

Abstract
We describe a statistical approach to the validation and improvement of molecular dynamics simulations of macromolecules. We emphasize the use of molecular dynamics simulations to calculate thermodynamic quantities that may be compared to experimental measurements, and the use of a common set of energetic parameters across multiple distinct molecules. We briefly review relevant results from the theory of stochastic processes and discuss the monitoring of convergence to equilibrium, the obtaining of confidence intervals for summary statistics corresponding to measured quantities, and an approach to validation and improvement of simulations based on out-of-sample prediction. We apply these methods to replica exchange molecular dynamics simulations of a set of eight helical peptides under the AMBER potential using implicit solvent. We evaluate the ability of these simulations to quantitatively reproduce experimental helicity measurements obtained by circular dichroism. In addition, we introduce notions of statistical predictive estimation for force-field parameter refinement. We perform a sensitivity analysis to identify key parameters of the potential, and introduce Bayesian updating of these parameters. We demonstrate the effect of parameter updating applied to the internal dielectric constant parameter on the out-of-sample prediction accuracy as measured by cross-validation.
INTRODUCTION
Computer simulation, especially molecular dynamics simulation, has become an important and widely used tool in the study of biomolecular systems (1–3). With the growing availability of high-speed desktop computers and cluster computing, simulations once requiring access to specialized supercomputers are now within the range of many individual laboratories. Nevertheless, simulation of macromolecules such as proteins and nucleic acids remains a computationally expensive, complicated process with many options and parameters that may significantly affect the results. An important step in the development of standardized simulation approaches for such problems is the study of predictive power—the ability of the simulation to reproducibly generate some externally validatable quantity such as a future experimental measurement. In fields such as physics and chemistry, as well as in macroscopic areas of engineering and astronomy, simulations are regularly used in lieu of physical experiment, due to their ability to accurately and consistently predict physical quantities. Currently macromolecular simulations are primarily used for exploratory and visualization purposes, rather than quantitative prediction. However, as computational resources grow and algorithms and theory improve, we can strive to develop truly accurate macromolecular computer experiments.
To do so, we must meet several challenges. First, molecular dynamics simulations of macromolecules typically generate configurations on a pico- or femtosecond timescale, due to limiting frequencies of bond vibration. Because we cannot experimentally observe the motions of protein atoms at such timescales, the quantities measured in experimental settings can only ever be time-averaged quantities. With the exception of single-molecule experiments, most experimental studies of macromolecules are also ensemble-averaged. Thus, we must be concerned primarily with the time- and ensemble-averaged behavior of our simulation model, which may be validated against real experimental observations. Because it cannot be compared against reality, more detailed information generated by a simulation such as specific atomic trajectories or kinetic pathways should be viewed with some skepticism. This distinguishes molecular dynamics simulations of macromolecules from common use of classical mechanics simulations in macroscopic engineering applications, where larger timescales allow for trajectories themselves to be predictively validated against observation.
Thus, to compare simulation with experiment requires the computation of ergodic average quantities under our theoretical model (molecular mechanics potential). As is well known, doing so requires adequate exploration of conformational space during the simulation, a difficult problem. However, modern simulation algorithms (4,5) have made it possible to achieve adequate sampling for small systems. We also require methods for determining when adequate sampling has been achieved. Finally, because sample path averages are only approximate due to finite simulation lengths, we must quantify the remaining uncertainty in computed quantities to properly compare them with experimental values. Some standard statistical methods for addressing these issues are described in Statistical Analysis of Simulation Output.
A grand challenge of macromolecular simulation is the simulation of protein folding (6). Adequate ensemble-averaging for large proteins remains beyond current computational resources; here we study helical peptide folding, which has been widely studied as a model system for protein folding both experimentally (e.g., (7), and the large body of subsequent literature summarized in (8)) and computationally (in (9–16)). For short helical peptides, using modern simulation algorithms and a cluster of computer processors, we are able to adequately address the sampling issue. We apply our approach to study eight helical peptides from the experimental literature, and compare results obtained from simulation to experimental data.
A common concern is whether existing force fields are adequate to simulate protein folding. We approach this question from a predictive perspective: all molecular mechanics potentials are “wrong,” but we can ask whether they are “good enough” to accurately predict specified experimental quantities of interest, just as we judge any other theoretical model. Due to the difficulties of comparing simulations against experiment described above, it has previously been very difficult to separate the question of force-field accuracy from that of adequate configurational sampling. Here we systematically and quantitatively address the latter, enabling us to focus on the former. In particular we ask the question: given reproducible, quantitative predictions of ensemble quantities (here, equilibrium helicities), how well do the force field and parameter values used predict experimental quantities (here, circular dichroism (CD) measurements)?
In taking a predictive perspective, we emphasize the need for a single set of energy parameters, which successfully predict experimental quantities of multiple different molecular systems. Recent work has evaluated simulation versus experiment for helicity and thermal melting of single peptides (11,15). However, the force fields and parameter settings chosen for simulation studies often vary significantly across studies, making generalizability difficult to assess: in this article, we show that the ranges of parameter values used in the literature provide widely different equilibrium values. In addition to choice of parameters, often the force field may be modified to improve reproduction of experimental values, an issue we address from a formal statistical perspective. A single set of parameters (or well-defined criteria for choosing) is critical for prediction of a new molecular system by simulation.
We emphasize that helicity is a coarse-grained measure of the equilibrium ensemble and thus provides only a first step in evaluating the simulation accuracy; in this manner our approach is meant to be demonstrative rather than exhaustive. However, even by looking only at helicity, we obtain important results about reproducibility, parameter sensitivity, and experiment predictive accuracy using a common set of parameters for simulations of multiple distinct peptide systems.
The outline of the remainder of the article is as follows. Replica-Exchange Molecular Dynamics describes the simulation algorithm (replica-exchange) used in our studies. Statistical Analysis of Simulation Output describes the statistical tools used to determine when the simulation has converged and to measure the accuracy of quantities computed from the simulation trajectories. Parameter Adaptation explores the critical issue of sensitivity of simulation quantities to the parameters of the simulation potential, and demonstrates the use of Bayesian statistics to estimate improved parameter values based on available experimental data. Results gives the results obtained from applying our approach to evaluate predictive accuracy for the eight peptides in Table 1.
TABLE 1.
Helical peptides studied by simulation in this article, along with original experimental characterization and conditions
| ID | N- | Peptide sequence | C- | Experimental helicity | Temp (K) | pH | Reference |
|---|---|---|---|---|---|---|---|
| DG | — | DGAEAAKAAAGR | Nhe | 0.196 | 273 | 7 | (46) |
| SA | Ace | SAEDAMRTAGGA | — | 0.168 | 273 | 7 | (47) |
| RD | — | RDGWKRLIDIL | — | 0.050 | 277 | 7 | (48) |
| ES | — | ESLLERITRKL | — | 0.217 | 277 | 7 | (48) |
| LK | Ace | LKEDIDAFLAGGA | Nhe | 0.150 | 298 | 7 | (49) |
| PS | Ace | PSVRKYAREKGV | Nhe | 0.097 | 298 | 7 | (49) |
| RE | Ace | REKGVDIRLVQG | Nhe | 0.134 | 298 | 7 | (49) |
| AE | Ace | AETAGAKFLRAHA | Nhe | 0.126 | 276 | 7 | (50) |
Peptides are either unblocked or have an N-terminal acetyl group (Ace) and/or a C-terminal amide group (Nhe). ID provides the peptide identifier used in other figures in this article.
THEORY AND METHODS
We have run extensive molecular dynamics simulations for eight helical peptides, some naturally occurring and some designed, which have been previously studied experimentally by CD and shown to have measurable helicity (mean θ222 ellipticity) in solution. Table 1 shows the peptides studied along with their original experimental characterization; these peptides were selected from a database of helical peptides (8) to obtain a range of helicities among native peptides at physiological pH.
Replica-exchange molecular dynamics
All simulations were performed using replica-exchange molecular dynamics (REMD) (17), an application of the parallel tempering method (18) to molecular dynamics (MD) simulation. REMD runs isothermal molecular dynamics simulations in parallel at a ladder of temperatures and attempts to swap chains between temperatures intermittently. Each replica was run under the AMBER94 force field using the AMBER 7 suite of programs (19) with a generalized Born model of implicit solvent (20,21) and a time step of 2 fs. SHAKE (22) was used (tolerance 5 × 10−5 Å) to constrain hydrogen atoms, and a weakly-coupled heat bath with coupling constant of
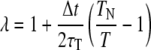 |
is used to maintain constant temperature (23), where TN is the fixed reference temperature and τT = 1.0 controls the strength of the coupling. The specific force field, solvent, and heat bath parameters used are given in Table 2, and are an attempt to replicate as closely as possible a protocol which has previously been successful in simulating helical peptide folding (11,24). A key question in the wider use of simulation techniques is whether such parameter sets that are successful in one instance are generalizable to other systems; in Results we explore this issue by evaluating the use of these parameters to predict experimentally measured helicities for the eight distinct peptides given in Table 1. A more detailed exploration of the effect of varying these parameter choices is described in Parameter Adaptation.
TABLE 2.
Force-field and simulation parameters used in the helical peptide replica-exchange simulations, and as default values for the parameter sensitivity analysis
| Simulation parameter | εext | εin | Salt concentration | Nonbonded cutoff | See | Snb | εdielc |
|---|---|---|---|---|---|---|---|
| Default value | 78.5 | 4.0 | 0.0 M | 8.0 Å | 1.2 | 1.0 | 1.0 |
Parameters values are those used previously for simulating a helical peptide (24).
Our REMD protocol utilizes 30 distinct MD simulations run in parallel at temperatures ranging from the target temperature T0 (273 Kelvin, 276 K, 277 K, or 298 K) to T29 = 624 K for each peptide simulation. Temperatures are spaced exponentially with Ti = ⌊T0 exp [ki]⌋, where k = ln(624/T0)/29 and i = 0, …, 29. During the REMD simulation, each replica is run at the assigned temperature for cycles of 1000 MD steps (2 ps), after which the translational and rotational motion of the center of mass is removed and 300 temperature-swapping moves attempted, as per a previous protocol (24). (Postsimulation analysis of swap acceptance rates indicated that approximately half as many replicas would have sufficed; this can be explained by fact that the protocol adopted from (24) was designed for use with both implicit and explicit solvent, the latter necessitating more replicas.)
Let xT = (p, q)T denote the coordinates (positions and momenta) of the replicate at temperature T. At each temperature-swap, two replicas xA and xB are chosen at random and a swap of their respective temperatures TA and TB is proposed, with acceptance probability given by the Metropolis criteria,
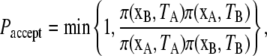 |
where π(x, T) ∝ exp[–E(x)/kBT] is the Boltzmann distribution over configurations at temperature T, and E is the total energy 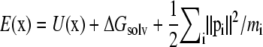 with U the potential function given by Eq. 4 and ΔGsolv the implicit solvent free energy term given by Eq. 5. When a swap is accepted, the two replicas exchange temperatures; otherwise, they remain at their respective temperatures. Associated velocities are rescaled to reflect the temperature swap before the next cycle of MD steps. This process of 1000 MD steps followed by 300 attempted temperature swaps is repeated until the convergence criteria described in Statistical Analysis of Simulation Output is reached.
with U the potential function given by Eq. 4 and ΔGsolv the implicit solvent free energy term given by Eq. 5. When a swap is accepted, the two replicas exchange temperatures; otherwise, they remain at their respective temperatures. Associated velocities are rescaled to reflect the temperature swap before the next cycle of MD steps. This process of 1000 MD steps followed by 300 attempted temperature swaps is repeated until the convergence criteria described in Statistical Analysis of Simulation Output is reached.
The above REMD protocol is used to conform as closely as possible to existing uses of REMD in protein simulation in the literature. The Metropolis criteria is used to guarantee invariance of (and therefore convergence to) the Boltzmann ensemble; however, recent theoretical analysis shows that corrections are needed to guarantee the proper invariant measure (25).
Statistical analysis of simulation output
As described above, molecular dynamics simulations of molecules differ somewhat from the use of classical mechanics simulations in macroscopic engineering applications, since detailed comparison of dynamical trajectories to experimental data is typically impossible. In fact, such trajectories are highly sensitive to starting conditions (26), parameterizations of the energy model, and other simulation details. Instead, it is the long-run, time-averaged behavior of the simulation that we can expect to produce observable macroscopic (thermodynamic) physical quantities, if the simulation model is adequate. To evaluate simulations against experimental data then, we must be able to accurately compute the long-run, time-averaged behavior implied by our theoretical model, specified by the molecular force field or potential. To do so, we rely on two important results. First, an ergodic theorem saying that if the dynamics of our simulation are ergodic (able to reach any region of the configuration space from any other region), then the time-averaged behavior of the simulation will converge to the configuration space integral representing the ensemble-averaged behavior for any (integrable) quantity of interest. Writing the quantity of interest as a function h(x) of configurations x in configuration space 𝒳, we have
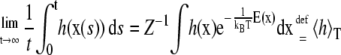 |
(1) |
for the canonical (constant N,V,T) ensemble, where 〈h〉 denotes the expectation or ensemble average of h(x) under the stationary Boltzmann distribution. Here h(x) is any quantity we wish to compute from a given configuration, and may be used to compute means (e.g., internal energy or helicity), variance-covariance matrices (for essential dynamics), indicator functions (for free energies), and so on. A major advantage of simulation-based methods is the ability to calculate a variety of such quantities from a single simulation. The right-hand integral yields the ensemble-averaged quantity under the theoretical model (force field); it is this quantity that can be compared with real-world experiments, which are themselves averaged over both time and molecules in solution.
Because we cannot run simulations infinitely long, we can only ever compute an approximation to the left-hand side of Eq. 1. Therefore, to use this result in practice, we need to know two things: how long must the simulation be run such that this approximation is “pretty good” (the convergence in Eq. 1 is approximately achieved), and how good is “pretty good” (error bounds on the computed quantities). Not only must the simulations have reached equilibrium, but they must also have run in equilibrium long enough to produce accurate approximations of the time/ensemble-averaged quantities of interest. From this perspective, MD simulation is simply a tool for computing the integral (Eq. 1), and often alternative numerical integration methods such as Monte Carlo sampling or replica-exchange dynamics may be more efficient than standard MD at this task. However, these methods often disrupt the kinetics of the process; interestingly, recently developed simulation methods, which do not guarantee proper ensemble sampling, may be useful in taking ensemble samples generated by methods such as MC or REMD and reconstructing the kinetics (27).
Another important result is statistical and provides guidance on these questions. It says that, for well-behaved functions h(x), the time-average of h computed from a simulation of N steps converges to the true value 〈h〉 as N → ∞. (Here “well-behaved” means h(x) has finite variance under Boltzmann distribution π(x), and the simulation dynamics are geometrically ergodic (28), a stronger assumption that can be difficult to verify in practice (B. Cooke and S. C. Schmidler, unpublished).) Moreover, this sample path average obeys a central limit theorem, converging in distribution to a normal random variable centered at the true value 〈h〉,
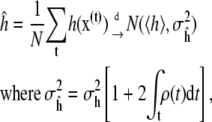 |
(2) |
where 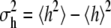 is the variance of h(x) under the Boltzmann distribution π(x), and
is the variance of h(x) under the Boltzmann distribution π(x), and 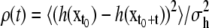 is the autocorrelation function for fluctuations in h of configurations at time separation t when the process is in equilibrium. Note that the Metropolis step in REMD creates a stochastic process (25), so we state results in those terms; central limit theorems for deterministic ergodic dynamical systems exist but are somewhat more delicate. Determinism of MD for molecules in solution is artificial and often replaced with stochastic (Langevin or Brownian) dynamics. In the stochastic case, however, ergodicity of the system is not an assumption, but can be shown directly.
is the autocorrelation function for fluctuations in h of configurations at time separation t when the process is in equilibrium. Note that the Metropolis step in REMD creates a stochastic process (25), so we state results in those terms; central limit theorems for deterministic ergodic dynamical systems exist but are somewhat more delicate. Determinism of MD for molecules in solution is artificial and often replaced with stochastic (Langevin or Brownian) dynamics. In the stochastic case, however, ergodicity of the system is not an assumption, but can be shown directly.
This theoretical result has important implications. It provides the distribution of errors obtained when we use the time average from a finite length simulation to approximate the theoretical ensemble average. This allows us to quantify uncertainty and produce error bars based on (100-α)% confidence intervals, which is critical for comparing the simulation output with experimental data. This in turn allows us to determine simulation time needed to approximate quantities to a predetermined level of accuracy. Failure to run a simulation long enough to adequately estimate quantities of interest is a common pitfall of molecular dynamics simulation (29).
In addition, knowledge that the errors are approximately normally distributed allows us to treat the simulation model as a (rather complicated) statistical model, and perform likelihood-based statistical inference on the simulation parameters, as described in Parameter Adaptation.
Interval predictions
A critical aspect of comparing simulation output with experiment is to account for the inherent variability of both the simulation output and the experimental measurement. As described above, variability in the simulation output can be characterized by a central limit theorem: the quantity ĥ approaches 〈h〉 in the limit of large N, with error 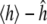 being normally distributed with variance
being normally distributed with variance 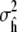 given by Eq. 2. This result allows us to construct normal-based confidence intervals for h of the form
given by Eq. 2. This result allows us to construct normal-based confidence intervals for h of the form 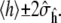 The variance of ĥ therefore determines how long we need to run a given simulation to obtain a predetermined level of accuracy. Since σh depends on the function h of interest, some quantities can converge significantly faster than others, a fact observed empirically (30); however, apparent convergence of some quantities while others have not converged can also be misleading. Theoretical guarantees on how long a simulation must be run are extremely difficult to come by, although recent progress has been made in this area for parallel tempering algorithms (31).
The variance of ĥ therefore determines how long we need to run a given simulation to obtain a predetermined level of accuracy. Since σh depends on the function h of interest, some quantities can converge significantly faster than others, a fact observed empirically (30); however, apparent convergence of some quantities while others have not converged can also be misleading. Theoretical guarantees on how long a simulation must be run are extremely difficult to come by, although recent progress has been made in this area for parallel tempering algorithms (31).
To determine this interval we require an estimate 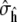 for
for 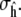 Direct estimates of the summed autocorrelation (Eq. 2) are inconsistent, but several other estimation methods exist (32). A common and relatively straightforward technique, which we use here, is the batch estimate, obtained by dividing the simulation of length N into a = N/M regions or batches of size M. Each batch is used to independently estimate 〈h〉,
Direct estimates of the summed autocorrelation (Eq. 2) are inconsistent, but several other estimation methods exist (32). A common and relatively straightforward technique, which we use here, is the batch estimate, obtained by dividing the simulation of length N into a = N/M regions or batches of size M. Each batch is used to independently estimate 〈h〉,
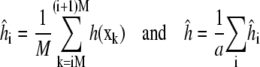 |
and M is chosen large enough to ensure the autocorrelation 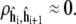 The batch estimates are then approximately independent samples whose empirical variance
The batch estimates are then approximately independent samples whose empirical variance
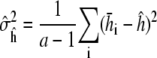 |
yields a simple estimate of the variance 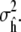 The quantile plot in Results (Fig. 4 a) indicates approximate normality is a reasonable assumption for our converged simulations.
The quantile plot in Results (Fig. 4 a) indicates approximate normality is a reasonable assumption for our converged simulations.
FIGURE 4.

Quantile plots of standardized residuals (left) 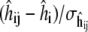 for the 8 × 4 = 32 individual REMD simulations, and (right)
for the 8 × 4 = 32 individual REMD simulations, and (right) 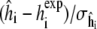 for the eight combined simulation peptide helicities versus experiment. The lack of significant deviation from the diagonal suggests the assumption of normally distributed noise is reasonable in each case.
for the eight combined simulation peptide helicities versus experiment. The lack of significant deviation from the diagonal suggests the assumption of normally distributed noise is reasonable in each case.
Monitoring convergence
The energy surface of proteins and polypeptides is characterized by large energy barriers and multiple local minima, making adequate exploration of configuration space a major challenge of protein simulation. While theoretical guarantees are very difficult to obtain for complex simulations, and observing the output of a simulation can never guarantee convergence, convergence diagnostics can be constructed to identify lack of convergence from simulation output. Our preferred approach is the use of multiple parallel simulations starting from diverse initial conditions to monitor the convergence by comparison of sample path quantities across distinct simulations. We use the multiple-chain approach (33) to assess convergence of our simulations by running multiple independent REMD simulations for each peptide in parallel starting from a diverse set of initial configurations, with each individual REMD simulation run according to the protocol of Theory and Methods. Let M denote the number of simulations and 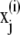 for j = 1, …, M the configuration of the jth simulation at time step i. Convergence of an observable quantity h(x) is by calculating
for j = 1, …, M the configuration of the jth simulation at time step i. Convergence of an observable quantity h(x) is by calculating
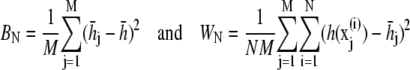 |
with 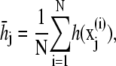 and
and 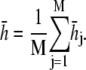 The value BN represents the between-chain variability and WN represents the within-chain variability. When multiple starting configurations are chosen to be widely dispersed throughout configuration space, early in the simulation the chains will be sampling distinct regions of phase space and the between-chain variance will be significantly higher than the within-chain. As the simulations converge to sampling from the same equilibrium Boltzmann distribution, these two quantities will converge. Comparison is based on techniques from the analysis of variance to determine whether significant differences remain. and convergence is monitored using the Gelman-Rubin shrink factor (37)
The value BN represents the between-chain variability and WN represents the within-chain variability. When multiple starting configurations are chosen to be widely dispersed throughout configuration space, early in the simulation the chains will be sampling distinct regions of phase space and the between-chain variance will be significantly higher than the within-chain. As the simulations converge to sampling from the same equilibrium Boltzmann distribution, these two quantities will converge. Comparison is based on techniques from the analysis of variance to determine whether significant differences remain. and convergence is monitored using the Gelman-Rubin shrink factor (37)
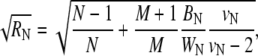 |
where 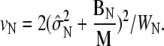 The quantity
The quantity 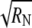 estimates the reduction in variance of the estimator ĥ if the simulation were to be run infinitely long, and converges to one as all of the parallel simulations converge to equilibrium.
estimates the reduction in variance of the estimator ĥ if the simulation were to be run infinitely long, and converges to one as all of the parallel simulations converge to equilibrium.
Once the chains have equilibrated, samples from all M = 4 independent simulations can be combined to obtain a pooled estimate of 〈h〉, with individual chain estimates combined inversely proportional their respective variances:
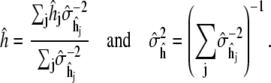 |
(3) |
Thus the effective trajectory length of the combined estimate is 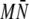 where
where 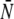 is the average production phase length; the only price paid for using multiple simulations compared to a single simulation is the replication of the equilibration phase. In our opinion, the advantage of being able to run in parallel and to obtain convergence diagnostics by inter-run comparisons far outweighs this cost in most situations. Note that combining the results of multiple simulations that have not been determined to have individually converged to the same stationary distribution, as is sometimes done in MD simulation, has no theoretical justification and can be badly misleading.
is the average production phase length; the only price paid for using multiple simulations compared to a single simulation is the replication of the equilibration phase. In our opinion, the advantage of being able to run in parallel and to obtain convergence diagnostics by inter-run comparisons far outweighs this cost in most situations. Note that combining the results of multiple simulations that have not been determined to have individually converged to the same stationary distribution, as is sometimes done in MD simulation, has no theoretical justification and can be badly misleading.
Numerous other convergence diagnostics have been developed in the statistics and operations research literature (34), including further developments of the approach used here (35,36). Note that no diagnostic based on simulation output can ever guarantee convergence, all such diagnostics can be fooled (34). However, theoretical bounds on simulation time are very difficult to obtain; although relevant work in this direction is ongoing (31).
Parameter adaptation
The energetics used in molecular dynamics simulations involve a large number of parameters that must be specified in advance. These include the parameters of the AMBER potential (19), given by the covalently-bonded and nonbonded terms,
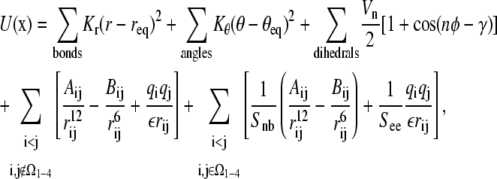 |
(4) |
where Ω1–4 is the set of atom pairs (i, j) which are separated by three bonds. For example, the parameter See weights the electrostatic interactions in Ω1–4, Snb weights the corresponding van der Waals interactions, and the dielectric constant ε affects all longer-range electrostatic terms.
In addition, the implicit-solvent model given by the generalized Born approximation (20,21) has associated parameters,
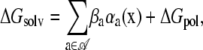 |
(5) |
where 𝒜 is the set of atom types, αa is the total solvent-accessible surface area of atoms of type a in configuration x, βa are solvation parameters, and the electrostatic polarization component of the free energy of solvation is given by
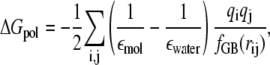 |
(6) |
which involves parameters such as the intramolecular dielectric constant εmol and the solvent dielectric εwater. (In AMBER, these parameters are specified as εin, εext, and εdielc, with εwater = εextεdielc, εmol = εinεdielc, and ε = εinεdielc.) Although in principle, these parameters represent physical quantities whose values can be known; in practice, they are approximations with values determined individually in empirical or theoretical studies.
The simulations of Results utilize a default set of parameters given in Table 2, chosen to comply with standard practice as described in Theory and Methods. Nevertheless, there is significant variation in the literature in values chosen for some of these parameters. Since the ensemble simulated is defined by these parameters, the simulation averages obtained and their comparison with experimental values will be a function of these parameter choices. It is therefore important to understand the how differences in these parameter values may be propagated into the resulting thermodynamic quantities estimated, and to determine the impact on the conclusions obtained. Sensitivity analysis of these parameters is described in Sensitivity Analysis.
Bayesian estimation of force-field parameters
Given the sensitivity of simulation results to certain force-field parameters as demonstrated in Sensitivity Analysis, we identify a standard set of values that could be used by various researchers to ensure consistency and comparability of simulations across different studies. A natural approach to determine such values is to optimize the parameter values with reference to experimental data. However, it is important to do so in such a way that the resulting parameter values are generalizable to other systems. A criticism commonly leveled at simulation research is that with the large number of parameters involved in specifying a potential energy function, a solvation model, and a simulation algorithm, the simulation may be adjusted to produce almost any behavior the investigator desires.
Such concerns can be addressed by standardized use of a common set of parameters, but the adaptation of these parameters to better match experimental observations remains important. The danger is that optimizing parameters on a specific set of data may provide good results, but generalize poorly to the study of other systems, a phenomenon known as overfitting. One solution to overfitting is the use of large datasets relative to the number of parameters, where the simultaneous adaptation to multiple experimental measurements ensures that no particular measurements are well described at the expense of others. We do have large quantities of experimental helicity data available (8), but we are currently limited by the fact that each data point requires parallel simulations at nanosecond or greater timescales for inclusion. However, overfitting is also avoided by a variety of parameter estimation and predictive validation techniques developed in statistics, such as Bayesian analysis and regularization. These techniques, which penalize large parameter changes when insufficient data is available to justify them, regularly allow the adaptation of complex, many-parameter models to relatively small data sets while avoiding overfitting and producing parameters that generalize well. In this article, we adopt a Bayesian approach, using prior information to adapt the parameters by Bayesian inference. Because computational considerations limit us to the use of the eight peptides in Table 1, we perform only a small example of this approach, adapting only one parameter at a time. Computational methodology for adaptation of many parameters simultaneously will be described elsewhere.
To evaluate generalizability of the parameters resulting from this adaptation approach, we apply the statistical method of cross-validation. This allows us to estimate out-of-sample prediction accuracy, i.e., how accurately we can expect these parameters to perform when simulating a new peptide to predict its experimental helicity.
We first specify a simple statistical error model for the experimental data, which says that the measured helicities may be described as a combination of the theoretical equilibrium helicity under our force field, plus some experimental noise:
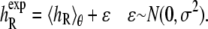 |
(7) |
Here 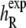 denotes the experimentally measured helicity of peptide R, and we now denote explicitly the dependence of 〈h〉 on the peptide sequence R as well as the force-field parameters θ. If 〈hR〉θ was a linear function of the parameters θ, then Eq. 7 would simply be a linear regression model. Instead, the ensemble helicity 〈hR〉θ is a complicated function given by the configuration integral under the Boltzmann distribution with potential function (Eq. 4) parameterized by θ. Similar statistical principles apply, however, allowing us to estimate the parameters θ from data. This has a slightly unusual aspect arising from the difficulty in calculating 〈hR〉θ, which can only be done approximately by the simulation average from a finite length simulation as described in Statistical Analysis of Simulation Output. The assumption of normally distributed noise can be justified by the previously described central limit theorem for
denotes the experimentally measured helicity of peptide R, and we now denote explicitly the dependence of 〈h〉 on the peptide sequence R as well as the force-field parameters θ. If 〈hR〉θ was a linear function of the parameters θ, then Eq. 7 would simply be a linear regression model. Instead, the ensemble helicity 〈hR〉θ is a complicated function given by the configuration integral under the Boltzmann distribution with potential function (Eq. 4) parameterized by θ. Similar statistical principles apply, however, allowing us to estimate the parameters θ from data. This has a slightly unusual aspect arising from the difficulty in calculating 〈hR〉θ, which can only be done approximately by the simulation average from a finite length simulation as described in Statistical Analysis of Simulation Output. The assumption of normally distributed noise can be justified by the previously described central limit theorem for 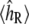 as well as standard usage for experimental noise; the quantile plot in Results (Fig. 4 b) shows that this assumption is quite reasonable.
as well as standard usage for experimental noise; the quantile plot in Results (Fig. 4 b) shows that this assumption is quite reasonable.
The Bayesian approach next specifies a prior distribution for the parameter P(θ), which captures any background or biophysical knowledge we may have about the parameter, to supplement the information contained in the experimental data. We then base our inference about the parameter on the posterior distribution,
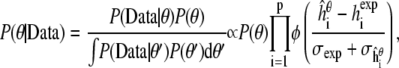 |
(8) |
where p is the number of peptides, φ is standard normal density function, and 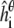 is the simulated helicity from Eq. 3 for peptide i run at parameter value θ. Note that
is the simulated helicity from Eq. 3 for peptide i run at parameter value θ. Note that 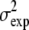 reflects the variance in
reflects the variance in 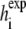 arising from experimental noise, and
arising from experimental noise, and 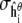 is the remaining simulation uncertainty in
is the remaining simulation uncertainty in 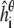 given in Eq. 3. We do use the estimated value σexp = 0.07, from Schmidler et al. (8), for convenience; more formally σexp could be estimated or integrated out to obtain the marginal posterior distribution (37).
given in Eq. 3. We do use the estimated value σexp = 0.07, from Schmidler et al. (8), for convenience; more formally σexp could be estimated or integrated out to obtain the marginal posterior distribution (37).
RESULTS
Predicting helicity of multiple distinct peptides by simulation with a single parameter set
REMD simulations were performed for the eight peptides given in Table 1. For each peptide, four REMD simulations were run in parallel, with each REMD simulation utilizing 30 temperatures according to the protocol of Theory and Methods. Initial configurations for the four REMD runs were generated as follows for each peptide: one ideal helix, one extended conformation, and two random configurations, one generated by uniformly sampling (φ, ψ) angles within the helical range and one generated by uniformly sampling (φ, ψ) outside of the helical range. Fig. 1 a shows the starting configurations for a particular peptide at T0. Initial velocities were generated randomly and independently for each configuration. Quantities monitored for convergence included backbone φ- and ψ-angles of each amino acid, helicity of the peptide, and total energy. Convergence to equilibrium was declared when the Gelman-Rubin shrink factor for these quantities reached 1.1, and the sample paths up to this time (equilibration phase) discarded. Sample paths from this time on (production phase) were included in computing time-averaged quantities ĥ. Fig. 2 shows plots of the Gelman-Rubin shrink factor for simulations of the eight peptides; for comparison, a standard MD simulation (without replica-exchange) of one of the peptides is shown.
FIGURE 1.

Three configuration snapshots from the four parallel REMD simulations of peptide SAEDAMRTAGGA. Shown are (a) the four starting configurations, (b) four configurations observed at time of convergence to equilibrium, and (c) four configurations from the production phase of the simulation.
FIGURE 2.

Convergence of REMD simulations of the eight peptides from Table 1, as measured by the Gelman-Rubin shrink factor (37) for helical content. Black lines represent the estimated shrink factor with dashed lines giving the boundary of the 95% confidence interval. Each plot represents convergence between four parallel simulations started from diverse initial configurations using parameters given in Table 2. Shown for comparison (lower right) is a convergence plot for four standard MD simulations of peptide AE simulated without replica-exchange.
Helicity of a peptide configuration was defined as the fraction of amino-acid (φ, ψ) pairs lying in a predefined helical range with the potential to form hydrogen bonds,
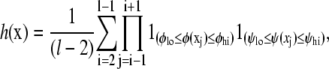 |
(9) |
for configuration x of a peptide of length l, where 1() is an indicator function. We use a standard range for defining helical angles: {φlo, φhi} = {−87, −27} and {ψlo, ψhi} = {−77, −17} (see Helical Backbone Angles for the effect of changing these boundaries on the resulting helicities).
The total simulation time required to reach convergence for each peptide is shown in Table 3. After equilibration, each simulation was continued until the estimated variance 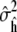 of the combined estimate of equilibrium helicity (Eq. 3) decreased to <0.001. The total simulation times required in the production phase to meet this criteria are also shown in Table 3.
of the combined estimate of equilibrium helicity (Eq. 3) decreased to <0.001. The total simulation times required in the production phase to meet this criteria are also shown in Table 3.
TABLE 3.
Time length of equilibration and production phases for REMD simulations of each peptide
| Peptide | DG | SA | RD | ES | LK | PS | RE | AE |
|---|---|---|---|---|---|---|---|---|
| Equilibration phase (ns) | 0.8 | 0.6 | 0.8 | 0.8 | 1.0 | 1.0 | 1.0 | 1.4 |
| Production phase (ns) | 1.0 | 0.8 | 1.4 | 1.6 | 2.4 | 2.4 | 1.6 | 2.9 |
Peptide identifiers are given in Table 1. Equilibration and production times were determined according to the statistical convergence criteria described in Statistical Analysis of Simulation Output. Due to the use of replica-exchange, equilibration is significantly faster than physical timescales (see Fig. 2).
Fig. 3 shows the simulated equilibrium helicities versus the published experimental helicities in Table 1. All experimental helicities are derived from mean-residue θ222 ellipticity measured by circular dichroism. Simulated helicities are shown with 95% confidence intervals obtained as described in Statistical Analysis of Simulation Output. For each peptide, we show the helicity interval obtained from each of the four independent REMD runs as well as the combined estimate ĥ (Eq. 3). Simulated helicities are correlated with the experimental helicities but are not within perfect agreement even within the sampling error of the simulations. Recent estimates place the standard deviation of experimental noise to be ∼0.05, so the simulations may agree within the tolerance of combined noise due to finite simulation sampling and experimental error. Fig. 4 shows quantile plots that validate the normality assumptions given in Theory and Methods.
FIGURE 3.

Peptide helicity as estimated from simulation versus experimentally measured helicity for the eight peptides in Table 1. The diagonal line y = x is shown as a reference. Simulation results are shown as 95% confidence intervals using standard errors estimates described in text, and are shown both for individual REMD runs and for the pooled estimates.
Sensitivity analysis
To address the questions of sensitivity of simulation results to choice of simulation parameters described in Parameter Adaptation, we performed a one-way sensitivity analysis of seven of the force-field parameters described there:
The external and internal dielectrics εext and εin.
The salt concentration constant used with implicit solvent, Salt.
The weight terms See and Snb, for atom pairs in Ω1–4.
The dielectric constant εdielc.
A simulation parameter for the nonbonded cutoff distance, Cutoff.
Ranges for these parameters were chosen based on the variation in use in published simulation studies and recommended values in force-field documentation.
Sensitivity analysis was performed via short REMD simulations of two peptides (DGAEAAKAAAGR and SAEDAMRTAGGA) starting from equilibrium states obtained from the longer simulations of Results. Parameters given in Table 2 were used as reference values, and each parameter was varied individually while holding the others constant, performing short REMD simulations of 200 ps for each peptide at 273 K. Four copies of each were run from different equilibrium starting configurations to monitor convergence as described in Monitoring Convergence. The resulting sensitivity of helicity to perturbations of these seven parameters is shown in Table 4. Of the parameters examined, the internal dielectric constant εin has the most dramatic effect on the helicity obtained from simulation.
TABLE 4.
One-way sensitivity analysis of helicity as a function of simulation parameters
| DGAEAAKAAAGR
|
SAEDAMRTAGGA
|
||||
|---|---|---|---|---|---|
| Parameter | Value | Mean | Variance | Mean | Variance |
| εext | 100 | 0.188 | 0.0006 | 0.170 | 0.0183 |
| 80 | 0.189 | 0.0024 | 0.209 | 0.0060 | |
| 78.5 | 0.170 | 0.0031 | 0.174 | 0.0183 | |
| 50 | 0.172 | 0.0006 | 0.232 | 0.0050 | |
| εin | 20 | 0.063 | 0.0002 | 0.091 | 0.0035 |
| 10 | 0.124 | 0.0003 | 0.138 | 0.0087 | |
| 4 | 0.168 | 0.0021 | 0.177 | 0.0191 | |
| 3 | 0.260 | 0.0003 | 0.226 | 0.0110 | |
| 1 | 0.388 | 0.0037 | 0.227 | 0.0112 | |
| Salt | 5.0 | 0.197 | 0.0015 | 0.219 | 0.0065 |
| 2.0 | 0.161 | 0.0005 | 0.146 | 0.0188 | |
| 1.0 | 0.173 | 0.0002 | 0.186 | 0.0204 | |
| See | 5.0 | 0.238 | 0.0004 | 0.262 | 0.0197 |
| 2.0 | 0.215 | 0.0010 | 0.235 | 0.0094 | |
| 1.5 | 0.179 | 0.0002 | 0.197 | 0.0184 | |
| 1.2 | 0.188 | 0.0016 | 0.176 | 0.0188 | |
| 1.0 | 0.161 | 0.0010 | 0.192 | 0.0155 | |
| Snb | 5.0 | 0.052 | 0.0001 | 0.039 | 0.0003 |
| 2.0 | 0.109 | 0.0016 | 0.104 | 0.0070 | |
| 1.5 | 0.171 | 0.0009 | 0.093 | 0.0043 | |
| 1.2 | 0.176 | 0.0018 | 0.158 | 0.0122 | |
| 1.0 | 0.202 | 0.0006 | 0.175 | 0.0188 | |
| Cutoff | 99.0 | 0.167 | 0.0007 | 0.167 | 0.0254 |
| 20.0 | 0.183 | 0.0008 | 0.173 | 0.0200 | |
| 15.0 | 0.177 | 0.0002 | 0.199 | 0.0230 | |
| 12.0 | 0.181 | 0.0004 | 0.228 | 0.0315 | |
| 10.0 | 0.171 | 0.0009 | 0.167 | 0.0160 | |
| 8.0 | 0.161 | 0.0002 | 0.209 | 0.0130 | |
| 5.0 | 0.111 | 0.0002 | 0.112 | 0.0052 | |
| εdielc | 100.0 | 0.059 | 0.0002 | 0.060 | 0.0015 |
| 80.0 | 0.052 | 0.0009 | 0.089 | 0.0046 | |
| 78.5 | 0.065 | 0.0010 | 0.125 | 0.0039 | |
| 50.0 | 0.093 | 0.0011 | 0.105 | 0.0072 | |
| 20.0 | 0.091 | 0.0003 | 0.067 | 0.0039 | |
| 5.0 | 0.074 | 0.0011 | 0.115 | 0.0071 | |
| 3.0 | 0.083 | 0.0011 | 0.119 | 0.0111 | |
Shown are mean helicity and variance obtained for two peptides DGAEAAKAAAGR and SAEDAMRTAGGA at a range of values for each parameter studied.
The results in Table 4 suggest that the variability among choices for both εin and Snb observed in the literature may significantly impact the thermodynamic quantities measured from these simulations. To evaluate the ability of this potential and solvent model to reproduce experimental helicities, we must obtain an appropriate consistent value for this and other parameters.
Effect of εin and Snb parameters
Variation of the two force-field parameters εin and Snb showed a significant effect impact on the helicity of the two peptides studied in the one-way sensitivity analysis. We focus on determining an appropriate value for εin in Bayesian Estimation of Internal Dielectric εin, but first we explore the manner in which these parameters affect helicity. The value εin affects both the solute-solvent electrostatic polarization term in Eq. 6 and nonbonded electrostatic interactions in Eq. 4. The ΔGpol term represents a difference in electrostatic interaction energy resulting from solvent screening of charges. As εin increases toward εext this difference shrinks, effectively increasing internal charge screening by making the interior of the molecule more polar. The electrostatic interactions in Eq. 4 also decrease as εin increases, reducing the favorability of hydrogen bonds formed in helix formation. Thus we expect that increasing εin will produce lower simulation helicity levels, as observed in Table 4. (Sensitivity to much larger changes in the structure of the solvation model has been previously reported (24,38), but our results show that, even for a given solvation model, the choice of parameter values may have a large impact.)
In contrast, Table 4 shows that as Snb increases, peptide helicity decreases. The quantity Snb scales the nonbonded van der Waals interactions in the potential energy U (Eq. 4). To interpret the effect of this parameter on helicity, we examined its effect on each of the 1-4 interactions along the peptide backbone, as well as the relation of these 1-4 distances to amino-acid helicity. The effect of the Snb parameter on most 1-4 interactions had little effect on helicity, with the notable exceptions of nitrogen-to-nitrogen (N-N) and hydrogen-to-carbon (H-C) distances, as pictured in Fig. 5. Equilibrium values of both of these distances (N-N and H-C) decrease as Snb increases, and in turn, lowers the helicity of the associated amino acid as shown in Fig. 6 in both peptides for which this sensitivity analysis was performed. Changes in other 1-4 atom pair distances induced by Snb increase had little effect on helicity; Cα-Cα is shown as an example.
FIGURE 5.

The N-N (dashed) and H-C (dotted) 1-4 interactions along the peptide backbone, which are most affected by changes in the Snb scaling constant in the AMBER potential. The effect of equilibrium distances for these atom pairs has a significant effect on the (φ,ψ) angles of their respective amino acids, and hence on peptide helicity.
FIGURE 6.

Effects of the nonbonded scaling parameter Snb on the equilibrium distances (in Å) of successive backbone nitrogen atoms (N-N), hydrogen-carbon atom pairs (H-C), and α-carbons (Cα-Cα). Line represents the ensemble-mean helicity for the ith amino acid as a function of the Ni-Ni+1 distance (plots a and b), Hi-Ci distance (c and d), or Cα-Cα distance (e and f) for the two peptides DGAEAAKAAAGR (a, c, and e) and SAEDAMRTAGGA (b, d, and f). Individually labeled points give the average N-N or H-C distance for simulations with Snb = {0.5, 1, 1.2, 1.5, 2, 3, 5}. Helicity changes in response to varying Snb can be explained by sensitivity to N-N and H-C distances; other 1-4 atom pairs have little effect on helicity as demonstrated here for Cα-Cα.
Bayesian estimation of internal dielectric εin
To demonstrate the parameter adaptation approach of Bayesian Estimation of Force-Field Parameters, we applied it to estimate the parameter εin shown in Table 4 to have the greatest impact on equilibrium helicity. (Computational considerations preclude simultaneous adaptation of many parameters using this approach, and as such, this example is intended to be illustrative. Computational methods for adapting many parameters simultaneously will be reported elsewhere.) To estimate an optimal value for internal dielectric we discretized this parameter into a set of plausible values εin ∈ {1, 2, 3, 4, 5} spanning the range of values that have been used previously in the literature (24,39). A noninformative uniform prior distribution was assigned for εin by giving each possible value of εin equal prior probability. To obtain the posterior distribution (Eq. 8) for εin, REMD simulations as described in Replica Exchange Molecular Dynamics were then run for each peptide at each discrete value of the dielectric constant with all other parameters fixed at their default values given in Table 2. The resulting posterior distribution over the discrete values of εin is shown in Fig. 7 a. Given this shape, we decided to refine the discretization of εin by adding another value at εin = 4.1 to obtain a more detailed look at the posterior in the high probability region. (Each such point requires eight peptides × 4 REMD runs × 30 MD simulations run to convergence to obtain equilibrium helicities, hence the coarseness of the original discretization.) The new posterior is shown in Fig. 7 b.
FIGURE 7.

Posterior distributions for the dielectric constant εin evaluated at discrete values, obtained using Bayesian parameter updating described in Bayesian Estimation of Force-Field Parameters under uniform prior. (Left) Posterior over εin ∈ {1, 2, 3, 4, 5}. (Right) An additional simulation was run at εin = 4.1 to help identify the mode of the posterior distribution.
Fig. 8 shows estimated helicities obtained by simulation versus experimental values for all eight peptides for the six different values of εin. It can then be seen that the highest posterior probability value of 4.1 is the one that gives the closest approximation to the set of experimental values. Fig. 9 plots the simulated helicity versus the experimental helicity individually for the eight peptides plotted at the different values of εin. Fig. 7 b shows that resulting posterior distribution is clearly peaked at 4.1. (Note that further refinement of the discretization may well lead to an improvement between 4.0 and 4.1 or 4.1 and 5.) As more experimental data and associated simulations are included, this posterior distribution can be updated to reflect the information in the larger data set, further refining the optimal value.
FIGURE 8.

Simulated helicity versus experimental helicity for the peptides in Table 1 evaluated at a range of values of the internal dielectric parameter εin.
FIGURE 9.

Helicity versus εin for each peptide in Table 1 at values of εin ∈ {1, 2, 3, 4, 4.1, 5}. The experimentally measured helicity for each peptide is plotted as a horizontal line.
Helical backbone angles
As another example, we consider the boundaries of the helical (φ, ψ) region that define a helical backbone conformation in Eq. 9. Although a general region may be defined based on Ramachandran plots to be φ ∈ {φmin, φmax} = {−87, −27} and ψ∈{ψmin, ψmax} = {−77, −17}, the exact region measured by CD at ellipticity θ225 is somewhat ambiguous. We may view the boundaries of this region to be parameters of the statistical mechanical model and estimate them by Bayesian inference as above. In this case, evaluation of the posterior at a range of 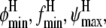 and
and 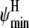 may be done more easily than for εin, since these are parameters of the statistical mechanical model for helicity but not of the force field that determines the simulation ensemble; thus, we need simply reanalyze the trajectories rather than resimulate for each value.
may be done more easily than for εin, since these are parameters of the statistical mechanical model for helicity but not of the force field that determines the simulation ensemble; thus, we need simply reanalyze the trajectories rather than resimulate for each value.
For simplicity, again we discretize and construct a four-dimensional grid for possible values of (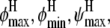 and
and 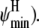 The marginal posterior distributions obtained for (
The marginal posterior distributions obtained for (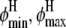 ) under a uniform prior are shown in Fig. 10 a and for (
) under a uniform prior are shown in Fig. 10 a and for (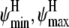 ) in Fig. 10 b. Peaks representing high probability values of (φmin, φmax) are seen at (−80, −50) and (−90, −40), with a ridge for φmin between −100 and −70. The joint distribution for (ψmin, ψmax) exhibits a sharp peak at (−60, −40) and a minor peak at (−50, −30). The ranges of dihedral angles with the largest peaks for both φ and ψ contain the values for an ideal helix (φ, ψ) = (−57, −47), but the joint mode of (φmin, φmax, ψmin, ψmax) = (−90, −40, −60, −40) yields a narrower range than that generally accepted for helical angles (−57 ± 30, −47 ± 30). Ridges are centered near ideal values of −57 for φ and −47 for ψ, further increasing the probability in these regions. The ridge suggests that the precise value of φmax is poorly identified, likely due to peptide backbone geometry where, for φ-values <−100 and ψ-values >−40, backbone steric clashes prevent configurations being sampled at all.
) in Fig. 10 b. Peaks representing high probability values of (φmin, φmax) are seen at (−80, −50) and (−90, −40), with a ridge for φmin between −100 and −70. The joint distribution for (ψmin, ψmax) exhibits a sharp peak at (−60, −40) and a minor peak at (−50, −30). The ranges of dihedral angles with the largest peaks for both φ and ψ contain the values for an ideal helix (φ, ψ) = (−57, −47), but the joint mode of (φmin, φmax, ψmin, ψmax) = (−90, −40, −60, −40) yields a narrower range than that generally accepted for helical angles (−57 ± 30, −47 ± 30). Ridges are centered near ideal values of −57 for φ and −47 for ψ, further increasing the probability in these regions. The ridge suggests that the precise value of φmax is poorly identified, likely due to peptide backbone geometry where, for φ-values <−100 and ψ-values >−40, backbone steric clashes prevent configurations being sampled at all.
FIGURE 10.

Marginal posterior distributions of boundaries of the helical angle region (a) φmin and φmax, and (b) ψmin and ψmax.
Cross-validation
To run further simulations, we must choose a particular value for the internal dielectric parameter; a natural choice is the mode of the posterior distribution at εin = 4.1. This value gives the best agreement between the experimental helicities and the simulated helicities for our observed peptides.
However, we wish to know how sensitive this result is to the particular set of peptides chosen, and thus how well we can expect this choice to generalize to accurately simulate the helicity of new peptides outside our data set. Simply taking the accuracy of εin = 4.1 in predicting these eight peptides will tend to overestimate this accuracy, because this value has been optimized to perform well on those peptides. Nevertheless, we can estimate the future (out-of-sample) predictive accuracy from the current set of peptides using the statistical method of cross-validation. We measure predictive accuracy via the mean-squared error (MSE) between the predicted and experimentally measure helicity values.
Cross-validation proceeds by removing one peptide (say the ith one) from the dataset, and using the other seven to estimate/optimize the parameter εin. Denote the resulting parameter value by 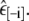 We then use this value to simulate the removed peptide and predict its helicity, calculating the squared error between predicted and experimental values. This process is then repeated to obtain similar predicted accuracies for each of the peptides in turn, always using the parameter value optimized over the other seven peptides, to calculate the overall estimated predictive accuracy:
We then use this value to simulate the removed peptide and predict its helicity, calculating the squared error between predicted and experimental values. This process is then repeated to obtain similar predicted accuracies for each of the peptides in turn, always using the parameter value optimized over the other seven peptides, to calculate the overall estimated predictive accuracy:
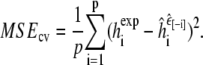 |
This procedure has well-established properties as an unbiased estimator of the out-of-sample predictive accuracy of our parameter adaptation method (40). Because the vast majority of our computational work is done upfront in running the simulations of each peptide at each value of εin, the expense of calculating the cross-validated prediction accuracy is negligible.
Table 5 shows the MSE for each value of εin and the cross-validated MSE; in this case the cross-validated MSE is equal to the MSE for εin = 4.1 since each 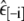 was equal to 4.1. Using the cross-validated MSE as an estimate of predictive variance, the predictive standard deviation is 3.6% and a standard interval prediction would contain simulated helicity ±7.2%.
was equal to 4.1. Using the cross-validated MSE as an estimate of predictive variance, the predictive standard deviation is 3.6% and a standard interval prediction would contain simulated helicity ±7.2%.
TABLE 5.
Mean-squared error (MSE) for each value of εin, along with estimated out-of-sample prediction accuracy given by MSE obtained from cross-validation
| εin | 1 | 2 | 3 | 4 | 4.1 | 5 | CV |
|---|---|---|---|---|---|---|---|
| MSE | 0.0405 | 0.0120 | 0.0025 | 0.0016 | 0.0013 | 0.0025 | 0.0013 |
It is important to note the limitations of this current predictive accuracy estimate due to data size and composition. In particular, the small dataset size leaves significant variability in the prediction accuracy estimate. In addition, cross-validation estimates the predictive accuracy for new data drawn from the same population as the observed data. Thus, the estimate of Table 5 is intended primarily as a demonstration of the approach; it should be interpreted as an estimated predictive accuracy for monomeric helical peptides, but may be less accurate for predominantly β-peptides, for example. A significantly enlarged and expanded dataset composition is needed to address these issues more generally.
CONCLUSIONS
Macromolecular simulation is becoming a widely used tool in structural and molecular biology. As usage grows and computational resources continue to accelerate, the development of true macromolecular computer experiments, which can accurately and reproducibly calculate thermodynamic or kinetic quantities that agree with experiment, is within sight. For researchers developing or utilizing macromolecular simulation, it is an exciting time.
Here we have attempted to focus attention on use of simulation in this quantitative, prediction fashion. We have described several statistical methods useful for addressing the challenges in doing so: quantitative measures of simulation convergence, construction of uncertainty intervals for simulated quantities, Bayesian and shrinkage estimation for parameter adaptation, and the use of cross-validation to evaluate predictive accuracy. We have also demonstrated the use of this approach to evaluating and improving molecular dynamics simulations of helical peptides, and explored the sensitivity of such simulations to small changes in parameter values. The tools described here are broadly applicable and we hope they will be adopted by other researchers and will help encourage further progress toward quantitative, predictive simulations of macromolecular systems.
The results described here are only a first step and may be improved in a number of ways. We have used relatively small amounts of data representing only equilibrium helicity in evaluating the predictive accuracy of our simulations, and more sophisticated comparisons to detailed experimental data will add significantly more information to evaluate and improve ensemble properties of simulations. Interesting recent examples have been applied to short timescale peptide kinetics (41,42) and comparison with nuclear magnetic resonance measurements (43,44). (Note that even when considering equilibrium helicity, Eq. 9 is crude, and may be more accurate if replaced by a calculation of ellipticity (45) or an entire CD spectrum for comparing with CD measurements; however, calculation of CD spectra from configurations is itself difficult.)
In addition, as pointed out above in Cross-Validation, our results may suffer from the use of helical peptides only, in our data set. An expanded study including β-peptides is warranted, as it is of significant interest to determine parameters appropriate for both α-helices and β-sheets. However, it is important to note that our emphasis on quantitative evaluation of equilibrium helical content (rather than, say, the minimum energy or most populated conformation) means that accuracy suffers if the force field either under- or overpredicts α-helix, and so this evaluation is sensitive to underprediction of β. Without direct measurements of β-content, however, we cannot resolve errors in the β-to-coil proportions, so inclusion of quantitative equilibrium data on β-content of β-hairpin peptides is of significant interest. Unfortunately we are currently limited by the lack of available experimental data in this area, due in no small part to difficulties in accurately quantifying equilibrium β-content from CD and nuclear magnetic resonance data.
In the presence of more detailed experimental information, the methods described in this article become even more important in enabling reliable quantitative comparisons, and for improving the predictive accuracy of force fields while avoiding overfitting.
Similarly, the purpose of a molecular simulation is rarely done simply to predict a single quantity such as helicity; and when such predictions are desired, statistical models may often be developed that are significantly more accurate across a wider range of input molecules (8). The advantage of a simulation is the ability to examine many different ensemble quantities calculated from a single simulation output. Nevertheless, predictive evaluation of measured quantities will help improve the underlying force fields and algorithms, thus improving the accuracy of, relevance of, and confidence in, other quantities obtained from simulation output.
Acknowledgments
S.C.S. and B.C. were supported in part by National Science Foundation grant No. DMS-0204690 (to S.C.S.). Computing resources were provided by National Science Foundation infrastructure grant No. DMS-0112340 (to S.C.S.).
Editor: Kathleen B. Hall.
References
- 1.Schlick, T. 2002. Molecular Modeling and Simulation. Springer-Verlag, Berlin, Germany.
- 2.Leach, A. R. 1996. Molecular Modeling: Principles and Applications. Addison Wesley Longman, Essex, UK.
- 3.Frenkel, D., and B. Smit. 1996. Understanding Molecular Simulation. Academic Press, San Diego, CA.
- 4.Hansmann, U. H. E. 1999. Computer simulation of biological macromolecules in generalized ensembles. Intl. J. Modern Phys. C. 10:1521–1530. [Google Scholar]
- 5.Mitsutake, A., Y. Sugita, and Y. Okamoto. 2001. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers. 60:96–123. [DOI] [PubMed] [Google Scholar]
- 6.Duan, Y., and P. A. Kollman. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 282:740–744. [DOI] [PubMed] [Google Scholar]
- 7.Scholtz, J. M., and R. L. Baldwin. 1992. The mechanism of α-helix formation by peptides. Annu. Rev. Biophys. Biomol. Struct. 21:95–118. [DOI] [PubMed] [Google Scholar]
- 8.Schmidler, S. C., J. Lucas, and T. G. Oas. 2007. Statistical estimation in statistical mechanical models: helix-coil theory and peptide helicity prediction. J. Comput. Biol. 14:1287–1310. [DOI] [PubMed] [Google Scholar]
- 9.Daggett, V., P. A. Kollman, and I. D. Kuntz. 1991. A molecular dynamics simulation of polyalanine: an analysis of equilibrium motions and helix-coil transitions. Biopolymers. 31:1115–1134. [DOI] [PubMed] [Google Scholar]
- 10.Brooks, C. L., and D. A. Case. 1993. Simulations of peptide conformational dynamics and thermodynamics. Chem. Rev. 93:2487–2502. [Google Scholar]
- 11.Garcia, A. E., and K. Y. Sanbonmatsu. 2002. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daura, X., B. Juan, D. Seebach, W. F. van Gunsteren, and A. E. Mark. 1998. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 280:925–932. [DOI] [PubMed] [Google Scholar]
- 13.Hansmann, U. H. E., and Y. Okamoto. 1999. Finite-size scaling of helix-coil transitions in poly-alanine studied by multicanonical simulations. J. Chem. Phys. 110:1267–1276. [Google Scholar]
- 14.Gnanakaran, S., H. Nymeyer, J. Portman, K. Y. Sanbonmatsu, and A. E. Garcia. 2003. Peptide folding simulations. Curr. Opin. Struct. Biol. 13:168–174. [DOI] [PubMed] [Google Scholar]
- 15.Jas, G. S., and K. Kuczera. 2004. Equilibrium structure and folding of a helix-forming peptide: circular dichroism measurements and replica-exchange molecular dynamics simulations. Biophys. J. 87:3786–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorin, E. J., and V. S. Pande. 2005. Empirical force-field assessment: the interplay between backbone torsions and noncovalent term scaling. J. Comput. Chem. 26:682–690. [DOI] [PubMed] [Google Scholar]
- 17.Sugita, Y., and Y. Okamoto. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151. [Google Scholar]
- 18.Geyer, C. J. 1991. Markov chain Monte Carlo maximum likelihood. In Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface. E. M. Keramidas, editor. 156–163.
- 19.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. R. Ross, I. T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structures and energies of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 20.Still, W. C., A. Tempczyk, R. Hawley, and T. Hendrickson. 1990. Semianalytic treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112:6127–6129. [Google Scholar]
- 21.Tsui, V., and D. A. Case. 2001. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 56:275–291. [DOI] [PubMed] [Google Scholar]
- 22.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 23.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 24.Nymeyer, H., and A. E. Garcia. 2003. Simulation of the folding equilibrium of α-helical peptides: a comparison of the generalized Born approximation with explicit solvent. Proc. Natl. Acad. Sci. USA. 100:13934–13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooke, B., and S. C. Schmidler. 2007. Preserving the Boltzmann ensemble in replica-exchange molecular dynamics. J. Chem. Phys. In press. [DOI] [PubMed]
- 26.Braxenthaler, M., R. Unger, A. Ditza, J. A. Given, and J. Moult. 1997. Chaos in protein dynamics. Proteins Struct. Funct. Genet. 29:417–425. [PubMed] [Google Scholar]
- 27.Pande, V. S., I. Baker, J. Chapman, S. P. Elmer, S. Khaliq, S. M. Larson, Y. M. Rhee, M. R. Shirts, C. D. Snow, E. J. Sorin, and B. Zagrovic. 2002. Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing. Biopolymers. 68:91–109. [DOI] [PubMed] [Google Scholar]
- 28.Tierney, L. 1994. Markov chains for exploring posterior distributions. Ann. Statist. 22:1701–1728. [Google Scholar]
- 29.van Gunsteren, W. F., and A. E. Mark. 1998. Validation of molecular dynamics simulation. J. Chem. Phys. 108:6109–6116. [Google Scholar]
- 30.Smith, L. J., X. Daura, and W. F. van Gunsteren. 2002. Assessing equilibration and convergence in biomolecular simulations. Proteins Struct. Funct. Genet. 48:487–496. [DOI] [PubMed] [Google Scholar]
- 31.Woodard, D., S. C. Schmidler, and M. Huber. 2007. Conditions for rapid mixing of parallel and simulated tempering on multimodal distributions. Ann. Appl. Probab. In press.
- 32.Geyer, C. J. 1992. Practical Markov chain Monte Carlo. Stat. Sci. 7:473–511. [Google Scholar]
- 33.Gelman, A., and D. B. Rubin. 1992. Inference from iterative simulation using multiple sequences. Stat. Sci. 7:457–511. [Google Scholar]
- 34.Cowles, M. K., and B. P. Carlin. 1996. Markov chain Monte Carlo convergence diagnostics: a comparative review. J. Am. Stat. Assoc. 91:883–904. [Google Scholar]
- 35.Brooks, S. P., and A. Gelman. 1998. General methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat. 7:434–455. [Google Scholar]
- 36.Fan, Y., S. P. Brooks, and A. Gelman. 2006. Output assessment for Monte Carlo simulations via the score statistic. J. Comput. Graph. Stat. 15:178–206. [Google Scholar]
- 37.Gelman, A., J. B. Carlin, H. S. Stern, and D. B. Rubin. 2004. Bayesian Data Analysis, 2nd Ed. Chapman & Hall/CRC, Boca Raton, FL.
- 38.Peng, Y., and U. H. E. Hansmann. 2002. Solvation model dependency of helix-coil transition in polyalanine. Biophys. J. 82:3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorin, E. J., and V. S. Pande. 2005. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 88:2472–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hastie, T., R. Tibshirani, and J. Friedman. 2001. The Elements of Statistical Learning. Springer-Verlag, New York.
- 41.Sporlein, S., H. Carstens, H. Satzger, C. Renner, R. Behrendt, L. Moroder, P. Tavan, W. Zinth, and J. Wachtveitl. 2002. Ultrafast spectroscopy reveals subnanosecond peptide conformation dynamics and validates molecular dynamics simulation. Proc. Natl. Acad. Sci. USA. 99:7998–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snow, C. D., N. Nguyen, V. S. Pande, and M. Gruebele. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 420:102–106. [DOI] [PubMed] [Google Scholar]
- 43.Daura, X., K. Gademann, H. Schafer, B. Juan, D. Seebach, and W. F. van Gunsteren. 2001. The β-peptide hairpin in solution: conformational study of a β-hexapeptide in methanol by NMR spectroscopy and MD simulation. J. Am. Chem. Soc. 123:2393–2404. [DOI] [PubMed] [Google Scholar]
- 44.Feenstra, K. A., C. Peter, R. M. Scheek, W. F. van Gunsteren, and A. E. Mark. 2002. A comparison of methods for calculating NMR cross-relaxation rates (NOESY and ROESY intensities) in small peptides. J. Biomol. NMR. 23:181–194. [DOI] [PubMed] [Google Scholar]
- 45.Shalongo, W., and E. Stellwagen. 1997. Dichroic statistical model for prediction and analysis of peptide helicity. Proteins Struct. Funct. Genet. 28:467–480. [PubMed] [Google Scholar]
- 46.Forood, B., E. J. Feliciano, and K. P. Nambiar. 1993. Stabilization of α-helical structures in short peptides via end capping. Proc. Natl. Acad. Sci. USA. 90:838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodman, E. M., and P. S. Kim. 1989. Folding of a peptide corresponding to the α-helix in bovine pancreatic trypsin inhibitor. Biochemistry. 28:4343–4347. [DOI] [PubMed] [Google Scholar]
- 48.Munoz, V., and L. Serrano. 1994. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1:399–409. [DOI] [PubMed] [Google Scholar]
- 49.Spector, S., M. Rosconi, and D. P. Raleigh. 1999. Conformational analysis of peptide fragments derived from the peripheral subunit-binding domain from the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus: evidence for nonrandom structure in the unfolded state. Biopolymers. 49:29–40. [DOI] [PubMed] [Google Scholar]
- 50.Strehlow, K. G., and R. L. Baldwin. 1989. Effect of the substitution Ala-Gly at each of five residue positions in the C-peptide helix. Biochemistry. 28:2130–2133. [DOI] [PubMed] [Google Scholar]