

Summary
Vaccinia virus (VACV) has been used as the vaccine to protect against smallpox, and recombinant VACVs have been used to develop vaccine candidates against numerous cancers and infectious diseases. Although relatively safe for use in humans, the strains of VACV that were used as smallpox vaccines led to several complications including, progressive infection in immune compromised individuals, eczema vaccinatum in individuals with a history of atopic dermatitis, and encephalitis and perimyocarditis in apparently healthy individuals. The work described in this paper focuses on attenuated strains of VACV that may have the potential for use as vaccine vectors with reduced pathogenicity. We have generated several VACV mutants in a WR background with specific mutations in the E3L gene that were at least a 1000-fold less pathogenic compared to wtVACV upon intra-nasal infection of mice. Many of these mutant viruses replicated to high titers in the nasal mucosa of mice following intra-nasal administration. Despite replication to high titers in the nose, there was little spread to other organs in infected animals. Intra-nasal vaccination with doses as low as 100–1000 pfu (plaque forming units) of these replicating VACV constructs were sufficient to protect the host from challenge with large doses of wtVACV. Similar constructs in a Copenhagen and a NYCBH background were highly attenuated, yet effective as vaccines in the mouse model. These recombinant VACV constructs may be promising vector candidates for use in vaccination strategies against smallpox and other pathogens.
Keywords: Vaccinia virus, E3L, Interferon-resistance, Smallpox vaccine
Introduction
Numerous products ranging from purified proteins and immunogenic peptides to DNA and viral vectors have been investigated as vaccine candidates [1]. Although protein and DNA vaccines are relatively safe the immune response they generate is not always protective [2]. A number of viruses including adeno- and herpes-virus have also been described as potential vectors for foreign antigens [3]. Among these viral vectors, poxviruses have been one of the most promising vectors for development of recombinant vaccines. Recombinant poxviruses expressing microbial antigens can induce protective immunity to parasitic, bacterial and viral infections [4].
The successful use of vaccinia virus (VACV) during the world wide program for the eradication of smallpox [9] in addition to the development of strategies to generate recombinant VACVs that efficiently express foreign proteins suggested the utility of this virus as a vaccine vector [5]. DNA inserts of up to 30 kb can be inserted into the genome of this virus by direct ligation [6]. Thus multiple genes for various pathogens can be expressed using this vector system to develop polyvalent vaccines. Apart from being heat stable and inexpensive to manufacture, these vectors offer the added benefits of a prolonged immune response and activation of both humoral and cell mediated branches of the immune system [7]. All these properties make VACV vectors promising candidates for vaccine development against numerous diseases.
Although these viral vectors offer several advantages, they also raise concomitant issues of safety [8–10]. While wtVACV is safe for use in the majority of people, there is a high complication rate in immune compromised individuals and people with a history of atopic dermatitis. In addition, apparently healthy individuals can rarely develop encephalitis and perimyocarditis post-vaccination [9,11,12]. Post-vaccinia encephalitis is most common in children under 1 year of age [13,14].
Due to the safety issues associated with the use of wtVACV most of the VACV currently employed in vaccine studies are non-replicating VACV vectors. These include modified Ankara strain (MVA), a mutant of the Lister strain of VACV deleted for the essential viral UDG gene [15], and NYVAC-based vectors which are unable to replicate in most mammalian cells [16–19]. Although non-replicating vectors have the advantage of being relatively safe, high doses of these vectors (107 to 108 pfu, plaque forming units) are often required to induce a strong immune response [18,20,21]. Most of these vaccinations also include multiple immunizations [22] often defeating the purpose of using live virus vaccines which usually induce potent immune response with a single dose.
Identification and characterization of VACV genes involved in virulence is a vital step in the development of more attenuated and immunogenic VACV vectors. E3L is a virulence gene of VACV that allows the virus to evade the innate immune responses of the host, including the interferon system. Both N- and C-terminal domains of the E3L gene products are required for pathogenicity of VACV in mouse models [23]. We have generated several E3L mutants of VACV that replicate to high titers in the nose following intra-nasal infection but do not spread to any other organs [24]. These mutants, which are over a 1000-fold less pathogenic than wtVACV, induce a strong immune response that is protective against wtVACV challenge. Therefore, these attenuated, replicating VACV mutants may be promising candidates for use as vectors in vaccine development.
Materials and methods
Plasmid and virus construction
The recombination plasmid pMPE3LΔgpt was used to insert genes into the E3L locus of vaccinia virus as previously described [25]. The various VACV mutants used in this study were generated by cloning the mutant genes into the recombination vector and inserting it by in vivo recombination in to VACVΔE3L. Construction of VACV mutants expressing E3LΔ54N, E3LΔ83N, E3LΔ7C and the E3L Orf virus E3L homologue have been previously described [23,26]. Unless otherwise indicated experiments were performed with viruses in a WR strain background. For experiments with the NYCBH and Copenhagen strains of VACV, wt strains were kindly provided by Acambis (ACAM2000) and Virogenetics (VC2), respectively. The E3L gene was deleted from these viruses using pMPE3LΔgpt as previously described [25].
Cell culture
RK-13 cells were maintained in Eagle's Minimum Essential Medium (MEM, Gibco, BRL) supplemented with 5% of fetal bovine serum (FBS, Irvine Scientific), 50 μg/ml of gentamycin and 0.1 mM non-essential amino acid solution (Gibco, BRL). BHK-21 cells were cultured in the same media supplemented with same amount of non-essential amino acids and gentamycin but FBS was added to a final concentration of 10%. HeLa S3 cells (ATCC) were grown in Dulbecco's modified-minimum essential medium (DMEM) (Gibco, BRL) supplemented with gentamycin (50 μg/ml) and 10% FBS. All cells were incubated at 37 °C in the presence of 5% CO2.
Virus infections and preparation of stocks
Stocks of recombinant viruses expressing E3L mutants were generated by amplification of a single plaque first in 35 mm dishes followed by 100 mm dishes of BHK-21 cells. For virus amplification from titrated stocks, the recombinant virus was used to infect 100 mm dishes of BHK-21 at an MOI of 0.01–0.1 and the amplification and titration procedure was repeated. All virus stocks were stored at −80 °C. Virus was purified through a 36% sucrose pad, as previously described [27,28].
All virus dilutions were done either in MEM containing 2% FBS or 1 mM Tris–HCl pH 8.8. 250 μl of virus suspension was used to infect a 100 mm dish and 100 μl of virus was used to infect a 60 mm as well as 35 mm dish. Infections of cell monolayers were performed after removing the cell culture media, adding virus diluted to the appropriate volume and incubating at 37 °C in the presence of 5% CO2 for 1 h. Infected cells were rocked at intervals of 10 min. Appropriate cell culture medium was overlaid on the monolayer following infection. All virus titrations were performed in RK-13 cell line.
Mice
C57BL/6 mice procured from Charles River were bred at the Arizona State University animal facility following the rules and regulations of the Institutional Animal Care and Use Committee. BALB/c and SCID (Fox Chase SCID mouse, CB17/lcr-PrKdcSCID/CrL) mice were obtained from the Jackson Laboratory and Charles River Laboratory, respectively. Four to five-week-old mice were used for all the experiments. Mice of either sex were used for all experiments except challenge studies. All the challenge studies were done using female mice after confirming that there was no difference in the susceptibility of the two sexes to VACV.
Intra-nasal infection of mice
C57BL/6 mice were anesthetized using a cocktail containing 7.5 mg/ml xylazine (Phoenix Pharmaceuticals, St. Joseph, MO), 2.5 mg/ml acepromazine maleate and 37.5 mg/ml ketamine (Fort Dodge Laboratories, Fort Dodge, IA) at the rate of 1 μl/g body weight administered by intramuscular injection. Five to six mice were then infected with serial dilutions of each virus in 1 mM Tris–HCl pH 8.8 by intra-nasal route. Numbers of deaths were recorded. The experiment was allowed to continue for 14 days following which all the surviving mice were euthanized using CO2.
Tissue distribution
Three C57BL/6 mice, 4 weeks of age, were anesthetized and infected with 106 pfu of each virus by intra-nasal route. Animals were euthanized 5 days post-infection using an overdose of halothane. The nasal turbinates, lungs and brains from these animals were harvested and quick-frozen in liquid nitrogen. The tissue samples were weighed and made into 10% tissue suspension in MEM containing 5% FBS and 100 μg gentamycin using a stomacher. The samples were subjected to three rounds of freezing at −80 °C and thawing at 37 °C and titrated in RK-13 cells. The limit of detection of virus in each tissue was calculated by adding known amounts of virus to processed tissues from uninfected mice, subjecting them to three rounds of freeze thaws. The supernatant was collected by centrifugation at 4 °C for 10 min at 1000 × g and titrated in RK-13 cells.
Intra-cranial infection of mice
Five to six C57BL/6 mice, 4 weeks of age were anesthetized using a cocktail containing 7.5 mg/ml xylazine (Phoenix Pharmaceuticals, St. Joseph, MO), 2.5 mg/ml acepromazine maleate and 37.5 mg/ml ketamine (Fort Dodge Laboratories, Fort Dodge, IA) at the rate of 1 μl/g body weight administered by intramuscular injection. The mice were then infected with serial dilutions of each virus by intra-cranial injection using a 1 ml tuberculin syringe and 27-gauge needle. The mice were weighed on alternate days to monitor weight loss. Deaths were recorded. The experiment was allowed to continue for 14 days following which all the surviving mice were euthanized using CO2.
For studies in newborn mice, pregnant CD1 mice were purchased from Charles River at 10 days gestation. The animals were housed one animal per cage. Intra-cranial infections, using a total volume of 10 μL, were conducted 48–72 h post-birth of the pups using a 27-gauge needle, as previously described [29]. Animals were monitored twice daily for 21 days for morbidity and mortality.
Challenge experiment
Five to six C57BL/6 or BALB/c female mice, approximately 4 weeks of age, were vaccinated intra-nasally with one of four dilutions of each of the viruses ranging from 10 to 10,000 pfu. All infections were performed by following the procedure described for intra-nasal infection. The mice were challenged by intra-nasal infection with 106 pfu of wtVACV 30 or 90 days after vaccination.
UV inactivation of virus
A new UV lamp (Philips G36Y6L) of UV wavelength 253.7 nm was installed in the virus hood and used for UV inactivation of virus. One thousand microliters of virus stock containing 107, 105 and 103 pfu per 100 μl of wtVACV was prepared by dilution of virus in Tris–Cl pH 8.8. Thirty-five millimeters dishes were placed on ice at a working distance of 5 in. from the UV lamp. Two hundred microliters of each virus stock was added to five dishes and exposed to UV irradiation for varying amounts of time (0, 1, 5, 10 and 20 min). All the stocks were titrated in RK-13 cells after UV irradiation. It is estimated that 5100 J/m2 of radiation is sufficient to kill 106 pfu of VACV in 5 min. Inactivation of virus was confirmed by plaque assay and the virus stocks containing completely inactivated virus were diluted and used for immunization at equivalent doses of 10, 100, 1000 and 10,000 pfu.
Statistical analysis
One-way ANOVA using the Tukey's method was run on SPSS software to compare the weight change pattern following vaccination with different doses of each mutant VACV. The weight of each mouse on day 0 of challenge was subtracted from weight on day 10 and the weight change observed following challenge were compared with mock vaccinated and challenged mice by statistical analysis. The error bars represent the standard error of the mean.
Results
Pathogenicity of E3L mutant VACVs in C57BL/6 mice
We have constructed several strains of VACV containing mutations in the E3L gene (see Fig. 1). These range from viruses that are nearly wt in cells in culture (VACVE3LΔ83N, VACVE3LΔ54N, VACVΔE3L∷Orf E3L) to viruses that are intermediate in their cell culture phenotype (VACVE3LΔ7C), to viruses that are quite compromised in their ability to replicate in cells in culture (VACVΔE3L) [30,31]. Both E3LΔ83N and E3LΔ54N are missing major portions of the N-terminal Z-nucleic acid (Z-NA)-binding domain, which is necessary for full pathogenesis in mice [23,24], and almost certainly do not bind to Z-form NA. In addition, E3LΔ54N has a very short half-life compared to wtE3L or E3LΔ83N (unpublished observations). VACVE3LΔ7C contains wt Z-NA and dsRNA-binding domains (the C-terminal deletion abuts just to the end of the consensus dsRNA-binding domain), but binds to dsRNA with a 100-fold lower affinity than wtE3L [32]. The Orf virus E3L homologue can complement deletion of E3L from VACV in cells in culture, but not in the animal model [26]. Failure to complement in the animal model maps to the C-terminal dsRNA-binding domain of the Orf virus E3L homologue [26]. In order to compare virulence of these different mutant viruses, 4–5-week-old C57BL/6 mice were infected intra-nasally and monitored for morbidity and mortality. wtVACV, WR strain has a LD50 of 4 × 103 pfu by the intra-nasal route of infection ([23] and Table 1). Of the mutants tested only VACVE3LΔ83N showed any pathogenicity, even at doses as high as 107 pfu (Table 1). Thus, all of the viruses tested were greater than 1000-fold less pathogenic than wtVACV (Table 1).
Figure 1.
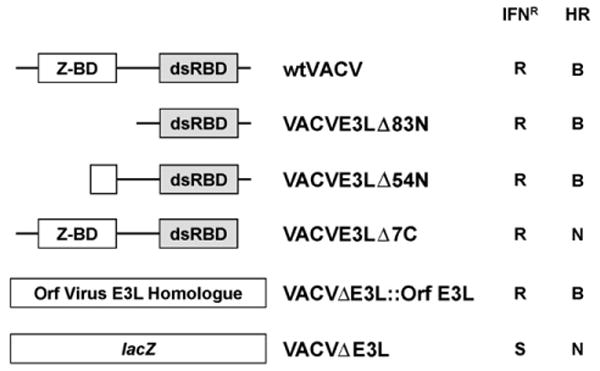
Cell culture phenotypes of vaccinia virus E3L mutants. Structures of the E3L gene of mutants used in this paper are presented. For partial deletion mutants the nomenclature used indicates the number of N-terminal or C-terminal amino acids deleted from wtE3L (i.e., VACVE3LΔ83N is a virus coding for an E3L protein deleted of 83 N-terminal amino acids). Z-BD, Z-NA-binding domain; dsRBD, dsRNA-binding domain; IFNR, sensitivity (S) or resistance (R) to interferon in RK-13 cells; HR, host range; B, broad or N, narrow.
Table 1.
Summary of results
| Virus | IC LD50 (pfu) | IN LD50 (pfu) | SCID pathogenesis | IN protective dose (pfu) | Efficacya |
|---|---|---|---|---|---|
| wt VACV | 102 | 4 × 103 | ++++ | nd | nd |
| VACVE3LA83N | 3 × 104 | 4 × 107 | +++ | 100 | 4 × 105 |
| VACVE3LA7C | 106 to 107 | >107 | ++ | 100–1000 | >104 to 105 |
| VACVE3LA54N | 8 × 106 | >107 | + | 20,000 | >5 × 102 |
| VACVAE3L∷Orf E3L | >107 | >107 | +++ | 100 | >105 |
| VACVAE3L | >106 | >107 | − | 1000 | >104 |
Efficacy is defined as IN LD50/IN protective dose.
Following intra-nasal administration, wtVACV, as well as most of the mutant viruses, replicated to high titers in the nasal turbinates. By 2 days post-infection, a large amount of wtVACV was detected in the lung and by 4 days post-infection this virus was detected in the brain of infected mice as seen in Fig. 2. While most of these mutants replicated to titers approaching wtVACV in the nose, they failed to efficiently spread to the lungs or brain (Fig. 2 and [23,24,26]). As described previously, VACVΔE3L replicated poorly even in the nasal mucosa and was not detected in the lungs or brain [23].
Figure 2.
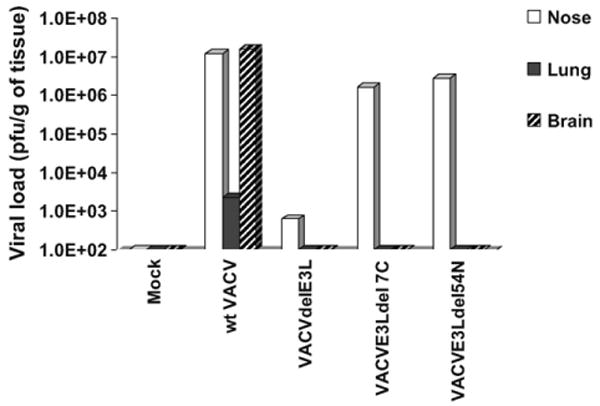
Tissue distribution of virus after intra-nasal infection. Groups of three C57BL/6 mice were infected intra-nasally with 106 pfu of wtVACV and the indicated mutant VACV constructs. Tissues were harvested at 4 days post-infection, processed and titrated in RK-13 cell line. The figure represents the average titer per gram of tissue of the three mice infected with each virus.
In order to test neurovirulence directly, animals were infected intra-cranially with each of these viruses. While these viruses had similar phenotypes after intra-nasal infection, clear differences in virulence could be detected after intra-cranial infection (Fig. 3). VACVE3LΔ83N is approximately 1000-fold less pathogenic than wtVACV [23,24]. VACVE3LΔ7C was approximately 10,000-fold less pathogenic, while VACVE3LΔ54N was approximately 100,000-fold less pathogenic than wtVACV. LD50s could not be determined for VACVΔE3L or VACVΔE3L∷Orf E3L because they failed to cause mortality in greater than 50% of infected animals [23,26], even at very high titers.
Figure 3.
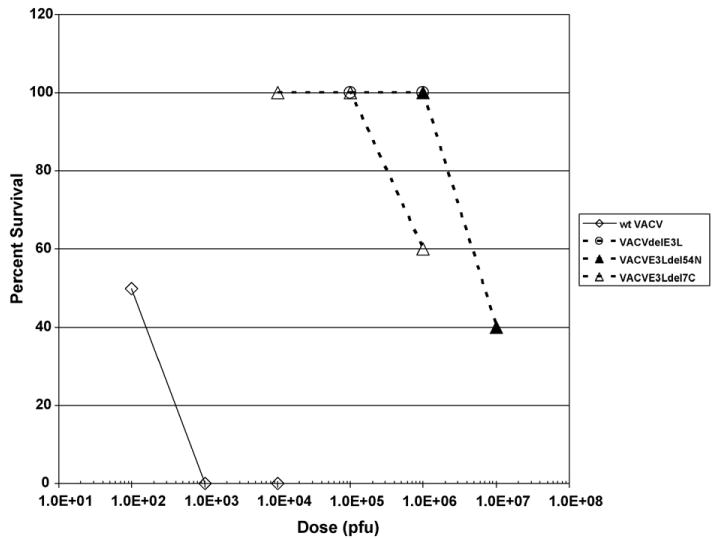
Survival of mice following intra-cranial infection with the various recombinant VACV. Groups of five C57BL/6 mice were infected intra-cranially with different doses of wtVACV and the indicated mutants of VACV. The infected mice were observed for 2 weeks following infection and all mortalities were recorded.
Pathogenicity of E3L mutant VACVs in SCID mice
Immune deficient people are often highly susceptible to virus infection. For this reason we tested these potential vaccine strains in SCID mice. SCID mice were infected intra-nasally with each of our virus constructs and the animals were followed for 1 month. wtVACV was highly pathogenic in SCID mice, having an LD50 of less than 10 pfu (Fig. 4A, open diamonds). Surprisingly, both VACVE3LΔ83N (open squares) and VACVΔE3L∷Orf E3L (closed circles) were quite pathogenic in SCID mice, having an LD50 of approximately 100 pfu (Fig. 4A). VACVE3LΔ54N (closed triangles) and VACVΔE3L (open circles) did not kill any SCID mice after intra-nasal infection with high doses of virus (106 to >107 pfu). Thus, these viruses are at least 10,000-fold less pathogenic in SCID mice than wtVACV. VACVE3LΔ7C led to a milder but protracted illness in mice as compared to wt (Fig. 4B). Animals infected with 106 pfu of VACVE3LΔ7C were euthanized by the end of the experiment for humane reasons. Neither VACVE3LΔ54N nor VACVΔE3L infection led to weight loss during the 1-month course of the experiment (Fig. 4B). To determine if VACVE3LΔ54N and VACVΔE3L might induce long-term pathogenesis, SCID mice were infected intra-nasally with these viruses and monitored for over 5 months (Fig. 5). Animals infected with VACVE3LΔ54N (Fig. 5A closed triangles) began losing weight by day 40 and by day 80 mortality started increasing with a mean time to death of 97.2 days (Fig. 5B, closed triangles), compared to a mean time to death of 8.6 days for animals infected with a similar dose of wtVACV (Fig. 5B, open diamonds). None of the animals infected with VACVΔE3L (Fig. 5A and B, open circles) showed any sign of pathogenesis or any weight loss throughout the nearly 6-month course of the experiment.
Figure 4.

Pathogenesis in SCID mice infected intra-nasally with wtVACV or VACV E3L mutants. Groups of five female SCID mice were infected intra-nasally with either wtVACV or the indicated VACV E3L mutants at the indicated doses. Mice were weighed every other day and monitored for 1 month for signs of illness. Percent survival is presented in panel A, and weight loss is presented in panel B. For panel B, lines ending prematurely indicate mortality.
Figure 5.
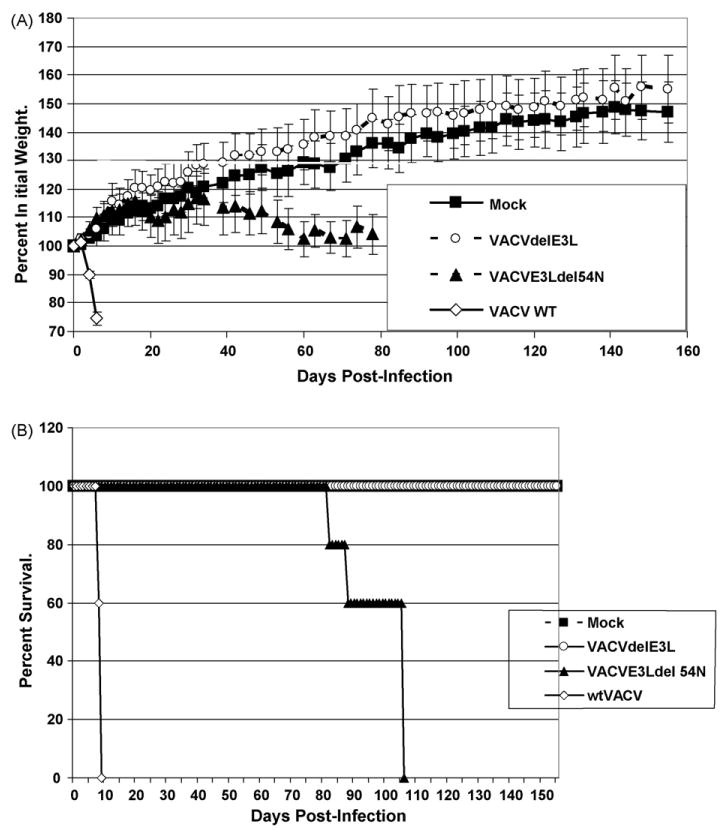
Long-term pathogenesis in SCID mice intra-nasally infected with wtVACV or VACV E3L mutants. Groups of five female SCID mice were infected intra-nasally with approximately 106 pfu of either wtVACV or the indicated VACV E3L mutants and monitored for weight loss (panel A) for 5 months after infection. Panel B represents survival of mice. For panel A, lines ending prematurely indicate mortality. Error bars indicated the standard error of the mean.
Intra-nasal vaccination experiments
Since several of these viruses replicated to nearly wt levels in the nasal mucosal we decided to determine if intra-nasal vaccination with these viruses could protect against a later challenge with wtVACV. C57BL/6 mice were immunized with doses ranging from 10 to 20,000 pfu of each of the viruses described in this paper. One month later they were challenged with 106 pfu of wtVACV. Weight loss, which has been demonstrated to be a reliable method to determine the relative virulence of poxviruses [33], was used as a parameter to assess morbidity in challenged mice. Mock vaccinated animals (Fig. 6A) lost 20% of their body weight over the 8-day course of the experiment. Morbidity in this model is age-dependent, and 8–9-week-old mice demonstrate moderate weight loss and little mortality, even when infected with high doses of virus. Immunization with 10 pfu of either VACVE3LΔ7C (Fig. 6A) or VACVE3LΔ54N (Fig. 6B) gave partial protection against a wtVACV challenge. Immunization with 100–20,000 pfu was required for full protection against wt challenge. Even the highest dose used for vaccination with these attenuated viruses, 20,000 pfu, was more than 1000-fold below the LD50 of these viruses. The minimal protective dose for each of the viruses tested in this paper is shown in Table 1.
Figure 6.
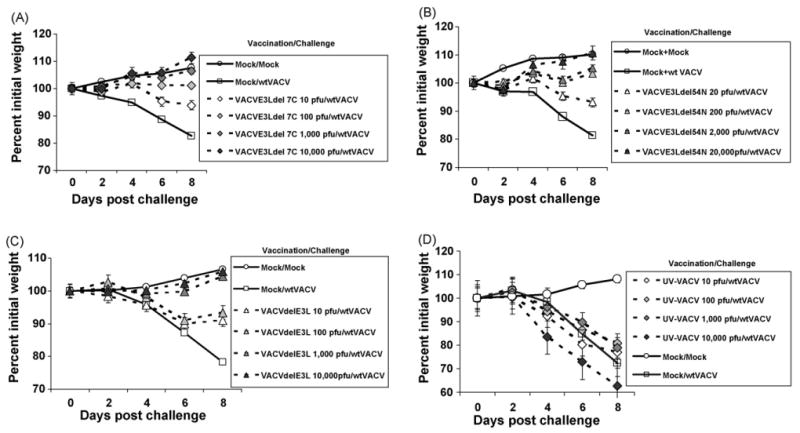
Intra-nasal vaccination. Groups of five C57BL/6 mice were immunized with different doses (ranging from 10 to 20,000 pfu) of the following recombinant VACVs: VACVE3LΔ7C, panel A; VACVE3LΔ54N, panel B; VACVΔE3L, panel C; UV-inactivated wtVACV, panel D. One month later the immunized mice and the unimmunized controls (mock) were challenged with a 106 pfu of wtVACV and monitored for weight loss for 8 days. The result depicted in the figure is the average of two independent experiments. Error bars represent standard error of the mean.
The data from Fig. 6A and B demonstrates that these replication-competent viruses can induce a protective immune response. We also tested VACVΔE3L for its ability to induce a protective immune response. As shown in Fig. 6C, immunization with 1000 pfu of VACVΔE3L gave full protection against challenge with wtVACV. This result was somewhat surprising in that VACVΔE3L appeared to replicate poorly, if at all, in the nasal mucosa (Fig. 2). To more carefully examine replication in the nasal mucosa, groups of mice were infected intra-nasally with 10, 100 or 1000 pfu of either VACVΔE3L or VACVE3LΔ83N (Fig. 7). For VACVE3LΔ83N, intra-nasal infection with 10 pfu (Fig. 7, open squares) led to a nasal viral load of 105 pfu, while intra-nasal infection with either 100 or 1000 pfu (shaded squares and closed squares, respectively) led to a nasal viral load of 106 pfu. Since immunization with 10 pfu of VACVE3LΔ83N was not sufficient to protect animals against a wtVACV challenge [24] this data suggests that a viral load of 106 pfu of VACVE3LΔ83N is required to induce enough antigen to produce a protective immune response. For VACVΔE3L no intra-nasal virus was detected when animals were infected with 10 pfu (open diamonds), and only very low titers were detected in animals infected with 100 pfu, and then only at 6 days post-infection (shaded diamonds). However, intra-nasal infection with 1000 pfu (closed diamonds) led to accumulation of 103 pfu of virus at days 4 and 6 post-infection. Since 1000 pfu of VACVΔE3L is sufficient to induce a protective immune response, these data suggest that a viral load of 103 pfu of VACVΔE3L is sufficient antigen to induce protection against challenge with the wt virus. This viral load is 1000-fold less than the viral load of VACVE3LΔ83N necessary to induce a similar protective immune response.
Figure 7.
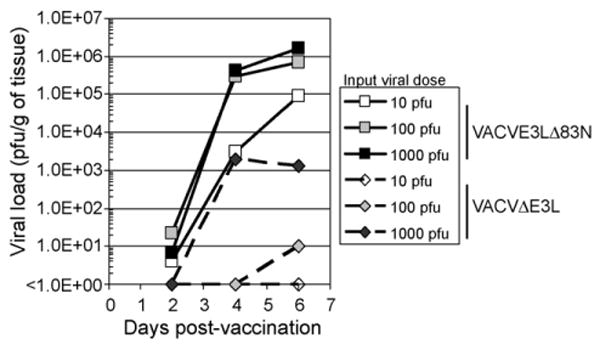
Virus replication in the nose of infected mice. VACVΔE3L (diamonds, dashed lines) or VACVE3LΔ83N (squares, solid lines) was administered intra-nasally to C57BL/6 mice (open symbols, 10 pfu; grey symbols, 100 pfu; closed symbols 1000 pfu). The noses of three mice infected with each dose of each virus were harvested on days 2, 4 and 6 post-infection. The harvested tissues were processed and titrated in RK-13 cells. The average viral load in the nose of mice infected with each virus on each of these days is shown.
One potential explanation for the apparent enhanced immunogenicity of VACVΔE3L is that this virus preparation contains more viral antigen or more contaminants that could act as pathogen-associated molecular patterns (PAMPs) than similar preparations of wtVACV. We believe that this is unlikely since both viruses grow to similar, high titers in permissive BHK cells, suggesting that there are not large numbers of defective particles in the VACVΔE3L preparation. We have also determined the specific infectivity of VACVΔE3L relative to wtVACV by performing a Western blot with anti-VACV serum on equal numbers of pfu. There were minor (less than three-fold) inconsistent difference in the specific infectivities of wtVACV preparations vs. VACVΔE3L preparations (data not shown), which suggests that minor differences in specific infectivities of virus preparations are not likely to be responsible for the large differences in viral load necessary to induce a protective immune response. We have also seen similar results whether we use crude virus preparations or virus purified by centrifugation through a sucrose pad (data not shown), suggesting that differences in cellular contaminants in our preparations are not leading to the increased immunogenicity of VACVΔE3L.
In order to determine whether a live virus was required for protection against wtVACV challenge, we inactivated VACV by UV irradiation. Complete inactivation of virus was confirmed by titration in RK-13 cell line. The results in Fig. 6D demonstrate that even 10,000 pfu of inactivated VACV was unable to induce an immune response that was protective against wtVACV challenge. This indicates that a live virus is required for efficient immunization.
In the previous experiments animals were challenged at 1 month post-vaccination, near the peak of the immune response. To determine if these vaccines could induce long-term protection vaccinated BALB/c mice were challenged at 3 months post-vaccination. As shown in Fig. 8, only animals vaccinated with VACVΔE3L (open diamonds) were protected at 3 months post-vaccination, while animals vaccinated with VACVE3LΔ7C (open triangles) or VACVE3LΔ54N (closed triangles) were not protected from disease.
Figure 8.
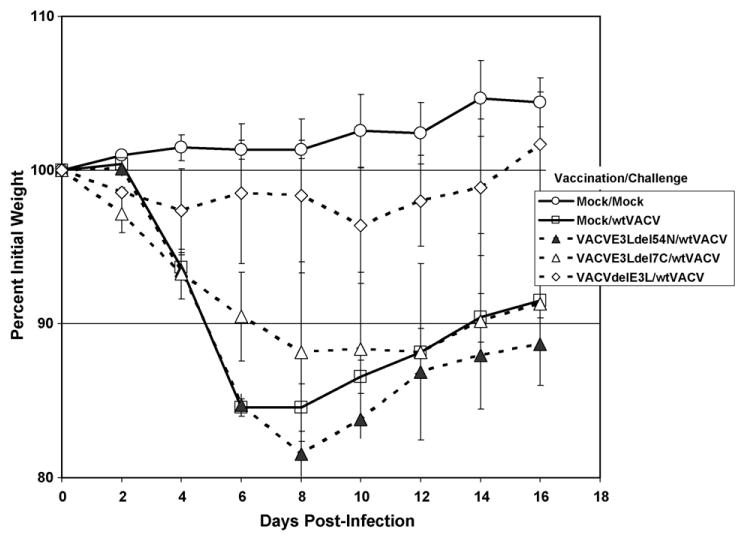
Long-term protection after intra-nasal vaccination. Animals were vaccinated with 103 pfu of the indicated viruses, as in Fig. 6, except BALB/c mice were used in this experiment. Animals were challenged as in Fig. 6 with 106 pfu of wtVACV, at 90 days post-vaccination.
Since the previous vaccination experiments were performed with the WR strain of VACV, a mouse-adapted neurovirulent strain, we wanted to determine if identical mutants in a background more appropriate for use in humans (either the Copenhagen strain of VACV, or the NYCBH strain of VACV) would be attenuated, but still induce a potent immune response. wtVACV(COP) did not cause disease in mice with a normal immune system after intra-nasal or intracranial infection [24]. However, SCID mice were susceptible to intra-cranially infection with wtVACV(COP) with an LD50 of approximately 10 pfu (Table 2). None of the SCID animals infected with up to 107 pfu of VACVΔE3L(COP) showed any signs of illness or weight loss compared to historic controls (data not shown), demonstrating that these mutations are attenuating in a Copenhagen background.
Table 2.
Virulence of vaccinia virus strains in multiple animal models
| IC LD50 BALB/c | IC LD50 SCID | IC LD50 newborn | ||||
|---|---|---|---|---|---|---|
| wt | ΔE3L | wt | ΔE3L | wt | ΔE3L | |
| WR | 102 | >107 | ||||
| COP | >105 | >105 | 101 | >107 | ||
| NYCBH | 105 | >107 | 103 | 106 | ||
Since the NYCBH strain of VACV is only marginally pathogenic in a SCID mouse, we assayed for pathogenicity of VACVΔE3L(NYCBH) in a more sensitive model, the IC newborn mouse model. The LD50 for wtVACV(NYCBH) in newborn mice is approximately 103 pfu (Fig. 9, open circles). The LD50 for VACVΔE3L(NYCBH) is approximately 106 pfu (Fig. 9, open squares), indicating that this virus is attenuated by approximately 3 logs, compared to wtVACV(NYCBH). The data for pathogenicity of the various viruses tested in these models is summarized in Table 2.
Figure 9.
Pathogenesis in newborn mice. Newly born CD1 mice were infected intra-cranially with the indicated dose of either wtVACV(NYCBH) or VACVΔE3L(NYCBH) and monitored twice daily for 21 days for signs of morbidity and mortality.
To assay for induction of protective immunity 4-week-old mice were vaccinated intra-nasally with a low dose (∼10 pfu), intermediate dose (∼1000 pfu) or high dose (∼100,000 pfu) of either wtVACV(COP) or VACVΔE3L(COP) (Fig. 10), or similar doses of wtVACV(NYCBH) or VACVΔE3L(NYCBH) (Fig. 11), and challenged 1 month later with 106 pfu of wtVACV(WR). Mice vaccinated with a low dose of wtVACV(COP) were unprotected against either weight loss or death (Fig. 10, open squares). Mice vaccinated with a low dose of VACVΔE3L(COP) (Fig. 10, closed squares) were fully protected from death (too few animals were used in this experiment to determine statistical significance), and partially protected from weight loss. Animals vaccinated intra-nasally with either the intermediate or high dose of either wtVACV(COP) or VACVΔE3L(COP) were fully protected from death and weight loss (Fig. 10). Thus, VACVΔE3L(COP) is at least as effective of a vaccine in this model as wtVACV(COP). For VACV(NYCBH), neither a low dose of wtVACV(NYCBH) nor VACVΔE3L(NYCBH) protected against a wtVACV(WR) challenge. Similar to VACV(COP), the intermediate and high doses of either wtVACV(NYCBH) or VACVΔE3L(NYCBH) fully protected against challenge.
Figure 10.
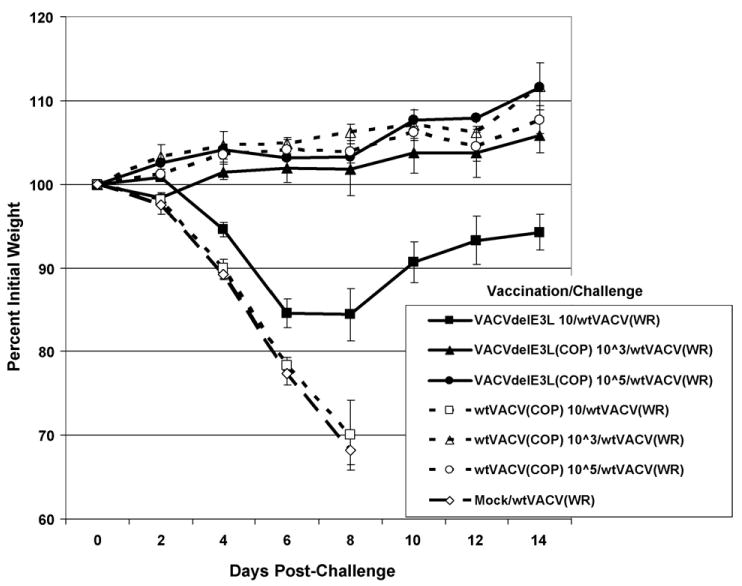
Intra-nasal vaccination with Copenhagen strain viruses. Groups of five female mice were vaccinated intra-nasally with either VACVΔE3L or wtVACV (both Copenhagen strain) at the indicated dose. One month after vaccination the mice were intra-nasally challenged with 106 pfu of wtVACV (WR strain). After challenge, mice were monitored for weight loss every other day and monitored every day for signs of illness. Mock/wtVACV mice were mock vaccinated with 1 mM Tris pH 8.8 and challenged with 106 pfu of wtVACV (WR strain). Error bars indicated standard error of the mean.
Figure 11.
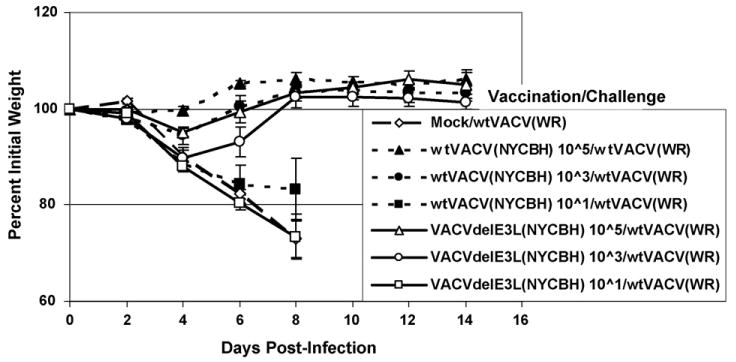
Intra-nasal vaccination with NYCBH strain viruses. Groups of five female mice were vaccinated intra-nasally with NYCBH strain VACVΔE3L or wtVACV at the indicated dose. Animals were challenged and monitored as described in Fig. 10.
Discussion
In this paper we demonstrate that strains of VACV containing mutations in the E3L virulence gene are highly attenuated in several mouse models, yet still induce potent protective immune responses. In the C57BL/6 intra-nasal model, these viruses are from 3 logs to greater than 5 logs less neurovirulent than wtVACV (WR strain). All of the viruses tested, with the exception of VACVΔE3L, replicated to high titers in the nasal mucosa, but failed to spread to the lungs or brain. Somewhat surprisingly two of the viruses that are highly attenuated in C57BL/6 mice (VACVE3LΔ83N and VACVΔE3L∷Orf E3L) were quite pathogenic in SCID mice. Since the defect in virulence for VACVΔE3L∷Orf E3L maps to its C-terminal dsRNA-binding domain [26], loss of pathogenicity for these two viruses and restoration of pathogenicity in SCID mice, appears to be mechanistically unlinked. Nonetheless, the data suggest that an intact immune response is responsible for attenuation of these viruses, but not for viruses that are highly attenuated in both models (VACVE3LΔ7C, Δ54N and ΔE3L). While VACVE3LΔ7C and Δ54N were highly attenuated in the SCID model, they did cause disease when used at high doses (>106 pfu IN), after long latent periods (30 days for VACVE3LΔ7C and 97 days for VACVE3LΔ54N). Both viruses established at least somewhat persistent infections in SCID mice, which in the case of VACVE3LΔ54N may have led to the selection of a more virulent virus. VACVE3LΔ54N encodes an unstable E3L protein, such that increased virulence could be attained by selection for a variant that encodes a stable E3L protein. VACVΔE3L was apathogenic for over 150 days even after infection of SCID mice with 106 pfu. Deletion of E3L in a Copenhagen or NYCBH vaccine strain background further attenuated these already attenuated human vaccine strains. Since VACV deleted for E3L either in a Copenhagen or a NYCBH background replicate to high titers in CEF cells, which are appropriate substrates for vaccine production, these viruses have the potential for development as a vaccine for use in humans.
All of the viruses tested here induced a potent protective immune response after vaccination intra-nasally with 100–20,000 pfu. Some of the viruses induced partial protection with intra-nasal immunization with as little as 10 pfu. These doses are over 3 logs lower than the IN LD50 for these viruses. For the viruses that replicate to high titers in the nasal mucosa this was not surprising, but we were surprised that VACVΔE3L, which does not replicate efficiently in the nasal mucosa, could induce a protective immune response. Immunohistochemical analysis of the nasal mucosa of mice infected with VACVΔE3L indicates that this virus can replicate efficiently in localized patches of the mucosa, unlike wtVACV, which essentially infects the entire mucosa (data not shown). Even this very limited replication appears to be necessary for vaccination, since UV-inactivated VACV does not induce a protective immune response. Induction of a protective immune response despite limited replication (a 3 log lower viral load than a fully replication-competent virus) suggests that VACVΔE3L has inherent adjuvancy activity that increases immunogenicity of this virus. We have shown that VACVΔE3L leads to activation of IRF3, ATF-2 and NFκB in infected cells [34]. This is likely at least partially due to the presence of unbound dsRNA in cells infected with VACVΔE3L [31]. Thus, the presence of this danger signal may act as a stimulator of the innate immune response, which may lead to the increased immunogenicity of this virus. VACVΔE3L was a potent inducer of protective immunity even in a Copenhagen and a NYCBH vaccine strain background when administered intra-nasally.
VACVs have proven to be one of the most promising vaccination strategies in infectious disease ranging from malaria [2] to HIV [22] in humans. Attenuated VACV are also being investigated for use as therapeutic vaccines against various tumors [35]. However due to safety issues associated with replicating VACV, most of these vaccination regimens use non-replicating recombinant VACV. When using non-replicating VACV vectors such as MVA, high doses ranging from 106 to 108 pfu are commonly employed for induction of immune response to the transgene [18]. Often augmentation of immunization protocols using attenuated strains of VACV with DNA or peptide vaccines in ‘prime-boost strategies’ yield better results than immunization with attenuated VACV alone. In fact this approach is being widely studied in potential vaccines against HIV and malaria. But the need for multiple inoculations [2,22] often defeats the purpose of using live vaccines, which are expected to provide protection after a single inoculation, an advantage that may offset the additional risks often associated with these vaccines. In this paper we demonstrate that highly attenuated vaccines can induce a protective immune response after vaccination intra-nasally with a single dose of as little as 100 pfu.
In contrast to non-replicating strains of VACV, a single dose of replicating VACV vector appears to be sufficient to induce a robust immune response against the transgene [36]. This is evident from the studies conducted by Ohishi et al. [48] using VACV encoding the haemagglutinin protein of rinderpest virus. They demonstrated that a single dose of this recombinant VACV expressing the H gene of rinderpest virus was sufficient to induce a protective immune response against rinderpest in cattle for over 3 years. Use of replicating VACV expressing rabies glycoprotein as an oral vaccine has led to elimination of sylvatic rabies from large areas of land in Europe and preliminary data from field trials in the United States indicate a significant reduction of rabies in vaccinated areas [37]. Replicating VACV was used globally for eradication of the deadly disease, smallpox.
Replicating VACV is also in clinical trials as vectors for tumor antigens against prostatic cancer [38], cervical carcinoma [39] and breast cancer [40]. Co-expression of cytokines like IL-4 and GM-CSF in vivo along with tumor-associated antigens using the VACV system enhances the anticancer activity of these vaccines [41,42]. However, safety has been a major concern involved in the use of these vectors.
Replication on mucosal surfaces, such as the intra-nasal epithelium, confers an added advantage to these vectors as it gives these viruses an opportunity to stimulate a mucosal immune response [43]. Since the vast majority of infections take place by a mucosal route the goal of developing a mucosal immune response is better achieved by administration of the vaccine by the intra-nasal route rather than parenterally [44]. Work done by Belyakov et al. [49] suggests that mucosal vaccination may overcome the barrier to recombinant VACV immunization caused by pre-existing poxvirus immunity. Apart from the efficacious immune response elicited by administration of immunogen by the intra-nasal route, ease of administration is also an added benefit.
For any live virus vaccine reversion of attenuation is a concern. All of the viruses tested here contain deletions for all or part of the E3L gene, which should make it difficult for reversion to occur. We have not succeeded in selecting for a phenotypic revertant of VACVΔE3L, despite repeated attempts (unpublished observations). Reversion by recombination with another poxvirus, which could donate a functional E3L gene, is a potential concern, as it would be with any strain containing a single attenuating mutation. However, not all poxvirus E3L genes can complement for deletion of VACV E3L [26], and several poxviruses contain N-terminal deletions in E3L [45–47] suggesting that reversion by recombination with another poxvirus may be limited.
In conclusion, we have characterized several highly attenuated strains of vaccinia virus containing mutations in the E3L interferon-resistance gene, that induce protective immune responses when delivered mucosally, either prior to or 1 day post-challenge. These viruses have the potential for being improved vaccines for protection against smallpox and improved vaccine vectors.
Acknowledgments
This work was supported by the following grants from the NIH: AI52347, AI66326 and AI 66501.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Li S, Rodrigues M, Rodriguez D, Rodriguez JR, Esteban M, Palese P, et al. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc Natl Acad Sci USA. 1993;90:5214–8. doi: 10.1073/pnas.90.11.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sedegah M, Jones TR, Kaur M, Hedstrom R, Hobart P, Tine JA, et al. Boosting with recombinant vaccinia increases immunogenicity and protective efficacy of malaria DNA vaccine. Proc Natl Acad Sci USA. 1998;95:7648–53. doi: 10.1073/pnas.95.13.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jolly D. Viral vector systems for gene therapy. Cancer Gene Ther. 1994;1:51–64. [PubMed] [Google Scholar]
- 4.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci USA. 1996;93:11341–8. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paoletti E. Application of poxvirus vectors to vaccination: an update. Proc Natl Acad Sci USA. 1996;93:11349–53. doi: 10.1073/pnas.93.21.11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merchlinsky M, M B. Introduction of foreign DNA into vaccinia virus genome by direct ligation: recombination-independent selection cloning vectors. Virology. 1992;190:522–6. doi: 10.1016/0042-6822(92)91246-q. [DOI] [PubMed] [Google Scholar]
- 7.Demkowicz WEJ, Littaua RA, Wang J, Ennis FA. Human cytotoxic T-cell memory: long-lived responses to vaccinia virus. J Virol. 1996;70:2627–31. doi: 10.1128/jvi.70.4.2627-2631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurvich EB, V IS. Vaccinia virus in postvaccinal encephalitis. Acta Virol. 1983;27:154–9. [PubMed] [Google Scholar]
- 9.Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. Smallpox and its eradication. Geneva, Switzerland: World Health Organization; 1988. [Google Scholar]
- 10.Kaplan C. Vaccinia virus: a suitable vehicle for recombinant vaccines? Arch Virol. 1989;106:127–39. doi: 10.1007/BF01311044. [DOI] [PubMed] [Google Scholar]
- 11.Arness MK, Eckart RE, Love SS, Atwood JE, Wells TS, Engler RJ, et al. Myopericarditis following smallpox vaccination. Am J Epidemiol. 2004;160:642–51. doi: 10.1093/aje/kwh269. [DOI] [PubMed] [Google Scholar]
- 12.Eckart RE, Love SS, Atwood JE, Arness MK, Cassimatis DC, Campbell CL, et al. Incidence and follow-up of inflammatory cardiac complications after smallpox vaccination. J Am Coll Cardiol. 2004;44:201–5. doi: 10.1016/j.jacc.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Aragon TJ, Ulrich S, Fernyak S, Rutherford GW. Risks of serious complications and death from smallpox vaccination: a systematic review of the United States experience, 1963–1968. BMC Public Health. 2003;3:1–12. doi: 10.1186/1471-2458-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cono J, Casey CG, Bell DM. Smallpox vaccination and adverse reactions. Guidance for clinicians. MMWR Recomm Rep. 2003;52:1–28. [PubMed] [Google Scholar]
- 15.Coulibaly S, Bruhl P, Mayrhofer J, Schmid K, Gerencer M, Falkner FG. The nonreplicating smallpox candidate vaccines defective vaccinia Lister (dVV-L) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology. 2005;341:91–101. doi: 10.1016/j.virol.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188:217–32. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia J, Cox WI, Pincus S, Paoletti E. Safety and immunogenicity of recombinants based on the genetically-engineered vaccinia strain, NYVAC. Dev Biol Stand. 1994;82:125–9. [PubMed] [Google Scholar]
- 18.Ramirez JC, Gherardi MM, Esteban M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol. 2000;74:923–33. doi: 10.1128/jvi.74.2.923-933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenwirth B, Kuhn EM, Heeney JL, Hurpin C, Tartaglia J, Bonnet MC, et al. Safety and immunogenicity of ALVAC wild-type human p53 (vCP207) by the intravenous route in rhesus macaques. Vaccine. 2001;19:1661–70. doi: 10.1016/s0264-410x(00)00416-3. [DOI] [PubMed] [Google Scholar]
- 20.Meseda CA, Garcia AD, Kumar A, Mayer AE, Manischewitz J, King LR, et al. Enhanced immunogenicity and protective effect conferred by vaccination with combinations of modified vaccinia virus Ankara and licensed smallpox vaccine Dryvax in a mouse model. Virology. 2005;339:164–75. doi: 10.1016/j.virol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Vollmar J, Arndtz N, Eckl KM, Thomsen T, Petzold B, Mateo L, et al. Safety and immunogenicity of IMVAMUNE, a promising candidate as a third generation smallpox vaccine. Vaccine. 2006;24:2065–70. doi: 10.1016/j.vaccine.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 22.Amara RR, Villinger F, Altman JD, Lydy SL, O'Neil SP, Staprans SI, et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001;292:69–74. doi: 10.1126/science.1058915. [DOI] [PubMed] [Google Scholar]
- 23.Brandt T, Jacobs B. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol. 2001;75:850–6. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt T, Heck M, Vijaysri S, Jentarra G, Cameron J, Jacobs B. The N-terminal domain of the vaccinia virus E3L-protein is required for neurovirulence, but not induction of a protective immune response. Virology. 2005;333:263–70. doi: 10.1016/j.virol.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Denzler KL, Jacobs BL. Site-directed mutagenic analysis of reovirus sigma 3 protein binding to dsRNA. Virology. 1994;204:190–9. doi: 10.1006/viro.1994.1523. [DOI] [PubMed] [Google Scholar]
- 26.Vijaysri S, Talasela L, Mercer AA, McInnes CJ, Jacobs BL, Langland JO. The Orf virus E3L homologue is able to complement deletion of the vaccinia virus E3L gene in vitro but not in vivo. Virology. 2003;314:305–14. doi: 10.1016/s0042-6822(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 27.Earl PL, Moss B. Generation of recombinant vaccinia viruses. New York: Green Publishing Associates and Wiley Interscience; 1991. [Google Scholar]
- 28.Blasco R, Sisler JR, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67:3319–25. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Rubin SA, Taffs RE, Merchlinsky M, Ye Z, Carbone KM. Mouse neurotoxicity test for vaccinia-based smallpox vaccines. Vaccine. 2004;22:1486–93. doi: 10.1016/j.vaccine.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Beattie E, Kauffman EB, Martinez H, Perkus ME, Jacobs BL, Paoletti E, et al. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes. 1996;12:89–94. doi: 10.1007/BF00370005. [DOI] [PubMed] [Google Scholar]
- 31.Langland J, Jacobs B. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- 32.Chang H, Jacobs BL. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double stranded RNA. Virology. 1993;194:537–47. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- 33.Bloom DC, Edwards KM, Hager C, Moyer RW. Identification and characterization of two nonessential regions of the rabbitpox virus genome involved in virulence. J Virol. 1991;65:1530–42. doi: 10.1128/jvi.65.3.1530-1542.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langland JO, Kash JC, Carter V, Thomas MJ, Katze MG, Jacobs BL. Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J Virol. 2006;80:10083–95. doi: 10.1128/JVI.00607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartaglia J, Bonnet MC, Berinstein N, Barber B, Klein M, Moingeon P. Therapeutic vaccines against melanoma and colorectal cancer. Vaccine. 2001;19:2571–5. doi: 10.1016/s0264-410x(00)00491-6. [DOI] [PubMed] [Google Scholar]
- 36.Cox WI, Tartaglia J, Paoletti E. Poxvirus recombinants as live vaccines Recombinant poxviruses. CRC Press; 1992. pp. 123–62. [Google Scholar]
- 37.Brochier B, Aubert M, Pastoret P. Field use of a vaccinia-rabies recombinant vaccine for the control of sylvatic rabies in Europe and North America. Rev Sci Tech. 1996;15:947–70. doi: 10.20506/rst.15.3.965. [DOI] [PubMed] [Google Scholar]
- 38.Eder JP, Kantoff PW, Roper K, Xu GX, Bubley GJ, Boyden J, et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res. 2000;6:1632–8. [PubMed] [Google Scholar]
- 39.Adams M, Borysiewicz L, Fiander A, Man S, Jasani B, Navabi H, et al. Clinical studies of human papilloma vaccines in pre-invasive and invasive cancer. Vaccine. 2001;19:2549–56. doi: 10.1016/s0264-410x(00)00488-6. [DOI] [PubMed] [Google Scholar]
- 40.Balloul JM, Acres RB, Geist M, Dott K, Stefani L, Schmitt D, et al. Recombinant MUC 1 vaccinia virus: a potential vector for immunotherapy of breast cancer. Cell Mol Biol. 1994;40:49–59. [PubMed] [Google Scholar]
- 41.Elkins KL, Ennist DL, Winegar RK, Weir JP. In vivo delivery of interleukin-4 by a recombinant vaccinia virus prevents tumor development in mice. Hum Gene Ther. 1994;5:809–20. doi: 10.1089/hum.1994.5.7-809. [DOI] [PubMed] [Google Scholar]
- 42.Qin HX, C SK. Construction of recombinant vaccinia virus expressing GM-CSF and its use as tumor vaccine. Gene Ther. 1996;3:59–66. [PubMed] [Google Scholar]
- 43.Gherardi MM, E M. Mucosal and systemic immune responses induced after oral delivery of vaccinia virus recombinants. Vaccine. 1999;17:1074–83. doi: 10.1016/s0264-410x(98)00324-7. [DOI] [PubMed] [Google Scholar]
- 44.Elson CO. In defense of mucosal surfaces. Regulation and manipulation of the mucosal immune system. Adv Exp Med Biol. 1997;412:373–85. [PubMed] [Google Scholar]
- 45.Cameron C, Hota-Mitchell S, Chen L, Barrett J, Cao JX, Macaulay C, et al. The complete DNA sequence of myxoma virus. Virology. 1999;264:298–318. doi: 10.1006/viro.1999.0001. [DOI] [PubMed] [Google Scholar]
- 46.Shchelkunov SN, Totmenin AV, Safronov PF, Mikheev MV, Gutorov VV, Ryazankina OI, et al. Analysis of the monkeypox virus genome. Virology. 2002;297:172–94. doi: 10.1006/viro.2002.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willer DO, McFadden G, Evans DH. The complete genome sequence of shope (rabbit) fibroma virus. Virology. 1999;264:319–43. doi: 10.1006/viro.1999.0002. [DOI] [PubMed] [Google Scholar]
- 48.Ohishi K, Inui K, Barrett T, Yamanouchi K. Long-term protective immunity to rinderpest in cattle following a single vaccination with a recombinant vaccinia virus expressing the virus haemagglutinin protein. J Gen Virol. 2000;81:1439–46. doi: 10.1099/0022-1317-81-6-1439. [DOI] [PubMed] [Google Scholar]
- 49.Belyakov IM, Moss B, Strober W, Berzofsky JA. Mucosal vaccination overcomes the barrier to recombinant vaccinia immunization caused by preexisting poxvirus immunity. Proc Natl Acad Sci USA. 1999;96:4512–7. doi: 10.1073/pnas.96.8.4512. [DOI] [PMC free article] [PubMed] [Google Scholar]