

Abstract
Adherence of bacterial pathogens to host tissues contributes to colonization and virulence and typically involves specific interactions between bacterial proteins called adhesins and cognate oligosaccharide (glycan) or protein motifs in the host that are used as receptors. A given pathogen may have multiple adhesins, each specific for a different set of receptors and, potentially, with different roles in infection and disease. This chapter provides strategies for identifying and analyzing host glycan receptors and the bacterial adhesins that exploit them as receptors, with particular reference to adherence of the gastric pathogen Helicobacter pylori.
Overview
Helicobacter pylori chronically infects the gastric (stomach) mucosa of billions of people worldwide. Infections tend to last for decades once established, despite host defenses such as mucosal shedding and immune and inflammatory responses. Histological inspection of gastric biopsy specimens generally shows most H. pylori cells in the thin mucus layer. Mucus matrix is formed by high-molecular-mass oligomeric glycoproteins known as mucins. The mucus layer protects the underlying epithelium from gastric acidity and infection by other microbes. In addition to the many H. pylori cells present in the mucin layer, others adhere directly to epithelial cell surfaces, and a few may enter and proliferate within epithelial cells. H. pylori is motile and chemotactic for particular host metabolites, including bicarbonate, arginine, and urea, and chemotaxis-driven motility probably allows efficient migration of the pathogen to preferred gastric mucosal sites. In one view, the primary benefit of adherence for H. pylori is in escaping clearance by mucosal shedding and peristalsis. Alternately, or in addition, much of adherence's value may come from placing H. pylori at or close to epithelial surfaces, for efficient scavenging of nutrients leached from host tissue. Adherence to epithelium also promotes efficient management of host responses by delivery of toxins and other signaling molecules. Therefore, tight adherence may be a mixed blessing for H. pylori, however, also potentially exposing it to intense, bactericidal inflammatory responses.
Two of H. pylori's adhesins have been characterized in terms of receptor interactions (BabA, specific for ABO and Lewis b [Leb] antigens; and SabA, specific for sialylated Lewis × [sLex] and sialylated Lewis a [sLea] antigens). Studies of numerous clinical isolates have shown that the BabA and SabA adhesins are diverse in terms of amino acid sequence, glycan specificity, and affinity, and each is expressed by only a subset of H. pylori strains. The genes encoding these two adhesins are members of a large multigene family of H. pylori outer membrane proteins (HOPs). Some proteins are probably porins, and some members probably encode adhesins, whose activities and regulation of expression have not yet been documented. It is noteworthy in this context that H. pylori is extremely diverse genetically as are its gastric mucosal habitats.
Gastric mucosal diversity is illustrated by the differences among people in glycan profiles because of genetic polymorphisms in underlying glycosyl transferase genes and by changes in a given person's glycosylation patterns caused by infection and host inflammatory response. It is in this context that one sees that H. pylori may well benefit from having multiple adhesins, differing in glycoprotein and glycolipid receptor specificities and affinities, and in how their expression is controlled.
Collectively, the constellation of adhesins and cognate receptors is postulated to affect how H. pylori resists mucosal shedding and peristalsis and how it signals target cell regulatory circuitry and captures nutrients leached from host tissue.
Characterization of Bacterial Adherence by In Vitro Binding to Host Tissue
The use of human biopsy materials and other preserved tissues in in vitro tissue binding assays allowed detection of tissue-specific adherence of H. pylori and other pathogens and characterization of the receptors used. Early studies showed that many H. pylori strains could bind fucosylated Lewis (or ABO histo blood group) antigens and the inflammation-associated sialylated antigens of the gastric epithelium (Aspholm-Hurtig et al., 2004; Boren et al., 1993; Mahdavi et al., 2002); that uropathogenic E. coli bound to globoseries glycolipids, which are abundant in kidney tissue (Roberts et al., 1994); and that Streptococcus pyogenes–bound protein antigens in human cutaneous tissue (Okada et al., 1994). In typical in vitro host tissue binding assays, bacterial cells are cultured, labeled, and overlaid on histotissue sections. The binding patterns observed give insight into expression, localization, and spatial distribution of receptors in target tissues. Further characterization of receptors can emerge from studies using various inhibitors of adherence (e.g., simple glycans to titrate adhesins; glycosidases or to disrupt potential receptors) in this in vitro tissue-binding assay. A general flowchart for initial identification of carbohydrate receptor structures is presented in the early review by Falk et al. (1994a).
Biopsy specimens from gastroscopy examinations and tissues from gastrointestinal surgery provide valuable material for adherence studies. It is often useful to choose tissues for analysis according to patient ABO and Lewis blood group status and disease condition (e.g., gastritis, peptic ulcer, gastric cancer). Also noteworthy are gender, age, medication, immune status, level of inflammation, and other associated infections, because these may also affect the types and tissue densities of various glycans that H. pylori can use as receptors and, thereby, vulnerability to infection or disease. The series of studies referred to previously also illustrates that H. pylori clinical isolates differ in types of glycans they can use as receptors and intensity of binding, reflecting diversity in their complement of adhesin genes, as noted earlier.
Tissues can be fixed by standardized formaldehyde treatment and embedded in paraffin blocks for later microtome sectioning. Most pathology laboratories use standardized protocols and robotic fixation and embedding instruments to ensure full reproducibility. Biopsy material can also be snap-frozen and embedded in cryopreservatives for later cryosectioning. There are pros and cons with both methods: Paraffin-embedded/fixed material usually cut in thinner sections than is cryopreserved material, and thus provides higher resolution images, and fixed material can be stored for years. However, cryosectioned materials retain more of the gastric mucin layer and conformational epitopes, and thereby allow better presentation of certain antigens for analyses of adherence or detection with monoclonal antibodies.
Bacterial cells used for initial in vitro tissue adherence studies are often FITC (green fluorochrome) labeled and then overlaid on tissue sections (Falk et al., 1993, 1994b; Borén et al., 1997; see also FITC-labeling in section 2: “Glycoprotein Array” and 8: “Identification of Bacterial Clones”). Red fluorochromes such as TRITC or Texas-Red can be used equivalently (Van de Bovenkamp et al., 2003), although TRITC tends to leach from bacterial cells and skew adherence profiles unless tissue sections are very thoroughly washed after the bacterial overlay. It is also important to take into consideration that bacterial surface labeling involves modification of basic amino acids and thus may diminish adherence by direct inactivation of an adhesin's binding domain or indirectly through steric hindrance. When this is a concern, fluorochrome-labeled bacterial cells can be tested by RIA or ELISA assays if defined receptors are available (see the next section). If no binding activity of fluorochrome-labeled H. pylori cells is detected, the possible influence of fluorolabeling can be tested using unlabeled cells for binding to tissue sections, followed by staining (1) with DNA intercalating agents such as acridine orange or (2) through enzymatic conversion of precursor substrate and fluorochrome-activation of bacterial cytoplasm (see Molecular Probes, http://www.probes.invitrogen.com), or (3) with antibodies against bacterial cells or their surface antigens.
Once H. pylori cells are successfully fluorochrome labeled they can be aliquoted and stored frozen at −20° for 6–12 mo. Use of aliquots from the same batch of labeled cells can contribute to consistency (e.g., when many tissues or receptors are being studied) and avoids complications from any possible growth-related variation in adhesin expression and presentation on cell surfaces. This procedure depends on intactness of bacterial cells and is compatible with most H. pylori strains. This said, there might be certain strains or species in which intrinsic fragility of bacterial cells or their adhesins, either naturally or during freezing and thawing, makes this protocol less suitable, such as Neisseria gonorrhoeae and N. meningitidis and various Borrelia species. An alternative could be to use minimal levels of DMSO as solvent for the FITC staining reagent. The in vitro tissue adherence assay is performed essentially as described by Falk et al. (1993, 1994b) and Borén et al. (1997), with modifications in duration and frequency of washing of the histotissue section overlaid with bacterial cells to fit bacterial affinity. Slides used to test for adherence by high-affinity interactions can be washed more extensively than those used to detect low-affinity adherence or if there are relatively few adhesin proteins per cell. Identification of optimal washing conditions may sometimes need careful titration and comparison with suitable references, such as derivative strains in which the adhesin gene of interest has been switched off in expression by phase variation or has been deleted.
The biopsy specimens used for the in vitro tissue adherence assay can alternately be cultured in vitro for 2–3 days and used for adherence analyses (i.e., by use of the in vitro explant culture [IVEC] technique) (Olfat et al., 2002). This is a most useful application for studies of the impact of adherence in terms of bacterial-host crosstalk, such as cytokine release and cellular signaling in host tissue. In addition, the IVEC technique also complements the use of primary cell cultures, because the cells in biopsies are most similar to the true in vivo conditions of the epithelium and in expression of naive glycosylation patterns. In addition, IVEC also ensures the integrity of the spatial distribution of cell lineages in the intact biopsy materials. However, use of human gastric mucosa requires pretty fluent collaboration with the gastrointestinal surgery department, especially because the gastric mucosa has to be in good and healthy condition, which excludes the use of most cases of dysplastic and cancer tissue obtained from eradication surgery. Nevertheless, representative biopsy material can sometimes be obtained from patients undergoing vertical banded gastroplasty operation (i.e., surgical removal of stomach tissue because of morbid obesity).
Conclusions
The in vitro tissue binding assay is valuable for initial characterization of microbial adherence to specific host cell lineages and receptors that the microbe exploits, and of variant strains, whether generated in the laboratory or recovered during the course of natural or experimental infection.
Glycoprotein Array for Screening and Identification of Adhesin Binding Properties
Various overlay techniques on immobilized glycolipids or glycoproteins had been developed on the basis of bacterial overlay on either thin-layer chromatography (Hansson et al., 1985; Karlsson and Stromberg, 1987) or nitrocellulose transfers of protein extracts separated by SDS-PAGE (Prakobphol et al., 1987) to identify molecules carrying carbohydrate receptors for bacterial lectin-like adhesins. Carbohydrate receptors are immobilized in these assays, which facilitates detection of low-affinity binding that depends on multivalent interactions between multiple adhesins on bacterial surfaces and clustered receptors on the solid-phase substrate. Soluble receptors that bind only weakly in RIA can be analyzed more robustly using solid-phase presentation on array-membranes. However, a disadvantage of the original array methods, as in the RIA assays (described in the following section “Analyses of Binding Activity”), is a requirement for radioactive labeling. As alternatives, the overlay technique was modified by tagging bacteria with biotin (Ruhl et al., 1996) or fluorescein isothiocyanate (FITC) (Walz et al., 2005). The bacterial overlay technique is helpful in searching for glycoprotein receptors on eukaryotic cell surfaces (Ruhl et al., 2000) and for identifying ligands for bacterial adhesins in complex body fluids (Murray et al., 1992; Ruhl et al., 2004). The method can also be used to determine adhesin-binding specificities using membranes that have been spotted with purified glycoproteins or neoglycoproteins carrying a defined oligosaccharide motif, detailed later. In addition, oligosaccharides on glycoproteins can be enzymatically modified once immobilized on nitrocellulose, before overlay with bacteria, which also makes it very useful for initial screening of adhesin specificities. For more detailed comparisons of binding strengths, serial dilutions of appropriate glycoconjugates can be spotted on the nitrocellulose and then used for bacterial overlay. For exact comparisons, the number and density of bound oligosaccharides per protein molecule must also be considered. As with in vitro tissue adherence to biopsy material, the glycan array method is most useful for analyzing effects of deletions or other mutations in adhesin genes (construction described later) on binding properties (Walz et al.,2005).
Method for Bacterial Overlay
Materials
Fluorescein-5-isothiocyanate (FITC, Molecular Probes, Eugene, OR)
Solutions
FITC stock solution: dissolve 1 mg FITC in 100 μl DMSO. Prepare freshly before use.
Tris-buffered saline (TBS): Tris-HCl buffer 20 mM, pH7.6, 150mM NaCl
Blocking buffer: TBS, 5% bovine serum albumin, immunoglobulin-free, fraction V, 1 mM CaCl2, 1 mM MgCl2.
Wash buffer: TBS, 0.05% Tween-20, 1 mM CaCl2, 1 mM MgCl2
Fluorescence Labeling of H. pylori
H. pylori J99 and its isogenic derivatives with deletion mutations in the sabA and/or babA genes are grown for 48–72 h at 37° in a microaerophilic atmosphere on Wilkins-Chalgren agar (Oxoid, Wesel, Germany) containing 10% horse blood, Dent supplement (Oxoid) and 0.4 g KNO3 per liter.
Harvest bacteria from plates by wiping off the colonies with a sterile cotton swab and wash bacteria twice in 20 mM phosphate-buffered saline, pH 7.2 (PBS).
Adjust the bacterial concentration to 108 cells/ml (equivalent to an optical density of 1–4) in PBS and label by incubation with FITC at 100 μg/ml (0.1 ml of FITC stock solution per 10 ml of bacterial suspension) for 30 min at RT. Please note that bacterial numbers estimated by optical density highly depends on the H. pylori strain used (a CFU [colony forming units]) analysis is recommended for calibration)
Recover labeled bacteria by centrifugation at 800g for 7 min (10 ml volumes), wash three times with PBS (until supernatant is free of yellow color), and resuspend in 10 ml blocking buffer.
Spotting of Glycoprotein Arrays
Spot dry nitrocellulose membranes (Schleicher und Schüll, Protran B85) with 1-μl volumes containing 1 μg of glycoproteins or neoglycoproteins of choice. Human serum albumin (HSA) or bovine serum albumin (BSA) should be included as negative (nonglycosylated) controls. Purified natural glycoproteins that can be used include fetuin (Calbiochem, Bad Soden, Germany), asialofetuin (Sigma), glycophorin A (Sigma), asialoglycophorin (Sigma), laminin (from human placenta, Sigma), transferrin (Sigma), fibronectin (from human plasma, Sigma), and lactoferrin (from human milk, Sigma).
Allow spots to dry before use in the overlay (membranes can be stored for at least a week at RT in a dry dust-free environment).
Pretreatment of Dot Blot Arrays (Optional)
For removal of terminal sialic acids, incubate membranes with 0.l U/ml of sialidase (from Clostridium perfringens, type X, Sigma) in TBS containing 1% BSA (fraction V, Sigma), 1 mM CaCl2, and 0.1% sodium azide at 37°. Wash three times with TBS to remove sialidase before overlay with bacteria.
For denaturation of spotted proteins, treat membranes with 0.1% SDS (Merck, Darmstadt, Germany) in PBS containing 50 mM beta-mercaptoethanol (Merck) for 5 min in a sealed plastic bag immersed in a cooking water bath.
For N-glycosidase F digestion, add 0.05 U/ml of recombinant Glyko N-glycanase from Chryseobacterium meningosepticum (PROzyme, San Leandro, CA) and 0.75% NP-40 (PROzyme) after prior denaturation (see earlier) and incubate overnight at 37°.
All enzymatic pretreatments of membranes should be performed in sealed plastic bags for purity and to save reagents.
Bacterial Overlay on Nitrocellulose Membranes
Block unspecific binding sites on membranes with TBS containing 5% BSA (fraction V, Sigma), 1 mM CaCl2, and 1mM MgCl2 for 2 h at 4°.
Add fluorescence-labeled bacteria to the membranes in a final concentration of 2.5 × 107 organisms per ml of blocking buffer (membranes should be covered with at least 0.6 ml of bacterial suspension per cm2 of nitrocellulose membrane).
Incubate overlaid membranes for 30 min at 4° without mixing to allow bacterial binding.
Wash three times for 5 min on a rotary shaker with TBS containing 0.05% Tween-20, 1 mM CaCl2, and 1 mM MgCl2 to remove unbound bacteria.
The fluorescence of bound bacteria can be detected by a fluorescence scanner (Typhoon 9200, GE Healthcare Biosciences, Freiburg, Germany).
Analyses of Binding Capacity by Use of RIA and Affinity according to Scatchard and, in Addition, a Nonradioactive Alternative Based on Fluorescent Glycoconjugates
As described previously, the in vitro tissue adherence analyses (Falk et al., 1993) and /or glycan arrays can be used to delimit the range of possible host receptor candidates. Radio immunoanalysis (RIA) can then be used for quantitative analyses of an adhesin's or cell's binding capacity and affinity, which together make up its binding activity. Once receptor structures have been better characterized, similar or related glycan substances or conjugates can be obtained or synthesized and analyzed by RIA. The main difference between the RIA and in vitro tissue adherence methods is the use of soluble receptors in RIA analyses vs. immobilized “solid-phase” receptors in histo tissue sections.
Method for RIA Assay Based on 125I-Labeled Receptor Conjugates
In RIA analysis, 1 ml of bacteria OD600 = 0.1 is allowed to react with 300 ng of radiolabeled glycoconjugate for 2 h at RT, and percent binding is calculated. For “strong binders,” most binding sites are occupied, and the percent binding corresponds to the total bacterial binding capacity. For “weak binders,, the percent binding will also reflect the affinity constant.
Solutions
10× PBS (phosphate-buffered saline): 250 mM phosphate, 850 mM NaCl
(6.81 g KH2PO4, 34.84 g K2HPO4, 49.67 g NaCl, 971 g H2O)
PBS-Tween: 25 mM phosphate, 85 mM NaCl, 0.05% Tween-20, pH 7.4
(106 g 10× PBS, 900 g H2O, 0.5 ml Tween-20
Blocking buffer: 1% BSA in PBS-Tween-20, filter through 0.22-μm filter.
Phosphate buffer: 50 mM phosphate, pH 7.4
1.66g KH2PO4, 6.59 g K2HPO4, H2O to 1 l)
125I Labeling of Glycoconjugate (Hunter and Greenwood, 1962)
To 2–10 μg of glycoconjugate diluted to 50 μl with phosphate buffer, 0.2 mCi 125I (7.4 MBq, carrier free) is added. The reaction is started by adding 20 μl of 0.6 mg/ml Chloramine-T. After 40 sec, the reaction is stopped by addition of 100 μl of 1 mg/ml sodium meta-bisulfite. Then, 200 μl of 9.6 mg/ml KI is added, and the sample is allowed to stand for at least 10 min before the radiolabeled glycoconjugate is separated from unbound 125I on a PD-10 column (GE Healthcare, Uppsala, Sweden). The column is equilibrated with 5 ml blocking buffer for 30 min, washed with PBS-Tween, and eluted with PBS-Tween, and 0.5 ml fractions are collected. The radioactivity of fractions is monitored with a GM counter and fractions that elute in the very first peak (“void fraction”) are pooled and kept frozen until used. It should be used within 2 mo, because iodinated material tends to self-destruct with concomitant release of free iodide (and loss of signal). If there is need to use iodinated conjugate of higher specific activity (labeling), less conjugate (<1 μg) is added to the labeling mixture for more focused 125I-labeling.
125I-labeled Conjugate Cocktail: 300 ng Conjugate/10 μl, 20.000 CPM/10 μl
Thirty micrograms of unlabeled “cold” conjugate and approximately 2,000,000 cpm of 125I-labeled “hot” conjugate are mixed and diluted to 1 ml of blocking buffer.
RIA Analysis
Ten microliters of cocktail is mixed with 1 ml bacteria (optical density at A600nm = 0.1). After 2 h incubation on a rocking table at RT, the bacterial cells are pelleted by a 5–15 min-centrifugation at 20,000g, and the supernatant and pellet are counted separately in a gamma scintillation counter. After subtracting the background count, the percent binding is calculated.
Applications
The RIA method is very useful for rapid tests of mutants, for scoring binding properties in collections of clinical isolates, and for quantitative analyses of variations in binding activities in response to culture conditions, such as limitation in nutrients, temperatures, and oxygen levels.
A Nonradioactive Alternative to Test for H. pylori Binding Properties in Solution: Fluorochrome-labeled Glycoconjugates
Materials
Fluorescein 5(6)-isothiocyanate (FITC, Sigma-Aldrich St Louis, MO) HSA-Glycoconjugate (Isosep, Tullinge, Sweden)
Solutions
Carbonate buffer: 0.15 M NaCl, 0.05 M carbonate, pH 9.0
FITC Labeling of Glycoconjugates
Dissolve 0.5 mg HSA-glycoconjugate in 0.1 ml carbonate buffer. Add 0.015 mg FITC solubilized in DMSO (do not store FITC dissolved, instead use fresh each time). Incubate the vial protected from light for 2 h with agitation. Remove excess of FITC by washing once with 0.5 ml carbonate buffer followed by three washes in PBS in a Microcon YM-50 centrifugal filter device (Millipore, Bedford, MA). The FITC-labeled glycoconjugates can be stored at −20° for >1 y. Aliquot the labeled glycoconjugate into single-use vials to avoid freeze–thaw cycles.
An alternative that provides options for use of multiple fluorochromes is to label the conjugates with the Alexa Fluor Monoclonal Antibody Labeling Kits (Molecular Probes, http://www.probes.invitrogen.com).
H. Pylori Binding Assay Using Fluorescent Glycoconjugates
Incubate 100 ul H. pylori (OD600 = 1.0) with 500 ng conjugate for 30 min in PBS containing 0.5% human serum albumin (HAS) and 0.05% Tween-20 in a 96-well round bottom plate. Wash three times by centrifugation (2200g) in 200 ul PBS containing 0.05% Tween-20 and then read fluorescence.
Several glycoconjugates and strains can be tested simultaneously, although some precaution is advised for use of clear-walled microtiter plates to avoid leakage of signal from one well to another. Thus, it is suggested to leave an empty well between each sample. It is also most useful to include a negative control (i.e., with no fluorochrome added) because H. pylori can emit autofluorescence.
Affinity Analysis by RIA According to Scatchard
The binding of receptor conjugates is often affected by the multivalent presentation of receptors on carrier molecules, as is seen with Leb-HSA (human serum albumin) conjugates (IsoSep), which carry 15–20 Lewis b antigen (Leb)—oligosaccharides attached to each HSA molecule. This allows low-affinity-interactions to be detected by adding saturating levels of soluble receptor conjugate to the bacterial suspension. The results obtained, however, can also be easily misinterpreted as “good/strong” binding to receptors. Scatchard analysis can be used for sensitive determination of binding-affinity (association constant [Ka]) and also total binding capacity (RT). Scatchard analysis is performed in a manner similar to that of RIA analysis, with minor modifications: For each sample, a number of test tubes with the same concentration of bacteria are prepared, but with different concentrations (a dilution series) of conjugate. In presenting data from a full series of binding experiments, conjugate bound to bacterial cells versus conjugate free in solution is depicted on the Y-axis; and total conjugate bound is depicted on the X-axis. The Y-axis' highest value is that obtained with only low levels of added conjugate (125I-labeled “hot” conjugate only, with no dilution of unlabeled “cold” carrier conjugate). In comparison, the other end of the curve, with saturating levels of receptor conjugate is bound to eventually cross the X-axis. This value provides information about the maximal number of receptor conjugates bound to each bacterial cell and, thereby, allows the number of cognate adhesins per bacterial cell to be estimated, see Fig. 1A (Scatchard, 1949; Rosenthal, 1967).
Fig. 1.
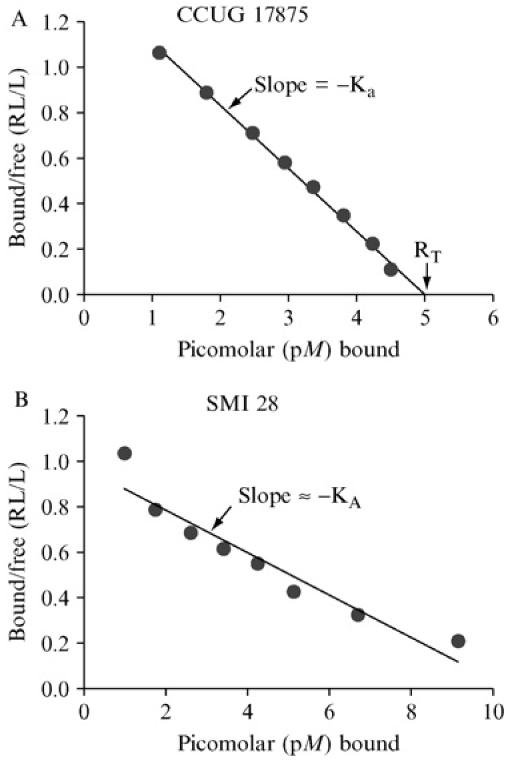
1. Affinity analyses according to Scatchard, essentially as described previously (Rosenthal, 1967; Scatchard, 1949). The equilibrium of glycoconjugate bound to bacterial cells versus conjugate free in solution (bound/free, i.e. pellet/supernatant) is depicted on the Y-axis, whereas total glycoconjugate bound is depicted on the X-axis. The maximal Y-axis value is achieved with only low levels of conjugate. The negative value of the slope derived by linear regression provides fairly good estimation of average KA. Thus, in such a Scatchard plot, the slope of the straight line is −Ka (i.e., the affinity constant). For bacterial strains with strong binding properties, high (saturating) levels of glycoconjugate make the line graph cross the X-axis and, by so doing, provides information about the total binding capacity of the glycoconjugate tested (RT). Thus, RT also tells us about the number of cognate adhesins per bacterial cell. (A) The affinity analysis is illustrated by H. pylori strain CCUG 17875 that produce an almost ideal linear slope, with an affinity (Ka)of 2.7 × 1011 M−1, whereas the H. pylori clinical isolate SMI 28 demonstrates a curved slope, with steeper slope and thus higher Ka during limiting conditions, followed by a more relaxed slope when the receptor glycoconjugate is added in excess (B). This gradual shift in Ka likely reflects heterogeneities in the adhesin complexes alternatively a mix of adhesin activities (proteins) that differ in binding properties and affinities for the receptor. Please note that in a curved slope, the total binding capacity of glycoconjugate (RT) cannot be deduced from the point where the line graph (eventually) crosses the X-axis but needs to be estimated by approximation (on the basis of the slope in the first part of the graph). Alternately, the total binding capacity can be functionally deduced by performing the RIA binding assay with receptor conjugate added in excess (i.e., RIA during saturating conditions). Fortunately, most BabA adhesins demonstrate high-affinity binding properties that allow for estimations of full binding capacity for interpretation of the corresponding numbers of bacterial adhesin molecules.
A constant number of bacterial cells (typically range of 107–109 cells/ml) is added in 900 μl to each of eight test tubes containing approximately 10,000 cpm of 125I-labeled conjugate and unlabeled conjugate (in 100 μl) with the relative concentrations 0, 4, 9, 14, 20, 30, 50, and 100. The bacterial numbers are adjusted so that the highest binding is approximately 50% (i.e., bound/free approximately = 1 on the Y-axis), and conjugate concentrations are selected so that the lowest binding is 10–20%. The samples are mixed and put on a rocking table until the reaction has reached equilibrium (17 h for binding H. pylori BabA adhesin to Leb-HSA-conjugate) and then the bacteria are pelleted by a 5–15-min centrifugation at 20,000g. The supernatant and the bacterial pellet are counted separately in a gamma scintillation counter. After correction for background cpm/counting, the bound/free (pellet/supernatant) ratio and concentration of bound conjugate (conjugate concentration * percent binding/100) is calculated for each sample and plotted as Y and X values, respectively. If all binding sites have the same affinity constant, this plot—a Scatchard plot—should produce a straight line, the slope of which is −Ka and the X-intercept represents RT (total binding capacity).
Practical Considerations
For detailed calculation of affinity, it is essential to achieve conditions of low unspecific binding. Ideally, a bacterial strain lacking all adhesins (e.g., an isogenic deletion derivative) would be the best negative reference. However, the cognate adhesin usually is not known, and thus an unrelated “nonbinding” strain is typically used as control to detect background or nonspecific binding. In the case of ABO/Leb-antigen binding, nonspecific binding is kept to a minimum using BSA/Tween blocking buffer. Alternatives could include complex protein mixtures such as serum, which also includes antibodies, or milk samples (bovine or human) that are rich in glycosylated conjugates and glycans. Typical biochemical parameters such as pH, ion strength, and temperature also need to be considered when optimizing a particular binding assay.
As described previously, bacterial suspensions should be diluted so that the highest binding is approximately 50%. If binding is much higher, most of the receptor conjugates are bound. This includes conjugates of lower multivalency, those damaged by iodination and/or irradiation, or contaminants from receptor purification or synthesis.
Despite these considerations, Scatchard diagrams are sometimes concave, with a steeper curve, indicating stronger affinity at low than at high receptor concentrations (see Fig. 1B). One likely explanation invokes two distinct adhesins that differ in affinity for the receptor used. An alternative explanation invokes heterogeneity in adhesin structure (e.g., if the adhesin consists of a supramolecular complex that also includes a variable number of regulatory or modulatory subunits).
ELISA Analysis of H. pylori Binding to Immobilized
Lewis b Glycoconjugate
Similar to assays using fluorescent glycoconjugates (described previously), ELISA can be used as an alternative to RIA for characterizing clinical strains, if routine use of 125I-labeling is not favored. The difference being that both RIA assays and assays using fluorescent glycoconjugates are based on the use of glycoconjugates free in solution, whereas ELISA assays are based on use of immobilized (solid-phase presented) glycoconjugates. For ELISA assays, freshly isolated bacterial strains are labeled with digoxigenin before testing them for adherence to Lewis b antigen (Leb)–coated microtiter plates. Covalently bound bacteria can then be detected using an anti-DIG-HRP antibody by measuring extinction in a microplate reader. This approach allows simultaneous examination of many strains, yielding highly reproducible results.
ELISA Methods
Materials
Digoxigenin-3-0-succinyl-aminocaproic acid-N-hydroxy-succinimide ester (DIG-NHS) (Roche Diagnostics, Mannheim, Germany).
Solutions
Carbonate buffer: mix 10 ml 0.2 M Na2CO3 with 90 ml 0.2 M NaHCO3: pH 9.2
PBST: PBS + 0.5% Tween-20
Blocking buffer: PBST + 0.5% non-fat dry milk
Surface Labeling of H. pylori Bacterial Cells
Grow clinical isolates for 48 h, harvest from agar plates with a cotton wool stick, and resuspend in 1 ml carbonate buffer.
Wash 2× in carbonate buffer, collect bacteria by centrifugation at 5000 rpm, 5 min. Surface label bacteria by incubation with 100 μg/ml Dig-NHS (freshly prepared in DMSO) in 1 ml carbonate buffer for 1 h at room temperature (RT) in the dark. Labeled bacteria are washed twice in PBST and collected by centrifugation (s.a.), dilute in PBST to a density of 1 OD A600 and freeze at −20° in 100-μl aliquots until use.
ELISA Binding Assay
For the binding assay we used 96-well Universal Covalent microtiter-plates (Corning Costar, Cambridge, MA), which contain photoactivatable linker used to covalently immobilize biomolecules by abstractable hydrogen using UV illumination resulting in a carbon–carbon bond. These plates are coated with human serum albumin (HSA) conjugated Leb or Lea (obtained from IsoSep AB, Tullinge, Sweden).
Dilute antigens at 50 ng/well in 0.2 M carbonate buffer to 1 ng/μl, add 50 μl of the solution (or 50 μl of buffer for controls) to each well.
Incubate for 1 h in the dark and remove liquid by carefully pipetting of the supernatants.
Expose plates to UV light for 30 sec in a Stratalinker (Stratagene, Germany) to immobilize the glycoproteins.
Block remaining binding sites on the plates by adding 100 μl blocking buffer to each well. After incubation for 1 h at RT, decant plates without washing.
Add 50 μl of bacterial suspension (diluted 1:1 in blocking buffer, including 10% FCS) and incubate for 1 h at RT in the dark with gentle agitation (100 rpm) to reduce nonspecific binding.
Remove unbound bacteria by vacuum aspiration using yellow micropipet tips.
Carefully wash each well three times with 200 μl PBS.
For detection, dilute anti-DIG-HRP-antibody 1:7000 in 2% BSA/50 mM TRIS.
Incubate for 30 min at RT and wash as above.
Add 50 μl ABTS-solution (Roche Diagnostics) to each well and incubate at 37° in the dark. After 15–30 min (depending on staining intensity versus the controls), extinction can be quantified by a microplate reader (Bio-Rad, Munich, Germany) at 405 nm and normalized to controls (uncoated wells).
Typically, all strains are tested in two Leb-HSA– or Lea-HSA–coated wells and two control (HSA-(nonglycosylated) coated wells). The extinction ratio is calculated from the mean of antigen coated/control values. Strains were considered positive if the ratio of ExLewis/Excontrol is >1.5.
This method was used to survey H. pylori strains from a German population for binding to fucosylated blood group antigens such as Leb (Gerhard et al., 1999). Most strains were readily assigned to either a “binder” or “nonbinder” group, and this classification correlated well with babA genotyping. “False-positive” binding (i.e., strains that lacked a functional babA gene) was not observed. However, some strains showed no binding to immobilized Leb despite presence of an apparently functional babA gene, which suggested allelic variation in binding specificity or mechanisms of gene regulation. In a further analysis with more geographically diverse strain set (Olfat et al., 2005), fewer of the strains from southern than from northern Europe exhibited strong Leb-binding.
These findings indicate that BabA expression can be regulated in individual strains. Because flexible regulatory mechanisms may interfere with in vitro adhesion properties, we strove to use early passages for determination of adhesion properties, thereby limiting the chance for changes in gene expression.
Receptor Activity–directed Affinity Tagging (Retagging) Technique for Adhesin Protein Identification
Receptor activity–directed affinity tagging (retagging) for selective affinity labeling of cognate adhesins is particularly valuable when cognate glycan receptors are available in conjugate form. For example, to identify carbohydrate-binding adhesins, the retagging technique can use albumin glycoconjugates with multivalently linked glycans. The retagging technique was developed and implemented for identification of H. pylori's blood group antigen binding adhesin (BabA) (Ilver et al., 1998) and further refined for identification of its sialic acid binding adhesin (SabA) (Mahdavi et al., 2002). The tri(multi)-functional Sulfo-SBED cross-linker was used for specific biotin (Re)tagging of the BabA and SabA adhesins. This cross-linker contains three reactive groups, an amine-reactive sulfo-NHS-ester, a photo reactive phenyl azide group, and a biotin moiety, each on a separate arm of the structure. The receptor glyco(albumin)conjugate is first labeled through Sulfo-SBED's sulfo-NHS-ester, which reacts with primary amines in the albumin molecule (procedure detailed later). Bacterial cells are then mixed with the cross-linker labeled glycoconjugate to allow for the bacterial adhesins to bind the glycan conjugate receptors, which brings the cross-linker in close proximity to the adhesin. The cross-linker is then activated by UV irradiation to photo cross-link the glycoconjugate and adhesin protein. A disulfide bond in the cross-linker's Sulfo-NHS-ester arm is cleaved with DTT or other reducing agents, a reaction that also removes cross-linker containing glycoconjugate but leaves a biotin-tag on the H. pylori hesins. The biotin group then provides a handle for streptavidin binding, applicable both for visualization and purification of adhesin protein (Fig. 2).
Fig. 2.

Retagging and adhesin purification, essentially as described (Ilver et al., 1998; Mahdavi et al., 2002). The multifunctional Sulfo-SBED cross-linker is chemically attached to the protein core of the cognate receptor glycoconjugate by the NHS group. The cross-linker– labeled glycoconjugate is next mixed with H. pylori and the bacterial adhesin proteins bind the glycoconjugate (left panel). The bacterial cells with bound cross-linker–tagged glycoconjugate are subjected to UV irradiation (mid panel), and the photoreactive azide group forms a covalent bond to structures in the immediate surrounding area (usually the adhesin protein). By addition of reducing conditions, the disulfide bond located in the cross-linker structure is cleaved, and the freed glycoconjugate can then usually be washed away (right panel). The retagged bacterial cells are fully solubilized with a harsh detergent to extract the adhesins, and retagged adhesin proteins are purified by adsorption to streptavidin-coated magnetic beads. The isolated adhesin protein is next subjected to MS analyses and identified by its unique peptide composition.
Preparation of Cross-linker–Labeled Glycoconjugate
Solutions
Phosphate-buffered saline (PBS): 150 mM NaCl, 10 mM phosphate, pH 7.6.
Wash buffer: PBS + 0.05% Tween-20
Tris-buffered saline (TBS): 150 mM NaCl, 10 mM Tris-HCl, pH 7.4
TBST: TBS + 0.05% Tween-20
Materials
Dimethylsulfoxide (DMSO)
Bovine serum albumin (BSA)
Glycoconjugate: A multitude of glycoconjugates is available from Isosep AB, Tullinge, Sweden.
Sulfo-SBED cross-linker (Pierce, Rockville, IL)
PD10 column (Amersham Pharmacia Biotech, Sweden)
Horseradish peroxidase (HRP)–streptavidin
Methods
The cross-linker is light sensitive, and the following steps should be carried out in reduced-light conditions.
Dissolve 1 mg of cross-linker in 100 μl DMSO (i.e., 10 μg/μl).
Apply 20 μg (2 μl) cross-linker solution per 100 μg of glycoconjugate in 1 ml PBS.
Incubate the reaction on a rotary table for 1 h at RT (cover sample with aluminum foil to protect it from light). In the meantime, prepare a PD10 column by washing it with 20 ml of wash buffer. Incubate it for 1 h with PBS-0.05% Tween-20 + 1% BSA and then wash it again with 20 ml of wash buffer.
Pass the glycoconjugate-Sulfo-SBED reaction through the PD-10 column to remove any excess cross-linker. Elute 0.5 ml flow-through fractions with wash buffer and use 25 μl from each fraction for analysis of protein content.
Pool fractions with the highest protein content (1.5–2 ml in total) and store them protected from light at −20° until use.
To assay success of cross-linker labeling, electrophorese 1 μl of cross-linker–labeled glycoconjugate in a sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) (7.5% gel) and blot proteins to a PVDF membrane (Bio-Rad, Hercules, CA). The PVDF membrane is blocked with 5% BSA in TBST over night at 4° and then incubated for 1 h with HRP-streptavidin diluted 1:10.000 in TBST + 1% BSA. After washing six times with TBST, the biotin-labeled glycoconjugate is visualized by enhanced chemiluminescence using ECL reagents (Amersham Biosciences, Uppsala, Sweden) according to manufacturer's instructions. The cross-linker–labeled conjugate is positive on the HRP-streptavidin immunoblot and has also gained somewhat in molecular mass, because of the SULFO-SBED substitutions.
Retagging Solutions
Wash buffer: PBS-0.05% Tween-20, protease inhibitors (1 mM EDTA and 10 mM benzamidine)
Reducing buffer: PBS-0.05% + Tween-20 protease inhibitors (1 mM EDTA and 10 mM benzamidine) and 50 mM dithiothreitol (DTT)
Laemmli buffer containing 5% beta-mercaptoethanol but without bromophenol blue staining
Materials
Sulfo-SBED cross-linker labeled glycoconjugate prepared as described previously.
UV-lamp, model UVL-56 (366 nm), Upland CA.
Method
The following protocol describes conditions used for photo-cross-linking and transfer of biotin tags to the H. pylori BabA adhesin. For use of retagging as an effective method for labeling other adhesin proteins, conditions such as the amount of glycoconjugate and incubation time may need to be optimized. This is illustrated by listing protocol modifications that led to successful isolation of the SabA adhesin, whose affinity is generally weaker than that of BabA for their cognate receptors. An albumin conjugate containing an oligosaccharide that is not bound by the bacterial adhesin can serve as a negative control in this retagging procedure, although a better negative control would be a glycoconjugate that is bound by a different type of adhesin.
Mix bacteria (A600 nm = OD 1.0) with cross-linker–labeled glycoconjugate and incubate the reaction on a rotary table for 2–4 h. For labeling the BabA adhesin, approximately 1 μg (about 80 μl) of cross-linker labeled glycoconjugate was used per ml bacteria A600 nm = OD 1.0.
Wash bacterial cells twice with wash buffer to remove unbound glycoconjugate.
Resuspend bacterial cells in wash buffer in a volume that covers the surface of a regular petri dish or other tray to be used for UV irradiation (300–366 nm) of the sample. A high surface/volume ratio increases cross-linking efficiency but also allows more fluid evaporation during UV irradiation.
Place the tray with bacterial suspension 5–10 cm from the UV lamp. Irradiate the sample for up to ∼15 h (over night). Titration of UV dose may be useful to optimize extent of cross-linking. Incubate in RT or in the cold room, depending on intensity of UV irradiation and heat output.
Wash bacterial cells twice for 5 min with reducing buffer to break the cross-linker's disulfide bond, leaving the biotin moiety on the adhesin.
Solubilize bacterial cells in Laemmli buffer, containing 5% beta-mercaptoethanol and boil the sample for 5 min.
Analyze a small sample of the crude cell extract by SDS-PAGE, blot to a PVDF membrane, and visualize the biotinylated proteins by probing the membrane with streptavidin-HRP, as described previously.
Materials for Magnetic-bead Enrichment of Biotin-retagged Adhesin
Magnetic Tube Holder
Streptavidin-coated magnetic beads (QIAGEN, Hilden, Germany)
Solutions
Wash buffer: PBS-0.05% Tween-20 + protease inhibitors (1 mM EDTA and 10 mM benzamidine)
Laemmli sample buffer with 5% beta-mercaptoethanol but without bromophenol blue staining, because the color makes it difficult to visualize the beads in the buffer.
Method for Magnetic-bead Enrichment of Biotin-labeled Adhesin
Dilute the bacterial crude cell extract (prepared as described earlier) 1:20 with wash buffer to lower the SDS concentration
Add streptavidin-coated magnetic beads that have been pretreated with wash buffer to the cell extract, and incubate for 3–4 h at 4° on a rotary table. Invert the tube at 30-min intervals to better disperse sedimented beads.
Separate magnetic beads from the bacterial cell extract using a dedicated magnetic tube holder, and wash the beads at least twice with wash buffer.
Resuspend the magnetic beads in 40–100 μl of Laemmli sample buffer containing 5% beta-mercaptoethanol, mix the beads gently, and boil them for 5 min to elute bound proteins. Separate the magnetic beads from the protein extract by using the magnetic tube holder. Repeat the procedure once (or several times), and pool the eluted fractions.
Separate a small fraction of the eluted proteins by SDS-PAGE, Western-transfer them to a membrane, and visualize the biotinylated proteins with streptavidin-HRP, as described earlier.
Identification of Retagged (Biotinylated) Adhesin Protein
Mass-spectrometry (MS) analysis is used for definite identification of the retagged protein. We use SDS-PAGE gels to separate the protein extract that has been eluted from the magnetic beads. The gels are stained with Coomassie Blue (avoid silver stain, because of lowered efficiency in subsequent MS analyses). With a clean razor blade, excise the band of interest that corresponds to the molecular mass detected with streptavidin-HRP, as described previously. In case your retagging is still not efficient enough to generate material for MS analysis, there is still the alternative to scale up the entire preparative procedure. The SDS gel separation can then be replaced with preparative SDS gel fractionation by the Prep-Cell system (Bio-Rad), which allows for large-scale preparative electrophoresis.
The band is normally digested with Trypsin (seq grade, Promega, USA) by the MS core facility, and peptides are identified by mass spectrometry on the basis of peptide masses and sequences. MALDI-TOF-MS on a Tof-Spec E mass spectrometer and ProFound (http://www.proteometrics.com) are used to match peptide masses to proteins in the NCBI database. Peptide identities can be validated by ESI-MS/MS sequencing on a Q-Tof instrument, using the nanospray source. The Mascot program (http://www.matrixscience.com) is recommended for identification of peptide sequences.
Practical Considerations of Retagging such as Modifications for SabA
For successful identification of adhesin proteins by retagging, proper choice of receptor glycoconjugates is critical, based both on high binding affinity and binding specificity. Thus, for efficient retagging, most receptor conjugates need to be tightly bound to the cognate adhesin protein. In addition, the conjugates should bind preferentially to only one type of adhesin protein. That is, well-targeted binding specificity is essential to minimize complications from unspecific and promiscuous binding and retagging. In essence, the probability of successful (Re)tagging of the adhesin is much enhanced by use of highest possible binding specificity and affinity receptors. However, to compensate for low-affinity binding adhesins, 10–100-fold higher concentrations of receptor conjugates can be used. Conjugate with higher level (density) of attached glycans can be used for better efficiency in binding and retagging, because increased numbers of receptor epitopes will increase the multivalency in binding and avidity, also described as the Velcro-effect.
The retagging UV exposure time can also be extended, because the Sulfo-SBED structure has turned out pretty stable. Thus, UV exposure can sometimes be successfully extended overnight, especially in case the receptor-ligand does not form a tight complex, and targeted transfer of biotin is less efficient. As described previously, retagging and transfer of biotin can be followed by immunoblots (SDS-PAGE separated proteins transferred to a membrane and retagged biotin residues are visualized by streptavidin, as described earlier.
When MS has tentatively identified the adhesin candidate, it is highly recommended to construct deletion mutants to verify the lack of phenotypic binding properties. This is especially important when several similar, often related, proteins are pinned down by the MS analyzes. The mutants are then analyzed by the series of binding assays described in this chapter, such as in vitro tissue binding, in vitro glycan array binding, RIA, and ELISA.
Phylogenetic Methods for Detecting Adaptive Change
Adaptive molecular evolution is fundamentally important for genomic innovation. Surveys of homologous protein sequences from bacterial populations often reveal substantial amino acid sequence variation both within and between species. A key question is what mechanisms produce and maintain the changes in amino acid sequences of proteins? It is well established that evolutionarily conserved regions of a protein are functionally “critical” and, therefore, generally refractory to amino acid change, amino acid-variability driven by “positive selection.” Such diversifying selection can contribute importantly to “fine-tuning” of protein function, in particular, when confronted with a new environment. Such adaptive evolution leading to a new or modified protein function is generally episodic in that it affects only few amino acids at select time points (Gillespie, 1991). The term “adaptive evolution” in the context of the bacterial adhesins studied here refers to changes in adhesin amino acid sequence or parameters such as rate of adhesin synthesis, stability, or localization that were selected by features of gastric physiology that change during the course of infection or that are manifest in some host individuals but not others. Selection for amino acids changes at different sites is often referred to as “diversifying selection” or as “positive selection.” Diversifying selection is expected to be common for adhesins, toxins, or other proteins that may be targeted by immune responses or that interact with host cells or factors that may themselves be variable in the population or over time (as exemplified by tissue glycosylation patterns). It should be uncommon in “housekeeping” genes whose encoded proteins act only internally (e.g., catalyzing particular steps in metabolic pathways).
Within proteins subject to diversifying selection, amino acid sequences will be better conserved in some domains than in others. Conserved domains may be critical for function and/or not directly involved in host interaction or subject to immune selection. Analyses of other, less conserved domains or motifs that may sometimes involve only a few amino acids is particularly important as a novel source of insights into protein function in variable or hostile host environments, and thus into mechanisms of infection or disease. Our analyses of babA evolutionary patterns illustrate approaches that may be suitable for many proteins involved in pathogen-host interaction.
Adaptive evolution can be detected by comparing synonymous (silent; dS) and nonsynonymous (amino acid-altering; dN) substitution rates in protein-coding DNA sequences (Nei and Kumar, 2000). The ratio, dN/dS = ω measures the difference between the two rates and provides an indication of selective pressures on a protein. Evolutionary theory predicts that if an amino acid change is functionally neutral (i.e., neither improves nor diminishes protein function), it will be fixed at the same rate as a synonymous change with ω = 1. A change that is deleterious will be fixed at lower rate than a synonymous change (ω < 1), and eventually purged from the population by selection against it (termed “purifying selection”). Only when an amino acid change offers a selective advantage is it fixed at a higher rate than a synonymous mutation (ω > 1). Thus, an ω ratio significantly greater than 1 provides a robust indication of adaptive evolution (Nei and Kumar, 2000).
Estimation of dN and dS between Homologous Sequences
Methods for estimation of dN and dS between two protein-coding DNA sequences can be broadly classified into two classes
The first class are those referred to as “approximate methods,” which involve the following steps: counting synonymous (S) and nonsynonymous (NS) sites, counting synonymous and nonsynonymous differences between the two sequences, and finally correcting for multiple substitution at the same site. Most models assume that mutations occur independently and at a constant rate and do not account for differences in the rates of transition (ti) and transversion (tv) base substitution changes. Synonymous mutations are most often tis rather than tvs. Ignoring the ti/tv bias can thus lead to underestimation of S and overestimation of NS (Fay and Wu, 2003). Codon usage bias, which may result from mutational bias or selection (e.g., for translational efficiency), often has the opposite affect to that seen with ti/tv bias: ignoring codon usage bias can lead to overestimation of S, underestimation of NS, and overestimation of ω (Fay and Wu, 2003). Finally, approximate methods average ω over all amino acid sites in the sequence and over time interval separating the two sequences. Because most amino acids are conserved and adaptive evolution is episodic, these methods lack the power to detect positive selection (i.e., they do not detect diversifying selection efficiently with dN/dS = ω ≫ 1 (Fay and Wu, 2003; Nei and Kumar, 2000).
The second class is the maximum likelihood (ML) method: the ML framework provides an explicit model of codon evolution and incorporates both ti/tv rate bias and codon-usage bias. Importantly, unlike approximate methods wherein ω is averaged across the entire protein sequence, the ML framework permits codons to evolve at heterogeneous rates (i.e., different rates at different sites) (Yang, 2004). For example, a fraction of codons may be constrained with ω < 1, a fraction are neutral, ω = 1, and a fraction are under positive selection wit ω ≫ 1.
Several codon-based models of sequence evolution have been developed for detecting heterogeneous selective pressures in protein coding sequences. These differ in the number of codon classes (neutrally, negatively, and positively evolving codons) incorporated. Positive selection is inferred when models that incorporate a class of codons under positive selection fit the data better than those that do not (Yang, 2004). A protein may experience different selective pressures in different phylogenetic lineages (often equivalent to different human populations in cases of H. pylori). By constructing a phylogeny and inferring the ancestral states of a protein (i.e., reconstruction of protein sequences at internal nodes of the phylogeny), ω can be determined for each lineage of a phylogeny. Models can then be constructed to test whether ω varies significantly among lineages of a phylogeny (Yang, 2004). For example, a model assuming a single ω for all lineages can be compared with a model that permits an independent ω for each lineage (free-ratio model). Bayesian statistics can then be used to determine the probability for each codon being associated with positive, neutral, or purifying selection. Models for assessing heterogeneous selective pressures along the length of the gene or in the phylogeny are implemented in the CODEML program of PAML version 3.14 software (http://www.abacus.gene.ucl.ac.uk/software/paml.html).
Molecular Adaptation in H. pylori Adhesin Gene, babA
Analyses of receptor specificities of many H. pylori strains from different human populations had distinguished “specialist” BabA adhesins that bound the simple Leb antigen (characteristic of people of blood group O) far better than bulkier ALeb and BLeb (characteristic of blood group A and B, respectively), vs. “generalist” BabA adhesins that also bound ALeb and BLeb antigens with high affinity (Aspholm-Hurtig et al., 2004). In this study, the ML method was applied to understand the origin and maintenance of specialist and generalist babA alleles in H. pylori lineages. A central ∼268 amino acid segment of BabA contains the domain that is predicted to make contact with the cognate receptor. The babA gene sequences encoding this domain were determined from representative strains, and three specific questions were asked: (1) Are the specialist and generalist babA alleles phylogenetically distinct? (2) Is the central variable region of babA subject to variable selective pressures? (3) Do BabA variants evolve at different rates in different H. pylori lineages?
babA phylogeny reconstructed and using ML methods showed that specialist generalist alleles did not form distinct clusters, which suggested that BabA binding specificity did not depend on large sequence differences. The average ω ranged from 04– .18, implying a relaxed selective constraint. ML estimates of tree length, ω, and average dN/dS were fairly homogeneous for all models under varying initialization parameters, thereby suggesting optimal convergence of ML algorithms and negligible impact of initialization parameters on model performance. Models that allowed for positive selection (M2, M3, and M8) fit the data better than those that did not (M01, M1, and M7); M3 and M8 both suggested that nearly 9% of codons in the central variable region of babA were under positive selection. For example, M3 models variable selective pressures using a discrete (statistical) distribution allowing for three codon site classes, each associated with its own ω. M3 parameter estimates suggested that 51.7% of codon sites were highly conserved (ω0 = 0.052), that 38.7% of sites moderately conserved and nearly neutral (ω1 = 0.869), and that 9.4% of sites were under positive selection (ω2 = 3.598) (Fig. 3). ω-ratios along individual branches in the babA phylogeny were determined using the free ratios model of codon evolution. The free-ratio model assumes an independent ω for each branch in the phylogeny, and was significantly better than the null model (M0), which assumes a single ω for the entire phylogeny (−InL(free-ratio) = 18641.945, −InL(M0) = 18750.405, ω2 = 216.92, d.f. 129, p < 0.0001), thereby suggesting babA evolves rapidly in different host lineages. Together, these data provided evidence of selection for amino acid sequence change at particular sites in BabA, which would be likely to affect glycan binding pocket structure. In one view, these sequence changes might simply fine-tune BabA activity for special conditions in different host populations.
Fig. 3.
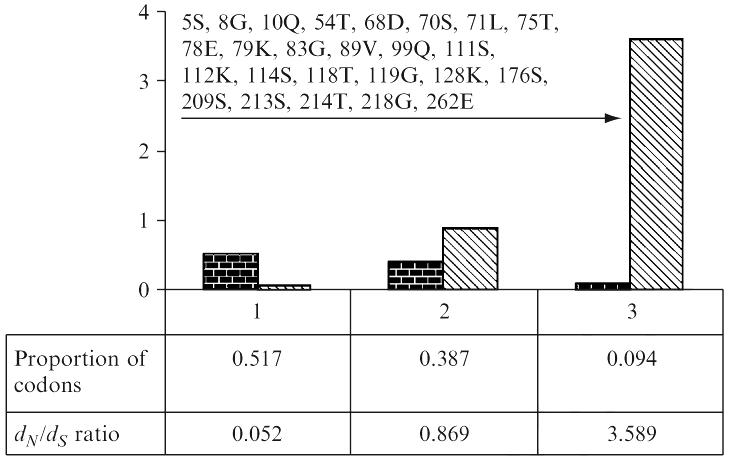
Histogram showing the frequency distribution of three codon classes and their associated dN/dS ratios computed under the M3 model. Arrow indicates positively selected codons. Codons identified to be under positive selection are shown. Alignment used this for analysis is available from GenBank [gi:49473301].
A variant model emerged in considering (1) the very similar distribution of receptor affinities of Leb-only specialists and generalists, (2) the many clinical isolates that seemed to naturally lack BabA adhesin function, and (3) a consideration that even though BabA adhesin-receptor interaction must often benefit H. pylori it may also be potentially detrimental, for example, at times or sites of intense inflammatory response. Alternating selection pressures for loss and then restitution of adherence would sometimes be satisfied by (1) an amino acid substitution that diminished BabA function, perhaps through change in the binding pocket, and (2) then “suppressor” mutations that restore binding activity through amino acid sequence elsewhere in the protein. Much of the diversifying selection at particular sites in BabA and the rapid emergence of specialist alleles of BabA uniquely in Amerindian Peruvians (peoples that are essentially all blood O) would then be ascribed to cycles of selection for mutations causing loss and restitution of function. Only restitution of Leb adherence would be selected in this population, almost devoid of blood group A or B individuals, and hence generalist binding would be lost by attrition, not direct selection for a tight binding pocket uniquely suited to the streamlined Leb structure.
Knock-out of Adhesin Gene: General Strategy for Genetic Constructions by PCR without Recombinant DNA Cloning
The ability to test the importance of particular proteins or residues in proteins by directed mutation is of immense value in analyses of many traits, bacterial adherence included. Most H. pylori strains are easily transformable and can undergo homology-based recombination between added DNAs (whether genomic, plasmid or PCR product) and corresponding chromosomal sequences. This makes it feasible to delete genes of interest and to replace one allele with another, especially if the allele is marked with a resistance determinant (e.g., ery, cat, or aph; resistances to erythromycin, chloramphenicol, or kanamycin, respectively).
Traditionally, the needed genetically marked alleles were usually made by recombinant DNA cloning of segments of interest into bacterial plasmids; digesting the recombinant plasmid with a restriction endonuclease that cleaved the cloned DNA at an appropriate site; ligating the linearized plasmid with a resistance gene-containing restriction fragment; selecting derivative plasmids containing the added resistance determinant by E. coli transformation; and further screening to identify plasmids with intended structures and orientation of the resistance determinant. Such genetic engineering, although immensely useful in many studies, remained limited, because useful restriction sites were only rarely exactly where they were needed.
PCR-based approaches freed experimentalists from limitations in distributions of useful restriction sites, and thus allowed genetic engineering with unprecedented precision because PCR primers could be designed to fix endpoints of PCR products, and thereby insertion or deletion endpoints of ensuing recombinant DNAs, at any specific template sequences of interest. PCR primers are also easily built with additional un-templated features, such as new or altered regulatory sites, as illustrated by our separation of a naturally occurring gene fusion into two component orfs, each well expressed in H. pylori (Raudonikiene et al., 1999).
Plasmid cloning can be laborious, particularly for many genes encoding membrane or secreted proteins or other factors that can be deleterious in E. coli. New PCR strategies also allow these problems to be avoided when seeking to engineer H. pylori or other transformable species. It had long been known that PCR is a discontinuous process, that DNA synthesis begun on one template can be halted, and then continued in a subsequent cycle on a different template to which the nascent strand's 3′ end may be complementary. In one application, this “crossover PCR” was used to map multiple sites of transposon insertion in a gene (Krishan et al., 1991).
Chalker et al. (2001), adapted this principle to the high throughput construction of deletion alleles of 42 H. pylori genes of to them, essentially as diagrammed in Fig. 4. Primers were developed (see list of primers below, primer number is indicated in bold) to amplify H. pylori DNA segments that were intended to flank each deletion (primers 1 and 2 for fragment A, and primers 5 and 6 for fragment C, in the nomenclature in this figure) and also for a resistance cassette (primers 3 and 4 for fragment B; see figure). The inside primers for the flanking segments (primers 2 and 5) were designed to allow crossover PCR: added on to the H. pylori specific sequences in their 3′ halves, were 5′ extensions that were complementary to primers 3 and 4 used for amplification of the resistance cassette (indicated by downward angled tails on primers 2 and 5 in Fig. 4). A second round of PCR amplification using a mixture of the left and right flanking DNAs (A, C) plus the resistance cassette (B) with just the exterior primers 1 and 6 usually results in the desired recombinant DNA fragment (“usual route” in Fig. 4).
Fig. 4.
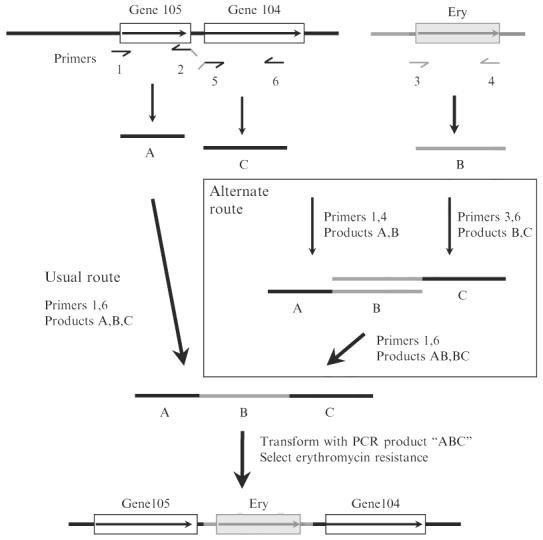
Construction of insertion deletion allele marked with a resistance determinant by PCR without need for recombinant DNA cloning, essentially as described (Chalker et al., 2001). In general, such constructions entail amplification of DNA segments to left and right of the intended site of insertion/deletion (products A and C, with primers 1 and 2 and with primers 5 and 6, respectively), and also a selectable resistance determinant (here resistance to erythromycin; designated fragment B with primers 3 and 4). The 5′ ends of primers 2 and 5 (denoted as downward slanted lines) are complementary to the primers used for amplification of the resistance determinant (fragment B). After mixing of separately amplified fragments A, B, and C, amplification with primers 1 and 6 generally results in an assembled PCR product containing the desired insertion/deletion allele as diagrammed at bottom, on the basis of “crossover PCR” at sites of overlap because of complementarity of 5′ ends of primers 2 and 3 and of primers 4 and 5. In some cases this simple three-fragment assembly fails as illustrated in Fig. 5 (gel lane marked ?? at bottom). In such cases, an alternate strategy (boxed) that entails separate two fragment assemblies of A and B, and of B and C, and then a final assembly of subassembly AB and BC often gives the desired product (rightmost experimental lane marked ABC at bottom). Gene105 and gene104 are arbitrary designations for orfs in the H. pylori genome. For detailed design of primer sequences for use with erythromycin and other resistance markers, see Tan and Berg (2004) and Tan et al. (2005).
It is important to recognize that this simple strategy for three-fragment assembly sometimes fails (an outcome that is easily scored by gel electrophoresis and absence of product of expected size) and that such failures can be reproducible. One such case is illustrated in Fig. 5 (the lane marked “??”). The mechanisms underlying such failures have not been examined but likely relate to distributions of alternative potential primer binding sites and/or secondary structures in DNAs to be amplified. More important, in our experience, such difficulty can be circumvented by first making two fragment subassemblies (AB with primers 1 and 4; and BC with primers 3 and 6), and then using these two subassemblies to make the final assembly, as diagrammed in Fig. 4, and with gel data in Fig. 5.
Fig. 5.

Agarose gel electrophoresis of products from construction of insertion/deletion allele diagrammed in Fig. 4. Primers 1 through 6 and simple PCR products A, B, C, and assembled products AB, BC, and ABC all correspond to those diagrammed in Fig. 4. Input DNA “gen” indicates genomic DNA. The product designated ?? is a PCR-generated deletion variant of the desired ABC assembly that came to predominate in the PCR product population because of its smaller size and, thus, replication advantage. It has not been characterized further. M indicates marker DNA, with sizes of characteristic fragments indicated at right.
The PCR products generated by either of these assembly routes are well suited for transformation of H. pylori. In our experience, all selected transformants contain the desired mutant allele in place of the original wild-type allele, unless the gene in question is essential for viability (Chalker et al., 2001; Tan and Berg, 2004; Tan et al., 2005).
PCR Condition for Initial DNA Segments (A, B, C) Amplification
Composition of PCR reaction mix: Template DNA (SS1 or X47 genomic DNA/pRH151 DNA) 1 μl, primer, forward (10 pmole) 1 μl, primer, reverse (10 pmole) 1 μl, 10× NH4 reaction buffer 5 μl, dNTP (2.5 mM each) 5 μl, MgCl2 (50 mM)1 μl, Biolase DNA polymerase (5 u/μl, Bioline, Germany) 0.4 μl, DW fill up to 50 μl (35.6 μl)
Cycling condition: 94°, 40 sec, 55°, 40 sec (50°, 40 sec for amplification of segment B, ermM), 72°, 1 min (30 cycles)
PCR Condition for DNA Fragment Assembly
Composition of PCR reaction mix: Template DNA (purified segments, 1 μl each) 2 or 3 μl. Primer, forward (10 pmole) 1 μl, primer, reverse (10 pmole) 1 μl, 10 μl NH4 reaction buffer 5 μl, dNTP (2.5 mM each) 5 μl, MgCl2 (50 mM) 1 μl, Biolase DNA polymerase (5 u/μl, Bioline, Germany) 0.4 μl, DW fill up to 50 μl (33.6 or 34.6 μl)
- Cycling condition
- Three fragments assembly (A + B + C): 94°, 40 sec, 55°, 40 sec (or 50°, 40 sec), 72°, 2 min 30 sec (25 cycles)
- Two fragments assembly (A + B or B + C) 94°, 40 sec, 55°, 40 sec, 72°, 2 min (30 cycles)
- Final assembly (AB + BC): 94°, 40 sec, 55°, 40 sec, 72°, 2 min 30 sec (25 cycles)
*Agarose gel: 1.2% (with 1-kb ladder DNA marker)
Primers (numbers in parenthesis refer to primers used in Figs. 4 and 5)
(6.) CF2 22mer 5′-ATCCTTGTCAAGCCGTTATTGG
(1.) CR2 23mer 5′-GGAGTTAAAAACATGAAAACACC
(2.) CF3 46mer5′-TACTGCAATCTGATGCGATTATTGTCAAACCCCCACTTCAGACCAC
(5.) CR3 52mer 5′-TTCAATAGCTATAAATTATTTAATAAGTAAGTGGTCTGAAGTGGGGGTTTGA
(3.) ermMF 24mer 5′-CAATAATCGCATCAGATTGCAGTA
(4.) ermMR 30mer 5′-TTACTTATTAAATAATTTATAGCTATTGAA
Identification of Bacterial Clones with Rare and/or Induced Receptor Binding Properties by Use of Glycoprobes
H. pylori is extremely diverse genetically both because of mutation and recombination, which can operate between different strains or between duplicate and divergent genes within a given lineage (Kersulyte et al., 1999; Suerbaum et al., 1998). Some of H. pylori's genes are particularly prone to genetic change, resulting in metastability or heterogeneity in bacterial phenotypes. For H. pylori, the genetic flexibility provides the prerequisites to adapt to certain environmental changes. Included among metastable genes are those for BabA and SabA adhesins, leading to ideas that an ability to cycle between adherent and nonadherent phenotypes contributes importantly to persistent H. pylori infection. FITC-labeled bacteria and semisynthetic glycoconjugates (i.e., glycoprobes) were used to study metastability in H. pylori Leb antigen binding phenotype (Bäckström et al., 2004). As one indication of metastability, individual cells proficient in binding were visualized by fluorescence microscopy of pure cultures of strains that had been found to have a nonbinding phenotype by RIA, albeit often at frequencies of only 1/50,000 bacterial cells or fewer “Bio-panning” was used to isolate rare clones with adherent phenotypes from these cultures (Aspholm-Hurtig et al., 2004; Bäckström et al., 2004). To accomplish this, bacterial cells were mixed with biotinylated receptor conjugate, and those proficient in binding were enriched for by use of streptavidin-coated magnetic beads. Individual clones that express adhesin protein and readily bind the receptor conjugate were then identified by membrane colony screening technique; biotinylated receptor conjugate was added to the membranes, and colonies that had gained proficiency in Leb antigen binding were detected with streptavidin staining.
Biotinylation of Receptor Conjugate
Materials
Biotin-XX-NHS (Calbiochem, San Diego, CA)
PD10 (Sephadex G-25) column (GE Healthcare Bio-sciences AB, Sweden)
Solutions
Dimethylsulfoxide (DMSO)
Carbonate buffer: 0.15 M NaHCO3, pH 9.0
1 M Tris-HCl, pH 9.0
PBS (phosphate-buffered saline): 150 mM NaCl, 10 mM phosphate, pH 7.6
Blocking buffer: PBS + 0.5% Tween-20 + 1% BSA
Elution buffer: PBS + 0.05% Tween-20 + 0.2 M Tris-HCl, pH 9.0
One milligram Biotin-XX-NHS (Calbiochem, San Diego, CA) dissolved in 50 μl dimethylsulfoxide (DMSO) is mixed with 10 μg of glyco-conjugate (for example, Leb-HSA, Isosep, Tullinge, Sweden) dissolved in carbonate buffer in a total volume of 1 ml. The sample is incubated with end-over-end mixing for 3 h at 23°; 260 μl 1 M Tris-HCl, pH 9.0, is added followed by further incubation for 30 min. Biotinylated receptor conjugate is purified on a column. Before addition of the sample, the PD10 column is washed with PBS + 0.05% Tween-20, blocked with blocking buffer for 30 min and washed with elution buffer. Samples are eluted with elution buffer and collected in aliquots of 0.5 ml. The void fraction, approximately 1.5 ml, is pooled, and the gel filtration is repeated once more to further purify the labeled Leb conjugate from the molar excess of biotin-XX-reagent.
FITC-Labeling of Bacterial Cells
Materials
FITC (Fluorescein isothiocyanate), (Sigma, St. Louis, MO)
Solutions
PBS (phosphate-buffered saline)
0.2 M NaHCO3
Washing buffer: PBS + 0.05% Tween-20
Blocking buffer: PBS + 0.5% Tween-20 + 1% BSA
Bacteria were grown for 40–45 h on Brucella agar medium supplemented with 10% bovine blood and 1% Iso Vitox (Svenska LABFAB, Ljusne, Sweden) at 37°, under 10% CO2 and 5% O2. A1-ml bacterial sample (A600 1.0) is washed in washing buffer, resuspended in 1 ml 0.2 M NaHCO3 and incubated for 30 min at 23°; 0.1 mg FITC, dissolved in 15 μl DMSO is added to the bacterial mixture and incubated end-over-end for 8 min. Samples are washed 4 × 8 min (or until the supernatant is clear) in washing buffer and finally resuspended in blocking buffer such that a cell density of A600 0.2 is achieved. Use the fluorolabeled bacterial cells directly or store them in aliquots at −20°. To get rid of lysed cells after storage, thawed samples are washed in blocking buffer followed by adjustment of cell density to A600 0.2.
Fluorescence Microscopy to Identify Clones of Rare Receptor Binding Phenotype
For fluorescence microscopy, a 1-ml sample of FITC-labeled bacteria (A600 0.2) is incubated with blocking buffer for 1 h, incubated with 2 μg of biotinylated receptor conjugate for 2 h at 23°, washed by centrifugation, resuspended in blocking buffer, and incubated with Cy-3-streptavidin (1:10.000) (Sigma) for 1 h at 23°. Samples are washed in washing buffer and resuspended in 200 μl blocking buffer. A 1–10-μl sample is analyzed by fluorescence microscopy (magnification ×400). The total number of cells in the sample is analyzed by counting cells in a Petroff-Hausser chamber (CA Hausser and Son, Philadelphia, PA) or similar.
Bio-panning for Clones with Certain Receptor Binding Phenotypes
A2-ml (A600 = 0.5) bacterial suspension is incubated with 10 μg of biotinylated receptor conjugate for 2 h at 37° in growth medium on a slow shaker. The bacterial-conjugate mixture is washed at least twice in growth medium, mixed with 100 μg of streptavidin magnetic beads (Qiagene, Inc. Hilden, Germany) for 2 h at 37° or other relevant growth conditions. Bacteria bound to the beads are recovered with a magnet and cultured on agar medium. Progeny bacterial cells can be used for additional rounds of enrichment.
Colony Blotting with Glycoprobes
Materials
Nitrocellulose membranes
Streptavidin-peroxidase (Roche, Stockholm, Sweden)
4-chloro-1-naphtol tablets (Sigma, MO)
Solutions
Washing buffer PBS + 0.05% Tween-20
TBS buffer: 50 mM Tris, pH 7.4, 150 mM NaCl + 0.05% Tween-20
Buffer I: TBS + 1.5% gelatin + 1% BSA
Bacteria are spread at a density of approximately 500 CFU per plate, incubated at appropriate growth conditions for single cell colonies. Bacteria are transferred to nitrocellulose membranes. Membranes must be organized such that positive clones can be identified on the master plate. Membranes are baked for 1 h at 70°, soaked and washed repeatedly in washing buffer, and blocked in buffer 1 overnight at 4°. The following day, 2 μg of biotinylated receptor conjugate in 5 ml buffer 1 is added to membranes, and the membranes are incubated for 2 h at 23°, washed in TBS buffer three × 10 min and thereafter incubated with streptavidin-peroxidase (Roche, Stockholm) (1:5.000) in buffer 1 for 1 h at 23°. Finally, membranes are washed in TBS buffer 3 × 10 min and exposed to 4-chloro-1-naphtol as recommended by the manufacturer (Sigma, MO). Receptor binding colonies are identified by blue color.
Characterization of Spatial Expression of H. pylori Virulence Products in Gastric Mucosa by In Situ Hybridization
Introduction
Transcripts from specific bacterial genes made during infection can be detected, localized, and quantified by in situ hybridization and a point-counting stereological method (Semino-Mora et al., 2003). This method is exquisitely sensitive and specific and is suitable for archived formalin-fixed specimens and biopsies. The specificity of the method could be further increased by use of multiprobe fluorescence in situ hybridization (FISH).
Methods
Biopsies
Biopsies are fixed in Z-fix (10% paraformaldehyde + 1% ionized Zn, Anatech LTD, Battle Creek, MI) within 1 min of harvesting, then dehydrated, and embedded in paraffin within the next 2 days. This timing is necessary to minimize risks of RNA degradation. Serial 5-μm sections are stained with hematoxylin–eosin or Genta stain (Genta et al., 1994) for grading of gastritis and atrophy according to the Sydney system (Dixon et al., 1997) (0 = none, 1 = mild, 2 = moderate, and 3 = marked). Additional serial sections are prepared for in situ hybridization as described later.
Oligonucleotide PROBES for H. pylori
Specific RNA and cDNA probes were designed using a sequence database for H. pylori 16S rRNA (Semino-Mora et al., 2003) or were as published in the case of cagA (Tummuru et al., 1993) and babA (Dubois et al., 2006).The 5′ end of the oligonucleotides is labeled with either biotin or digoxigenin-3-0-methylcarbonyl-e-aminocaproic acid-N-hydroxy-succinimide ester (DIG-NHS ester) (Roche Diagnostics, Indianapolis, IN). In initial experiments, it is important to use both RNA and cDNA probes concurrently and to confirm that the two probes can detect the same structures. In addition, it is critical to demonstrate that detection is abolished by RNase but not DNase (to demonstrate that these probes specifically target mRNA). Having validated the cDNA antisense probes in one's system, RNase and DNase should be included as controls in each run. Once the method is specific, the use of RNA probes can be discontinued, especially because they are more susceptible to degradation and more delicate and costlier to prepare.
In Situ Hybridization. All procedures are performed as described in the following for paraffin-embedded tissues (Semino-Mora et al., 2003; Wilkinson, 1998) and at RT except as specified:
Section pretreatment: Sections are first deparaffinized in xylene, rehydrated by a series of washes in graded ethanol (100%, 95%, 80%, 70%, and 50%), and finally washed in DEPC-treated water and PBS. To improve subsequent probe penetration, sections are treated with proteinase K (Roche Diagnostic, Indianapolis, IN; 10 μg/ml in 100 mM Tris-HCl buffer, pH 8.0, containing 5 mM EDTA) for 10 min, and then washed twice for 5 min each with 1 × standard saline citrate solution (SSC, Quality Biological, Inc, Gaithersburg, MD).
Pre-hybridization: Sections are covered for 5 min with hybridization mixture prepared as follows: 200 μl Denhardts solution (2% bovine serum albumin [BSA], 2% Ficoll, and 2% polyvinylpyrrolidone [PVP] in water), 5 ml formamide (Roche Diagnostic, Indianapolis, IN), 600 μl 5 M NaCl, 2 ml dextran sulfate (0.8 g/8 ml) heated at 65° for 20 min, 20 μl EDTA (0.29 g/10 ml), 2.5 ml hybridization solution (phenol-chloroform extracted salmon testis DNA and SSC), 20 μl 1.0 M Tris-HCl buffer, pH 7.5, and 660 μl DEPC-treated water). Sections are then covered for 20 min with blocking solution (0.2 g BSA fraction V, 0.5 ml normal horse serum [Jackson ImmunoResearch, Inc.]), and 10 ml of immuno-Tris buffer (50 ml of 1 M Tris-HCl buffer, pH 7.5, and 25 ml of 5 M NaCl in 1000 ml water, pH 7.5).
Hybridization: The probes listed in “DNA probes used” are denatured by heating for 10 min at 65° and subsequent chilling on ice for 3 min. Each unstained section is layered with 70–100 μl of the probe solution (4 pmol/μl), cover-slipped, and placed horizontally on filter paper soaked with water in an airtight box wrapped with aluminum foil. Separate boxes are used for different probes and for negative controls. Each box is incubated overnight at 37°.
Post-hybridization: After 18 h of incubation, the unbound probe is removed by successive washes in decreasing concentrations of SSC (2 × SSC 30 min, 1 × SSC 10 min, 0.5 × SSC 10 min at RT, and 0.1 × SSC 15 min at 60°).
Detection of H. pylori Gene Expression
Bright-field ISH: Anti-digoxigenin antibody-conjugated or streptavidin-conjugated alkaline phosphatase (AP; Roche Diagnostic, Indianapolis, IN) is used to detect the digoxigenin-labeled (cagA) or the biotin-labeled (16S rRNA or babA2) probe, respectively (see, DNA probes used). Sections are incubated for 2 h with anti-digoxigenin-AP or streptavidin-AP respectively, 1:500 dilution in blocking solution. Unbound antibody-alkaline phosphatase is washed gently with immuno-Tris buffer (3 times, 5 min each), using a different Coplin jar for each probe and negative controls. Bound alkaline phosphatase is detected by covering the slides with a chromogenic substrate (5-bromo-4-chloro-3indolyl phosphate/nitroblue tetrazolium BCIP/NBT kit, Vector Labs, Burlingame, CA) and keeping them in the dark for 20 min. After washing in distilled water for 3 min, slides are counterstained with Nuclear Fast Red (Vector, Burlingame, CA) for 15 min, washed in water, dehydrated, cleared in xylene, briefly air dried, and mounted with Permount (Biomedia Corporation, Foster City, CA).
Fluorescence ISH (FISH) dual localization (Jaju et al., 1999; Semino-Mora et al., 2003; Wilkinson, 1998): The hybridization mixture contains both a biotin-labeled probe recognizing H. pylori 16S rRNA (Semino-Mora et al., 2003) and a digoxigenin-labeled probe recognizing cagA (see, DNA probes used). After incubation as described previously, the slides are covered with the following three layers: first layer: 1 μl avidin-Texas red + 1.5 μl mouse monoclonal anti-digoxigenin; second layer: 10 μl biotin-anti-avidin + 1 μl rabbit anti-mouse-IgG-fluorescein isothiocyanate (FITC) conjugated; third layer: 1 μl avidin-Texas red + 10 μl monoclonal anti-rabbit-FITC conjugated. All antibodies are prepared in 1 ml of blocking solution. Slides are incubated with each antibody layer for 30 min at 37°, in a humid chamber, and protected from the light and washed 3 times 5 min each. After air-drying, the sections are mounted in Vectashield (Vector Labs, Burlingame, CA).
Controls
H. pylori pure cultures are streaked onto precleaned microscope slides and used as positive controls. E. coli and S. flexneri cultures and gastric biopsies from a 62-year-old H. pylori–negative patient with dyspepsia are used as negative controls. Control for nonspecific binding is performed by using: (1) sense instead of antisense probe; (2) hybridization buffer instead of antisense probe; (3) unlabeled antisense probe; (4) digoxigenin or biotin-labeled probe for scorpion Buthus martensi Karsch neurotoxin sequence [5′-GGC CAC GCG TCG ACT AGT AC-3′] (Lan et al., 1999); (5) RNase A pretreatment (Roche); (6) DNase I pretreatment (Roche); and (7) RNase + DNase I pretreatment.
DNA probes used (RNA probes are identical except for substitution of U for T):
| 16S rRNA (Semino-Mora et al., 2003) | antisense: 5′-TACCTC TCC CACACTCTAG AATAGTAGTTTCAAATGC-3′ |
| sense: 5′-CTA TGA CGG GTA TCC GGC-3′ | |
| cagA (Tummuru et al., 1993) | antisense: 5′-CTG CAA AAG ATT GTT TGG CAG A-3′ |
| sense: 5′ GAT AAC AGG CAA GCT TTT GAG G-3′ |
Application of Laser Tweezers Technique for Analyzes of Binding Strength
Optical tweezers (OT) is a laser-based tool for nonintrusive manipulation of particles on the micrometer scale, including living cells and bacteria. It consists basically of a microscope objective with high numerical aperture in which a continuous laser beam is focused. A small object, like a micrometer-sized bead with a refractive index larger than that of the surrounding medium, experiences a restoring force in all spatial directions in the focal region, thus becoming trapped in the laser beam focus. The OT-technique is being used as a sensitive way of measuring weak molecular interactions in biological systems (forces typically in the lower picoNewton range) (Ashkin, 1992; Ashkin, et al.,1990; Simmons et al., 1996). The object under study (e.g., a bacterial cell) is approached and allowed to bind to a bead trapped in the focus. By slowly retracting the cell from the trapped bead, a force is exerted on the system, and the position of the bead is shifted. The shift of the bead in the trap constitutes a measure of the exerted force. We recently developed an assay for in situ force measurements of specific interactions between the H. pylori BabA adhesins and the blood group antigen Lewis b (Leb) presented in solid (surface) phase (Björnham et al., 2005). Moreover, physical properties of E.coli P pili, associated with stretching and contracting of the pilus structure, were experimentally assessed by OT force measurements. Three characteristic elongation regions were identified while loading the pili under constant rate (Jass et al., 2004). It was found that pili extension is fully elastic, because the pilus structure exhibited equivalent reciprocal forces during unloading and loading (Fällman et al., 2005). In the following, a short review is presented of recent OT experiments for probing the H. pylori BabA-Leb interaction.
Materials and Methods
Instrumentation
The optical tweezers system is constructed around an inverted microscope (Olympus IX70) with a high numerical aperture oil-immersion objective (Zeiss CP-Achromat 100× N.A. 1.25). argonion laser–pumped continuous-wave titanium-sapphire laser, operating at a wavelength of 810 nm, is used for optical trapping. A complete description of the optical system for controlling the laser beam that forms the optical trap and modifications made to the microscope to realize the OT system is reported in Fällman and Axner (1997) and Fällman et al. (2004). The displacement of a small bead held in the optical trap constitutes a measure of the force that is exerted on the bead. To monitor the displacement, a weak probe laser beam (HeNe) is conveyed into the microscope. Figure 6 illustrates the optical trap and the position detection principle. The probe beam is focused a short distance below the trapped bead, whereby the trapped bead acts as a lens and refracts the beam, in such a way that a well-defined light spot is produced in the far field. A position sensitive detector (PSD) is used to monitor the lateral position of this spot. If an object (e.g., a bacterial cell) bound to the trapped bead is slowly moved (in the horizontal direction), the trapped bead will be shifted a short distance from the focal point, resulting in a shifted position of the probe laser spot on the PSD. The output voltage signal of the PSD is directly proportional to the distance the trapped bead is shifted. A calibration procedure based on the Brownian motion of the trapped bead in the liquid is used to transform the position data to force data in units of pN (Fällman et al., 2004).
FIG. 6.
Position detection of a bead confined in the optical trap. T, trapping laser beam (Ti-sapphire); P, probe laser beam (HeNe); PSD, position sensitive detector.
Biological Model System
The model system for probing the Leb-BabA interaction contained the following components suspended in phosphate-buffered saline (PBS): H. pylori cells, a functionalized large bead that served as immobilization tool for the bacterium and a small Leb-coated bead that served both as a surface hosting the receptors and as a force indicator for the OT. The H. pylori used were the 17875Leb strain and the 17875babA1::kanbabA2::cam (denoted the BabA-mutant) (Mahdavi et al., 2002). Bacteria were grown for 40–45 h on Brucella agar medium supplemented with 10% bovine blood and 1% Iso Vitox (Becton Dickinson, US) at 37°, under 10% CO2 and 5% O2, and suspended in a density of ∼50. 106 cells per ml. The large functionalized polystyrene beads with carboxyl surface groups (Interfacial Dynamics Corp, USA) had a diameter of 9.6 μm and were immobilized on the coverslip. The carboxyl surface group beads were activated to allow covalent attachment of bacteria by use of a two-step protocol (Staros et al., 1986). The small carboxyl polystyrene beads (Interfacial Dynamics Corp, USA), with a diameter of 3.2 μm, were coated with Leb HSA (human serum albumin) conjugate (IsoSep AB, Tullinge, Sweden) (Björnham et al., 2005). The sample mixture consisted of a ∼1.106 bacterial cells, large beads with activated surface carboxyl groups (∼500.106ml), all suspended in a droplet of ∼25μl PBS. The sample mixture was placed in a sealed chamber, which was mounted on a piezo stage (Physik Instrumente, GmbH) that allowed precise translation of the sample at a controlled speed (0.5 μm/s) relative to the microscope objective.
Measurement Procedure
A typical force measurement is illustrated in Fig. 7. A free-floating individual bacterial cell was first trapped by the OT, approached to the large bead, and allowed to bind covalently to the surface. Next, the large bead with the bacterium was brought nearer to the small Leb bead held in the optical trap. The initial bacterium-bead contact allowed specific bind- ing between the BabA-adhesins and the Leb-receptors. After ∼5 sec, the bacterium was retracted from the Leb bead by a moving the piezo stage at constant rate (0.5 μm/s) (Fig. 7A). The force in the system increased until the binding suddenly breached (Fig. 7B), whereby the Leb bead returned instantaneously to the center of the trap. The last position of the Leb bead before breakage is thus a measure of the maximal binding force.
Fig. 7.
Illustration of the measurement procedure. (A) The large bead with the bacterium pulls the small Leb-coated bead away from the center of the optical trap. (B) The force in the system increases until the binding breaks, whereby the Leb bead returns to the center.
Conclusions
A typical force-versus-distance graph from a measurement on an individual bacterial cell of the 17875Leb strain is displayed in Fig. 8A. The graph can be seen as consisting of two characteristic regions. The first region consists of a linear force-versus-distance dependence, which demonstrates adhesion and originates from the stress response of the biological system under elongation. The negative force data at the beginning demonstrate an initial contact between the bacterial cell and the Leb bead. The force increases with elongation until the binding breaks, whereby the Leb bead detaches from the bacterium and returns instantaneously to the center of the optical trap. The second part of the data, which constitutes of a zero force region, indicates thereby that the binding has ruptured. The distribution of rupture forces from in total 61 measurements (performed in eight series) on the 17875Leb strain is displayed in the form of a histogram (Fig. 8B). The histogram intervals are chosen as multiples of 12.5 pN, with a width of ±6.25 pN. Apparently, most measurements (46 measurements of 61) gave rise to rupture forces that appear in the even intervals marked with filled bars, which occurs at multiples of 25 pN. A substantially smaller fraction of all measurements (15 measurements) gave rise to forces that appear in the odd intervals (marked with unfilled bars). Thus, a pronounced high-low staggering distribution of the histogram is apparent. Such a distribution of forces suggests the existence of an elemental force in the system, FLeb. The even intervals represent, thereby, multiple bindings with a force of NFLeb, with N being an integer, whereas the odd intervals correspond to intermediate force intervals, centered around (N − 0.5)FLeb. It was found that the strongest high-low staggering appearance occurred at an elemental force of FLeb = 25 pN. Thus, the measurement data suggest a rupture force of a single BabA-Leb binding that amounts to ∼25 pN.
Fig. 8.
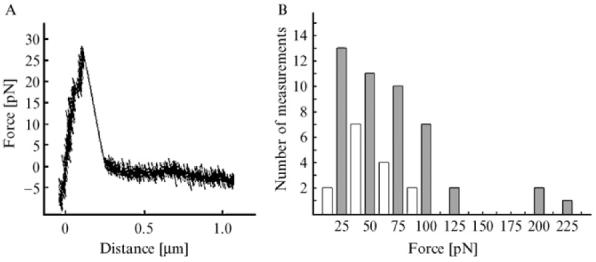
(A) Typical force-versus-distance graph from a measurement on a single bacterium of the 17875Leb strain. The force increases linearly force until the binding breaks. (B) Histogram of the distribution of rupture forces from in total 61 measurements on the 17875Leb strain. The intervals are centered at multiples of 12.5 pN with a width of ±6.25 pN. Most of the rupture forces appear in the even intervals (marked with filled bars). This suggests the existence of an elemental force in the system of ∼25 pN, which is interpreted as the rupture force of a single BabA-Leb binding.
Negative control measurements on the BabA-mutant strain were performed to test the inferred specific BabA-Leb binding. It was found that the rupture force of BabA-mutants was 75% lower than that of 17875Leb. This difference showed specific BabA-Leb binding, in agreement with previous findings (Borén et al., 1993). Also, the data from the BabA-mutant do not exhibit a high-low staggering feature when plotted in a histogram, which, in contrast to the 17875Leb, suggests the absence of an elemental force in the control system.
Further investigations will include assessment of the rupture force under different pH conditions (acidic compared with neutral), different loading rates, and influence of shear forces (shear flow), which all are assumed to be important factors for a general characterization of the adhesion properties of H. pylori.
Analyses of Nonopsonic Bacterial–Cell Interactions by Chemiluminescence Measurements of Released Oxidative Burst Metabolites
Role of Adhesins in Activation of Human Neutrophils
Certain H. pylori strains have a nonopsonic (i.e., nonantibody mediated) neutrophil activating capacity and are found significantly more in patients with peptic ulcer disease than gastritis only (Rautelin et al., 1993). To test whether this H. pylori–induced neutrophil activation involves lectino-phagocytosis, H. pylori strains were incubated with oligosac- charides, and effects on the oxidative burst of subsequently challenged neutrophils were measured by luminol-enhanced chemiluminescence (CL) and flow cytometry (FC) (Teneberg et al., 2000). By both methods a reduced response was obtained by incubation of H. pylori with sialic acid–terminated oligosaccharides, whereas lactose had no effect. This suggested that this H. pylori–induced neutrophil activation entails bacterial adhesin recognition of sialylated glycoconjugates on neutrophil cell surfaces, leading to phagocytosis and the oxidative burst reaction.
After identification of SabA and characterization of sialyl-dimeric-Lex as a high-affinity receptor (Mahdavi et al., 2002), variant gangliosides structurally related to the sialyl-dimeric-Lex nonaglycosylceramide were tested in binding studies (Roche et al., 2004). SabA also bound sialylα3-neolacto-tetraosyl-ceramide, sialylα3-neolacto-hexaosyl-ceramide, sialylα3- neolacto-octaosyl-ceramide, the VIM-2 ganglioside, and sialyl-dimeric- Lex glycosphingolipids, complex gangliosides also present in human neutrophils (Müthing et al., 1996; Stroud et al. 1996a,b). This led to investigation of SabA's contribution to human neutrophil activation (Unemo et al., 2005). Wild-type and mutant strains deficient of SabA had no neutrophil-activating capacity, showing that H. pylori binding to sialylated receptors on neutrophils contributes to bacterial adherence and phagocytosis.
Methodological Considerations
CL measures both activation of plasma membrane NADPH oxidase (usually referred to as the external oxidative burst) and intracellular NADPH oxidase (internal oxidative burst). The external burst can be checked by quenching (and chemical elimination) with catalase, and the internal by quenching with horseradish peroxidase and azide or by use of isoluminol instead of luminol (Dahlgren and Karlsson, 1999; Dahlgren and Lock, 1988). Flow cytometry of hydroethidine-loaded neutrophils measures only the internal burst (Danielsson and Jurstrand, 1998; Teneberg et al., 2000). A biphasic response is usually obtained when H. pylori– induced neutrophil activation is measured by luminol enhanced CL. Most H. pylori strains induce a strong initial phase within the first minutes, mostly because of activation of the plasma membrane NADPH oxidase. The second phase, which develops in the following 5–30 min, represents activation of both plasma membrane and intracellular NADPH oxidases.
There is variation among strains that express SabA in the ability to induce the oxidative burst manifested by differences in peak values (mV), time to reach peak (min), and absence of a recognizable biphasic response. Luminol-enhanced CL offers some advantages in this respect, because it can be used to measure kinetics, as well as detecting external and internal oxidative bursts.
Isolation of Human Neutrophil Granulocytes
Neutrophils are prepared from heparinized blood from healthy blood donors by Ficoll-Paque (Amersham Biosciences Ab, Uppsala, Sweden) centrifugation as described (Böyum, 1974), with slight modifications (Rautelin et al., 1993). For each series of experiments on a particular day neutrophils were prepared and pooled from three blood donors of the same blood group (A rh+ or O rh+). Neutrophils were thus obtained from different blood donors at each experiment. They were suspended in PBS supplemented with MgCl2,CaCl2, glucose, and gelatin (PBS-GG) (Rautelin et al., 1993). The purity and viability of the neutrophils exceeded 95%.
Culture Conditions
The H. pylori strains were grown in a microaerophilic atmosphere at 37° on GC agar plates (GC II agar base; BBL, Cockeysville, MD) supplemented with 1% bovine hemoglobin (BBL), 10% horse serum, and 1% IsoVitaleX enrichment (BBL), without antibiotics for the wild-type strains, and with appropriate antibiotics for mutant strains. After 48 h, bacteria were collected in PBS and used in CL experiments.
Chemiluminescence (CL) Experiments
The oxidative burst of neutrophils challenged with nonopsonized whole and live H. pylori organisms was measured with luminol-enhanced CL as previously described (Rautelin et al., 1993). To each test tube (LKB, Bromma, Sweden) was added 300 μl of PBS supplemented with MgCl2 and CaCl2, 100 μl of neutrophils (5 × 106/ml), 50 μl of 10−5 M luminol (Sigma, St. Louis, MO), and finally 50 μl of nonopsonized H. pylori (5 × 108/ml).
For oligosaccharide inhibition experiments, 50 μl of H. pylori (5 × 108/ml) were mixed with 50 μl of oligosaccharides at final concentrations of 0.1–1.0 mM in the CL, for 15 min at 37°, and 100 μl of the mixture was transferred to a test tube for CL measurement.
The oxidative bursts of the neutrophils were measured as luminol-enhanced CL with a luminometer (LKB Wallac 1251, Turku, Finland), starting within 1 min of adding the bacterial suspension. The assays were performed at 37°, and CL from each sample was measured at 60–90-sec intervals during a period of 30–60 min. Data were stored in a computer for computerized calculations. Strain H. pylori NCTC 11637 (sabA+: provides a strong and rapid CL response) and C-7050 (sabA−: inducing no CL response) (Rautelin et al., 1993) were included in each series of experiments as positive and negative controls, respectively.
References
- Ashkin A, Schutze K, Dziedzic JM, Euteneuer U, Schliwa M. Force generation of organelle transport measured in vivo by an infrared laser trap. Nature. 1990;348:346–348. doi: 10.1038/348346a0. [DOI] [PubMed] [Google Scholar]
- Ashkin A. Forces of a single-beam gradient laser trap on a dielectric sphere in the ray optics regime. Biophys. J. 1992;61:569–582. doi: 10.1016/S0006-3495(92)81860-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikström S, Sjöström R, Linden S, Bäckström A, Lundberg C, Arnqvist A, Mahdavi J, Nilsson UJ, Velapatino B, Gilman RH, Gerhard M, Alarcon T, Lopez-Brea M, Nakazawa T, Fox JG, Correa P, Dominguez-Bello MG, Perez-Perez GI, Blaser MJ, Normark S, Carlstedt I, Oscarson S, Teneberg S, Berg DE, Borén T. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- Bäckström A, Lundberg C, Kersulyte D, Berg DE, Borén T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc. Natl. Acad. Sci. USA. 2004;101:16923–16928. doi: 10.1073/pnas.0404817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnham O, Fällman E, Axner O, Nilsson U, Borén T, Schedin S. Measurements of the binding force between the Helicobacter pylori adhesin BabA and the Lewis b blood group antigen using optical tweezers. J. Biomedical Optics. 2005;10:044024–0444024-9. doi: 10.1117/1.1989227. [DOI] [PubMed] [Google Scholar]
- Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- Borén T, Wadström T, Normark S, Gordon JI, Falk PG. Methods for the Identification of H. pylori Host Receptors. In: Clayton CL, Mobley HLT, editors. “Helicobacter pylori Protocols”. Humana Press Inc; Totowa, NJ: 1997. pp. 205–224. Chapter 8. [DOI] [PubMed] [Google Scholar]
- Böyum A. Separation of blood leukocytes, granulocytes and lymphocytes. Tissue Antigens. 1974;4:269–274. [PubMed] [Google Scholar]
- Chalker AF, Minehart HW, Hughes NJ, Koretke KK, Lonetto MA, Brinkman KK, Warren PV, Lupas A, Stanhope MJ, Brown JR, Hoffman PS. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 2001;183:1259–1268. doi: 10.1128/JB.183.4.1259-1268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren C, Lock R. The limitation of the human neutrophil chemiluminescence response by extracellular peroxidase is stimulus dependent: Effect of added horseradish peroxidase on the response induced by both soluble and particulate stimuli. J. Clin. Lab. Immunol. 1988;26:49–53. [PubMed] [Google Scholar]
- Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J. Immunol. Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- Danielsson D, Jurstrand M. Nonopsonic activation of neutrophils by Helicobacter pylori is inhibited by rebamipide. Dig. Dis. Sci. 1998;43(9 Suppl):167S–173S. [PubMed] [Google Scholar]
- Dixon MF, Genta RM, Yardley JH, Correa P. Histological classification of gastritis and Helicobacter pylori infection: An agreement at last? The International Workshop on the Histopathology of Gastritis. Helicobacter. 1997;2(Suppl 1):S17–S24. doi: 10.1111/j.1523-5378.1997.06b09.x. [DOI] [PubMed] [Google Scholar]
- Dubois A, Liu H, Semino-Mora C, Mog S, Boren T. Role of H. pylori adhesins in colonization of the primate stomach. Gastroenterology. 2006;130(4):938. (145) [Google Scholar]
- Falk P, Roth KA, Borén T, Westblom TU, Gordon JI, Normark S. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc. Natl. Acad. Sci. USA. 1993;90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk P, Borén T, Normark S. Characterization of microbial host receptors. Methods Enzymol. 1994a;236:353–374. doi: 10.1016/0076-6879(94)36027-8. [DOI] [PubMed] [Google Scholar]
- Falk P, Borén T, Haslam D, Caparon M. Bacterial adhesion and colonization assays. Methods Cell. Biol. 1994b;45:165–192. doi: 10.1016/s0091-679x(08)61851-8. [DOI] [PubMed] [Google Scholar]
- Fällman E, Axner O. Design for fully steerable dual-trap optical tweezers. Appl. Opt. 1997;36:2107–2113. doi: 10.1364/ao.36.002107. [DOI] [PubMed] [Google Scholar]
- Fällman E, Schedin S, Jass J, Andersson M, Uhlin BE, Axner O. Optical Tweezers based force measurement system for quantitating binding interactions: System design and application for the study of bacterial adhesion. Biosensors Bioelectronics. 2004;19:1429–1437. doi: 10.1016/j.bios.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Fällman E, Schedin S, Jass J, Uhlin BE, Axner O. The unfolding of the P pili quaternary structure by stretching is reversible, not plastic. EMBO Repo. 2005;6 doi: 10.1038/sj.embor.7400310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Sequence divergence, functional constraint, and selection in protein evolution. Ann. Rev. Genomics Hum. Genet. 2003;4:213–235. doi: 10.1146/annurev.genom.4.020303.162528. [DOI] [PubMed] [Google Scholar]
- Genta RM, Robason GO, Graham DY. Simultaneous visualization of Helicobacter pylori and gastric morphology: A new stain. Hum. Pathol. 1994;25:221–226. doi: 10.1016/0046-8177(94)90191-0. [DOI] [PubMed] [Google Scholar]
- Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. USA. 1999;96:12778–127783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JH. The Causes of Molecular evolution. Oxford University Press; NY: 1991. [Google Scholar]
- Hansson GC, Karlsson KA, Larson G, Strömberg N, Thurin J. Carbohydrate-specific adhesion of bacteria to thin-layer chromatograms: A rationalized approach to the study of host cell glycolipid receptors. Anal. Biochem. 1985;146:158–163. doi: 10.1016/0003-2697(85)90410-5. [DOI] [PubMed] [Google Scholar]
- Hunter WM, Greenwood FC. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature. 1962;194:495–496. doi: 10.1038/194495a0. [DOI] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ögren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- Jaju R, Haas OA, Neat M, Harbott J, Saha V, Boultwood J, Brown JM, Pirc-Danoewinata H, Krings BW, Muller U, Morris SW, Wainscoat JS, Kearney L. A new recurrent translocation, t(5;11)(q35;p15.5), Associated with del(5q) in childhood acute myeloid leukemia. Blood. 1999;94:773–780. [PubMed] [Google Scholar]
- Jass J, Schedin S, Fällman E, Ohlsson J, Nilsson U, Uhlin BE, Axner O. Physical properties of Escherichia coli P Pili measured by optical tweezers. Biophys. J. 2004;87:4271–4283. doi: 10.1529/biophysj.104.044867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson KA, Strömberg N. Overlay and solid-phase analysis of glycolipid receptors for bacteria and viruses. Methods Enzymol. 1987;138:220–232. doi: 10.1016/0076-6879(87)38019-x. [DOI] [PubMed] [Google Scholar]
- Kersulyte D, Chalkauskas H, Berg DE. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol. Microbiol. 1999;31:31–43. doi: 10.1046/j.1365-2958.1999.01140.x. [DOI] [PubMed] [Google Scholar]
- Lan Z-D, Dai L, Zhuo X-L, Feng J-C, Xu K, Chi CW. Gene cloning and sequencing of BmK AS and BmK AS-1, two novel neurotoxins from the scorpion Butus martensi Karsch. Toxicon. 1999;37:815–823. doi: 10.1016/s0041-0101(98)00221-9. [DOI] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Ångström J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadström T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarström L, Borén T. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PA, Prakobphol A, Lee T, Hoover CI, Fisher SJ. Adherence of oral streptococci to salivary glycoproteins. Infect. Immun. 1992;60:31–38. doi: 10.1128/iai.60.1.31-38.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müthing J, Spanbroek R, Peter-Katalinic J, Hanisch FG, Hanski C, Hasegawa A, Unland F, Lehmann J, Tschesche H, Egge H. Isolation and structural characterization of fucosylated gangliosides with linear poly-N-acetyllactosaminyl chains from human granulocytes. Glycobiology. 1996;6:147–156. doi: 10.1093/glycob/6.2.147. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford University Press; NY: 2000. [Google Scholar]
- Okada N, Pentland AP, Falk P, Caparon MG. M protein and protein F act as important determinants of cell-specific tropism of Streptococcus pyogenes in skin tissue. J. Clin. Invest. 1994;94:965–977. doi: 10.1172/JCI117463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olfat FO, Näslund E, Freedman J, Boren T, Engstrand L. Cultured human gastric explants: A model for studies of bacteria-host interaction during conditions of experimental Helicobacter pylori infection. J. Infect. Dis. 2002;186:423–427. doi: 10.1086/341459. [DOI] [PubMed] [Google Scholar]
- Olfat FO, Zheng Q, Oleastro M, Voland P, Boren T, Karttunen R, Engstrand L, Rad R, Prinz C, Gerhard M. Correlation of the Helicobacter pylori adherence factor BabA with duodenal ulcer disease in four European countries. FEMS Immunol. Med. Microbiol. 2005;44:151–156. doi: 10.1016/j.femsim.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Prakobphol A, Murray PA, Fisher SJ. Bacterial adherence on replicas of sodium dodecyl sulfate-polyacrylamide gels. Anal. Biochem. 1987;164:5–11. doi: 10.1016/0003-2697(87)90359-9. [DOI] [PubMed] [Google Scholar]
- Raudonikiene A, Zakharova N, Su WW, Jeong JY, Bryden L, Hoffman PS, Berg DE, Severinov K. Helicobacter pylori with separate beta- and beta′-subunits of RNA polymerase is viable and can colonize conventional mice. Mol. Microbiol. 1999;32:131–138. doi: 10.1046/j.1365-2958.1999.01336.x. [DOI] [PubMed] [Google Scholar]
- Rautelin H, Blomberg B, Fredlund H, Järnerot G, Danielsson D. Incidence of Helicobacter pylori strains activating neutrophils in patients with peptic ulcer disease. Gut. 1993;34:599–603. doi: 10.1136/gut.34.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Möllby R, Winberg J, Normark S. The Gal(alpha1-4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc. Natl. Acad. Sci. USA. 1994;91:11889–11893. doi: 10.1073/pnas.91.25.11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche N, Ångström J, Hurtig M, Larsson T, Borén T, Teneberg S. Helicobacter pylori and complex gangliosides. Infect. Immun. 2004;72:1519–1529. doi: 10.1128/IAI.72.3.1519-1529.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal HE. A graphic method for the determination and presentation of binding parameters in a complex system. Anal. Biochem. 1967;3:525–532. doi: 10.1016/0003-2697(67)90297-7. [DOI] [PubMed] [Google Scholar]
- Ruhl S, Sandberg AL, Cole MF, Cisar JO. Recognition of immunoglobulin A1 by oral actinomyces and streptococcal lectins. Infect. Immun. 1996;64:5421–5424. doi: 10.1128/iai.64.12.5421-5424.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Cisar JO, Sandberg AL. Identification of polymorphonuclear leukocyte and HL-60 cell receptors for adhesins of Streptococcus gordonii and Actinomyces naeslundii. Infect. Immun. 2000;68:6346–6354. doi: 10.1128/iai.68.11.6346-6354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Sandberg AL, Cisar JO. Salivary receptors for the proline-rich protein-binding and lectin-like adhesins of oral actinomyces and streptococci. J. Dent. Res. 2004;83:505–510. doi: 10.1177/154405910408300614. [DOI] [PubMed] [Google Scholar]
- Scatchard G. The attraction of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949;51:660–672. [Google Scholar]
- Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of H. pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J. Infect. Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons RM, Finer JT, Chu S, Spudich JA. Quantitative measurements of force and displacement using an optical trap. Biophys. J. 1996;70:1813–1822. doi: 10.1016/S0006-3495(96)79746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staros O, Wright RW, Swingle DM. Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal. Biochem. 1986;156:220–222. doi: 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
- Stroud MR, Handa K, Salyan MEK, Ito K, Levery SB, Hakomori S.-i., Reinhold BB, Reinhold VN. Monosialogangliosides of human myelogenous leukemia HL60 cells and normal human leukocytes. 1. Separation of E-selectin binding from nonbinding gangliosides, and absence of sialosyl-Lex having tetraosyl to octaosyl core. Biochemistry. 1996a;35:758–769. doi: 10.1021/bi951600r. [DOI] [PubMed] [Google Scholar]
- Stroud MR, Handa K, Salyan MEK, Ito K, Levery SB, Hakomori S.-i., Reinhold BB, Reinhold VN. Monosialogangliosides of human myelogenous leukemia HL60 cells and normal human leukocytes. 2. Characterization of E-selectin binding fractions, and structural requirements for physiological binding to E-selectin. Biochemistry. 1996b;35:770–778. doi: 10.1021/bi952461g. [DOI] [PubMed] [Google Scholar]
- Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Berg DE. Motility of urease-deficient derivatives of Helicobacter pylori. J Bacteriol. 2004;186:885–888. doi: 10.1128/JB.186.3.885-888.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Fraley CD, Zhang M, Dailidiene D, Kornberg A, Berg DE. Diverse phenotypes resulting from polyphosphate kinase gene (ppk1) inactivation in different strains of Helicobacter pylori. J. Bacteriol. 2005;187:7687–7695. doi: 10.1128/JB.187.22.7687-7695.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneberg S, Jurstrand M, Karlsson KA, Danielsson D. Inhibition of Helicobacter pylori-induced activation of human neutrophils by sialylated oligosaccharides. Glycobiology. 2000;10:1171–1181. doi: 10.1093/glycob/10.11.1171. [DOI] [PubMed] [Google Scholar]
- Tummuru MK, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: Evidence of linkage to cytotoxin production. Infect. Immun. 1993;61:1799–1809. doi: 10.1128/iai.61.5.1799-1809.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unemo M, Aspholm-Hurtig M, Ilver D, Borén T, Bergström J, Danielsson D, Teneberg S. The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J. Biol. Chem. 2005;280:15390–15397. doi: 10.1074/jbc.M412725200. [DOI] [PubMed] [Google Scholar]
- Van de Bovenkamp JH, Mahdavi J, Korteland-Van Male AM, Buller HA, Einerhand AW, Boren T, Dekker J. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter. 2003;8:521–532. doi: 10.1046/j.1523-5378.2003.00173.x. [DOI] [PubMed] [Google Scholar]
- Walz A, Odenbreit S, Mahdavi J, Borén T, Ruhl S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology. 2005;15:700–708. doi: 10.1093/glycob/cwi049. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG. A Practical Approach,”. 2nd Ed. Oxford University Press; B.D., Hames: 1998. “In Situ hybridization; pp. 23–66. [Google Scholar]
- Yang Z. Inference of selection from multiple species alignments. Curr. Opin. Genet. Dev. 2004;12:688–694. doi: 10.1016/s0959-437x(02)00348-9. [DOI] [PubMed] [Google Scholar]
- Krishnan BR, Kersulyte D, Brikun I, Berg CM, Berg DE. Direct and crossover PCR amplification to facilitate Tn5supF-based sequencing of lambda phage clones. Nucleic Acids Res. 1991;19:6177–6182. doi: 10.1093/nar/19.22.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]