

Abstract
Here it is reported that aggrecan, the highly negatively charged macromolecule in the cartilage extracellular matrix, undergoes Ca2+-mediated self-adhesion after static compression even in the presence of strong electrostatic repulsion in physiological-like solution conditions. Aggrecan was chemically end-attached onto gold-coated planar silicon substrates and gold-coated microspherical atomic force microscope probe tips (end radius R ≈ 2.5 μm) at a density (∼40 mg/mL) that simulates physiological conditions in the tissue (∼20–80 mg/mL). Colloidal force spectroscopy was employed to measure the adhesion between opposing aggrecan monolayers in NaCl (0.001–1.0 M) and NaCl + CaCl2 ([Cl−] = 0.15 M, [Ca2+] = 0 – 75 mM) aqueous electrolyte solutions. Aggrecan self-adhesion was found to increase with increasing surface equilibration time upon compression (0–30 s). Hydrogen bonding and physical entanglements between the chondroitin sulfate-glycosaminoglycan side chains are proposed as important factors contributing to aggrecan self-adhesion. Self-adhesion was found to significantly increase with decreasing bath ionic strength (and hence, electrostatic double-layer repulsion), as well as increasing Ca2+ concentration due to the additional ion-bridging effects. It is hypothesized that aggrecan self-adhesion, and the macromolecular energy dissipation that results from this self-adhesion, could be important factors contributing to the self-assembled architecture and integrity of the cartilage extracellular matrix in vivo.
INTRODUCTION
It has long been hypothesized that intra- and intermolecular electrical double-layer and steric (e.g., entropic, excluded volume) repulsive interactions between the densely packed, highly negatively charged glycosaminoglycans (GAGs) of aggrecan (1) (Fig. 1, a–c) are critical determinants of the unique biomechanical properties of cartilage tissue, in particular its compressive (2) and shear (3) stiffness. This hypothesis was initially developed based on the known molecular structure and chemical composition of aggrecan (first determined in the 1970s (4) and recently directly imaged at the single-molecule level by Ng et al. (5)) and then confirmed by the measurement of tissue-level biomechanical properties of cartilage with varying GAG contents (6) and in different electrolyte solution conditions (3,7). It has been suggested that aggrecan also plays a critical role within connective tissues by protecting the collagen fibrillar network within which it is embedded from proteolytic degradation (8) as well as mechanical overload (9). Only with recent nanotechnological advances have the local molecular interactions between aggrecan and their constituent GAG chains been directly quantified using biomimetic model systems, new surface patterning methodologies, high-resolution force spectroscopy (HRFS) instrumentation, and Poisson-Boltzmann-based electrostatic molecular-level continuum and atomistic-level modeling (10–15). These studies have elucidated the molecular origins of biomechanical properties by showing that nanoscale trends in the biomimetic aggrecan systems are reflected in macroscale tissue-level behavior (7,16,17), e.g., the dependence of nanomechanical properties on bath ionic strength (IS), calcium ion concentration, and displacement rate. Previous work has focused on the effects of molecular-level repulsive forces during compression and shear of aggrecan (10–13).
FIGURE 1.

Overview of the cartilage aggrecan macromolecule and experimental setup, and results of aggrecan self-adhesion. (a) Schematic of major load-bearing constituents of the ECM, including the type II collagen network and proteoglycan (aggrecan). Aggrecan macromolecules are attached to hyaluronan and stabilized via link protein. (b) Schematic of the cartilage aggrecan cylindrical brush-like structure (contour length, Lcontour ∼400 nm, molecular mass ∼3 MDa) (5), which is composed of a core protein backbone (cp) containing three globular domains (G1, G2, G3), grafted CS-GAG chains (Lcontour ∼40 nm, shown as C4S-GAG) with interchain spacing of ∼2–4 nm along the cp, and keratan sulfate GAG chains; N = N-terminal, C = C-terminal. (c) Tapping-mode AFM height image of two-dimensional closely packed fetal epiphyseal aggrecan on an atomically flat mica surface at a density that is ∼40× less than its physiological concentration in cartilage (adapted from Ng et al. (5), height scale <1 nm). (d) Contact-mode AFM two-dimensional (left, in 0.001 M NaCl, pH ∼5.6) and three-dimensional (right, in 0.1 M NaCl, pH ∼5.6) height image of a microcontact printed aggrecan-functionalized (inside the hexagonal pattern) and OH-SAM-functionalized (outside the hexagonal pattern) substrate imaged with an OH-SAM colloidal tip at 3 nN normal force (adapted from Dean et al. (11)). This image reflects a sample with the same aggrecan packing density as the nanomechanical experiments carried out in this study. (e) Schematics of colloidal force spectroscopy experiments reported in this work depicting the interactions between a functionalized planar substrate with end-grafted aggrecan and a hydroxyl-terminated monolayer (OH-SAM)-functionalized probe tip or an aggrecan end-grafted colloidal probe tip. (f) Comparison of colloidal force spectroscopy force-distance curves obtained via OH-SAM- and aggrecan-functionalized colloidal probe tips on aggrecan end-grafted planar substrates (1.0 M NaCl aqueous solution, surface dwell time t = 30 s, maximum compressive force, Fmax ≈ 45 nN, z-piezo displacement rate, z = 4 μm/s). Data for different experiments carried out at 10 different locations are shown for each probe tip. (Inset) Definition of the adhesive interaction distance, Dad, the maximum adhesion force, Fad, and the adhesion energy, Ead, for each pair of approach-retract force-distance curves. Statistically significant differences were observed for Fad of OH-SAM compared to aggrecan-functionalized probe tips (ANOVA, p < 0.001). (g) Histogram of Ead obtained via OH-SAM and aggrecan-functionalized colloidal probe tips on aggrecan end-grafted planar substrates (1.0 M NaCl aqueous solution, surface dwell time, t = 30 s, Fmax ≈ 45 nN, z-piezo displacement rate, z = 4 μm/s).
We have discovered and report here direct experimental evidence that aggrecan macromolecules can also undergo self-adhesion if they are compressed together for a sufficient amount of time, thereby sustaining tensile interactions that lead to energy dissipation, even in the presence of strong electrostatic repulsion. These results are consistent with previous nuclear magnetic resonance (NMR) studies on hyaluronan, keratan, dermatan, and chondroitin sulfate (CS) GAG chains in solution, which showed intramolecular hydrogen bonding and supramolecular organization that were suggested to result in helical, complementary antiparallel structures (18). These NMR findings (18) were extended to hypothesize that the small proteoglycan, decorin, could bridge between collagen fibrils in connective tissues via interactions between the single GAG chain present on each of two neighboring decorin core proteins (19). Here, we focus on the large aggregating proteoglycan, aggrecan, and propose a new biological function, i.e., that self-adhesion between aggrecan and the resulting macromolecular energy dissipation can be a factor in the assembly, organization, and physiological function of the pericellular and interterritorial regions of the cartilage extracellular matrix (ECM). This conclusion is based on the experimental quantification of the self-adhesion forces and energies between two well-defined, chemically end-grafted aggrecan layers at physiologically relevant packing densities using colloidal force spectroscopy as a function of displacement rate, compression time, bath IS (NaCl), and calcium concentration.
MATERIALS AND METHODS
Purified fetal bovine epiphyseal aggrecan was chemically end-functionalized with thiol groups and end-attached on a planar gold-coated silicon substrate via microcontact printing as described previously (10,11) (Fig. 1, d and e). The possible chemical binding sites along aggrecan core protein exist mostly at the N-terminal or the G1, G2, and G3 globular domains of the core protein (10). A polydimethylsiloxane (PDMS) stamp with hexagonal holes (∼10 μm side) was immersed in a 5 mg/mL ethanol solution of hydroxyl-terminated self-assembled monolayer (OH-SAM, 11-mecaptoundecanol, HS(CH2)11OH; Aldrich, St. Louis, MO) for ∼30 min. The stamp was brought into contact with a 1 cm × 1 cm freshly cleaned gold-coated substrate (via Piranha solution, v3:1 of 98% H2SO4 and 30% H2O2) for ∼30 s to form an OH-SAM layer in the region outside of the hexagons. The hexagonal region was then filled with aggrecan by incubating 50 μL of 1 mg/mL thiol-aggrecan solution in a humidity chamber for 48 h. The sample was thoroughly rinsed with deionized water (low salt concentration, large electrostatic double-layer intermolecular repulsion) before experimentation to remove any possible physisorbed molecules. The presence of a monolayer was verified by ensuring that the aggrecan layer height at low salt concentrations (measured by contact mode AFM) was consistent with the known contour length from biochemical and single-molecule AFM imaging (5), as well as by checking for reproducible and spatially uniform nanomechanical data using nanoscale probe tips (10,11). Gold-coated spherical colloidal AFM probe tips (end radius R ≈ 2.5 μm, spring constant k ≈ 0.12 N/m; BioForce Nanosciences, Ames, IA) were functionalized with the same OH-SAM layer, or end-attached aggrecan layer (Fig. 1 e).
Colloidal force spectroscopy was performed in bathing electrolyte solutions of known ionic content, i.e., NaCl at various concentrations (0.001–1.0 M) in Milli-Q water. The bath pH, measured to be ∼5.6, was kept approximately constant by controlling the ambient temperature, humidity, and CO2 concentration. The pH of grade 0 cartilage typically is ∼7.1 and can range down to 5.5 for osteoarthritic cartilage (20,21). For GAGs, the pKa of the sulfate group is ∼2–2.5 (22) and that of the carboxyl group is 3–4 (23,24) Hence, for pH > 4.5, the GAG charge density remains constant (as verified experimentally in previous studies (20,21)). GAG-GAG electrostatic interactions are thus pH independent in this range (pH > 4.5) as well (10). Experiments were conducted via a Nanoscope IV MultiMode atomic force microscope with a PicoForce piezo (Veeco, Santa Barbara, CA). The sample was moved toward the cantilever probe tips (approach) and then, after contact, was moved away from the cantilever probe tip (retract) using both the OH-SAM and aggrecan-functionalized tips while the force versus tip-sample separation distance was recorded (Fig. 1 f). The surface interaction area during the colloidal force spectroscopy experiment (25) contained ∼103 aggrecan molecules. Retraction of the z-piezo was carried out after a surface dwell time (∼0–30 s) at a constant normal force (Fmax ≈ 50 nN) and z-piezo displacement rate (z = 4 μm/s). In another series of experiments, Fmax was varied between ∼10 and 60 nN at t = 5 s and z = 4 μm/s; z was also varied between 0.1 and 10 μm/s at t = 1 s and Fmax ≈ 50 nN. Similar experiments were performed using the aggrecan-functionalized tip with NaCl + CaCl2 aqueous solutions at varying desired [Ca2+] concentrations, but varying [Na+] to maintain a constant [Cl−] = 0.15 M.
Since the height of OH-SAM is negligible (∼1–2 nm (26)) compared to that of the end-attached aggrecan layer, the height difference between the aggrecan and OH-SAM regions measured via contact mode AFM imaging was taken as the aggrecan layer height, as described previously (11). The absolute value of tip-surface separation distance was obtained by offsetting the force-distance curve by the “incompressible” layer height of aggrecan measured via contact mode AFM at the force equivalent to the maximum force applied in the force-distance curve measurement (Fig. 1, d–f). Hence, the adhesive interaction distance Dad (the distance from D = 0 to the unbinding length on retract), the maximum adhesion force, Fad, and adhesive interaction energy, Ead (area under the force-distance curve on retract), were measured for each experiment (Fig. 1, f and g). Nonspecific substrate adhesion was negligible because it takes place at short distances (less than a few nanometers) and would not affect the long-range (hundreds of nanometers) macromolecular bioadhesion values measured in the aggrecan experiments. Control experiments show that the force-distance data on approach are highly reproducible for ∼1000 consecutive experiments at the same location (standard deviations less than the size of the data points), which confirms the reversibility of the deformation (i.e., indicating negligible molecular rearrangement) and the stability of the chemical end-attachment.
After the nanomechanical experiments were conducted, aggrecan molecules were detached from the gold-coated substrate via 15 min boiling in 5 mL deionized water. The solution was then lyophilized and adjusted to 200 μL using deionized water. The amount of sulfate-functional groups in the solution was assessed using the dimethylmethylene blue dye assay (10,27). Based on the measured amount of sulfate and the surface area of the gold-coated sample, the aggrecan packing density was calculated to be ∼25 nm intermolecular spacing, equivalent to ∼40 mg/mL at 0.1 M IS, within the range of 20–80 mg/mL physiological aggrecan concentration in cartilage tissue.
RESULTS AND DISCUSSION
Aggrecan self-adhesion was observed given a sufficient surface dwell or equilibration hold time (approximately seconds) at a constant compressive load for IS values ranging from 0.001 to 1.0 M NaCl, pH ∼5.6 (including near-physiological solution conditions of 0.1 M NaCl, pH ∼5.6) for both neutral hydroxyl-terminated self-assembled monolayer (OH-SAM) and aggrecan-functionalized colloidal probe tips versus aggrecan-functionalized planar surfaces. Fig. 1 f shows typical force versus tip-sample separation distance data sets on approach (i.e., loading: colloidal probe tip advancing toward the planar surface) and retract (i.e., unloading: colloidal probe tip moving away from the planar surface) for both the OH-SAM- and aggrecan-functionalized probe tips at 1.0 M NaCl, pH ∼5.6 (the solution conditions that exhibited the strongest and longest-range adhesive interactions due to salt screening of the electrostatic double-layer repulsion forces). Here, a long-range nonlinear attractive force profile was observed on retract with jagged contours (Fig. 1 f), indicating extension of multiple bridging macromolecular chains, where the range of these adhesive forces is observed to be much greater than the expected range for van der Waals interactions (28). The maximum force on loading, Fmax, was ∼10–60 nN, which imparted compressive molecular strains of ∼20–50% (relative to the initial layer height at ∼0 nN compression (12)). This range of compressive strain corresponds to the aggrecan molecular compaction found within nonloaded cartilage in vivo (11,29). The average maximum adhesive interaction distance, Dad (Fig. 1, f and g), measured by the OH-SAM-functionalized colloidal probe tip (1.0 M, pH ∼5.6) was 410 ± 100 nm, which agrees well with the known contour length of fetal epiphyseal aggrecan, Lc = 398 ± 57 nm (5). Dad obtained using the aggrecan-functionalized colloidal probe tip (1.0 M, pH ∼5.6) was measured to be 755 ± 145 nm ≈ 2 × Lc of aggrecan, demonstrating that the adhesive interactions were in fact between two opposing end-attached aggrecan layers, as opposed to the tethering of aggrecan to the underlying substrate.
For the OH-SAM-functionalized probe tip, adhesion may originate from short-range noncovalent binding between functional groups along aggrecan molecular segments and the OH-SAM, e.g., hydrogen bonding between GAG –OH, –COOH, –COO−, –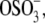 and the OH-terminal groups on the SAM, and/or van der Waals interactions. The increased net adhesion with aggrecan-functionalized probe tips relative to OH-SAM-functionalized probe tips (Figs. 1, f and g, and 2) can be attributed to both chemical and physical factors. First, there is stronger hydrogen bonding between the two aggrecan layers (–COO− versus –COOH, –COOH versus –COOH, –
and the OH-terminal groups on the SAM, and/or van der Waals interactions. The increased net adhesion with aggrecan-functionalized probe tips relative to OH-SAM-functionalized probe tips (Figs. 1, f and g, and 2) can be attributed to both chemical and physical factors. First, there is stronger hydrogen bonding between the two aggrecan layers (–COO− versus –COOH, –COOH versus –COOH, –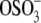 versus –COOH) (30) compared to that between the aggrecan layer and OH-SAM probe tip. Second, it is possible for interpenetration between opposing aggrecan macromolecules and their GAG branches to occur during compression (5,11), which results in a larger number of molecular contacts. Additional physical adhesive interactions for the two opposing aggrecan layers include macromolecular entanglements between the two layers.
versus –COOH) (30) compared to that between the aggrecan layer and OH-SAM probe tip. Second, it is possible for interpenetration between opposing aggrecan macromolecules and their GAG branches to occur during compression (5,11), which results in a larger number of molecular contacts. Additional physical adhesive interactions for the two opposing aggrecan layers include macromolecular entanglements between the two layers.
FIGURE 2.

Dependence of aggrecan self-adhesion on surface dwell time and bath IS. (a) Maximum adhesion forces, Fad (open symbols), and (b) total adhesion energy, Ead (closed symbols), as a function of surface dwell time, t, between an OH-SAM- and an aggrecan-functionalized planar substrate; (c) Fad (open symbols) and (d) Ead (closed symbols) as a function of surface dwell time, t, between two opposing aggrecan end-grafted layers (c and d) in 0.001–1.0 M NaCl, pH ∼5.6 (Fmax ≈ 45 nN, z ≈ 4 μm/s; mean ± SE, n ≥ 30 for each surface dwell time, t, at each IS). Statistically significant differences were observed for Fad and Ead at different surface dwell times, t, and different IS values measured by both OH-SAM and aggrecan probe tips (two-way ANOVA, pt < 0.0001, pIS < 0.0001).
Fig. 2 shows the dependence of aggrecan self-adhesion on the surface dwell (equilibration) hold time, t, at a constant compressive load and salt concentration (NaCl) for both the OH-SAM- and aggrecan-functionalized probe tips versus the aggrecan-functionalized planar surfaces. The nonlinearly increasing adhesion force and energy with t for both the OH-SAM and aggrecan-functionalized probe tips (Fig. 2) is attributed to the time-dependent increase in the number of molecular interactions, which may be facilitated by the configurational rearrangement of aggrecan macromolecules. These changes with time observed in Fig. 2 have been observed previously for many different nonmacromolecular systems (e.g., silica versus silicon, and silica versus mica); however, the equilibration time to reach plateau values of Fad and Ead in those systems is typically ∼1–2 s (31), which is shorter than that observed for aggrecan (∼5 s). The times required to reach plateau values of Fad and Ead were found to be similar for the OH-SAM-functionalized and aggrecan-functionalized probe tips, indicating that the introduction of the second macromolecular layer (on the colloidal probe tip) did not delay the formation of adhesive interactions. As observed in Fig. 2, a and b, there is minimal adhesion between the OH-SAM-functionalized probe tip and aggrecan at the lower IS values (0.001–0.01 M NaCl) in the presence of strong electrostatic double-layer repulsion. The larger adhesion observed between two opposing aggrecan layers at these lower salt concentrations (Fig. 2, c and d) could be the result of increased regions of molecular contact between two aggrecan layers, stronger molecular interactions, and the presence of physical adhesive interactions, e.g., molecular entanglements. As the IS increases, the electrostatic double-layer repulsion is screened, as reflected in a decrease in the electrical Debye length, and both result in a decrease in CS-GAG molecular spacing (11).
The net aggrecan self-adhesion is expected to be a fairly complex balance between a host of possible attractive and repulsive interactions, similar to most biomacromolecular systems (Fig. 3). Regarding attractive interactions, first, physical linkages are likely to result from chain diffusion, interpenetration, and interdigitation (32) during the initial compressive and surface dwell phases. The presence of GAG-GAG interdigitation is supported by the observation that interdigitation between GAGs can occur even at the aggrecan packing density ∼10 × less than its physiological concentration (as shown schematically in Fig. 3 a) (5). Second, attractive chemical secondary intermolecular interactions (e.g., van der Waals contacts, hydrophobicity, and hydrogen bonding (18,33)) are expected to be present, as are interactions with multivalent cations (if present) (34). Each CS-GAG chain contains ∼40–50 disaccharide units (5) in which both polar groups (three hydroxyl, one carboxyl, and one sulfate) and nonpolar groups (one methyl and two sugar rings) are present (4,35) (Fig. 1 b). Hydrogen bonds could occur between the –OH, –COOH, and –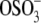 groups (pKa of GAG carboxyl ∼3 in aqueous solutions (36)); NMR shows the presence of hydrogen bonding between the acetamido =NH and –COO− groups (37). In addition, nonpolar patches along CS-GAG chains can lead to possible van der Waals and hydrophobic interactions (Fig. 3 c) (38). Since the interaction energy of hydrogen bonding is usually larger compared to the hydrophobic or van der Waals contacts, among these possible interactions, hydrogen bonding between different functional groups is expected to be a critical contributing factor to the observed aggrecan self-adhesion in the absence of multivalent ions.
groups (pKa of GAG carboxyl ∼3 in aqueous solutions (36)); NMR shows the presence of hydrogen bonding between the acetamido =NH and –COO− groups (37). In addition, nonpolar patches along CS-GAG chains can lead to possible van der Waals and hydrophobic interactions (Fig. 3 c) (38). Since the interaction energy of hydrogen bonding is usually larger compared to the hydrophobic or van der Waals contacts, among these possible interactions, hydrogen bonding between different functional groups is expected to be a critical contributing factor to the observed aggrecan self-adhesion in the absence of multivalent ions.
FIGURE 3.

Schematics of (a) two opposing aggrecan macromolecules undergoing interpenetration and entanglement in vivo; (b) network structure between aggrecan via self-adhesion due to Ca2+-mediated ion-bridging and molecular entanglements between GAG chains, and energy dissipation of the Ca2+-ion-bridges upon mechanically induced GAG molecular elongation (not drawn on scale); and (c) possible hydrogen bonding (dashed arrows), hydrophobic interaction (could occur between the methyl groups and carbon rings), and Ca2+-mediated ion-bridging between CS-GAG chains in the presence of water molecules. Water bridges could exist between the hydrogen bonding donors and acceptors.
Regarding repulsive interactions, electrostatic double-layer, configurational, translational, and rotational entropic penalties; hydration effects; and local steric hindrance (39,40) may all contribute. It is known that GAG chains have an extended conformation (persistence length ≈ 20 nm (5)) due to the limited mobility present in the glycosidic linkage torsions and intermolecular electrostatic interactions (15,41–43), and hence the conformational entropic penalty is expected to be minimal (relative to a random coil) and could be counterbalanced by the favorable enthalpic decrease from other attractive interactions (e.g., hydrogen bonding (37), van der Waals, hydrophobicity, etc.). Carbohydrate-protein interactions between GAGs and the aggrecan core proteins may also contribute to the aggrecan self-adhesion (4); however, since they are surrounded by densely packed GAGs, binding to the core protein is likely not preferable. Hence, electrostatic double-layer interactions are expected to be the dominant repulsive contribution.
In the presence of Ca2+, the adhesive interaction energy Ead increased by ∼4-fold from 0 to 75 mM CaCl2 at fixed [Cl−] = 0.15 M (Fig. 4). This significant increase, even at the near-physiological [Ca2+]= 2 mM, could be due to ion-bridging effects associated with the presence of multivalent ions (44), as it is known that one Ca2+ can bind electrostatically between two monovalent negative charges on the GAG side chains (45,46), or between the GAG chain and the core protein (47) from two opposing aggrecan molecules (Fig. 3 c). This Ca2+-mediated binding mechanism is also known to occur between other biological molecules. For example, the carbohydrate-carbohydrate interactions between trisaccharide Lewis X is relevant to cell-cell adhesion (48), and the intra- and intermolecular adhesion for type I collagen and other noncollagenous biomacromolecules (e.g., osteopontin) is an essential contributor to the mechanical properties of bone (49). Even a small amount of Ca2+ ([Ca2+] ≈ 1–2% of [Na+]) can lead to large energy dissipation (49) upon mechanical deformation-induced molecular strain of the aggrecan network (Fig. 3 b) corresponding to the physiological range (∼70% increase in Ead at 2 mM; Fig. 4 b). The Ca2+-mediated bridging effect is reversible, as replacing the CaCl2 + NaCl solution with 0.15 M NaCl resulted in a significant drop in adhesion to the same level observed before the addition of Ca2+, similar to what is observed in Ca2+-mediated binding of osteopontin in bone (34).
FIGURE 4.

Dependence of aggrecan self-adhesion on Ca2+ concentration. (a) Maximum adhesion forces, Fad (open symbols), and (b) total adhesion energy, Ead (solid symbols), as a function of surface dwell time, t, between two opposing aggrecan layers in NaCl + CaCl2 solutions, [Cl−] = 0.154 M at varying [Ca2+], pH ≈ 5.6 (z ≈ 4 μm/s, Fmax ≈ 45 nN; mean ± SE, n ≥ 30 for each surface dwell time, t, at each IS). Statistically significant differences were observed for Fad and Ead at different [Ca2+] (two-way ANOVA test, p < 0.0001).
There was no significant effect of maximum compressive force Fmax in the range of ∼10–60 nN (corresponding to the molecular conformation of aggrecan in vivo (11)) on the measured adhesion energy for both the OH-SAM-functionalized (data not shown) and aggrecan-functionalized colloidal probe tips (Fig. 5 a). In addition, the loading-unloading z-piezo displacement rate (0.1–10 μm/s) did not significantly affect the measured adhesion energy (Fig. 5 b) (both verified by a one-way analysis of variance (ANOVA), p > 0.05), suggesting that aggrecan behaves in an elastic-like (non-rate-dependent) manner in this range of loading rates, consistent with previous measurements of aggrecan compressive properties in the same range of displacement rates (11). Since the energy dissipation arising from extension and relaxation of the interfacial bonds highly depends on the thermal state of the system (50), this negligible dependence of adhesion on the displacement rate suggests that the applied unloading rates (∼0.5–50 pN/s per pair of aggrecan, estimated from the average binding force per aggrecan (= Fad/number of pairs of aggrecan) divided by the experimental time) are within the range of quasi-equilibrium conditions. In the unloading rate range far from equilibrium, rupture forces measured in interaction force experiments depend logarithmically on loading rates (51,52), as observed in other similar systems that test the binding forces between single or a few biological molecules, such as hyaluronan versus hyaluronan binding protein (53), and biotin versus streptavidin (52). Independence of the unloading rate has been observed for other molecules in experiments that were carried out under quasi-equilibrium conditions (54–57) (e.g., unbinding of ureido-4(1H)-pyrimidinone dimers (UPy)2 in hexadecane at 330 K at ∼104–105 pN/s unloading rate (54) and 6-ferrocenylhexanethiol (C6Fc) and thiol-functionalized 2-mercaptoethanol (C2OH) in Milli-Q water at ∼103–106 pN/s (56)). The z-piezo displacement rates used in the study presented here (0.1–10 μm/s) correspond to the range that is consistent with the known quasi-equilibrium macroscopic behavior of cartilage (11), suggesting that aggrecan self-adhesion can occur within cartilage under quasi-equilibrium conditions.
FIGURE 5.

Dependence of aggrecan self-adhesion on maximum compression and displacement rate. Total adhesion energy, Ead, as a function of (a) maximal compressive force, Fmax (t = 5 s, z ≈ 4 μm/s; mean ± SE, n ≥ 30 for each Fmax, one-way ANOVA p > 0.05) and (b) z-piezo displacement rate between two opposing aggrecan layers in 0.1 M NaCl, pH ∼5.6 (t = 1 s, Fmax = 45 nN; mean ± SE, n ≥ 30 for each z, one-way ANOVA, p > 0.05). The trends observed at other IS values are similar.
Our experimental setup was designed to be as close as possible to physiological conditions. The two-dimensional chemical end-grafting of aggrecan onto a planar gold surface simulates the three-dimensional binding of aggrecan to hyaluronan via its G1 domain, the corresponding strain and aggrecan packing density at Fmax ≈ 10–60 nN are consistent with aggrecan molecular concentration in vivo (11), the loading-unloading rate (0.1–10 μm/s, or ∼10–1000 ms) covers the timescale of joint loading (within 10–1000 ms) (62), and the aqueous solution (IS = 0.15 M, [Ca2+] = 2 mM) mimics in vivo ion concentrations (IS = 0.15 M, [Ca2+] ≈ 2–4 mM) (6). The self-adhesion forces, Fad, and energies, Ead, reported in this study, which involved large, well-defined assemblies of ∼103 aggrecan macromolecules, were highly repeatable (Figs. 2, 4, and 5) and did not have the additional complexities that need to be considered in single-molecule binding measurements, such as nonspecific surface interactions, multibridging macromolecules, and the effect of molecular linkers (59).
The estimated average binding force between one pair of opposing aggrecans at IS = 0.15 M, [Ca2+] = 2 mM, is ∼1 pN; however, since adhesion may only occur between a fraction of the total aggrecan in the ensemble, this value should be taken as a lower limit. It should be noted that the estimated aggrecan-aggrecan ∼1 pN binding force is much weaker compared to experimentally measured single-molecule binding forces involving other ECM proteoglycans, e.g., hyaluronan/aggrecan G1 core protein domain (40 ± 11 pN) (53), decorin/decorin (16.5 ± 5.1 pN), type IX collagen/biglycan (∼15 pN) (60), type I collagen/decorin(core protein) (54.5 ± 20 pN), and type I collagen/decorin (GAG side chain) (31.9 ± 12.4 pN) (61). Two differences between those experiments, which measured single- or double-molecular interactions, and the data presented here on adhesion between two opposing aggrecan layers are as follows: first, aggrecan-aggrecan intermolecular interactions within each of the layers could affect local binding interactions; second, the maximum compressive load for a single pair of aggrecan monomers within the densely packed layers is much less than the single-molecular interactions on lower grafting density surfaces.
Aggrecan self-adhesion could be an important factor contributing to the self-assembled architecture and integrity of the cartilage pericellular and interterritorial matrix in vivo. Recent studies have recognized the importance of binding interactions between aggrecan with a variety of ECM and pericellular matrix macromolecules, including the collagens, small proteoglycans, and other glycoproteins, in the organization and stability of the aggrecan-rich matrix (62,63). Even though the magnitude of the self-adhesion forces is at least one order of magnitude smaller than the repulsive forces arising from the electrostatic double layers between aggrecan GAGs, the multiplicity of interactions existing within the aggrecan moiety results in a large reservoir for energy dissipation through the rupture of reversible self-adhesion interactions (49), and hence could protect the fine collagen meshwork upon repeated joint loading or impact. This hypothesis is supported by the self-assembly of glyconanoparticles in vitro due to similar carbohydrate-carbohydrate interactions (64). Additionally, adhesive interactions between ECM constituents are critical for ECM assembly and thus can play essential roles in overall tissue organization and proper tissue physiological function (65). For example, the reversible sacrificial bonding of osteopontin in bone is thought to act as an adhesive layer between the mineralized collagen fibrils and to be responsible for significant amounts of tensile strength at specific locations (34).
CONCLUSIONS
In this study we have presented direct experimental evidence of aggrecan self-adhesion in the presence and absence of Ca2+, including the kinetics of formation and resultant energy dissipation. To summarize, we have discovered that two opposing aggrecan layers, when compressed together for a sufficient amount of time (seconds), can undergo self-adhesion (up to 5 nN forces or ∼1 pN per aggrecan pair), which enables the extension of the macromolecular chains and, correspondingly, a large energy dissipation (up to 1.5 fJ) even in the presence of strong electrostatic double-layer repulsion. Aggrecan self-adhesion was found to be highly dependent on the time of compression (0–30 s), as well as [NaCl] = 0.001–1 M and [Ca2+] = 0–75 mM concentrations, but independent of the displacement rate (0.1–10 μm/s) and compressive load (10–60 nN) in the ranges tested. Our results are consistent with the known characteristics of other carbohydrate-carbohydrate interactions, a time-dependent formation of interactions, and a strong dependency on divalent cations [Ca2+] (66,67). Aggrecan self-adhesion may have both chemical (e.g., van der Waals, H-bonding) and physical (i.e., molecular entanglements) contributions. In the presence of Ca2+ (also present in vivo), ion bridging may be another important factor that contributes to self-adhesion. It is hypothesized that aggrecan self-adhesion may play an important role in the structural and mechanical integrity of the cartilage tissue.
Acknowledgments
The authors thank the Institute for Soldier Nanotechnologies at MIT, funded through the U.S. Army Research Office, for use of instruments.
This work was supported by the National Science Foundation, the Nanoscale Interdisciplinary Research Team (grant 0403903), the National Institutes of Health (grant AR45779), and a Whitaker Foundation Fellowship (D.D.).
The content does not necessarily reflect the position of the government and no official endorsement should be inferred.
Editor: Peter Hinterdorfer.
References
- 1.Hardingham, T. E., and A. J. Fosang. 1992. Proteoglycans: many forms and many functions. FASEB J. 6:861–870. [PubMed] [Google Scholar]
- 2.Williamson, A., A. Chen, and R. Sah. 2001. Compressive properties and function-composition relationships of developing bovine articular cartilage. J. Orthop. Res. 19:1113–1121. [DOI] [PubMed] [Google Scholar]
- 3.Jin, M. S., and A. J. Grodzinsky. 2001. Effect of electrostatic interactions between glycosaminoglycans on the shear stiffness of cartilage: A molecular model and experiments. Macromolecules. 34:8330–8339. [Google Scholar]
- 4.Muir, I. H. M. 1979. Biochemistry. In Adult Articular Cartilage. M. A. R. Freeman, editor. Pitman Medical, Kent, UK. 145–214.
- 5.Ng, L., A. J. Grodzinsky, P. Patwari, J. Sandy, A. Plaas, and C. Ortiz l. 2003. Individual cartilage aggrecan macromolecules and their constituent glycosaminoglycans visualized via atomic force microscopy. J. Struct. Biol. 143:242–257. [DOI] [PubMed] [Google Scholar]
- 6.Maroudas, A. 1979. Physicochemical properties of articular cartilage. In Adult Articular Cartilage. M. A. R. Freeman, editor. Pitman Medical, Kent, UK. 215–290.
- 7.Eisenberg, S. R., and A. J. Grodzinsky. 1985. Swelling of articular cartilage and other connective tissues: electromechanochemical forces. J. Orthop. Res. 3:148–159. [DOI] [PubMed] [Google Scholar]
- 8.Pratta, M. A., W. Yao, C. Decicco, M. D. Tortorella, R.-Q. Liu, R. A. Copeland, R. Magolda, R. C. Newton, J. M. Trzaskos, and E. C. Arner. 2003. Aggrecan protects cartilage collagen from proteolytic cleavage. J. Biol. Chem. 278:45539–45545. [DOI] [PubMed] [Google Scholar]
- 9.Hardingham, T. E., H. Muir, M. K. Kwan, W. M. Lai, and V. C. Mow. 1987. Viscoelastic properties of proteoglycan solutions with varying proportions present as aggregates. J. Orthop. Res. 5:36–46. [DOI] [PubMed] [Google Scholar]
- 10.Dean, D., L. Han, C. Ortiz, and A. J. Grodzinsky. 2005. Nanoscale conformation and compressibility of cartilage aggrecan using microcontact printing and atomic force microscopy. Macromolecules. 38:4047–4049. [Google Scholar]
- 11.Dean, D., L. Han, A. J. Grodzinsky, and C. Ortiz. 2006. Compressive nanomechanics of opposing aggrecan macromolecules. J. Biomech. 39:2555–2565. [DOI] [PubMed] [Google Scholar]
- 12.Han, L., D. Dean, C. Ortiz, and A. J. Grodzinsky. 2007. Lateral nanomechanics of cartilage aggrecan macromolecules. Biophys. J. 92:1384–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han, L., D. Dean, P. Mao, C. Ortiz, and A. J. Grodzinsky. 2007. Nanoscale shear deformation mechanisms of opposing cartilage aggrecan macromolecules. Biophys. J. 93:L23–L25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bathe, M., G. C. Rutledge, A. Grodzinsky, and B. Tidor. 2005. Osmotic pressure of aqueous chondroitin sulfate solution: a molecular modeling investigation. Biophys. J. 89:2357–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bathe, M., G. C. Rutledge, A. J. Grodzinsky, and B. Tidor. 2005. A coarse-grained molecular model for glycosaminoglycans: application to chondroitin, chondroitin sulfate, and hyaluronic acid. Biophys. J. 88:3870–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mak, A. F. 1986. The apparent viscoelastic behavior of articular cartilage—the contributions from the intrinsic matrix viscoelasticity and interstitial fluid-flows. J. Biomech. Eng. 108:123–130. [DOI] [PubMed] [Google Scholar]
- 17.Grodzinsky, A. J., V. Roth, E. Myers, W. D. Grossman, and V. C. Mow. 1981. The significance of electromechanical and osmotic forces in the nonequilibrium swelling behavior of articular cartilage in tension. J. Biomech. Eng. 103:221–231. [DOI] [PubMed] [Google Scholar]
- 18.Scott, J. E. 2001. Structure and function in extracellular matrices depend on interactions between anionic glycosaminoglycans. Pathol. Biol. 49:284–289. [DOI] [PubMed] [Google Scholar]
- 19.Scott, J. E., and R. A. Stockwell. 2006. Cartilage elasticity resides in shape module decoran and aggrecan sumps of damping fluid: Implications in osteoarthrosis. J. Physiol. 574:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konttinen, Y. T. J. Mandelin, T.-F. Li, J. Salo, J. Lassus, M. Liljestrom, M. Hukkanen, M. Takagi, I. Virtanen, and S. Santavirta 2002. Acidic cysteine endoproteinase cathepsin k in the degeneration of the superficial articular hyaline cartilage in osteoarthritis. Arthritis Rheum. 46:953–960. [DOI] [PubMed] [Google Scholar]
- 21.Frank, E. H., A. J. Grodzinsky, S. L. Phillips, and P. E. Grimshaw. 1990. Physicochemical and bioelectrical determinants of cartilage material properties. In Biomechanics of Diarthrodial Joints. V. C. Mow, A. Ratcliffe, and S. L.-Y. Woo, editors. Springer-Verlag, New York. 261–282.
- 22.Kuettner, K., and A. Lindenbaum. 1965. Analysis of mucopolysaccharides in partially aqueous media. Biochim. Biophys. Acta. 101:223–225. [DOI] [PubMed] [Google Scholar]
- 23.Freeman, W. D. S. C., and A. Maroudas. 1975. Charged group behaviour in cartilage proteoglycans in relation to pH. Ann. Rheum. Dis. 34:44–45. [Google Scholar]
- 24.Cleland, R. L. 1991. Electrophoretic mobility of wormlike chains. I. Experiment: hyaluronate and chondroitin 4-sulfate. Macromolecules. 24:4386–4390. [Google Scholar]
- 25.Rixman, M. A., D. Dean, C. E. Macias, and C. Ortiz. 2003. Nanoscale intermolecular interactions between human serum albumin and alkanethiol self-assembling monolayers. Langmuir. 19:6202–6218. [Google Scholar]
- 26.Schreiber, F. 2000. Structure and growth of self-assembling monolayers. Prog. Surf. Sci. 65:151–257. [Google Scholar]
- 27.Farndale, R. W., D. J. Buttle, and A. J. Barrett. 1986. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta. 883:173–177. [DOI] [PubMed] [Google Scholar]
- 28.Israelachvili, J. N. 1992. Intermolecular and Surface Forces. 2nd ed. Academic Press, London.
- 29.Wight, T. N., D. K. Heinegard, and V. C. Hascall. 1991. Proteoglycans: structure and function. In Cell Biology of the Extracellular Matrix. E. D. Hey, editor. Plenum Press, New York. 45–78.
- 30.van der Vegte, E. W., and G. Hadziioannou. 1997. Scanning force microscopy with chemical specificity: an extensive study of chemically specific tip-surface interactions and the chemical imaging of surface functional groups. Langmuir. 13:4357–4368. [Google Scholar]
- 31.Kapple, M., and H.-J. Butt. 2002. The colloidal probe technique and its application to adhesion force measurements. Part. Part. Syst. Charact. 19:129–143. [Google Scholar]
- 32.Voyutskii, S. S. 1963. Autoadhesion and Adhesion of High Polymers. Wiley InterScience, New York.
- 33.Kinloch, A. J. 1980. The science of adhesion: I. Surface and interfacial aspects. J. Mater. Sci. 15:2141–2166. [Google Scholar]
- 34.Fantner, G. E., J. Adams, P. Turner, P. J. Thurner, L. W. Fisher, and P. K. Hansma. 2007. Nanoscale ion mediated networks in bone: osteopontin can repeatedly dissipate large amounts of energy. Nano Lett. 7:2491–2498. [DOI] [PubMed] [Google Scholar]
- 35.Spillmann, D., and M. M. Burger. 2000. Carbohydrate-carbohydrate interactions. In Carbohydrates in Chemistry and Biology. B. Ernst, G. W. Hart, and P. Sinay, editors. Wiley-VCH, Weinheim, Germany. 1061–1091.
- 36.Cleland, R. L., J. L. Wang, and D. M. Detweiler. 1982. Polyelectrolyte properties of sodium hyaluronate. II. Potentiometric titration of hyaluronic acid. Macromolecules. 15:386–395. [Google Scholar]
- 37.Scott, J. E., and F. Heatley. 1982. Detection of secondary structure in glycosaminoglycans via the 1H n.m.r. signal of the acetamido NH group. Biochem. J. 207:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott, J. E. 1992. Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J. 6:2639–2645. [PubMed] [Google Scholar]
- 39.Derjaguin, B. V., Y. P. Toporov, V. M. Muller, and I. N. Aleinikova. 1977. On the relationship between the electrostatic and molecular component of the adhesion of elastic particles to a solid surface. J. Colloid Interface Sci. 58:528–533. [Google Scholar]
- 40.Rojo, J., J. C. Morales, and S. Penades. 2002. Carbohydrate-carbohydrate interactions in biological and model systems. Top. Curr. Chem. 218:45–92. [Google Scholar]
- 41.Fouissac, E., M. Milas, M. Rinaudo, and R. Borsali. 1992. Influence of the ionic strength on the dimensions of sodium hyaluronate. Macromolecules. 25:5613–5617. [Google Scholar]
- 42.Hayashi, K., K. Tsutsumi, F. Nakajima, T. Norisuye, and A. Teramoto. 1995. Chain stiffness and excluded volume effects in solutions of sodium hyaluronate at high ionic strength. Macromolecules. 28:3824–3830. [Google Scholar]
- 43.Esquenet, C., and E. Buhler. 2002. Aggregation behavior in semidilute rigid and semirigid polysaccharide solutions. Macromolecules. 35:3708–3716. [Google Scholar]
- 44.de la Cruz, M. O., L. Belloni, M. Delsanti, J. P. Dalbiez, O. Spalla, and M. Drifford. 1995. Precipitation of highly-charged polyelectrolyte solutions in the presence of multivalent salts. J. Chem. Phys. 103:5781–5791. [Google Scholar]
- 45.MacGregor, E. A., and J. M. Bowness. 1971. Interaction of proteoglycans and chondroitin sulfates with calcium or phosphate ions. Can. J. Biochem. 49:417–425. [DOI] [PubMed] [Google Scholar]
- 46.Hunter, G. K., K. S. Wong, and J. J. Kim. 1988. Binding of calcium to glycosaminoglycans: an equilibrium dialysis study. Arch. Biochem. Biophys. 260:161–167. [DOI] [PubMed] [Google Scholar]
- 47.Saleque, S., N. Ruiz, and K. Drickamer. 1993. Expression and characterization of a carbohydrate-binding fragment of rat aggrecan. Glycobiology. 3:185–190. [DOI] [PubMed] [Google Scholar]
- 48.de la Fuente, J. M., P. Eaton, A. G. Barrientos, M. Menendez, and S. Penades. 2005. Thermodynamic evidence for Ca2+-mediated self-aggregation of Lewis X gold glyconanoparticles. A model for cell adhesion via carbohydrate-carbohydrate interaction. J. Am. Chem. Soc. 127:6192–6197. [DOI] [PubMed] [Google Scholar]
- 49.Fantner, G. E., T. Hassenkam, J. H. Kindt, J. C. Weaver, H. Birkedal, L. Pechenik, J. A. Cutroni, G. A. G. Cidade, G. D. Stucky, D. E. Morse, and P. K. Hansma. 2005. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat. Mater. 4:612–616. [DOI] [PubMed] [Google Scholar]
- 50.Ghatak, A., K. Vorvolakos, H. She, D. L. Malotky, and M. K. Chaudhury. 2000. Interfacial rate processes in adhesion and friction. J. Phys. Chem. B. 104:4018–4030. [Google Scholar]
- 51.Evans, E. 2001. Probing the relation between force, lifetime, and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- 52.Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- 53.Liu, X., J. Q. Sun, M. H. Heggeness, M.-L. Yeh, and Z.-P. Luo. 2004. Direct quantification of the rupture force of single hyaluronan/hyaluronan binding protein bonds. FEBS Lett. 563:23–27. [DOI] [PubMed] [Google Scholar]
- 54.Zou, S., H. Schonherr, and G. J. Vancso. 2005. Force spectroscopy of quadruple h-bonded dimers by AFM: dynamic bond rupture and molecular time-temperature superposition. J. Am. Chem. Soc. 127:11230–11231. [DOI] [PubMed] [Google Scholar]
- 55.Auletta, T., M. R. de Jong, A. Mulder, F. van Veggel, J. Huskens, D. N. Reinhoudt, S. Zou, S. Zapotoczny, H. Schonherr, G. J. Vancso, and L. Kuipers. 2004. β-Cyclodextrin host-guest complexes probed under thermodynamic equilibrium: thermodynamics and AFM force spectroscopy. J. Am. Chem. Soc. 126:1577–1584. [DOI] [PubMed] [Google Scholar]
- 56.Zapotoczny, S., T. Auletta, M. R. de Jong, H. Schonherr, J. Huskens, F. van Veggel, D. N. Reinhoudt, and G. J. Vancso. 2002. Chain length and concentration dependence of β-cyclodextrin ferrocene host guest complex rupture forces probed by dynamic force spectroscopy. Langmuir. 18:6988–6994. [Google Scholar]
- 57.Schonherr, H., M. W. J. Beulen, J. Bugler, J. Huskens, F. van Veggel, D. N. Reinhoudt, and G. J. Vancso. 2000. Individual supramolecular host-guest interactions studied by dynamic single molecule force spectroscopy. J. Am. Chem. Soc. 122:4963–4967. [Google Scholar]
- 58.Shepherd, D. E. T., and B. B. Seedhom. 1997. A technique for measuring the compressive modulus of articular cartilage under physiological loading rates with preliminary results. Proc. Inst. Mech. Eng. [H]. 211:155–165. [DOI] [PubMed] [Google Scholar]
- 59.Willemsen, O. H., M. M. E. Snel, A. Cambi, J. Greve, B. G. D. Grooth, and C. G. Figdor. 2000. Biomolecular interactions measured by atomic force microscopy. Biophys. J. 79:3267–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen, C.-H., M.-L. Yeh, M. Geyer, G.-J. Wang, M.-H. Huang, M. H. Heggeness, M. Hook, and Z.-P. Luo. 2006. Interactions between collagen IX and biglycan measured by atomic force microscopy. Biochem. Biophys. Res. Commun. 339:204–208. [DOI] [PubMed] [Google Scholar]
- 61.Yeh, M.-L., X. Liu, and Z.-P. Luo. 2005. Quantification of the binding strength of type I collagen and decorin using optical tweezers. Proc. Orthopaedic Research Society, 51st, Washington, DC. 922.
- 62.Dudhia, J. 2005. Aggrecan, aging and assembly in articular cartilage. Cell. Mol. Life Sci. 62:2241–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindahl, U., and M. Hook. 1978. Glycosaminoglycans and their binding to biological macromolecules. Annu. Rev. Biochem. 47:385–417. [DOI] [PubMed] [Google Scholar]
- 64.de la Fuente, J. M., and S. Penades. 2006. Glyconanoparticles: types, synthesis and applications in glycoscience, biomedicine and material science. Biochim. Biophys. Acta. 1760:636–651. [DOI] [PubMed] [Google Scholar]
- 65.Gumbiner, B. M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 84:345–357. [DOI] [PubMed] [Google Scholar]
- 66.Popescu, O., I. Checiu, P. Gherghel, Z. Simon, and G. N. Misevic. 2003. Quantitative and qualitative approach of glycan-glycan interactions in marine sponges. Biochimie. 85:181–188. [DOI] [PubMed] [Google Scholar]
- 67.Haseley, S. R., H. J. Vermeer, J. P. Kamerling, and J. F. G. Vliegenthart. 2001. Carbohydrate self-recognition mediates marine sponge cellular adhesion. Proc. Natl. Acad. Sci. USA. 98:9419–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]