

Abstract
Objectives
Analyze the distribution of polymorphism in the dopamine receptor D3 (DRD3) gene, which was previously reported as a susceptibility risk for essential tremor (ET), in a large cohort of ET.
Methods
The role of 312G>A DRD3 polymorphism was analyzed using linkage analysis, association study and transmission disequilibrium test in a group of 433 ET patients, and two unrelated control groups with 121 and 151 individuals.
Results
Allelic frequencies of glycine and serine forms of the DRD3 gene did not differ between patients and both control groups, and were in Hardy-Weinberg equilibrium. Linkage analysis identified obligatory recombinants in every large pedigree, even in those with relatively high frequency of glycine allele, thus excluding the linkage to this locus. Both alleles were transmitted with an equal likelihood to affected offspring. We also failed to replicate the relationship between glycine homozygosity and an earlier age of onset or more severe tremor course.
Conclusions
Our comprehensive genetic analysis in a large ET cohort strongly argues against the role of the DRD3 gene in ET pathogenesis.
Keywords: Essential tremor, DRD3, Genetics
The genetics of essential tremor (ET) is fraught with controversies, and in spite of the high ET prevalence, the understanding of its genetic causes lags behind less common inherited movement disorders, such as ataxias or Parkinson disease [1–3]. Although an autosomal dominant (AD) mode of inheritance is widely accepted as the most plausible genetic model, the possibility of complex inheritance has been suggested [4]. Linkage analysis has identified four genetic loci on chromosome 3q13, 2p24.1, 6p23 and 5q31.1–q33.1, with additional evidence for further genetic heterogeneity [5–8]. However, no disease-causing mutations have been identified within these loci. Two single nucleotide polymorphisms (SNP) have been suggested as susceptibility factors, 828C>G in the HS1-BP3 gene mapped to the ETM2 locus on chromosome 2p, and 312G>A in the dopamine receptor 3 (DRD3) gene mapped to the ETM1 locus on chromosome 3q [9, 10]. The SNP in the HS1-BP3 gene is likely to be a benign polymorphism rather than an ET susceptibility factor, as studies attempting to replicate this finding did not find any differences in its frequency between ET patients and matched controls [11].
Polymorphism 312G>A (rs6280) in the DRD3 gene, resulting in a non-synonymous Ser9Gly amino acid change, is mapped within the ETM1 locus on chromosome 3q13 [5, 10]. A recent analysis of 30 French families found that the glycine allele segregated with the disease in 23 families. Furthermore, patients who were homozygous for this variant had an earlier age of onset and more severe tremor. This was further supported by an association study using a cohort of American patients with ET that found a higher frequency of the glycine allele in ET cases [10].
We have analyzed the occurrence of the DRD3 polymorphism in a group of 433 patients with ET using a complex approach with a combination of linkage analysis, transmission disequilibrium test and genetic association studies. Our findings were unable to replicate the previously reported role of the glycine and serine forms of the DRD3 gene.
Material and Methods
Clinical studies
We included 433 individuals who were diagnosed with definitive ET. The first group of subjects consisted of 237 unrelated ET patients who were selected for the case-control part of the study. These subjects were either selected as random probands from small pedigrees (less than five affected individuals available) or were the only affected subjects from a family. This proband subgroup reported a family history of ET in 157 (66%) patients with the remainder having apparently sporadic ET. The second group consisted of an additional 196 familial cases that were evaluated as a part of family-based methods. Large pedigrees with more than 10 affected individuals spanning at least two consecutive generations were evaluated using linkage analysis method, while smaller pedigrees were evaluated for the rate of transmission of the DRD3 SNP, 312G>A.
We obtained informed consent, approved by the Institutional Review Board at Vanderbilt University, from every participating subject. All participants were examined by a neurologist who specializes in the diagnosis and treatment of movement disorders (PH); the examination was focused on the tremor assessment and presence or absence of additional extrapyramidal signs. Clinical data including age of onset, tremor severity and distribution, as well as types of activities associated with tremor were collected. We also collected a detailed family history and obtained extended pedigrees.
Definite ET was diagnosed in subjects who had bilateral postural and kinetic arm tremor with or without asymmetry for more than five years, absence of additional neurologic abnormalities, no history of exposure to tremorogenic drugs before the onset of symptoms, and no history or examination suggestive of psychogenic tremor or sudden onset with a stepwise deterioration [12, 13]. Subjects with prominent resting tremor were required to have no signs of hypokinetic-rigid syndrome on examination and a history of failed dopaminergic therapeutic challenge.
Tremor was quantified using the NIH ET consortium grading [14]. A disabling tremor with excursions >4 cm was classified as severe tremor, partially disabling with excursions 2–4 cm as marked, and noticeable tremor with excursions < 2 cm and only partially disabling as moderate. Patients with slight tremor (barely noticeable) were not included. Postural tremor was assessed in two positions: with arms held in an outstretched position and with elbows flexed and shoulders abducted (wing-beating position); both positions were evaluated without any load and after the patient held a nearly full 12-ounce cup filled with water. Kinetic tremor was also assessed by sampling of handwriting, drawing of Archimedes spiral, and attempts to thread a needle in patients with moderate tremor. Furthermore, the degree of disability was also judged by self-reporting of questions adapted from the Tremor disability questionnaire [15]. The age of onset of tremor was self reported by the affected individuals and we did not include the date obtained from other sources. We rounded the age of onset to the nearest decade rather than using the exact stated age in years.
We included two independent control groups; the first consisted of 121 unrelated individuals older than 60 years whose neurologic screening examination did not show any tremor, who did not report any previous episodes of tremor lasting for more than seven days, a positive family history of ET, or Parkinson disease in their first degree relatives. The second control group with 151 individuals was recruited from married-in spouses who, similarly to the first control group, did not have any individual or family histories of tremor or parkinsonism; we included individuals older than 45 years in this control group. All groups were matched in regards to the ethnic origin with the ET group; therefore we included only Caucasian individuals.
Demographic characteristics between cases and controls were compared using Student t-test. We also tested the previously suggested association between DRD3 polymorphisms with the age of tremor onset and the severity of the disease using the Spearman rank order correlation test.
Genetic studies
The patients were classified as definitely affected or unaffected prior to genetic analysis. DNA was extracted from peripheral blood using previously published methods and was available from all subjects who agreed to participate. Every affected and control subject was genotyped for the presence of 312G>A polymorphism, resulting in Ser9Gly amino acid change in the DRD3 gene. For this, we used a previously described assay that initially required amplification of the 388 bp DNA segment containing the first exon with forward primer 5’-TTCTGTCTCCTCACAGGAAGC and reverse primer 3’-GCCAGAACTCAGGGAAGACA [4]. This amplicon contains four restriction sites recognized by the enzyme MscI, and one restriction site abolished in the A allele, that encodes for glycine. Thus, individuals homozygous for the serine allele have four bands, homozygotes for the glycine allele have three bands, and a heterozygote has five bands of different sizes. The fragments of DNA after overnight digestion following the manufacturer protocol (New England BioLabs, Beverly, MA) were separated on a 6% polyacrylamide gel and visualized by silver staining. The accuracy of the genotyping assay was verified by sequencing ten randomly selected individuals for each genotype; there was complete agreement between restriction digestion and sequencing results for these samples.
The potential role of this DRD3 polymorphism was investigated using three different methods of genetic analysis. The first was a case control study approach comparing the distribution of DRD3 genotypes within the group of 237 unrelated probands to both control groups. We then determined whether observed genotypes were in Hardy-Weinberg equilibrium. Allele frequency comparisons between patients and control groups were determined using a chi-squared test. The second approach was transmission disequilibrium test (TDT), which was determined in informative meioses in a subset of patients with familial ET. We included only affected individuals and required that the glycine allele was only inherited from an affected parent; pedigrees where glycine was inherited from a clinically unaffected parent were not included. TDT was analyzed using Genehunter v2.0 software. The third method of analysis was a linkage analysis using large pedigrees with at least 10 affected individuals from at least two generations, consistent with an autosomal mode (AD) of inheritance. We studied whether inheritance of the glycine allele segregated with the disease and determined the presence of obligatory cross-over events within these pedigrees. Two-point linkage to the DRD3 locus was calculated with the FASTLINK program assuming an AD mode of disease inheritance, observed allele frequencies as stated in the Results and included only affected individuals. We assigned a penetrance of 0.90 and 0.80 for LOD score calculations. We also analyzed the presence and inheritance of the studied DRD3 polymorphism in eight large pedigrees with an unrelated neurodegenerative condition, hereditary spastic paraplegia (HSP). We selected HSP pedigrees with at least 10 affected individuals and spanning at least three generations. Every affected HSP patient had a pure (uncomplicated) HSP phenotype without tremor and also married-in spouses had normal neurologic examination and no history of tremor in first-degree relatives [16] Genetic analysis of the DRD3 locus in HSP families was done in a similar fashion as in ET pedigrees.
Results
Frequency of genotypes and calculated allele frequency detected in ET unrelated probands (100 men, 137 women, average age 47±19.77 years), unrelated controls (58 men and 63 women, average age 66.33±5.34 years) and family-based controls (65 men and 86 women, average age 54±12.67 years) are summarized in Table 1. Overall, there were no statistically significant differences between allele frequencies and the observed genotypes did not deviate from expected Hardy-Weinberg distribution.
Table.
Genotypic frequencies in analyzed groups.
| Demographics | Ser/Ser | Gly/Gly | Gly/Ser | Ser allele | Gly allele | |
|---|---|---|---|---|---|---|
| ET unrelated probands |
100M/137F AA: 47±19.77 AO: 43±17.69 |
92 (0.38) | 34 (0.14) | 111 (0.48 | 295 (0.62) | 179 (0.38) |
| Unrelated controls |
58M/63W AA: 66.33±5.34 |
55 (0.46) | 19 (0.15) | 47 (0.39) | 157 (0.65) | 85 (0.35) |
| Family controls |
65M/86W AA: 54±12.67 |
48 (0.32) | 27 (0.18) | 76 (0.50) | 172 (0.57) | 130 (0.43) |
M – males, F – females, AA – average age (in years), AO – age of onset (in years)
We were able to analyze 47 informative transmissions of the glycine allele and this putative risk factor was transmitted to affected offspring in 21 cases and did not get transmitted in 26 informative meioses (difference not statistically significant); furthermore, we identified nine additional instances where the glycine allele was inherited from an apparently unaffected parent.
Linkage analysis suggested a possible linkage to the DRD3 locus in five smaller pedigrees consisting of five or less affected individuals and spanning only two generations; the highest detected two-point lod score was 0.78 at θ=zero (small pedigrees not shown). However, the high likelihood of chance segregation of the relatively common glycine allele with the disease was demonstrated on analysis of eight multigenerational ET pedigrees. Only one family with a predicted highest two-point lod score > 3 did not contain a single affected individual with the glycine allele. The incidence of the glycine allele in affected individuals from the remainder of the rest of analyzed large kindreds varied from 20% to 80% but we did not observe a complete segregation with the disease in any of these pedigrees. The frequency of the glycine allele was highest in three large pedigrees (Figure). The high frequency of individuals with the glycine alleles in the oldest analyzed generations, especially in pedigree 1 and 2, is likely a result of glycine homozygosity in their affected parents. However, we were able to identify obligatory recombinants in definitely affected individuals in successive generations, strongly arguing against the linkage to the DRD3 locus; two-point lod scores were < −2, further indicating an exclusion of this gene as a cause of ET in these families. Genotypes in pedigree 2 also demonstrate a relatively high frequency of the glycine allele in married-in spouses who did not have any individual or family history of ET. Genotyping of eight control pedigrees with HSP also showed that each of these families had several individuals with the glycine allele and the highest frequency of this putative ET risk factor was 75–80% in three of these families (data not shown).
Figure.
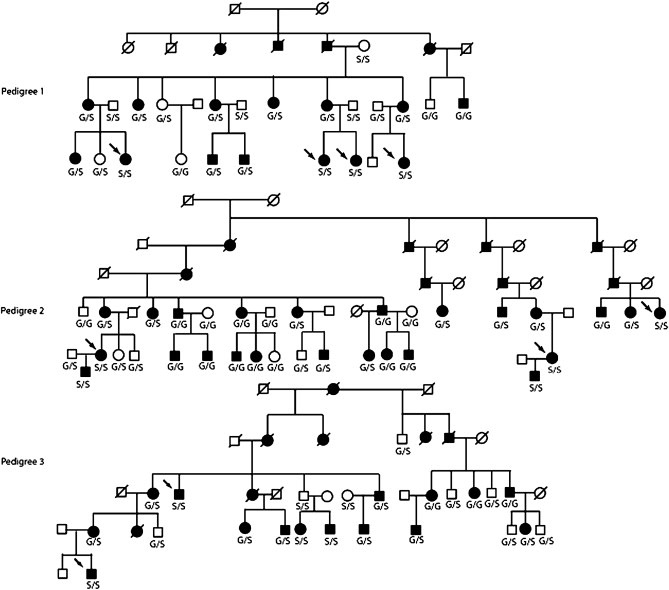
Three large multigenerational pedigrees with the highest frequency of the glycine allele. Obligatory cross-over events are denoted with an arrow.
The age of ET onset did not differ between gly/ser heterozygous and gly/gly homozygous individuals (r=0.33) and between genotypes and the severity of the disease (r=0.47). The comparison of these patients with ser/ser homozygotes was also not statistically significant (r=0.41 and r=0.29, respectively).
Discussion
Our data, obtained from a large cohort of ET patients and using a combination of three genetic analysis methods, strongly argues against 312G>A DRD3 polymorphism as a cause of ET or a significant susceptibility factor in its pathogenesis. Genetic association studies are fraught with difficulties and the literature is laden with several failures to replicate studies claiming positive associations between risk for a disease-phenotype and certain genetic variations. For example, the same DRD3 variant has been previously suggested to increase the risk for schizophrenia or modify the therapeutic response to neuroleptics; however, several studies failed to confirm these associations [17–20].
What are likely explanations of our observations? Glycine at the ninth codon is a common variant and the published frequency of this allele varies in the Caucasian population from 0.30 to 0.85 (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=6280). The conclusion that American ET patients show a relatively weak positive association (p = 0.039) in the original report of DRD3 role by Jeanneteau et al., [10] is based on rather low frequency 0.27 of the A allele (glycine) in their control group. Population stratification, caused by a systemic difference in ancestry between cases and controls, is the most common problem, which may result in erroneous allele frequencies affecting the outcome of genetic analysis [21, 22]. It is more likely to be present in populations with a mixed origin, such as North American Caucasian population. Hidden population stratification may be present even if both studied groups are carefully matched for their ethnic backgrounds [23]. To minimize this possibility, we selected two independent control groups for our comparisons. Furthermore, another study recently failed to replicate this association studying patients of Asian ethnic background, and the glycine allele frequency 0.33 in their group was very similar to our two sets of Caucasian controls [24].
We used additional methods of genetic analysis to evaluate the role of the DRD3 gene in ET to reduce problems seen in association studies. The 312G>A DRD3 polymorphism is relatively common and association study may underestimate the effect of this allele on the development of ET. However, if this SNP indeed influences the risk of ET, then it is expected to be preferentially transmitted to affected offspring. However, we did not observe any increased transmission of the glycine allele to affected offspring with essentially random inheritance of either allele in affected offspring. Furthermore, detected allelic frequencies were in Hardy-Weinberg equilibrium in all three analyzed groups of individuals.
It is still unknown whether this DRD3 variant segregates with the disease in the original Icelandic families used to identify ETM1 locus [5] Linkage analysis was reported as confirming the linkage of French families to the DRD3 locus [10]. However, these families are relatively small with a limited number of informative meioses, and given the high allelic frequency of presumed disease allele, it may occur by chance. This argument is supported by genotyping of several large families in this report, in which genotyping of incomplete pedigrees could be easily interpreted as evidence for linkage to the DRD3 locus. However, each kindred had several obligatory recombinants in otherwise typically affected individuals with postural and action tremor who did not have any other abnormal findings. We also analyzed kindreds with an unrelated neurodegenerative disorder HSP, whose phenotype does not include tremor, to explore whether the glycine allele may seemingly segregate with the disease in large multigenerational families. High frequency of this possible ET risk factor in several HSP pedigrees indicates the high likelihood of a chance occurrence of the glycine allele in some families. This approach with inclusion of control multigenerational families, whose members do not suffer from the studied disease, may be useful in testing the specificity of any putative genetic risk factors, especially in common genetic variants.
In vitro analysis of both DRD3 receptor variants identified a significantly increased affinity to dopamine when the glycine amino acid residue was present. This is consistent with a gain-of-function mechanism and glycine homozygous patients had more severe tremor with an earlier disease-onset in the same study [10]. However, we were also unable to replicate this finding, similar to a previous failure in ET patients with a different ethnic background.
The confirmed role of the DRD3 gene in ET pathogenesis would support the use of DRD3-specific antagonists in its therapy. However, our analysis strongly argues against a significant role of the DRD3 gene in ET etiology and it is likely that the linkage to the ETM1 locus is not due to this gene.
Acknowledgements
We thank the members of the families described here, whose help and participation made this work possible. This work was supported by NIH K08NS42743 to PH, and in part by GCRC Grant RR00095 to the VUMC General Clinical Research Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain. 2007;130:1456–1464. doi: 10.1093/brain/awm018. [DOI] [PubMed] [Google Scholar]
- 2.Louis ED. Essential tremor. Lancet Neurol. 2005;4:100–1010. doi: 10.1016/S1474-4422(05)00991-9. [DOI] [PubMed] [Google Scholar]
- 3.Tanner CM, Goldman SM, Lyons KE, Aston DA, Tetrud JW, Welsh MD, et al. Essential tremor in twins: an assessment of genetic vs. environmental determinants of etiology. Neurology. 2001;57:1389–1391. doi: 10.1212/wnl.57.8.1389. [DOI] [PubMed] [Google Scholar]
- 4.Ma S, Davis TL, Fang JY, Blair MA, Haines JL, Hedera P. Familial essential tremor with apparent autosomal dominant inheritance: Should we also consider other inheritance modes? Mov Disord. 2006;21:1368–1374. doi: 10.1002/mds.20950. [DOI] [PubMed] [Google Scholar]
- 5.Gulcher JR, Jonsson P, Kong A, Kristjánsson K, Frigge ML, Kárason A, et al. Mapping of a familial essential tremor gene, FET1 to chromosome 3q13. Nature Genet. 1997;17:84–87. doi: 10.1038/ng0997-84. [DOI] [PubMed] [Google Scholar]
- 6.Higgins JJ, Pho LT, Nee LE. A gene (ETM) for essential tremor maps to chromosome 2p22–p25. Mov Disord. 1997;12:859–864. doi: 10.1002/mds.870120605. [DOI] [PubMed] [Google Scholar]
- 7.Shatunov A, Sambuughin N, Jankovic J, Elble R, Lee HS, Singleton AB, et al. Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain. 2006;129:2318–2331. doi: 10.1093/brain/awl120. [DOI] [PubMed] [Google Scholar]
- 8.Hedera P, Blair Ma, Ma S, Bradford Y, Fang YY, Haines JL, et al. Identification of a novel locus for autosomal dominant essential tremor on chromosome 5q. Mov Disord. 2006;21:S708. doi: 10.1002/mds.20950. [DOI] [PubMed] [Google Scholar]
- 9.Higgins JJ, Lombardi RQ, Pucilowska J, Jankovic J, Tan EK, Rooney JP. A variant in the HS1-BP3 gene is associated with familial essential tremor. Neurology. 2005;64:417–421. doi: 10.1212/01.WNL.0000153481.30222.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeanneteau F, Funalot B, Jankovic J, Deng H, Lagarde JP, Lucotte G, et al. A functional variant of the dopamine D3 receptor is associated with risk and age-at-onset of essential tremor. PNAS. 2006;103:10753–10758. doi: 10.1073/pnas.0508189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng H, Le WD, Guo Y, Huang MS, Xie WJ, Jankovic J. Extended study of A265G variant of HS1BP3 in essential tremor and Parkinson disease. Neurology. 2005;65:651–652. doi: 10.1212/01.wnl.0000173033.32535.23. [DOI] [PubMed] [Google Scholar]
- 12.Jankovic J. Essential tremor: clinical characteristics. Neurology. 2000;54(Suppl):S21–S25. [PubMed] [Google Scholar]
- 13.Jankovic J. Essential tremor: a heterogeneous disorder. Mov Disord. 2002;17:638–644. doi: 10.1002/mds.10221. [DOI] [PubMed] [Google Scholar]
- 14.Brain PG, Findley LJ, Atchinson P. Assessing tremor severity. J Neurol Neurosurg Psychiatry. 1993;56:868–873. doi: 10.1136/jnnp.56.8.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis ED, Yousefzadeh E, Barnes LF, Yu Q, Pullman SL, Wendt KJ. Validation of a portable instrument for assessing tremor severity in epidemiologic field studies. Mov Disord. 2000;15:95–102. doi: 10.1002/1531-8257(200001)15:1<95::aid-mds1015>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 16.Fink JK, Hedera P. Hereditary spastic paraplegia: genetic heterogeneity, known genes and genotype-phenotype correlation. Sem Neurol. 1999;19:301–309. doi: 10.1055/s-2008-1040846. [DOI] [PubMed] [Google Scholar]
- 17.Crocq M-A, Mant R, Asherson P, Williams J, Hode Y, Mayerova A, et al. Association between schizophrenia and homozygosity at the dopamine D3 receptor gene. J Med Genet. 1992;29:858–860. doi: 10.1136/jmg.29.12.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanko S, Sasaki T, Fukuda R, Hattori M, Dai XY, Kazamatsuri H, et al. A study of the association between schizophrenia and the dopamine D3 receptor gene. Hum Genet. 1993;92:336–338. doi: 10.1007/BF01247330. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzo CV, Baca-Garcia E, Hernandez MD, Martin CB, Perez-Rodriguez MM, Saiz-Gonzalez MD, et al. No association between the Ser9Gly polymorphism of the dopamine D3 receptor gene and schizophrenia in a Spanish sample. Am J Med Genet B Neuropsychiatr Genet. 2007;144:344–346. doi: 10.1002/ajmg.b.30331. [DOI] [PubMed] [Google Scholar]
- 20.Fathalli F, Rouleau GA, Xiong L, Tabbane K, Benkelfat C, Deguzman R, et al. No association between the DRD3 Ser9Gly polymorphism and schizophrenia. Schizophr Res. 2007 Aug 13; doi: 10.1016/j.schres.2007.07.002. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet. 2003:361865-872. doi: 10.1016/s0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- 22.Cardon LP, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 23.Freedman ML, Reich D, Penny KL, McDonald GJ, Mignault AA, Patterson N, et al. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 24.Tan EK, Prakash KM, Fook-Chong S, Yih Y, Chua E, Lum SY, et al. DRD3 variant and risk of essential tremor. Neurology. 2007;68:790–791. doi: 10.1212/01.wnl.0000256773.87498.2f. [DOI] [PubMed] [Google Scholar]