

Abstract
Electrostatic interactions between negatively charged membranes and basic peptides/protein domains have been implicated as the driving force for several important processes, often involving membrane aggregation, fusion, or phase separation. Recently, acidic lipids were reported to both catalyze amyloid fiber formation by amyloidogenic proteins/peptides and induce formation of “amyloid-like” fibrils by nonamyloidogenic proteins. This study aims to characterize the structure of the aggregates of a basic protein (lysozyme) and negatively charged membranes (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine 4:1 mixture) at the molecular level, using Förster resonance energy transfer. It is concluded that lysozyme induced formation of a “pinched lamellar” structure, with reduced interbilayer distance in the regions where there is bound protein and increased interbilayer distance (stabilized by hydration repulsion) outside these areas.
INTRODUCTION
Acidic phospholipids often promote membrane binding of several protein modules via electrostatic interactions between the negatively charged lipid headgroups and clusters of basic groups on the proteins (1). Common phenomena coupled with this association are formation of lateral domains in mixed lipid membranes (2) and membrane aggregation and/or fusion (3). On the other hand, binding to membrane lipids has been recognized increasingly as an important step in the aggregation and cytotoxicity of several amyloidogenic proteins (4–6). Membranes have been implicated both as the targets of toxicity, via membrane disruption, and as the catalysts that facilitate protein aggregation. The lipid composition of the membrane, particularly its anionic phospholipid content, also seems to play an important role in this process: the establishment of electrostatic interactions with acidic lipids favors membrane-induced protein misfolding and subsequent aggregate nucleation, as well as prefibrillar aggregate interaction with the membranes, by both amyloidogenic peptides (e.g., (7,8)) and proteins (e.g., (9–11)). In addition, it has been recently reported that membranes containing negatively charged phospholipids, like phosphatidylserine, can also trigger rapid amyloidlike fiber formation by a variety of several nonamyloidogenic proteins, such as cytochrome c, lysozyme, and myoglobin (12–14). These soluble proteins, as well as the islet amyloid polypeptide (15) have been described as producing mature fibrils containing both protein and acidic lipid components. However, details regarding the arrangement at the molecular level of protein and lipid in these supramolecular assemblies are still missing. In an effort to further elucidate the structure of these protein-membrane aggregates, we chose to investigate the interaction of natively folded lysozyme with phosphatidylserine-containing lipid vesicles using Förster resonance energy transfer (FRET) methodologies.
Lysozyme is a stable, highly basic small globular protein with a well characterized three-dimensional structure (16). This protein, widely distributed in biological fluids and tissues, and best known for its ability to hydrolyze the β(1–4) linkage between N-acetylmuramic acid and N-acetylglucosamine (muramidase activity), leading to degradation of peptidoglycan in the cell wall and rapid killing of Gram-positive bacteria, has also often been used as a model to study lipid-protein interactions (e.g., (17–20)). Lysozyme is known to exhibit very weak nonspecific binding to lipid bilayers of phosphatidylcholine (PC) and strong binding to PC containing negatively charged phospholipids (17,18). This binding was also shown to be strongly dependent upon the protein net charge, pH, and ionic strength of the medium, and the molar fraction of negatively charged phospholipids (17–20), as expected for a predominantly electrostatics-mediated interaction. It is interesting to note that several recent studies have implicated not only the catalytic but also the lipid-binding properties of lysozyme in its bactericidal action. It has been found recently that the denatured (21) and mutated forms of the protein (22,23), which lack enzymatic activity, are still bactericidal. Furthermore, several studies have shown that naturally occurring proteases like pepsin and trypsin are capable of breaking down lysozyme to release antimicrobial peptides (24,25), and several synthetic peptides derived from this enzyme can give rise to membrane permeabilization and loss of viability of bacterial cells (26,27).
Because of its large sensitivity to distances in the 1- to 10-nm scale, FRET is particularly suited to the study of structural effects of membrane-active peptides or proteins on the organization of lipid bilayers (28). Carefully designed experiments allow detection of lateral phase separation (and estimation of the size of nanodomains (29,30) or measurement of relevant distances in the direction normal to the bilayer plane (31–33)). Most of the past work using FRET spectroscopy in biophysical applications has been carried out in photostationary conditions (steady-state measurement). However, to make the most of the technique's potential, measurement should be made of the actual fluorescence-decay kinetics of the FRET donor in the presence of the acceptor, combined with a careful analysis based on a meaningful model. This was illustrated recently in our FRET study of the effect of the model basic peptide hexalysyltryptophan (K6W) on the structure of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)/1,2-dipalmitoyl-sn-glycero-3-phosphoserine (DPPS) mixtures (34). In this work, we showed that the enhancement of FRET between PC-mimicking fluorescent lipids observed in the fluid-lipid mixture in the presence of K6W was due to the formation of a multilamellar geometry rather than peptide-induced phase separation.
This study aims to characterize the structure of the aggregates formed by a basic protein (lysozyme) and negatively charged membranes (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPC/POPS) 4:1 mixture) at the molecular level, using FRET methodologies. FRET experiments between lipid-linked donor and acceptor molecules and a protein-bound donor and a membrane probe were both able to detect lysozyme-mediated aggregation of the anionic lipid vesicles. It was concluded that lysozyme influenced the intermembrane separation distances in the protein-lipid supramolecular assemblies formed, yielding a reduced interbilayer distance for the regions where the protein and lipid membranes associate, and thus giving rise to localized “pinched regions.”
MATERIALS AND METHODS
Materials
POPC, POPS, and N-(lissamine-rhodamine B)-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (Rh-PE) were obtained from Avanti Polar Lipids (Birmingham, AL). 2-(4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (BODIPY-PC), 1,6-diphenyl-1,3,5-hexatriene (DPH), and Alexa-488 (carboxylic acid, succinimidyl ester, mixed isomers, dilithium salt) were obtained from Invitrogen (Carlsbad, CA). Lysozyme from chicken egg white was purchased from Fluka Biochemika (Buchs, Switzerland). 4-(2-hydroxyethyl)-1-piperazine sulfonic acid (HEPES), KOH, and EDTA (all from Merck, Darmstatdt, Germany) were used to prepare the buffer solution, 20 mM HEPES, 0.1 mM EDTA, pH 7.4. All organic solvents were of spectroscopic grade and came from Merck. All the above materials were used without further purification. Distilled water (>18 MΩ·cm), produced using a Millipore (Billerica, MA) system, was used throughout the work. The concentrations of stock solutions of the probes were determined spectrophotometrically using the molar absorption coefficient values ɛ(DPH, 358 nm, in CHCl3) = 8.06 × 104 M−1 cm−1 (35), ɛ(Rh-PE, 560 nm, in MeOH) = 7.5 × 104 M−1 cm−1 (36), and ɛ(BODIPY-PC, 509 nm, in EtOH) = 8.6 × 104 M−1 cm−1 (36).
Labeling of lysozyme with Alexa-488
Lysozyme labeling was carried out essentially according to the instructions provided by the manufacturer. Briefly, lysozyme and the carboxylic acid succinimidyl ester of Alexa488 were dissolved in 0.1 M sodium bicarbonate buffer, pH 8.3, and dimethylsulphoxide (DMSO), respectively. Covalent labeling reactions were allowed to proceed in the dark for 2 h at room temperature under constant stirring at a molar dye/protein ratio of 2. The labeling reaction was stopped using 1.2 M hydroxylamine, pH 8.5, followed by a further incubation for 1 h at room temperature. Alexa-488-labeled lysozyme (A488-lysozyme) was separated from unreacted free probe by gel filtration through a Sephadex G-25 column equilibrated in 50 mM sodium phosphate buffer, pH 7.4. The final protein concentration and the bound label/protein stoichiometry were determined spectrophotometrically, using the molar absorptivities ɛ495 nm = 7.1 × 104 M−1 cm−1 and ɛ280 nm = 7.8 × 103 M−1 cm−1 for A488-lysozyme (36), and ɛ280 nm = 37,680 M−1 cm−1 for lysozyme.
Sample preparation
Large unilamellar vesicles (LUVs, ∼100 nm in diameter), containing phospholipids POPC and POPS (molar ratio 4:1) and the desired amount of BODIPY-PC, Rh-PE, or DPH (in the experiments that used these probes; see FRET measurements subsection below) were prepared by extrusion of lipid dispersions through 100-nm pore diameter polycarbonate membranes as previously described (37). The resulting lipid dispersions were stored at 4°C and used within 24 h of preparation. The desired amount of lysozyme was added to LUVs from a fresh stock solution. The concentrations of phospholipid stock solutions were determined using phosphate analysis (38).
Liposome aggregation measurements
Protein-induced aggregation of liposomes was determined from the change in absorbance that follows the increase in turbidity of the liposome suspension upon addition of lysozyme or A488-lysozyme. Absorbance was measured at 360 nm in a spectrophotometer using 0.5-cm quartz cuvettes. The lipid suspensions were prepared independently in the presence and in the absence of protein and were allowed to stand for at least 1 h at room temperature before the measurements were carried out after extensive mixing of the samples.
FRET measurements
All energy transfer experiments were carried out at room temperature after addition of an adequate amount (varied between 0.2 and 4 mol %) of lysozyme to POPC/POPS 4:1 LUVs. For the BODIPY-PC/Rh-PE FRET pair, the donor/lipid molar ratio was kept at 1:1000. For the A488-lysozyme/Rh-PE and lysozyme/DPH FRET pairs, the donor concentration was given by the protein content (multiplied by the dye/protein labeling ratio (D/P) of 0.44 for A488-lysozyme). The acceptor/lipid molar ratios used were 1:400, 1:400, and 1:200 for the BODIPY-PC/Rh-PE, A488-lysozyme/Rh-PE, and lysozyme/DPH FRET pairs, respectively.
The critical parameter in FRET is the Förster radius, R0. It is the distance at which the transfer efficiency is 50% for an isolated donor-acceptor pair and was calculated as
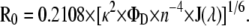 |
(1) |
In Eq. 1, κ2 is the orientation factor (assumed in this study as 2/3, the dynamical isotropic limit value; see (39,40) for detailed discussions on the validity of this assumption for fluid bilayers), ΦD is the donor quantum yield in the absence of acceptor, n is the refractive index, λ is the wavelength, and J(λ) is the spectral overlap integral between the emission spectrum of the donor, ID(λ), and the molar absorption spectrum of the acceptor, ɛA(λ), given by
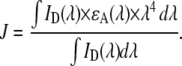 |
(2) |
In Eq. 1, if the λ units used are nanometers, the calculated R0 has Å units. The quantum yield of the donor, ΦD, was estimated in the presence of LUVs for A488-lysozyme, D/P = 0.44 (ΦD = 0.47), taking into account the variation of lifetime-weighted quantum yield relative to A488-lysozyme in buffer (for the latter, Φ = 0.71 was measured using fluorescein in NaOH, 0.1 M, as reference, Φ = 0.92 (41)). For BODIPY-PC, the published value in ethanol (ΦD = 0.90) was used (36). Using these values, together with the measured spectra, R0 values of 5.5 nm and 4.9 nm were obtained for the BODIPY-PC/Rh-PE and A488-lysozyme/Rh-PE FRET pairs, respectively. For lysozyme/DPH, the value R0 = 3.3 nm, reported for Trp(nicotinic acetylcholine receptor)/DPH (42), was used.
FRET modeling
To obtain quantitative topological and topographical information regarding the interaction of lysozyme with the model membranes, two donor-acceptor arrangements were considered: 1), FRET between two membrane probes (Figs. 1, A and B), and 2), FRET between lysozyme and a convenient membrane probe acceptor (Fig. 1, C and D). For each arrangement, different FRET formalisms, corresponding to different geometries of donor/acceptor relative distribution, were addressed, as briefly presented below.
FIGURE 1.

Schematic representations of the FRET models used for both experimental setups explored in this work: FRET between two membrane probes (A and B), and where donors are located in the protein and acceptor is a membrane probe (C and D). A and C illustrate topological models for protein interaction with a single lipid bilayer, whereas B and D describe a multibilayer arrangement with protein molecules sandwiched between adjacent bilayers. Only two bilayers are depicted, because FRET to further acceptor planes is negligible. h1, h2, and h3 are the distances between planes of donors and acceptors taken into account in the FRET models.
FIGURE 2.

(A) Aggregation of liposomes induced by lysozyme. The indicated protein concentrations were added to POPC/POPS 4:1 lipid vesicles (total lipid 215 μM), and the extent of light scatter by the suspensions was measured by absorbance at 360 nm. The line is a mere guide to the eye. Error bars are ± SD for triplicate measurements (lipid vesicles only, lipid vesicles plus donor, and lipid vesicles plus donor and acceptor). (B) Efficiency of FRET (Eq. 11) between BODIPY-PC (BODIPY-PC/total lipid = 1:1000) and Rh-PE (Rh-PE/total lipid = 1:400) in 4:1 POPC/POPS LUVs (total lipid 215 μM) as a function of the concentration of added lysozyme. The line is a mere guide to the eye. The error bars' extremes are the results of two different measurements.
Fig. 1 A depicts the situation of FRET between membrane probes without vesicle aggregation. Each donor fluorophore senses only acceptors located either in the same bilayer leaflet (at a transverse distance h1) or in the opposing leaflet (at a transverse distance h2), but not acceptors located in other bilayers. In this case, the donor decay in the presence of uniformly distributed acceptor is given by
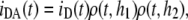 |
(3) |
where
 |
(4) |
is the donor decay in the absence of acceptor (given by a sum of exponentials), and the FRET contributions are calculated from (32)
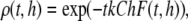 |
(5) |
where
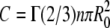 |
(6) |
and
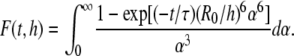 |
(7) |
In Eqs. 5 and 6, 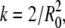 whereas n is the surface density of acceptors, τ is the average donor lifetime in the absence of acceptor (34), and Γ is the complete gamma function.
whereas n is the surface density of acceptors, τ is the average donor lifetime in the absence of acceptor (34), and Γ is the complete gamma function.
If h1 ≪ R0 (identical transverse locations of donor and acceptor fluorophores), the corresponding FRET contribution is simplified, leading to
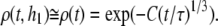 |
(8) |
Fig. 1 B depicts the situation of protein-mediated bilayer aggregation. In this case, for each donor, a new acceptor plane becomes available at distance h3, leading to an additional term in the donor decay law,
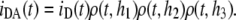 |
(9) |
Fig. 1 C refers to FRET from donors bound to proteins adsorbed on a lipid bilayer, without bilayer aggregation, to acceptor membrane probes. In the depicted situation, there would be a single acceptor plane available for FRET, either because all acceptors have the same transverse location (e.g., the center of the bilayer) or because the acceptors located in the furthest leaflet are too distant from donors for efficient FRET to occur. The donor decay would then be given simply by
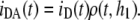 |
(10) |
Of course, if protein-mediated bilayer aggregation should occur, there would be an additional acceptor plane available, at a distance h2. In case the protein-bound donor is located in a symmetrical position between the two leaflets, we would have h2 ≈ h1, as depicted in Fig. 1 D. In this situation, Eq. 10 is still valid, but now the numerical value of the acceptor concentration is the double of the previous scenario (as two acceptor planes at the same distance are equivalent to a single plane with doubled acceptor density).
A common observation in FRET experiments is FRET efficiency, E, which is calculated by
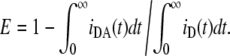 |
(11) |
Instrumentation
Absorption spectroscopy was carried out with a Shimadzu UV-3101PC spectrophotometer (Shimadzu Scientific Instruments, Columbia, MD). Steady-state fluorescence measurements were carried out with an SLM-Aminco 8100 Series 2 spectrofluorimeter (Spectronics, Rochester, NY) with double excitation and emission monochromators (MC-400) in a right-angle geometry. The light source was a 450-W Xe arc lamp and the reference was a Rhodamine B quantum counter solution. Correction of emission spectra was performed using the correction software of the apparatus. Quartz cuvettes 5 × 5 mm were always used.
Fluorescence decay measurements were carried out with time-correlated single-photon counting systems described elsewhere (29,39). Excitation and emission wavelengths, respectively, were 290 nm and 340 nm for wild-type lysozyme, 460 nm and 515 nm for A488-lysozyme, and 360 nm and 515 nm for BODIPY-PC. Timescales were chosen for each sample to observe the decay through two to three intensity decades. Instrumental response functions for deconvolution were generated from a scattering dispersion (silica, colloidal water suspension, Aldrich, Milwaukee, WI). Data analysis was carried out using a nonlinear least-squares iterative convolution method based on the Marquardt algorithm. The goodness of fit was judged from the experimental χ2 values, weighted residuals, and autocorrelation plots.
RESULTS AND DISCUSSION
FRET between lipid probes senses repeat distance of 9–10.5 nm
It has been reported that lysozyme binding to anionic liposomes is accompanied by aggregation and, sometimes, visible precipitation of liposomes (17), a result that was confirmed here. To evaluate the morphological transformation of POPC/POPS LUVs with molar ratio 4:1 upon lysozyme binding, the turbidity of liposome suspensions at a total lipid concentration of 215 μM was monitored in the presence of increasing amounts of lysozyme (up to 9 μM). As illustrated in Fig. 2 A, lysozyme caused an increase in light scattering of the lipid vesicles, which reached a plateau at a protein concentration of ∼2 μM, confirming that the binding of this protein to the lipid vesicles promoted its extensive aggregation and/or fusion. Direct visual inspection of the samples prepared with protein concentrations >2 μM revealed an extensive flocculation of the lysozyme-liposome dispersions that caused its progressive precipitation in the cuvettes during the measurements carried out in the absence of magnetic stirring. In addition, the inclusion of the lipid-linked donor (BODIPY-PC with molar ratio 1:1000) and acceptor (Rh-PE with molar ratio 1:400) in the composition of the liposomes did not have an appreciable effect upon the lysozyme's ability to cluster the phosphatidylserine-containing liposomes (Fig. 2 A).
To further study the effect of lysozyme on the structure of anionic lipid-containing membranes, time-resolved FRET experiments were carried out and analyzed using the formalisms presented above in the FRET modeling subsection. In a first arrangement, FRET between appropriate donor and acceptor membrane probes was measured. To this goal, a FRET pair with large R0 (5.5 nm) was used, with BODIPY-PC as donor and Rh-PE as acceptor. Upon increasing the concentration of lysozyme, a small but significant increase in FRET efficiency is observed. As shown in Fig. 2 B, this occurs for 1.0 μM < [lysozyme] < 2.0 μM, coincident with the lysozyme concentration range where this protein caused an extensive increase in the light scattering of the lipid suspension (Fig. 2 A). Despite being a useful indicator, FRET efficiency alone cannot resolve the changes in membrane lipid organization responsible for the observed variation, as exemplified and discussed in detail elsewhere (34). To this effect, a more rigorous test is model fitting to time-resolved FRET data, using the formalisms outlined above (see FRET modeling subsection).
To decrease the number of freely optimized fitting parameters, we draw here brief considerations regarding the relative donor and acceptor transverse locations in the system at hand. The location of the fluorophore in Rh-PE near the lipid/water interface (protruding ∼3.5 Å into the water phase) is known (43). Regarding BODIPY-PC, from quenching experiments, the existence of two probe populations, one with fluorophore looping to the interface and another with fluorophore deeply embedded within the bilayer, has been suggested (44). The first scenario corresponds to the schematic representation in Fig. 1, A and B, allowing us to take h1 ≈ 0 and use Eq. 8 in the calculation of the cis FRET component. The second scenario is similar, but with h1 ≈ h2 ≈ half the bilayer width. If BODIPY-PC existed in both conformations inside the bilayer, the actual decay kinetics would be a linear combination of the kinetics corresponding to these two topologies, lying between them. Looping of the BODIPY group would imply very efficient FRET to a plane of acceptors at a very near transverse location, and fitting this model to the decay measured from a heterogeneous population would lead to an overestimation of h2 and h3. Therefore, FRET data analysis with this model (pictured in Fig. 1, A and B) is expected to overestimate the repeat distance, h2 + h3. Conversely, a lower limiting value of repeat distance would be recovered from a model assuming no looping, with all BODIPY groups in the bilayer middle plane. The actual value is expected to lie between these two extremes, but given that the BODIPY-PC population with fluorophore looping to the interface seems to be the dominant one (44), the repeat distance recovered from this model geometry will probably be the most significant one.
Table 1 compares the fitting parameters recovered for the single-bilayer (Fig. 1 A and Eq. 3) and aggregated-bilayer (Fig. 1 B and Eq. 9) models assuming BODIPY looping, namely, the two lifetimes τ1 and τ2 and the amplitude ratio A2/A1; C, which is proportional to the acceptor concentration in both models (see Eq. 5) and 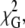 the global chi-square considering both donor-only and donor-plus acceptor samples for a given protein concentration. For the multibilayer model, the recovered h3 distance is also shown (h2 = 3.8 nm is recovered from the sample with no lysozyme, in accordance with the expected bilayer width, and fixed for all other samples).
the global chi-square considering both donor-only and donor-plus acceptor samples for a given protein concentration. For the multibilayer model, the recovered h3 distance is also shown (h2 = 3.8 nm is recovered from the sample with no lysozyme, in accordance with the expected bilayer width, and fixed for all other samples).
TABLE 1.
Comparison of the fitting parameters recovered for single-bilayer and aggregated-bilayer models
| Single bilayer*
|
Aggregated bilayers*
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [Lysozyme]/μM | τ1/ns | τ2/ns | A2/A1 | C | 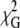 |
τ1/ns | τ2/ns | A2/A1 | h3/nm | C | 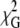 |
| 0 | 2.24 | 6.26 | 13.6 | 0.346 | 1.067 | 2.25 | 6.26 | 13.6 | >100 | 0.348 | 1.065 |
| 0.5 | 2.39 | 6.30 | 12.9 | 0.354 | 1.119 | 2.27 | 6.31 | 12.6 | 11.2 | 0.344 | 1.116 |
| 1.0 | 2.35 | 6.26 | 15.1 | 0.357 | 1.056 | 2.19 | 6.27 | 14.5 | 10.4 | 0.344 | 1.049 |
| 1.5 | 2.47 | 6.23 | 14.0 | 0.372 | 1.132 | 2.21 | 6.23 | 13.2 | 9.2 | 0.352 | 1.113 |
| 2.0 | 2.42 | 6.16 | 15.6 | 0.399 | 1.153 | 1.93 | 6.18 | 13.6 | 7.4 | 0.352 | 1.068 |
| 4.5 | 3.96 | 6.21 | 7.58 | 0.410 | 1.273 | 2.29 | 6.14 | 13.7 | 6.2 | 0.334 | 1.104 |
| 6.0 | 4.08 | 6.17 | 8.41 | 0.410 | 1.228 | 2.14 | 6.11 | 16.1 | 6.7 | 0.346 | 1.073 |
| 9.0 | 3.32 | 6.13 | 12.1 | 0.414 | 1.182 | 2.15 | 6.12 | 14.0 | 6.7 | 0.350 | 1.039 |
Parameters were obtained from pairwise global analysis of the time-resolved decays of BODIPY-PC (donor) in the absence and presence of Rh-PE (acceptor) for POPC/POPS 4:1 LUVs at different lysozyme concentrations (total lipid concentration = 215 μM, Rh-PE:total lipid = 1:400). The time-resolved FRET data were analyzed assuming BODIPY-PC looping (h1 ≈ 0; h2 = 3.8 nm was recovered from fitting the single-bilayer model to the data obtained from the sample with no lysozyme (see text for more details)). The two lifetimes τ1 and τ2, and the amplitude ratio A2/A1 (see Eq. 4) were linked in the global analysis of the donor-only and donor-plus-acceptor decays obtained with a given protein concentration; C is a parameter proportional to the acceptor concentration in both models (see Eq. 5).
Monomeric BODIPY fluorophore displays a single-exponential decay in polar environments such as methanol, acetonitrile, and mixtures of these solvents with water, with τ ≈ 6 ns (45). For all systems under study, the measured BODIPY-PC decay was biexponential, clearly dominated by an ∼6-ns component (93–94% amplitude; see Table 1). The residual ∼2-ns component (6–7% amplitude) could be ascribed to a small population of fluorophores located in a very apolar environment, in which the fluorescence lifetime of BODIPY fluorophore is markedly shorter (45). In the context of the latter hypothesis, this population would probably represent the donor population with the fluorophore buried in the bilayer center, whereas the unquenched component would reflect a population located in a more polar environment, possibly the water/lipid interface (the looping dominant population).
Comparing the two FRET models now under consideration, although the fitting statistics obtained with the single-bilayer model are satisfactory, there is a significant improvement (>10%) in the samples with the most protein when allowing for an additional acceptor plane, which would become available through bilayer aggregation. Fig. 3 shows decays of donor only and donor in the presence of acceptor for [lysozyme] = 9.0 μM and illustrates this improvement, which is especially evident from the autocorrelation of residuals plot (Fig. 3, lower). In addition, incorporation of multibilayer formation in the model naturally leads to the invariance of the recovered C with lysozyme concentration, whereas the single-bilayer model predicted an unphysical steady increase of this parameter, which could only be rationalized on the basis of an unclear decrease in bilayer area (20% for the [lysozyme] = 9.0 μM relative to the sample without protein). In a similar way, whereas the analysis with the single-bilayer formalism (Fig. 1 A and Eq. 3) leads to variations in the values recovered for the donor decay parameters (τ1, τ2, and A2/A1), mainly for the samples with higher lysozyme concentration (τ1 tends to increase, whereas A2/A1 tends to decrease), the use of the aggregated-bilayer (Fig. 1 B and Eq. 9) model results in a much increased overall consistency in values of these parameters. This is expected, since BODIPY-PC should not interact with the protein or be located in close proximity to the latter, and therefore, its decay should be unaffected by the increase in lysozyme concentration.
FIGURE 3.

(Upper) BODIPY-PC fluorescence decays in absence (donor only (D)) and presence (donor + acceptor (DA)) of Rh-PE (1:1000 and 1:400 relative to total lipid for donor and acceptor, respectively) in 4:1 POPC/POPS LUVs (total lipid 215 μM), in the presence of 9.0 μM lysozyme. Also shown are the global fits using the multibilayer model of Fig. 1 B and Eq. 9 (smooth gray lines) and the instrument response function (IRF). (Middle) Weighted residual plots for the fits of the single bilayer (left; see Fig. 1 A and Eq. 3) and multibilayer (right; see Fig. 1 B and Eq. 9) models to the DA decay shown in the upper panel. (Lower) Autocorrelation plots for the fits of the single bilayer (left; see Fig. 1 A and Eq. 3) and multibilayer (right; see Fig. 1 B and Eq. 9) models to the DA decay shown in the upper panel.
The aggregation process should involve more than two bilayers, and evolve into formation of multilamellar structures with protein sandwiched between adjacent bilayers, as recently verified in a simpler system (34). Because the interplanar distances are larger in this study, the contribution of acceptors in nonadjacent bilayers to the quenching of a given donor is negligible. The parameters recovered from the multibilayer model with the BODIPY donor group in the middle of the bilayer are similar to those obtained for the model with looping, with slightly worse statistics. For example, taking h1 = h2 = 2.0 nm, one recovers h3 = 7.0 and 7.1 nm, C = 0.305 and 0.311, and 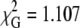 and 1.048 for [lysozyme] = 6.0 and 9.0 μM, respectively. The fact that better statistics are obtained with BODIPY looping than with BODIPY in the center of the bilayer agrees with the abovementioned observation of Kaiser and London (44) that the looping population is the dominant one for these probes.
and 1.048 for [lysozyme] = 6.0 and 9.0 μM, respectively. The fact that better statistics are obtained with BODIPY looping than with BODIPY in the center of the bilayer agrees with the abovementioned observation of Kaiser and London (44) that the looping population is the dominant one for these probes.
The range of lysozyme concentrations for which 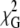 starts to worsen in the single-bilayer model is 1.0 μM < [lysozyme] < 2.0 μM, identical to the FRET efficiency increase seen in Fig. 2 B. Analysis with a formalism allowing for domain formation (34) (one of the possible justifications for the FRET efficiency increase) does not lead to any improvement (results not shown), ruling out the hypothesis of extensive protein-induced phase separation. The small magnitude of the measured effects is due to the fact that R0, which is a measure of the distance range probed by FRET with a given Förster pair, is smaller than the interplanar distance between donors and the newly available acceptors, h3. Due to limited availability of membrane probe FRET pairs, it was not possible to further increase R0 and thus increase sensitivity. The superiority of time-resolved fluorescence measurements coupled with global decay analysis allows the observation of small changes in FRET efficiency that are not accessible in a steady-state experiment. The clear trends obtained in the analysis allow the quantitative estimation of the lamellar repeat distance, which is given by h2 + h3 = 10.5 nm. These data also show that most structural changes accessible to this FRET pair occur in the range 1.0 μM < [lysozyme] < 2.0 μM for the studied lipid system.
starts to worsen in the single-bilayer model is 1.0 μM < [lysozyme] < 2.0 μM, identical to the FRET efficiency increase seen in Fig. 2 B. Analysis with a formalism allowing for domain formation (34) (one of the possible justifications for the FRET efficiency increase) does not lead to any improvement (results not shown), ruling out the hypothesis of extensive protein-induced phase separation. The small magnitude of the measured effects is due to the fact that R0, which is a measure of the distance range probed by FRET with a given Förster pair, is smaller than the interplanar distance between donors and the newly available acceptors, h3. Due to limited availability of membrane probe FRET pairs, it was not possible to further increase R0 and thus increase sensitivity. The superiority of time-resolved fluorescence measurements coupled with global decay analysis allows the observation of small changes in FRET efficiency that are not accessible in a steady-state experiment. The clear trends obtained in the analysis allow the quantitative estimation of the lamellar repeat distance, which is given by h2 + h3 = 10.5 nm. These data also show that most structural changes accessible to this FRET pair occur in the range 1.0 μM < [lysozyme] < 2.0 μM for the studied lipid system.
FRET between protein and membrane probes reports reduced repeat distance
In a second arrangement, pictured in Fig. 1, C and D, FRET was measured between a protein-bound donor and a membrane probe. The first FRET pair used in this setup was A488-lysozyme/Rh-PE. The Alexa Fluor 488 dye contains an amine-reactive succinimidyl ester group and is able to specifically modify the amino groups in proteins (46). Although seven amino groups on lysozyme, including the ɛ-amino group in lysine residues and the α-amino group at the N-terminus, could be modified by the Alexa succinimidyl ester, the reactivity of lysine residues has been shown to be primarily dependent on their relative surface accessibilities on the protein (47). The major modification sites detected in several studies were Lys97 and Lys33, followed by the amino group on the N-terminus and Lys1 (47–49). Therefore, the first two residues are also the best candidates for the modification site of the dye, given the fact that the conjugation reaction is based on the same chemistry (49). Furthermore, since for each Alexa 488 molecule covalently bound to lysozyme, the net charge of the protein is reduced by 3, it was important to check first whether the membrane binding ability of the enzyme was affected by the introduction of this label, because electrostatic interactions between anionic lipids and cationic lysozyme play a dominant role in this process. Using turbidimetry measurements, we verified that this difference in net positive charge between wild-type and derivatized lysozyme does not reflect significantly on their ability to induce vesicle aggregation in our experimental conditions (Fig. 4 A). In addition, and in accordance with the data presented above, Fig. 4 A shows that the phosphatidylserine-containing vesicles become saturated with lysozyme at ∼2 μM total protein concentration.
FIGURE 4.

(A) Influence of lysozyme labeling on protein-mediated aggregation of liposomes. The indicated protein concentrations of lysozyme and A488-lysozyme (D/P = 0.44) (open and solid symbols, respectively) were added to POPC/POPS 4:1 lipid vesicles (total lipid 215 μM), and the extent of light scatter by the suspensions was measured by absorbance at 360 nm. The lines are mere guides to the eye. Error bars are ± SD for duplicate measurements (lipid vesicles only and lipid vesicles plus acceptor). (B) Efficiency of FRET (Eq. 11) between A488-lysozyme (D/P = 0.44) and Rh-PE (Rh-PE/total lipid = 1:400) in 4:1 POPC/POPS LUVs (total lipid 215 μM) as a function of the concentration of added protein. The line is a mere guide to the eye.
Fig. 4 B shows that for this pair, significant FRET is observed up to 1.5 μM A488-lysozyme (D/P = 0.44), which constitutes direct evidence for lipid/A488-lysozyme association. For higher protein concentrations, i.e., upon exceeding the maximal lysozyme binding capacity of the liposomes, the energy transfer efficiencies between A488-lysozyme and Rh-PE progressively decreases, reflecting the presence of an increasing fraction of free protein molecules in the aqueous phase (Fig. 4 B). Whereas this is not a problem regarding the BODIPY-PC/Rh-PE pair discussed above, the same is not true when the FRET donor is attached to the protein as a covalently bound label. The presence of fluorescent donor unbound to the vesicles (and therefore unable to take part in FRET) leads to a decrease in FRET efficiency and effectively masks energy transfer between bound protein and membrane probe, as is clearly visible in Fig. 4 B. As a consequence, meaningful analysis of the data was only possible within the range where the vesicles are not saturated with protein.
The acceptor densities obtained from analysis of the decays of the samples with [A488-lysozyme] = 0.5 μM and [total lipid] = 215 μM show agreement with the theoretical expectation for protein adsorption to a single bilayer (Fig. 5 A). However, a clear increase in the recovered density to twice this value is observed for [A488-lysozyme] ≈ 1.0–1.5 μM, as illustrated in Fig. 5 A. This protein concentration range is similar to that for which there is an increase in FRET efficiency in the BODIPY-PC/Rh-PE pair (Fig. 2 B). The fact that there is no absolute coincidence between them, with the FRET increase occurring for higher protein concentrations for the BODIPY-PC/Rh-PE pair, will be discussed below. The rise in recovered acceptor concentration is due to multibilayer formation, with the protein sandwiched between two adjacent bilayers, resulting in double the number of acceptors sensed by each donor.
FIGURE 5.

(A) Acceptor numerical density, C, recovered from global analysis of fluorescence decays of FRET donor A488-lysozyme (D/P = 0.44) in the absence and presence of FRET acceptor Rh-PE (1 Rh-PE:400 total lipid) in 4:1 POPC/POPS vesicles (total lipid 215 μM). Experimental values (circles) are compared with theoretical expectations for single-bilayer (Fig. 1 C) and multibilayer (Fig. 1 D) models (dashed lines). (B) Global χ2 for the A488-lysozyme/Rh-PE pair samples, with [lysozyme] = 1.5 μM, as a function of the donor-acceptor interplanar distance (h1). The dashed lines are mere guides to the eye. (C and D) Same as A and B, but for the FRET pair lysozyme/DPH (1 DPH:200 total lipid).
As shown in Fig. 5 B for the A488-lysozyme/Rh-PE FRET pair ([A488-lysozyme] = 1.5 μM), model fitting has low sensitivity to the distance between donor and acceptor planes (h1), a result that is probably due to heterogeneity in donor transverse location. As discussed above, lysozyme labeling with Alexa-488 was not site-specific; and in addition, this protein can eventually assume several concentration-dependent orientations within the lipid bilayers. Both these effects are expected to contribute to broaden the range of possible donor plane-acceptor plane distances recovered in the data analysis. Even taking this into account, it is interesting to note that for this FRET pair the multilamellar repeat distance is considerably smaller than those obtained from FRET between two membrane probes described in the previous subsection. For the A488-lysozyme/Rh-PE pair, the absolute 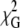 minima are obtained for donor-acceptor interplanar distance h1 ≈ 0.5–1.5 nm, corresponding to a multilamellar repeat distance of ∼5–7 nm (approximately 2h1 plus the thickness of one bilayer), which—it is interesting to note—is not larger than the sum of the thicknesses of one bilayer and one protein molecule.
minima are obtained for donor-acceptor interplanar distance h1 ≈ 0.5–1.5 nm, corresponding to a multilamellar repeat distance of ∼5–7 nm (approximately 2h1 plus the thickness of one bilayer), which—it is interesting to note—is not larger than the sum of the thicknesses of one bilayer and one protein molecule.
The second FRET pair explored in the experiments conducted with a protein-bound donor and a membrane probe was lysozyme (tryptophan (Trp) residues)/DPH. There are six tryptophan residues in wild-type lysozyme, and in our FRET experiment, any of them can be directly excited. In addition, homo-FRET among them cannot be excluded. The complexity of this system, containing multiple intrinsic tryptophan residues (donors) at different positions in the protein, strongly limited the quality of model fitting to the experimental data, as is evident from analysis of Fig. 5, C and D. However, apart from minor differences, the results presented the same overall trend as those described previously for the A488-lysozyme/Rh-PE pair, namely, the approximate doubling of the acceptor density obtained from the analysis of donor and donor + acceptor decays of the samples prepared with 0.5 < [lysozyme] < 1.5 μM. In addition, h1 was again ill defined from the fitting of the model to the data (Eq. 10), with 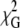 starting to increase significantly only for h1 > 1.5 nm. Considering that in this case the acceptors (DPH molecules) are located near the center of the bilayer, this would correspond to a repeat distance similar to the actual bilayer width (∼2h1 in this case). To explain these results, one must conclude that the protein partially penetrates the bilayer. These observations agree with recent studies that reveal a certain degree of penetration of lysozyme in 1,2-dioleoyl-sn-glycero-3-phosphocholine, from both x-ray reflectivity and Trp fluorescence (50), and of FRET from lysozyme to anthrylvinyl-labeled phospholipids (51), with the Trp residues located at the interface between the hydrocarbon core and the headgroup region of the bilayer. In addition, a bactericidal domain has been mapped to the C-terminal region of chicken lysozyme, where lysozyme contains a conserved helix-loop-helix motif (residues Arg87–Arg114 from hen egg white lysozyme) that may serve as a possible membrane anchor for this protein (27).
starting to increase significantly only for h1 > 1.5 nm. Considering that in this case the acceptors (DPH molecules) are located near the center of the bilayer, this would correspond to a repeat distance similar to the actual bilayer width (∼2h1 in this case). To explain these results, one must conclude that the protein partially penetrates the bilayer. These observations agree with recent studies that reveal a certain degree of penetration of lysozyme in 1,2-dioleoyl-sn-glycero-3-phosphocholine, from both x-ray reflectivity and Trp fluorescence (50), and of FRET from lysozyme to anthrylvinyl-labeled phospholipids (51), with the Trp residues located at the interface between the hydrocarbon core and the headgroup region of the bilayer. In addition, a bactericidal domain has been mapped to the C-terminal region of chicken lysozyme, where lysozyme contains a conserved helix-loop-helix motif (residues Arg87–Arg114 from hen egg white lysozyme) that may serve as a possible membrane anchor for this protein (27).
In a very recent study, Gorbenko et al. (51) studied the interaction of lysozyme with POPC/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-glycerol (POPG) bilayers. It is interesting to note that these authors rationalized their steady-state FRET data (between lysozyme and anthrylvinyl-labeled PC and PG) essentially on the basis of the model underlying Fig. 1 C above, without needing an additional acceptor plane. This is probably a consequence of the fact that the two tryptophan residues that dominate lysozyme fluorescence (Trp62 and Trp108) penetrate one of the adjacent bilayers, and thus are necessarily somewhat more distant from the other one. This would be unnoticed in our experiments (because of the high R0 values, implying that small differences in donor-acceptor interplanar distance are not important), but due to the smaller tryptophan-anthrylvinyl R0 value (<2.5 nm (52)), the extent of FRET measured by these authors would essentially reflect the contribution due to the closest acceptor plane, and the contribution due to the more distant plane would effectively be masked. This masking effect occurs commonly when measuring FRET to multiple acceptor planes: if R0 is not high enough, only the closer ones can be resolved. For example, even in this work, our first choice of labeled phospholipid/labeled phospholipid donor/acceptor pair was 1-palmitoyl-2-[3-(diphenylhexatrienyl)propanoyl]-sn-glycero-3-phosphocholine (DPH-PC)/1-palmitoyl-2-[12-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)aminododecanoyl]-sn-glycero-3-phosphocholine (NBD-PC), the same one we used in a previous study (34), which has R0 = 4.0 nm. FRET using this pair was insensitive to lysozyme concentration (results not shown), and only when we switched to the BODIPY-PC/Rh-PE pair (R0 = 5.5 nm) were we able to resolve the additional plane of acceptors available at higher lysozyme concentrations.
The two sets of results can be explained on the basis of a “pinched lamellar” model
The scenario of two nonoverlapping sets of repeat distances described above can be rationalized by recognizing that FRET between protein-bound donor and a membrane probe allows estimation of the distance between adjacent bilayers in the regions where protein is located, whereas the value obtained from FRET between two membrane probes is probably an average of the distances between the two bilayers in the protein-excluded regions. In this way, we propose a “pinched lamellar” model for the lipid/lysozyme aggregates, similar to that described by Subramanian et al. (53) for the oppositely charged dimethylammonium bromide/1,2-dilauroyl-sn-glycero-3-phosphocholine/poly-L-glutamic acid system. In this structure, depicted in Fig. 6, lysozyme would connect two adjacent bilayers, probably causing short-range sequestration of acidic lipids (no extensive protein-induced phase separation is observed, even in the nanometer range), in agreement with Gorbenko et al. (51), who estimated that the size of the POPG-rich lysozyme-induced microdomains does not extend beyond the projected area of one protein molecule. In this region, there is a reduced lamellar repeat distance of no more than ∼5–7 nm (as revealed by protein/membrane probe FRET). The region between these “pinched regions” contains large pockets of water stabilized by hydration repulsion, and larger lamellar repeat distances (∼9–10.5 nm) are measured (by FRET between two membrane probes).
FIGURE 6.

Schematic representation of the “pinched lamellar model” for the interaction between lysozyme (blue) and lipid bilayers containing anionic lipids (red). The model is drawn to scale taking into account the repeat distances obtained using FRET. For the sake of clarity, only three bilayers are shown of this multilamellar structure.
It is now timely to address the observation alluded to above, that changes reported by FRET occur for lower concentrations when the donor is in the protein than when the donor is a membrane probe. We believe that this is in fact consistent with (and actually reinforces) our pinched lamellar model. For low protein concentrations (≤0.5 μM in the studied system (Fig. 7 A)), the protein may bind to one vesicle without bridging adjacent bilayers, or even if such bridges occur, they will be rare and the overall interbilayer distance, as recovered by FRET from both A488-lysozyme (D1) and BODIPY-PC (D2) to Rh-PE (A) is expected to be large. For intermediate protein concentrations (between 0.5 μM and 1.5 μM in the studied system (Fig. 7 B)), bridging occurs at the protein “pinches”. These are still not widespread in the membranes' surface, and therefore FRET from D2 to A still senses a large interbilayer distance overall. However, because D1 is necessarily located at the pinches, FRET from D1 to A already senses a greatly reduced interbilayer distance, and acceptors from both bilayers are felt in more or less equal measure (even though only acceptors on the top bilayer are depicted in the figure for the sake of clarity), which accounts for the acceptor concentration doubling. This explains the difference in behavior between the two FRET pairs. For higher protein concentrations (≥2.0 μM in the studied system (Fig. 7 C)), bridging at the pinches is so widespread (because of the higher protein surface concentration) that even the interbilayer distance sensed by FRET from D1 to A is now somewhat reduced (due to obvious restriction in the extent of membrane bending between neighboring pinches), though not nearly as much as that reported by FRET from D2 to A. In addition, for these higher concentrations, it is expected that protein inserts into the bilayer to some extent, which is also schematically depicted in the figure.
FIGURE 7.

Schematic representation of how the FRET pairs A488-lysozyme/Rh-PE (D1/A) and BODIPY-PC/Rh-PE (D2/A) probe differently the lamellar repeat distance in a pinched bilayer arrangement, as verified from the experimental data. (A) Low protein concentration (≤0.5 μM in the studied system). The protein binds to one vesicle without bridging adjacent bilayers. Repeat distances are high, and the recovered acceptor concentration is close to the single-bilayer expectation. (B) Intermediate protein concentrations (between 0.5 and 1.5 μM). Bridging occurs at the protein “pinches”. They are already reported by the protein donor D1 (because of its location at the pinches) but not by the derivatized lipid donor D2 (preferentially located away from these regions). (C) High protein concentrations (≥2.0 μM in the studied system). Bridging at the pinches is widespread and even the interbilayer distance sensed by FRET from D2 to A is reduced, though not as much as that reported by FRET from D1, for which a doubled acceptor concentration is sensed. D1 also inserts into the bilayer to some extent. Each thick solid line represents a lipid/water interface of a different bilayer, whereas molecules capable of undergoing FRET are united by dashed and dotted lines for the D1/A and D2/A pairs, respectively.
The pinched lamellar structure depicted in Fig. 6 is likely to be the structure at the molecular level of the recently reported putative “amyloid-like” fibrils. In an atomic force microscopy study of fibers formed by cytochrome c upon interaction with negatively charged membranes (14), protofibers of 3–4 nm width and lateral spacing of ∼10 nm were observed, among other structures. It is interesting that these distances are close to the thickness of a single lipid bilayer and to the multibilayer repeat distance, respectively, recovered in this work. These aggregates are probably organized in a closed form compatible with their amphipathic character, and work is under progress to further characterize them, as well as the aggregation state and secondary structure of the protein (in particular, if β-sheet aggregates are formed) inside these structures.
Acknowledgments
Funding from POCI and PPCDT projects of the Fundação para a Ciência e a Tecnologia, Portugal, is acknowledged.
Editor: Alberto Diaspro.
References
- 1.Mulgrew-Nesbitt, A., K. Diraviyam, J. Wang, S. Singh, P. Murray, Z. Li, L. Rogers, N. Mirkovic, and D. Murray. 2006. The role of electrostatics in protein-membrane interactions. Biochim. Biophys. Acta. 1761:812–826. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin, S., and D. Murray. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 438:605–611. [DOI] [PubMed] [Google Scholar]
- 3.Diraviyam, K., and D. Murray. 2006. Computational analysis of the membrane association of group IIA secreted phospholipase A2: a differential role for electrostatics. Biochemistry. 45:2584–2598. [DOI] [PubMed] [Google Scholar]
- 4.Kayed, R., Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall, and C. G. Glabe. 2004. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- 5.Gorbenko, G. P., and P. K. J. Kinnunen. 2006. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 141:72–82. [DOI] [PubMed] [Google Scholar]
- 6.Munishkina, L. A., and A. L. Fink. 2007. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta. 1768:1862–1885. [DOI] [PubMed] [Google Scholar]
- 7.Knight, J. D., and A. D. Miranker. 2004. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 341:1175–1187. [DOI] [PubMed] [Google Scholar]
- 8.Bokvist, M., F. Lindström, A. Watts, and G. Gröbner. 2004. Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 335:1039–1049. [DOI] [PubMed] [Google Scholar]
- 9.Hou, X., S. J. Richardson, M.-I. Aguilar, and D. H. Small. 2005. Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry. 44:11618–11627. [DOI] [PubMed] [Google Scholar]
- 10.Hou, X., A. Mechler, L. L. Martin, M.-I. Aguilar, and D. H. Small. 2008. Cholesterol and anionic phospholipids increase the binding of amyloidogenic transthyretin to lipid membranes. Biochim. Biophys. Acta. 1778:198–205. [DOI] [PubMed] [Google Scholar]
- 11.Stöckl, M., P. Fischer, E. Wanker, and A. Herrmann. 2008. α-Synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol. 375:1394–1404. [DOI] [PubMed] [Google Scholar]
- 12.Zhao, H., E. K. J. Tuominen, and P. K. J. Kinnunen. 2004. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 43:10302–10307. [DOI] [PubMed] [Google Scholar]
- 13.Zhao, H., A. Jutila, T. Nurminen, S. A. Wickström, J. Keski-Oja, and P. K. J. Kinnunen. 2005. Binding of endostatin to phosphatidylserine-conatining membranes and formation of amyloid-like fibers. Biochemistry. 44:2857–2863. [DOI] [PubMed] [Google Scholar]
- 14.Alakoskela, J.-M., A. Jutila, A. C. Simonsen, J. Pirneskoski, S. Pyhäjoki, R. Turunen, S. Marttila, O. G. Mouritsen, E. Goormaghtigh, and P. K. J. Kinnunen. 2006. Characteristics of fibers formed by cytochrome c and induced by anionic phospholipids. Biochemistry. 45:13447–13453. [DOI] [PubMed] [Google Scholar]
- 15.Sparr, E., M. Engel, D. Sakharov, M. Sprong, J. Jacobs, B. de Kruijff, J. Höppener, and J. A. Killian. 2004. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 577:117–120. [DOI] [PubMed] [Google Scholar]
- 16.Blake, C. C. F., D. F. Koeing, G. A. Mair, A. C. T. North, D. C. Phillips, and V. R. Sarma. 1965. Structure of hen egg-white lysozyme. Nature. 206:757–761. [DOI] [PubMed] [Google Scholar]
- 17.Bergers, J. J., M. H. Vingerhoeds, L. van Bloois, J. N. Herron, L. H. M. Janssen, M. J. E. Fischer, and D. J. A. Crommelin. 1993. The role of protein charge in protein-lipid interactions. pH-dependent changes of the electrophoretic mobility of liposomes through adsorption of water-soluble, globular proteins. Biochemistry. 32:4641–4649. [DOI] [PubMed] [Google Scholar]
- 18.Pap, E. H. W., M. C. Houbiers, J. S. Santema, A. van Hoek, and A. J. W. G. Visser. 1996. Quantitative fluorescence analysis of the adsorption of lysozyme to phospholipid vesicles. Eur. Biophys. J. 24:223–231. [Google Scholar]
- 19.Zschornig, O., G. Paasche, C. Thieme, N. Korb, and K. Arnold. 2005. Modulation of lysozyme charge influences interaction with phospholipid vesicles. Colloids Surf. B Biointerfaces. 42:69–78. [DOI] [PubMed] [Google Scholar]
- 20.Gorbenko, G. P., V. M. Ioffe, and P. K. J. Kinnunen. 2007. Binding of lysozyme to phospholipid bilayers: evidence for protein aggregation upon membrane association. Biophys. J. 93:140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ibrahim, H. R., S. Higashiguchi, M. Koketsu, L. R. Juneja, M. Kim, T. Yamamoto, Y. Sugimoto, and T. Aoki. 1996. Partially unfolded lysozyme at neutral pH agglutinates and kills Gram-negative and Gram-positive bacteria through membrane damage mechanism. J. Agric. Food Chem. 44:3799–3806. [Google Scholar]
- 22.Ibrahim, H. R., T. Matsuzaki, and T. Aoki. 2001. Genetic evidence that antibacterial activity of lysozyme is independent of its catalytic function. FEBS Lett. 506:27–32. [DOI] [PubMed] [Google Scholar]
- 23.Nash, J. A., T. N. S. Ballard, T. E. Weaver, and H. T. Akinbi. 2006. The peptidoglycan-degrading property of lysozyme is not required for bactericidal activity in vivo. J. Immunol. 177:519–526. [DOI] [PubMed] [Google Scholar]
- 24.Mine, Y., F. Ma, and S. Lauriau. 2004. Antimicrobial peptides released by enzymatic hydrolysis of hen egg white lysozyme. J. Agric. Food Chem. 52:1088–1094. [DOI] [PubMed] [Google Scholar]
- 25.Ibrahim, H. R., D. Inazaki, A. Abdou, T. Aoki, and M. Kim. 2005. Processing of lysozyme at distinct loops by pepsin: a novel action for generating multiple antimicrobial peptide motifs in the newborn stomach. Biochim. Biophys. Acta. 1726:102–114. [DOI] [PubMed] [Google Scholar]
- 26.During, K., P. Porsch, A. Mahn, O. Brinkmann, and W. Gieffers. 1999. The non-enzymatic microbicidal activity of lysozymes. FEBS Lett. 449:93–100. [DOI] [PubMed] [Google Scholar]
- 27.Ibrahim, H. R., U. Thomas, and A. Pellegrini. 2001. A helix-loop-helix peptide at the upper lip of the active site cleft of lysozyme confers potent antimicrobial activity with membrane permeabilization action. J. Biol. Chem. 276:43767–43774. [DOI] [PubMed] [Google Scholar]
- 28.Loura, L. M. S., R. F. M. de Almeida, and M. Prieto. 2001. Detection and characterization of membrane microheterogeneity by resonance energy transfer. J. Fluoresc. 11:197–209. [Google Scholar]
- 29.de Almeida, R. F. M., L. M. S. Loura, A. Fedorov, and M. Prieto. 2005. Lipid rafts have different sizes depending on membrane composition: a time-resolved fluorescence resonance energy transfer study. J. Mol. Biol. 346:1109–1120. [DOI] [PubMed] [Google Scholar]
- 30.Towles, K. B., A. C. Brown, S. P. Wrenn, and N. Dan. 2007. Effect of membrane microheterogeneity and domain size on fluorescence resonance energy transfer. Biophys. J. 93:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valenzuela, C. F., P. Weign, J. Yguerabide, and D. A. Johnson. 1994. Transverse distance between the membrane and the agonist binding sites on the Torpedo acetylcholine receptor: a fluorescence study. Biophys. J. 66:674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davenport, L., R. E. Dale, R. H. Bisby, and R. B. Cundall. 1985. Transverse location of the fluorescent probe 1,6-diphenyl-1,3,5-hexatriene in model lipid bilayer membrane systems by resonance excitation energy transfer. Biochemistry. 24:4097–4108. [DOI] [PubMed] [Google Scholar]
- 33.Wong, A. P., and J. T. Groves. 2002. Molecular topography imaging by intermembrane fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 99:14147–14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loura, L. M. S., A. Coutinho, A. Silva, A. Fedorov, and M. Prieto. 2006. Structural effects of a basic peptide on the organization of dipalmitoylphosphatidylcholine/dipalmitoylphosphatidylserine membranes: a fluorescent resonance energy transfer study. J. Phys. Chem. B. 110:8130–8141. [DOI] [PubMed] [Google Scholar]
- 35.Lentz, B. R. 1988. Membrane “fluidity” from fluorescence anisotropy measurements. In Spectroscopic Membrane Probes, Vol. I. L. M. Loew, editor. CRC, Boca Raton, FL. 13–41.
- 36.Haugland, R. P. 1996. Handbook of Fluorescent Probes and Research Chemicals, 6th ed. Molecular Probes, Eugene, OR.
- 37.Coutinho, A., and M. Prieto. 2003. Cooperative partition model of nystatin interaction with phospholipid vesicles. Biophys. J. 84:3061–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McClare, C. W. 1971. An accurate and convenient organic phosphorus assay. Anal. Biochem. 39:527–530. [DOI] [PubMed] [Google Scholar]
- 39.Loura, L. M. S., A. Fedorov, and M. Prieto. 1996. Resonance energy transfer in a model system of membranes: application to gel and liquid crystalline phases. Biophys. J. 71:1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van der Meer, B. W., G. Coker III, and S.-Y. S. Chen. 1994. Resonance Energy Transfer: Theory and Data. VCH Publishers, New York.
- 41.Weber, G., and F. W. J. Teale. 1957. Determination of the absolute quantum yield of fluorescent solutions. Trans. Faraday Soc. 53:646–655. [Google Scholar]
- 42.Poveda, J. A., J. A. Encinar, A. M. Fernández, C. R. Mateo, J. A. Ferragut, and J. M. González-Ros. 2002. Segregation of phosphatidic acid-rich domains in reconstituted acetylcholine receptor membranes. Biochemistry. 41:12253–12262. [DOI] [PubMed] [Google Scholar]
- 43.Medhage, B., E. Mukhtar, B. Kalman, L. B.-Å. Johansson, and J. G. Molotkovsky. 1992. Electronic energy transfer in anisotropic systems. Part 5.—Rhodamine-lipid derivatives in model membranes. J. Chem. Soc., Faraday Trans. 88:2845–2851. [Google Scholar]
- 44.Kaiser, R. D., and E. London. 1998. Determination of the depth of BODIPY probes in model membranes by parallax analysis of fluorescence quenching. Biochim. Biophys. Acta. 1375:13–22. [DOI] [PubMed] [Google Scholar]
- 45.Mikhalyov, I., N. Gretskaya, F. Bergström, and L.-B. Å. Johansson. 2002. Electronic ground and excited state properties of dipyrrometheneboron difluoride (BODIPY): dimers with application to biosciences. Phys. Chem. Chem. Phys. 4:5663–5670. [Google Scholar]
- 46.Brinkley, M. 1992. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 3:2–13. [DOI] [PubMed] [Google Scholar]
- 47.Suckau, D., M. Mak, and M. Przybylski. 1992. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. USA. 89:5630–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnaible, V., and M. Przybylski. 1999. Identification of fluorescein-5′-isothiocyanate-modification sites in proteins by electrospray-ionization mass spectrometry. Bioconjug. Chem. 10:861–866. [DOI] [PubMed] [Google Scholar]
- 49.Teske, C. A., R. Simon, A. Niebisch, and J. Hubbuch. 2007. Changes in retention behaviour of fluorescently labelled proteins during ion-exchange chromatography caused by different protein surface labelling positions. Biotechnol. Bioeng. 98:193–200. [DOI] [PubMed] [Google Scholar]
- 50.Yuan, B., L.-L. Xing, Y.-D. Zhang, Y. Lu, Y.-Y. Luo, Z.-H. Mai, and M. Li. 2007. Penetration and saturation of lysozyme in phospholipids bilayers. J. Phys. Chem. B. 111:6151–6155. [DOI] [PubMed] [Google Scholar]
- 51.Gorbenko, G. P., V. M. Ioffe, J. G. Molotkovsky, and P. K. J. Kinnunen. 2008. Resonance energy transfer study of lysozyme-lipid interactions. Biochim. Biophys. Acta. 1778:1213–1221. [DOI] [PubMed] [Google Scholar]
- 52.Lakowicz, J. R. 2006. Principles of Fluorescence Spectroscopy, 3rd ed. Springer, New York.
- 53.Subramanian, G., R. P. Hjelm, T. J. Deming, G. S. Smith, Y. Li, and C. R. Safinya. 2000. Structure of complexes of cationic lipids and poly(glutamic acid) polypeptides: a pinched lamellar phase. J. Am. Chem. Soc. 122:26–34. [Google Scholar]