

Summary
Removal of TRF2, a telomere shelterin protein, recapitulates key aspects of telomere attrition including the DNA-damage response and cell-cycle arrest [1]. Distinct from the response of proliferating cells to loss of TRF2 [2, 3], in rodent non-cycling cells, TRF2 inhibition promotes differentiation and growth [4, 5]. However, the mechanism that couples telomere gene-silencing features [6-8] to differentiation programs has yet to be elucidated. Here we describe an extra-telomeric function of TRF2 in epigenetic regulation of neuronal genes mediated by the interaction of TRF2 with repressor element 1 silencing transcription factor (REST), a master repressor of gene networks devoted to neuronal functions [9-12]. TRF2-REST complexes are readily detected by co-immunoprecipitation assays and are localized to aggregated PML-nuclear bodies in undifferentiated pluripotent human NTera2 stem cells. Inhibition of TRF2, either by a dominant-negative mutant or by RNA interference, dissociates TRF2-REST complexes resulting in ubiquitin-proteasomal degradation of REST. Consequentially, REST targeted neural genes (L1CAM, β3-tubulin, synaptophysin and others) are derepressed resulting in acquisition of neuronal phenotypes. Notably, selective damage to telomeres without affecting TRF2 levels causes neither REST degradation nor cell differentiation. Thus, in addition to protecting telomeres, TRF2 possesses a novel role in stabilization of REST that is required for controlling neural tumor and stem cell fate.
Keywords: REST/NRSF, TRF2, ubiquitin-proteasome, PML-nuclear bodies, neuronal genes
Results and Discussion
Telomere repeat factor 2 (TRF2) is essential for telomere integrity [1]. To investigate whether TRF2 is also involved in epigenetically regulating neuronal differentiation (cessation of cell division, the expression of neuron-specific genes and the outgrowth of neurites), we first expressed a dominant-negative mutant of TRF2 (DN-TRF2, also known as TRF2ΔBΔM) [2] in human SH-SY5Y neuroblastoma and NTera2 cells, a pluripotent embryonal carcinoma stem cell line. Cells expressing DN-TRF2 exhibited neuron-like morphology with extension of neurite processes in SH-SY5Y cells (Figure 1B), and an enrichment of the neuronal marker β3 tubulin [13] in NTera2 cells (Figure 1A). A significant increase of total neurite length occurred and was maintained for at least 15 days in cells expressing DN-TRF2 compared to vector-controls (Figure 1D). Similarly, selective depletion of endogenous TRF2 using shRNA interference promoted neurite outgrowth (Figure 1E and Figure S3A). These results from inhibition of TRF2 by two independent approaches suggest that TRF2 is required for the maintenance of neural progenitors and stem cells in an undifferentiated state. Thus, we further examined the effects of overexpression of wild type (WT) TRF2 in SH-SY5Y cells. As expected, stable expression of WT-TRF2 prevented neurite outgrowth upon exposure to terminal differentiation reagents [14] (Figure. S1A). In addition, TRF2 inhibition-induced neurite elongation was coupled to cell cycle arrest, whereas the effect of WT-TRF2 on cell cycle progression from at least 6 different clones was transient and returned to levels of vector-transfected control cells within two weeks (Figure S2A and 2B ).
Figure 1. Inhibition of TRF2 Promotes Neuronal Differentiation.

(A) NTera2 cells stably expressing either empty vector or dominant negative (DN)-TRF2 were exposed to PA6 cell conditioned differentiation medium for 21 days. The representative micrographs show β3 tubulin immunoreactivity (a neural marker) (red) and DAPI staining (blue), and the graph shows the percentage of β3 tubulin positive cells from 10 randomly chosen fields (n=3, error bars ± SE). **p<0.005 compared to the vector value. (B-C) Total neurite length was measured in SH-SY5Y expressing GFP vector, DN-TRF2 (with GFP reporter) and in combination with flag-REST. The representative micrographs and the graph show the GFP-positive processes and average of total neurite length (65-100 cells, n= 3), respectively. **p<0.0001 compared DN-TRF2 to the vector, *p<0.001 compared either flag-REST to vector, or flag REST plus DN-TRF2 to DN-TRF2. Bar = 20 μm. (D) SH-SY5Y cells stably transfected with either empty vector or DN-TRF2 for 10 days, were stained for the senescence associated β-galactosidase (SA-βGal). The arrow points to long neurites of differentiated cells and the arrowheads point to SA-βgal positive (blue color) cells. *p<0.001 (40-48 cells, n=4). (E) SH-SY5Y cells expressing a TRF2 shRNA vector or empty vector were selected with 1ug/ml puromycin for 10 days, and were then stained for SA–βgal. *p<0.01 (60-100 cells, n= 4).
To understand the molecular basis of the cellular actions of DN-TRF2, we initially performed an unbiased gene array analysis to evaluate gene expression changes resulting from inhibition of TRF2 in SH-SY5Y cells. Upon TRF2 inhibition, 61 genes were significantly affected. The distribution of these genes in the genome was scattered among chromosomes with no obvious relationship to telomere location (Supplemental Table 1). This suggests that modulation of gene expression by TRF2 dysfunction is unlikely to involve a telomere positional effect, in which telomeres are capable of exerting a silencing effect on adjacent genes [6]. Notably, gene ontology analysis indicated that 34% of the genes up-regulated in response to DN-TRF2 expression were involved in functions of differentiated neurons (Figure S2C and Supplemental Table 1).
REST is a master repressor that negatively regulates a network of neuronal genes by binding to a conserved 23 bp motif known as repressor element 1 (RE1) [9]. Several hundred potential REST target genes have been identified, including many involved in excitability, synaptic plasticity, cell adhesion and neurotrophic signaling [10, 11]. Because TRF2 inhibition induced the expression of many genes associated with neuronal functions, we explored a potential role for REST in this process by co-expressing both DN-TRF2 and REST in SH-SY5Y cells. Overexpression of REST prevented DN-TRF2-induced neurite outgrowth (Figure 1C), suggesting that REST blocks the ability of DN-TRF2 to induce differentiation. The latter observation prompted us to investigate if TRF2 and REST co-exit in a functional complex in human pluripotent ES cells before and after exposure to the differentiation stimuli. In undifferentiated NTera2 cells, endogenous TRF2 and REST were concentrated in numerous nuclear foci (Figure 2A). The majority of TRF2 and REST double-positive foci were distinctively colocalized in PML-nuclear body (PML-NB) aggregates. In contrast, differentiated NTera2 cells (β3 tubulin-positive) exhibited diffuse nuclear TRF2 and REST immunoreactivities, with little or no concentration in PML-NB. Consistent with these observations, REST protein in NTera2 cells was decreased following 28 days of differentiation (Figure 2B). These results agree with recent reports that degradation of REST is required for differentiation of stem cells into neurons [9, 12].
Figure 2. TRF2 and REST are Concentrated in PML-Nuclear Bodies (NB) in Undifferentiated ES Cells, and are Displaced from Those Sites in Response to Neuronal Differentiation Signals or to TRF2 Inhibition.

(A) Representative confocal micrographs show that REST (green), TRF2 (red) and PML-NB (purple) are colocalized in nuclear foci in undifferentiated NTera2 cells, but not in NTera2 cells being differentiated into β3 tubulin positive neuron upon exposure to conditioned medium for 28 days. Scale bar = 5 μm. Large arrows point to examples of foci containing REST, TRF2 and PML. The small arrow points to PML- and TRF2-positive foci exhibiting little or no REST immunoreactivity. **p<0.005 (50-75 cells per culture, n=4). (B) Lysates from undifferentiated and differentiated NTera2 cells were analyzed by immunoblots using antibodies against the indicated proteins. (C) Numerous nuclear foci of REST and TRF2 are colocalized in PML-NB in NTera2 cells expressing WT-TRF2 (top). The concentration of REST and TRF2 in PML-NB was markedly reduced in NTera2 cells expressing DN-TRF2 (bottom). Large arrows and small arrowheads point to examples of foci with or without the association of REST, TRF2 and PML, respectively. Scale bar = 5 μm. **p<0.005 (50-75 cells per culture, n=4).
Next, we asked whether inhibition of TRF2 mimics the effects of differentiation medium on the subcellular localization of TRF2 and REST. Overexpression of WT-TRF2 resulted in increased numbers of nuclear foci of REST and TRF2 that colocalized in PML-NB. In contrast, overexpression of DN-TRF2 resulted in decreased localization of TRF2 and REST in PML-NB and a diffuse distribution of these proteins throughout the nucleus, although a few TRF2 and REST foci were present (Figure 2C).
PML-NB are critical for varied cellular functions including DNA transcription/repair, apoptosis/senescence and tumor suppression. By orchestrating the recruitment of the ubiquitin-proteasome system [15] or the small ubiquitin-like modifier (SUMO)-ylation system [16, 17] PML-NB play a key role in regulating nuclear proteins dynamics. The association of SUMOylated TRF2 with PML-NB is observed in a subtype of telomerase-negative ALT (alternative lengthening of telomeres) cells [18]. Moreover, early totipotent embryos also exhibit high levels of ALT-like activity reminiscent of pluripotent human cancer cells [19]. Consistently, we observed a more extended association between REST, TRF2 and PML-NB in pluripotent NTera2 cells compared to SH-SY5Y neuroblastoma cells (data not shown).
Recent evidence demonstrates that SCF β-TRCP, an E3 ubiquitin ligase, is responsible for REST degradation during neuronal differentiation [12]. However, the regulatory factor that prevents REST from being degraded by the ubiquitin-proteasome system is unknown. To further characterize the interaction of TRF2 with REST, we performed a reciprocal co-immunoprecipitation analysis with whole-cell lysates of NTera2 cells (data not shown) and Hela cells using antibodies against TRF2 and REST proteins. The REST and TRF2 immunoactivities were readily detected in their reciprocal immunoprecipitates, suggesting that endogenous TRF2 and REST interact (Figure 3A). We also observed that the interaction of REST with TRF2 in the nucleus is enhanced by expressing WT-TRF2, and noticeably reduced by expressing DN-TRF2 (Figure 3B). We next determined which region of the TRF2 protein interacts with REST. Lysates from Cos-7 cells, expressing GST alone or GST fused either to full-length TRF21-500 or to three different TRF2 truncated mutants were subjected to GST pulldown analysis. Co-precipitation of endogenous REST was achieved with either GST-TRF2 1-500 or GST-TRF45-446, but not with GST-TRF22-68 or GST-TRF2447-500 (Figure 3C), indicating both WT-TRF2 and DN-TRF2 bind REST.
Figure 3. REST Binds Both WT-TRF2 and DN-TRF2, and Binding to DN-TRF2 is Associated with REST Degradation via the Ubiquitin-Proteasome Pathway.
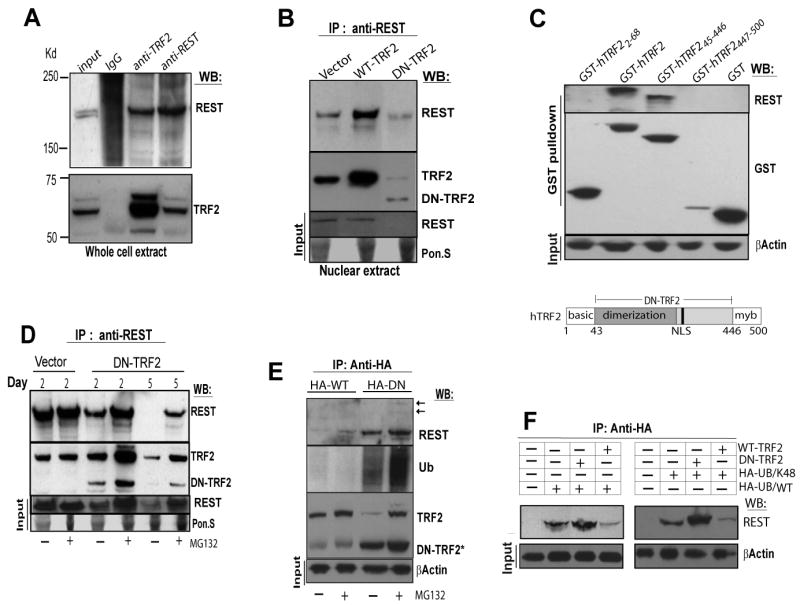
(A) HeLa cell extracts were co-immunoprecipitated reciprocally with polyclonal antibodies to either REST or TRF2. Input protein (120 μg) and immunoprecipitates were subjected to immunoblot analysis with indicated antibodies. (B) Nuclear extracts from HeLa cells were immunoprecipitated with a polyclonal antibody to REST and blots were probed with the indicated antibodies. Equal loading for input proteins was evaluated by ponceau S (Pon.S) stain. (C) GST fusion proteins containing full-length hTRF2, amino acids 2-68 of hTRF2 (basic region), amino acids 45-446 of hTRF2 (DN-TRF2) or amino acids 447-500 of hTRF2 (myb domain) were expressed in Cos-7 cells. Cells were subjected to GST pulldown and the precipitates were then analyzed by immunoblot using antibodies against REST and GST. A schematic representation of the domains in human TRF2 protein is shown at the bottom. (D) HeLa cells expressing vector or DN-TRF2 for 2 or 5 days were treated without (-) or with (+) the proteasome inhibitor MG132 (10 μM) for 5 hours. Cell extracts were subjected to immunoprecipitation with an anti-REST antibody and blots were probed with the indicated antibodies. (E) HeLa cells were co-transfected with either HA-tagged WT-TRF2 or HA-tagged DN-TRF2 in combination with Myc-tagged ubiquitin, then treated without (-) or with (+) 10 μM MG132 for 5 hours. Cell extracts were subjected to immunoprecipitation with anti-HA affinity matrix. REST immunoblot was reprobed with ubiquitin antibody. Arrows point to slow migration species of REST. *DN-TRF2 overlays with IgG heavy chain. (F) HeLa cells were co-transfected with either HA-tagged wild-type ubiquitin (HA-UB/WT) or an ubiquitin mutant (HA-UB/K48), in combination with either WT-TRF2 or DN-TRF2. Ubiquitin-conjugated proteins from cell extracts were captured with anti-HA affinity matrix, and were subjected to immunoblot analysis with anti-REST antibody.
DN-TRF2 expression reduced REST protein levels in nuclear or whole cell extracts (Figure 3B, 3D). We therefore tested the possibility that TRF2 inhibition facilitates REST degradation via the ubiquitin-proteasome pathway. Upon loss of TRF2, REST protein from its immunoprecipitates was gradually reduced (Figure 3D). However, after treatment with the proteasome inhibitor MG132, this reduction was completely prevented at post-transfection day 2, but was partially restored by post-transfection day 5. Moreover, the slower migrating species of REST was associated with an increase of ubiquitinated proteins in the same molecular-weight range in HeLa cell co-expressing HA-tagged DN-TRF2 and Myc-tagged ubiquitin following MG132 treatment (Figure 3E). These results suggested that polyubquitin attachment to REST is increased in response to loss of TRF2. Consistently, DN-TRF2 expression in SH-SY5Y cells also caused a significant decrease in REST levels within 2 days (Figure S3B), which could be rescued by blocking proteasome activity. Nevertheless, upon TRF2 inhibition, p53 levels were increased, whereas REST levels were decreased (Figure S3B), indicating that DN-TRF2 has a selective effect on REST degradation. In contrast, elevation of TRF2 in SH- SY5Y cells attenuated REST depletion during the course of neuronal differentiation (Figure S1B).
Because conjugation of proteins to lys48-linked polyubiquitin chains is a common signal for proteasomal degradation, we next investigated whether DN-TRF2 promotes the binding of Lys48-linked ubiquitin chains to REST. The REST conjugation to Lys48-linked ubiquitin was enhanced in cells co-expressing DN-TRF2 (Figure 3F) with a HA-tagged ubiquitin mutant in which all lysine residues except Lys48 were mutated to arginine (HA-UB/K48) [12, 20] (Figure 3F). In contrast, expression of WT-TRF2 reduced the conjugation of ubiquitin to REST (Figure 3E-F). These results suggest that loss of TRF2 expedites lys48-linked polyubiquitination and proteasomal degradation of REST, and that the interaction of TRF2 with REST maintains the stability of REST.
We next asked whether the reduction of REST levels in response to TRF2 inhibition was directly responsible for the observed changes in neural-specific genes. Analysis of the DN-TRF2 target genes for the consensus REST binding sequence (NTYAGMRCCNNRGMSAG) [10] revealed that 12 out of 61 genes contained at least one putative RE1 element within 50 kb of the transcriptional start site (Supplemental Table 1, Figure S2C).
To further validate the differentiation-inducing effect of TRF2 inhibition on bona fide REST target genes, SH-SY5Y cells were infected with adenovirus expressing DN-TRF2, WT-TRF2 or control (β-galactosidase) for 5 days, and REST binding was measured using chromatin immunoprecipitation (ChIP). We found that REST bound to 9 of the 14 tested RE1 elements, but not to any of the 6 promoters. Expression of DN-TRF2 for 5 days reduced REST binding at most, if not all loci tested (Figure 4A). REST binding was detected at neuronal gene loci (BDNF, L1CAM, miR-124a2, SCG10, SCN3A, synaptophysin, LSAMP and synapsin ) suggesting a direct role of REST in regulation of these genes, including one microarray target (β3-tubulin). But REST binding was not detected at the predicted RE1 elements of two other microarray targets (GATA3 and GAP43), suggesting they were regulated indirectly by REST in the system we tested. The observed REST binding was 5 to 45 times higher than the background from ChIP with control IgG (data not shown), suggesting the binding was specific.
Figure 4. Evidence for Telomere-Independent Inhibition of REST Degradation and Repression of Neuronal Genes and Cell Differentiation by TRF2.

(A) Chromatin immunoprecipitation was used to measure REST binding in SH-SY5Y cells infected with adenovirus bearing DN-TRF2, WT-TRF2 or control (β-galactosidase) for 5 d. Binding to each locus was determined relative to the input DNA. RE1 elements (RE1) and promoters (p) were tested. Binding to control IgG averaged 0.0049%, ± 0.0045. This experiment is representative of three independent replicates. (B) Realtime RT-PCR measurements of mRNA from SH-SY5Y cells that had been infected for 6 days with adenovirus bearing either DN-TRF2 or β-galactosidase (control). The relative expression level of REST target genes was normalized against GAPDH and control (n=3, error bars ± SD). This result is representative of three independent experiments. (C) SH-SY5Ycells were exposed to either 40 μM of T-oligo or the complementary antisense oligonucleotide (AS). Cells treated with both oligos for 8 d exhibited morphologies characteristic of undifferentiated cells (left). Arrowheads point to SA-βgal positive cells. bar = 50 μm. Naive SH-SY5Y cells (control) and cells were treated with T-oligo or AS-oligo for 2 d. Cell lysates were subjected to immunoblot analysis using antibodies against the indicated proteins. (D) Model: In human neural progenitors and pluripotent stem cells, the formation of TRF2-REST complex stabilizes REST in PML-NB, which silences neuronal genes in heterochromatin region to suppress neuronal differentiation. Removal of TRF2 disassociates the TRF2-REST complex, facilitating polyubiquitination-proteasomal degradation of REST in a telomere-independent manner. Consequentially REST targeted neural genes are derepressed, resulting in acquisition of neuronal phenotypes.
We next performed quantitative RT-PCR and immunoblot analysis to confirm that reduction of REST binding via TRF2 inhibition results in derepression of REST target genes. Relative mRNAs levels of several REST target genes including L1CAM, SNAP25, synaptophysin, synapsin 1 and SCN3A were increased two- to four-fold in SH-SY5Y cells expressing DN-TRF2 in comparison with control (Figure 4B). The expression level of β3-tubulin was increased 1.5-fold (data not shown). Within 3-5 days of adenoviral infection, SH-SY5Y cells expressing DN-TRF2, but not vector or WT-TRF2, also exhibited increased levels of L1CAM, β3 tubulin and GAP43 (Figure S3C), three well-known neurite outgrowth-regulating proteins [21, 22]. Consistently, interfering with TRF2 expression using shRNAs also resulted in REST degradation and derepression of the REST target gene L1CAM (Figure S3A).
We next performed quantitative RT-PCR and immunoblot analysis to confirm that reduction of REST binding via TRF2 inhibition results in derepression of REST target genes. Relative mRNAs levels of several REST target genes including L1CAM, SNAP25, synaptophysin, synapsin 1 and SCN3A were increased two- to four-fold in SH-SY5Y cells expressing DN-TRF2 in comparison with control (Figure 4B). The expression level of β3-tubulin was increased 1.5-fold (data not shown). Within 3-5 days of adenoviral infection, SH-SY5Y cells expressing DN-TRF2, but not vector or WT-TRF2, also exhibited increased levels of L1CAM, β3 tubulin and GAP43 (Figure S3C), three well-known neurite outgrowth-regulating proteins [21, 22]. Consistently, interfering with TRF2 expression using shRNAs also resulted in REST degradation and derepression of the REST target gene L1CAM (Figure S3A).
We and others recently reported that in non-dividing cells TRF2 dysfunction promotes differentiation/growth instead of cell cycle arrest [4, 5]. Thus, we predict that in proliferating neuronal cells, the differentiation-inducing effects of TRF2 inhibition may proceed independently of telomere damage responses that typically involve the accumulation of γH2AX at the site of DNA damage (Figure S3C) and cell senescence (Figure 1D and 1E) [4]. To test this hypothesis, we employed a previously established telomere-decapping model, in which cells are treated with a telomere G-strand oligonucleotide (T-oligo) that mimicks the exposure of unprotected telomeres, thus triggering DNA damage responses [23]. As expected, cells treated with T-oligo, but not those treated with the anti-sense (AS) oligo, exhibited features of a DNA damage response including increased levels of γH2AX, p53 and p53 phosphorylated on serine 15 (Figure 4C). However, despite these DNA damage responses, cells treated with T-oligo or with AS-oligo TRF2 levels were unchanged and neither a reduction in REST levels nor neurite outgrowth occurred (Figure 4C). Similar results were obtained in neuroblastoma cells treated with telomestatin, a chemical that causes selective damage to telomeres [24] (Figure S4). Taken together, our data suggest that it is not telomere DNA damage per se, but rather, inhibition of TRF2 binding to REST that triggers proteolytic degradation of REST and so neuronal differentiation.
Our findings reveal a non-telomeric role for TRF2 in regulating cell proliferation and differentiation through an interaction with REST in PML-NB. The TRF2-REST complex may serve to protect REST from ubiquitin-proteasome degradation, thereby maintaining neuronal progenitors and stem cells in an undifferentiated stage. Removal of TRF2 not only induces telomere DNA damage responses, but also expedites proteolytic degradation of REST in a telomere-independent manner thereby de-repressing the expression of neuronal genes such as L1CAM, β3-tubulin, BDNF, synaptophysin and synapsin 1, resulting in the morphological and functional differentiation of neurons (Figure 4D). Our data support the telomere-looping-effect theory from yeast studies [7, 8], in which telomeres are juxtaposed with distal gene loci via a transient interaction between telomere binding proteins and transcription factors, permitting long range repression. Although details of the molecular interactions remain to be established (e.g. whether the TRF2 and REST interaction could bridge telomere region close to REST targets (RE-1 locus)), our results provide evidence that the telomere shelterin protein TRF2 is involved in the regulation of heterochromatin silencing domains by interaction with the master neuronal repressor REST. These findings expand an understanding of the function of a telomere-associated protein in the epigenetic modulation of cell proliferation and differentiation. Our findings from studies of human neuroblastoma cells further show that by maintaining REST binding at RE1 elements TRF2 maintains tumor cells in a proliferative state, and that inhibition of the TRF2 –REST interaction can suppress cancer cell growth.
Supplementary Material
Experimental procedures, 4 figures, and one table are available at http://www.cerrent-biology.com.
Acknowledgments
We thank G. Mandel for REST plasmids, F. Dantzer for GST-TRF2 plasmids and V. M. Vishva Dixit for HA-ubiquitin plasmids; Dr. Kazuo Shin-ya for telomestatin; C. Morris for help with flow cytometry analysis; V. Bohr, Y. Liu, H. Tahara and O. Taeko for valuable comments on the paper and technical support. This research was supported by the National Institute on Aging Intramural Research Program.
References
- 1.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 2.Van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 3.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 4.Zhang P, Furukawa K, Opresko PL, Xu X, Bohr VA, Mattson MP. TRF2 dysfunction elicits DNA damage responses associated with senescence in proliferating neural cells and differentiation of neurons. J Neurochem. 2006;97:567–581. doi: 10.1111/j.1471-4159.2006.03779.x. [DOI] [PubMed] [Google Scholar]
- 5.Lazzerini Denchi E, Celli G, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev. 2006;20:2648–2653. doi: 10.1101/gad.1453606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Q, Reynolds GE, Innes L, Pedram M, Jones E, Junabi M, Gao DW, Ricoul M, Sabatier L, Van Brocklin H, Franc BL, Murnane JP. Telomeric Transgenes are Silenced in Adult Mouse Tissues and Embryo Fibroblasts, but are Expressed in Embryonic Stem Cells. Stem Cells. 2007;25:3085–3092. doi: 10.1634/stemcells.2007-0478. [DOI] [PubMed] [Google Scholar]
- 7.Zaman Z, Heid C, Ptashne M. Telomere looping permits repression “at a distance” in yeast. Curr Biol. 2002;12:930–933. doi: 10.1016/s0960-9822(02)00865-5. [DOI] [PubMed] [Google Scholar]
- 8.Babu MM, Balaji S, Iyer LM, Aravind L. Estimating the prevalence and regulatory potential of the telomere looping effect in yeast transcription regulation. Cell Cycle. 2006;5:2354–2363. doi: 10.4161/cc.5.20.3386. [DOI] [PubMed] [Google Scholar]
- 9.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 12.Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, Leng Y, Maehr R, Shi Y, Harper JW, Elledge SJ. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–374. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz CM, Spivak CE, Baker SC, McDaniel TK, Loring JF, Nguyen C, Chrest FJ, Wersto R, Arenas E, Zeng X, Freed WJ, Rao MS. NTera2: a model system to study dopaminergic differentiation of human embryonic stem cells. Stem Cells Dev. 2005;14:517–534. doi: 10.1089/scd.2005.14.517. [DOI] [PubMed] [Google Scholar]
- 14.Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Ceña V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- 15.Lafarga M, Bercian MT, Pena E, Mayo I, Castaño JG, Bohmann D, Rodrigues JP, Tavanez JP, Carmo-Fonseca M. Clastosome: a subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome. Mol Biol Cell. 2002;13:2771–2782. doi: 10.1091/mbc.E02-03-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2006;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 17.Matunis MJ, Zhang XD, Ellis NA. SUMO: the glue that binds. Dev Cell. 2006;11:903. doi: 10.1016/j.devcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007;14:570–571. doi: 10.1038/nsmb1259. [DOI] [PubMed] [Google Scholar]
- 19.Liu L, Bailey SM, Okuka M, Muñoz P, Li C, Zhou L, Wu C, Czerwiec E, Sandler L, Seyfang A, Blasco MA, Keefe DL. Telomere lengthening early in development. Nat Cell Biol. 2007;9:1436–1441. doi: 10.1038/ncb1664. [DOI] [PubMed] [Google Scholar]
- 20.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 21.Avwenagha O, Campbell G, Bird MM. Distribution of GAP-43, beta-III tubulin and F-actin in developing and regenerating axons and their growth cones in vitro, following neurotrophin treatment. J Neurocytol. 2003;32:1077–1089. doi: 10.1023/B:NEUR.0000021903.24849.6c. [DOI] [PubMed] [Google Scholar]
- 22.Dihne M, Bernreuther C, Sibbe M, Paulus W, Schachner M. A new role for the cell adhesion molecule L1 in neural precursor cell proliferation, differentiation, and transmitter-specific subtype generation. J Neurosci. 2003;23:6638–6650. doi: 10.1523/JNEUROSCI.23-16-06638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li GZ, Eller MS, Firoozabadi R, Gilchrest BA. Evidence that exposure of the telomere 3′ overhang sequence induces senescence. Proc Natl Acad Sci U S A. 2003;100:527–531. doi: 10.1073/pnas.0235444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MY, Gleason-Guzman M, Izbicka E, Nishioka D, Hurley LH. The different biological effects of telomestatin and TMPyP4 can be attributed to their selectivity for interaction with intramolecular or intermolecular G-quadruplex structures. Cancer Res. 2003;63:3247–3256. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, 4 figures, and one table are available at http://www.cerrent-biology.com.