrease (or do not alter) the inner pore diameter during the simulations. For the same bilayer topology, the channels have slightly larger water pores in the presence of anionic lipids, indicating that the pore is more accessible to solvent. On the other hand, the shape of the water pore evolves significantly from a straight cylindrical tube in the initial channels (Fig. 2) to a jagged tube in the relaxed channels (Fig. 3). The inhomogeneous shapes of the water pores suggest that the channel is not a rigid body as a whole, and that each subunit in the channel has an independent motion in the lipid bilayer. In general, the channels have similar shapes and dimensions in both zwitterionic and anionic lipid bilayers. We suggest that the effects of anionic lipids are less important once the channel has formed, but anionic lipids significantly contribute to the PG-1 insertion into the lipid bilayer. Without anionic lipids, PG-1 aggregates into β-sheets on the bilayer surface, whereas PG-1 induces significant bilayer thinning with anionic lipids, producing bilayer defects (20). The peptides easily insert into the bilayer from the defects and form an oligomeric structure (11).
The averaged outer diameters ∼3.56 and ∼3.58 nm for the antiparallel and parallel β-sheet channels, respectively, are slightly less than the ∼4.2 nm obtained from solid-state NMR spectroscopy for the PG-1 β-barrel in POPE/POPG membrane (11). The inconsistency in the outer size could result from the different number of peptides: the PG-1 β-barrel contains eight to 10 peptides, which is larger than (or at least equal to) the simulated PG-1 channels involving only eight peptides. The different sizes of the PG-1 oligomers may also lead to the discrepancy in the size of the water pore. In the simulations, the average diameters of the water pores are ∼0.93 and ∼0.96 nm for the antiparallel and parallel β-sheet channels, respectively, whereas experiment estimates the PG-1 β-barrel diameter to be ∼2.1 nm (11). The experimental estimation of the pore diameter derives from observed size-dependent blockage of PG-1 pores by PEG across the membrane: PEG molecules with hydrodynamic radii up to 0.94 nm crossed the membrane; on the other hand, those with 1.05 nm were blocked by the PG-1 water pore. This indirect estimation of the pore size is based on the assumption that the PG-1 β-barrel is a rigid body. The simulations directly measure the pore size from the atomic-level channel structures.
The subunit morphology observed here for the β-sheet channels of PG-1 is shared by other β-sheet channels (Fig. 9 a). Recent simulations of the Alzheimer amyloid Aβ channels have shown that these channels also abandon the perfect annular morphology. Instead, the annular Alzheimer channels divide into three to six localized β-sheet subunits (Fig. 9 b) (32,33). The Aβ channel dimensions, shape, and subunit organization were in good agreement with high-resolution AFM images of other amyloid channels (Fig. 9 c) (26,27), validating it as a viable candidate channel model. Consistent with the Aβ channels, in all simulated membrane environments, the PG-1 channels do not retain the perfect annular channel shape, breaking into subunit organization with several localized β-sheets (Fig. 9 a). The antiparallel β-sheet channels divide into four localized β-sheet subunits. Each β-sheet subunit is an NCCN dimer favoring the C-C dimer interface. As observed in the previous PG-1 dimer simulations, the NCCN dimer is held tightly by at least two intermolecular backbone H-bonds between two cysteine residues at the C-terminal strands from each monomer (21). The cysteine is relatively rigid due to the disulfide bond. The N-N dimer interface in the antiparallel β-sheet channels is broken due to the repulsive force between Arg residues in the loop (Arg9 and Arg10) from one monomer, and in the N-terminal strand (Arg1 and Arg4) from the other monomer, destabilizing intermolecular backbone H-bonds between the N-strands (Fig. 1 a). However, in the C-C dimer interface, the repulsive force between Arg residues in the loop (Arg10 and Arg11) and in the C-terminal strand (Arg18) is weak, because their location is relatively distant. Antiparallel NCCN packing was also found for a PG-1 dimer in DPC micelles by solution NMR (48).
FIGURE 9.

Side-by-side comparison between the PG-1 and Aβ channels. All channels are viewed from the top leaflet of the lipid bilayer. (a) The simulated PG-1 channels show the antiparallel (Aba-pc, Ato-pc, Aba-pg, and Ato-pg channels from left) and parallel (Pba-pc, Pto-pc, Pba-pg, and Pto-pg channels from left) β-sheet channels. The PG-1 channels are depicted in a cartoon representation with a transparent surface. Each subunit in the channels is colored in a different color. The discontinuous β-sheet network determines the boundary between the subunits in the channels. Antiparallel β-sheet channels of PG-1 contain four subunits. Each subunit is a β-sheet dimer (peptides 1 and 2 (green), peptides 3 and 4 (blue), peptides 5 and 6 (red), and peptides 7 and 8 (yellow)). Parallel β-sheet channels contain four to five subunits. The shapes of the subunits vary, with β-hairpin monomers, and β-sheet dimer or trimer; there are four subunits in the Pba-pc channel (peptide 1 (green), peptides 2–4 (blue), peptide 5 (red), and peptides 6–8 (yellow)), five subunits in the Pto-pc channel (peptides 1–8 (green), peptides 2 and 3 (blue), peptides 4 and 5 (red), peptide 6 (yellow), and peptide 7 (cyan)), four subunits in the Pba-pg channel (peptides 1–8 (green), peptides 2 and 3 (blue), peptides 4 and 5 (red), and peptides 6 and 7 (yellow)), and five subunits in the Pto-pg channel (peptides 1–8 (green), peptides 2 and 3 (blue), peptide 4 (red), peptide 6 (yellow), and peptide 7 (magenta); here peptide 5 is disordered). (b) The simulated channels show the Aβ17-42 and Aβ9-42 channels (24-mer). (Taken from Jang et al. (32). Permission obtained.) (c) AFM images show the Aβ1-40 and Aβ1-42 channels. (Taken from Quist et al. (26) and Lin et al. (27). Permission obtained.)
The PG-1 parallel β-sheet channels also divide into four to five β-sheet subunits. However, unlike the antiparallel β-sheet channels, the shapes of the subunits vary, with β-hairpin monomers, and β-sheet dimer or trimer. The parallel β-sheet channels favor the CNNC dimer formation, indicating that the N-N dimer interface is more stable than the C-C interface. During the simulations, we observed that the N-N interface has an additional intermolecular backbone H-bond between Gly2-Gly2, whereas the C-C dimer interface loses the H-bond between Arg11-Cys13 due to their long distance. This suggests that with more intermolecular backbone H-bond interactions, the N-N interface is more stabilized. However, the simulation result for the parallel β-sheet channels favoring the CNNC dimer formation is inconsistent with the experimental PG-1 β-barrel with NCCN dimers (11,12). Experiment suggests that the N-N dimer interface is unstable due to the cationic interaction between Arg4 residues. In the N-N interface, there is no repulsive force between the Arg4 side chains from neighboring monomers, because they are oriented opposite to each other: one Arg4 side chain points to the interior water pore, whereas the other one points to lipids. In contrast to the antiparallel β-sheet of PG-1 dimer with the common NCCN packing mode, the intermolecular parallel packing of a PG-1 β-hairpin may fluctuate depending on the surrounding environments, e.g., lipid composition, bilayer structure, and peptide location. Solid-state aggregates of PG-1 show the parallel NCCN packing mode (14). NMR distance measurements indicate a parallel NCCN dimer of PG-1 in the zwitterionic POPC bilayer (15) and parallel β-barrel with NCCN packing in the toroidal pore in anionic POPE/POPG bilayer (11,12). At the amphipathic interface of POPC bilayer, the parallel NCCN dimers of PG-1 are more active, but have less stable C-C intermolecular interface as compared to the antiparallel NCCN dimers (21). Our simulations show that parallel β-sheet channels favor the CNNC packing in the anionic POPC/POPG bilayer and in the toroidal pore membranes, whereas they have semistable NCCN packing in the POPC bilayer with barrel-stave setting. We speculate that the competition between the intermolecular interactions at the C-C and N-N interfaces could determine the peptide packing in the PG-1 channels in the different environments.
PG-1 β-barrel is reported to induce a toroidal membrane pore (11). Our simulation results also show that PG-1 channels preserve a toroidal membrane pore. However, the intrinsic barrel-stave membrane pore does not support PG-1 channels, because the initially flattened two lipid leaflets bend toward each other, producing significant thinning of the bilayer near the channel edges. Although the lipid bending is similar to that observed in the toroidal membrane pore, the two lipid leaflets do not merge due to the hydrophobic residues in the middle of the channel, preventing the polar headgroups from penetrating into the bilayer center, inducing a semitoroidal membrane pore. The conformation of the PG-1 channel is well supported by the semitoroidal membrane pore, and the channels interact more strongly with lipids in the semitoroidal pore as compared to the perfect toroidal membrane pore.
Our simulation results provide important information for the PG-1 channel activity against bacteria. Mechanistically, PG-1 presumably forms weak anion-selective channels in planar phospholipids bilayers, causing potassium leakage from liposomes (5). In the simulations, the selective anion conductance can be observed in the channels, indicating that the channels provide low free energy profile for Cl− in the pore, whereas the channels prevent Na+ from crossing the pore by a high free energy barrier. The unregulated anionic flow across the membrane might trigger cation leakage leading to disruption of cellular ionic homeostasis. The proposed mechanism is similar to the Alzheimer Aβ (32,33) and other amyloid channels (26,27) where the β-sheet channels are nonphysiological, leaking ions through channel pores that are enclosed by several β-sheet subunits. The computational model of the PG-1 channel suggests that the antiparallel β-sheet channels provide the double wells profile in the free energy changes for anion crossing, whereas the parallel β-sheet channels exhibit the single well profile in the free energy changes suggesting higher anion activity. This is consistent with experimental observations that the parallel PG-1 dimer is biologically more active, inserting into POPC membrane (13–15), and that parallel PG-1 β-barrel forms a water pore in the anionic POPE/POPG membrane (11).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
This project has been funded in whole or in part with funds from the National Cancer Institute, National Institutes of Health (contract number N01-CO-12400). This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. It was also supported by the Extramural Research Program of the National Institutes of Health (R.L.). This research was funded in part by the US Army Medical Research Acquisition Activity (grant W81XWH-05-1-0002) (B.M.). This study used the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Editor: Jose Onuchic.
References
- 1.Sitaram, N., and R. Nagaraj. 2002. Host-defense antimicrobial peptides: importance of structure for activity. Curr. Pharm. Des. 8:727–742. [DOI] [PubMed] [Google Scholar]
- 2.Hancock, R. E., and A. Rozek. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206:143–149. [DOI] [PubMed] [Google Scholar]
- 3.Kourie, J. I., and A. A. Shorthouse. 2000. Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell Physiol. 278:C1063–C1087. [DOI] [PubMed] [Google Scholar]
- 4.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature. 415:389–395. [DOI] [PubMed] [Google Scholar]
- 5.Sokolov, Y., T. Mirzabekov, D. W. Martin, R. I. Lehrer, and B. L. Kagan. 1999. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta. 1420:23–29. [DOI] [PubMed] [Google Scholar]
- 6.Yang, L., T. M. Weiss, R. I. Lehrer, and H. W. Huang. 2000. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 79:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250. [DOI] [PubMed] [Google Scholar]
- 8.Fahrner, R. L., T. Dieckmann, S. S. Harwig, R. I. Lehrer, D. Eisenberg, and J. Feigon. 1996. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 3:543–550. [DOI] [PubMed] [Google Scholar]
- 9.Kokryakov, V. N., S. S. Harwig, E. A. Panyutich, A. A. Shevchenko, G. M. Aleshina, O. V. Shamova, H. A. Korneva, and R. I. Lehrer. 1993. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 327:231–236. [DOI] [PubMed] [Google Scholar]
- 10.Miyasaki, K. T., and R. I. Lehrer. 1998. Beta-sheet antibiotic peptides as potential dental therapeutics. Int. J. Antimicrob. Agents. 9:269–280. [DOI] [PubMed] [Google Scholar]
- 11.Mani, R., S. D. Cady, M. Tang, A. J. Waring, R. I. Lehrer, and M. Hong. 2006. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA. 103:16242–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang, M., A. J. Waring, and M. Hong. 2007. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 129:11438–11446. [DOI] [PubMed] [Google Scholar]
- 13.Buffy, J. J., A. J. Waring, and M. Hong. 2005. Determination of peptide oligomerization in lipid bilayers using 19F spin diffusion NMR. J. Am. Chem. Soc. 127:4477–4483. [DOI] [PubMed] [Google Scholar]
- 14.Tang, M., A. J. Waring, and M. Hong. 2005. Intermolecular packing and alignment in an ordered β-hairpin antimicrobial peptide aggregate from 2D solid-state NMR. J. Am. Chem. Soc. 127:13919–13927. [DOI] [PubMed] [Google Scholar]
- 15.Mani, R., M. Tang, X. Wu, J. J. Buffy, A. J. Waring, M. A. Sherman, and M. Hong. 2006. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 45:8341–8349. [DOI] [PubMed] [Google Scholar]
- 16.Herce, H. D., and A. E. Garcia. 2007. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA. 104:20805–20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mecke, A., D. K. Lee, A. Ramamoorthy, B. G. Orr, and M. M. Banaszak Holl. 2005. Membrane thinning due to antimicrobial peptide binding: an atomic force microscopy study of MSI-78 in lipid bilayers. Biophys. J. 89:4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen, F. Y., M. T. Lee, and H. W. Huang. 2003. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys. J. 84:3751–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heller, W. T., A. J. Waring, R. I. Lehrer, T. A. Harroun, T. M. Weiss, L. Yang, and H. W. Huang. 2000. Membrane thinning effect of the beta-sheet antimicrobial protegrin. Biochemistry. 39:139–145. [DOI] [PubMed] [Google Scholar]
- 20.Jang, H., B. Ma, T. B. Woolf, and R. Nussinov. 2006. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 91:2848–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang, H., B. Ma, and R. Nussinov. 2007. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gidalevitz, D., Y. Ishitsuka, A. S. Muresan, O. Konovalov, A. J. Waring, R. I. Lehrer, and K. Y. Lee. 2003. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA. 100:6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sitaram, N., and R. Nagaraj. 1999. Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim. Biophys. Acta. 1462:29–54. [DOI] [PubMed] [Google Scholar]
- 24.Hancock, R. E., and R. Lehrer. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16:82–88. [DOI] [PubMed] [Google Scholar]
- 25.Ludtke, S. J., K. He, Y. Wu, and H. W. Huang. 1994. Cooperative membrane insertion of magainin correlated with its cytolytic activity. Biochim. Biophys. Acta. 1190:181–184. [DOI] [PubMed] [Google Scholar]
- 26.Quist, A., I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso, and R. Lal. 2005. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 102:10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, H., R. Bhatia, and R. Lal. 2001. Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15:2433–2444. [DOI] [PubMed] [Google Scholar]
- 28.Lau, T. L., E. E. Ambroggio, D. J. Tew, R. Cappai, C. L. Masters, G. D. Fidelio, K. J. Barnham, and F. Separovic. 2006. Amyloid-beta peptide disruption of lipid membranes and the effect of metal ions. J. Mol. Biol. 356:759–770. [DOI] [PubMed] [Google Scholar]
- 29.Lashuel, H. A., D. Hartley, B. M. Petre, T. Walz, and P. T. Lansbury Jr. 2002. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 418:291. [DOI] [PubMed] [Google Scholar]
- 30.Kawahara, M., and Y. Kuroda. 2000. Molecular mechanism of neurodegeneration induced by Alzheimer's beta-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res. Bull. 53:389–397. [DOI] [PubMed] [Google Scholar]
- 31.Arispe, N., H. B. Pollard, and E. Rojas. 1994. beta-Amyloid Ca(2+)-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 140:119–125. [DOI] [PubMed] [Google Scholar]
- 32.Jang, H., J. Zheng, R. Lal, and R. Nussinov. 2008. New structures help the modeling of toxic amyloidbeta ion channels. Trends Biochem. Sci. 33:91–100. [DOI] [PubMed] [Google Scholar]
- 33.Jang, H., J. Zheng, and R. Nussinov. 2007. Models of beta-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 93:1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirakura, Y., and B. L. Kagan. 2001. Pore formation by beta-2-microglobulin: a mechanism for the pathogenesis of dialysis associated amyloidosis. Amyloid. 8:94–100. [DOI] [PubMed] [Google Scholar]
- 35.Hirakura, Y., R. Azimov, R. Azimova, and B. L. Kagan. 2000. Polyglutamine-induced ion channels: a possible mechanism for the neurotoxicity of Huntington and other CAG repeat diseases. J. Neurosci. Res. 60:490–494. [DOI] [PubMed] [Google Scholar]
- 36.Lin, M. C., T. Mirzabekov, and B. L. Kagan. 1997. Channel formation by a neurotoxic prion protein fragment. J. Biol. Chem. 272:44–47. [DOI] [PubMed] [Google Scholar]
- 37.Castano, S., B. Desbat, and J. Dufourcq. 2000. Ideally amphipathic beta-sheeted peptides at interfaces: structure, orientation, affinities for lipids and hemolytic activity of (KL)(m)K peptides. Biochim. Biophys. Acta. 1463:65–80. [DOI] [PubMed] [Google Scholar]
- 38.Bechinger, B. 2000. Understanding peptide interactions with the lipid bilayer: a guide to membrane protein engineering. Curr. Opin. Chem. Biol. 4:639–644. [DOI] [PubMed] [Google Scholar]
- 39.Zakharov, S. D., E. A. Kotova, Y. N. Antonenko, and W. A. Cramer. 2004. On the role of lipid in colicin pore formation. Biochim. Biophys. Acta. 1666:239–249. [DOI] [PubMed] [Google Scholar]
- 40.Allende, D., S. A. Simon, and T. J. McIntosh. 2005. Melittin-induced bilayer leakage depends on lipid material properties: evidence for toroidal pores. Biophys. J. 88:1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang, H., N. Michaud-Agrawal, J. M. Johnston, and T. B. Woolf. 2008. How to lose a kink and gain a helix: pH independent conformational changes of the fusion domains from influenza hemagglutinin in heterogeneous lipid bilayers. Proteins. 72:299–312. [DOI] [PubMed] [Google Scholar]
- 42.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 43.Smart, O. S., J. M. Goodfellow, and B. A. Wallace. 1993. The pore dimensions of gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen, T. W., O. S. Andersen, and B. Roux. 2006. Molecular dynamics - potential of mean force calculations as a tool for understanding ion permeation and selectivity in narrow channels. Biophys. Chem. 124:251–267. [DOI] [PubMed] [Google Scholar]
- 46.de Groot, B. L., and H. Grubmuller. 2001. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- 47.Leontiadou, H., A. E. Mark, and S. J. Marrink. 2007. Ion transport across transmembrane pores. Biophys. J. 92:4209–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roumestand, C., V. Louis, A. Aumelas, G. Grassy, B. Calas, and A. Chavanieu. 1998. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett. 421:263–267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.