

Abstract
We exploited the fact that leukemic cells utilize significantly higher
levels of S-adenosylmethionine (SAMe) than normal lymphocytes and
developed tools that selectively diminished their survival under physiologic
conditions. Using RNA interference gene silencing technology, we modulated the
kinetics of methionine adenosyltransferase-II (MAT-II), which catalyzes SAMe
synthesis from ATP and l-Met. Specifically, we silenced the
expression of the regulatory MAT-IIβ subunit in Jurkat cells and
accordingly shifted the 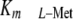 of the enzyme
10–15-fold above the physiologic levels of l-Met, thereby
reducing enzyme activity and SAMe pools, inducing excessive apoptosis and
diminishing leukemic cell growth in vitro and in vivo. These
effects were reversed at unphysiologically high l-Met (>50
μm), indicating that diminished leukemic cell growth at
physiologic l-Met levels was a direct result of the increase in
MAT-II
of the enzyme
10–15-fold above the physiologic levels of l-Met, thereby
reducing enzyme activity and SAMe pools, inducing excessive apoptosis and
diminishing leukemic cell growth in vitro and in vivo. These
effects were reversed at unphysiologically high l-Met (>50
μm), indicating that diminished leukemic cell growth at
physiologic l-Met levels was a direct result of the increase in
MAT-II 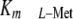 due to MAT-IIβ ablation
and the consequent reduction in SAMe synthesis. In our NOD/Scid
IL-2Rγnull humanized mouse model of leukemia, control
shRNA-transduced Jurkat cells exhibited heightened engraftment, whereas cells
lacking MAT-IIβ failed to engraft for up to 5 weeks post-transplant.
These stark differences in malignant cell survival, effected by MAT-IIβ
ablation, suggest that it may be possible to use this approach to disadvantage
leukemic cell survival in vivo with little to no harm to normal
cells.
due to MAT-IIβ ablation
and the consequent reduction in SAMe synthesis. In our NOD/Scid
IL-2Rγnull humanized mouse model of leukemia, control
shRNA-transduced Jurkat cells exhibited heightened engraftment, whereas cells
lacking MAT-IIβ failed to engraft for up to 5 weeks post-transplant.
These stark differences in malignant cell survival, effected by MAT-IIβ
ablation, suggest that it may be possible to use this approach to disadvantage
leukemic cell survival in vivo with little to no harm to normal
cells.
Leukemia are among the deadliest and most common cancers. Despite advancements in novel individual and combination drug treatment modalities, mortality rates remain high, and some medications have serious adverse effects (2, 3). Our goal has been to develop novel approaches that exploit physiological differences in metabolic needs between normal and leukemic cells to generate tools that would selectively diminish tumor cell growth in vivo, with minimal harm to normal host cells. Specifically, we sought to exploit significant differences in S-adenosylmethionine (SAMe)2 metabolism between normal and leukemic T cells (4, 5). SAMe is an essential molecule in the metabolism of every living species (6–10). As the main methyl group donor, it methylates DNA, RNA, fatty acids, proteins, and small molecules and regulates several transcription and translation processes, protein function, and membrane integrity. SAMe is also involved in DNA mismatch repair, chromatin modeling, epigenetic modifications and imprinting, cell replication, neurotransmission, and signaling (11). Additionally, SAMe is an important precursor of the polyamines and a major player in biological trans-sulfuration as well as folic acid and one-carbon metabolism (8, 9, 11, 12).
The importance of SAMe, together with the fact that its metabolism is constitutively elevated in malignant versus normal cells, has for years made it an attractive target for cancer chemotherapy (13–20). Unfortunately, chemical inhibitors of SAMe synthesis have been difficult to generate in quantities needed for clinical use, and most were either unstable, reversible, nonspecific, or highly toxic because no cell can survive total inhibition of SAMe synthesis. To this end, our approach has been to take advantage of the dependence on higher SAMe levels in leukemic cells to diminish rather than totally block their ability to synthesize the needed amount of SAMe and thereby selectively halt their growth while sparing normal cells.
The advent of novel biotools that can selectively silence protein
expression has made it possible to initiate studies to target the regulatory
subunit of methionine adenosyltransferase (MAT), which catalyzes the synthesis
of SAMe from l-Met and ATP. All living organisms have at least one
MAT enzyme (5,
13). Mammals have
liver-specific MAT-I/III and another isozyme, MAT-II, that is expressed in all
tissues (21,
22). MAT-I/III are
tetramer/dimer forms of a catalytic α1 subunit, and they differ
considerably in their kinetic and physical properties. It is believed that
this differential oligomerization of MAT-α1 is an important adaptation
to cope with special metabolic requirements in the liver, where SAMe levels
need to be maintained at a certain range inasmuch as a deficiency or excess of
SAMe has been associated with serious pathology
(23–25).
In livers of healthy subjects, MAT-III, which has a high
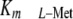 (80–100 μm), is
the major isoform. By contrast, MAT-II is a hetero-oligomer that has a
catalytic α2 subunit and a regulatory β subunit with a
(80–100 μm), is
the major isoform. By contrast, MAT-II is a hetero-oligomer that has a
catalytic α2 subunit and a regulatory β subunit with a
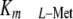 of 4–20 μm
(21,
26,
50). The α2 subunit,
which has 84% sequence identity to α1, undergoes post-translational
modifications resulting in expression of α2 (53 kDa) and α2′
(51 kDa) forms (21). In fetal
liver and certain adult liver diseases, including hepatocellular carcinoma,
α1 subunit expression is diminished and replaced by α2, along with
the induction of MAT-IIβ expression
(27,
28).
of 4–20 μm
(21,
26,
50). The α2 subunit,
which has 84% sequence identity to α1, undergoes post-translational
modifications resulting in expression of α2 (53 kDa) and α2′
(51 kDa) forms (21). In fetal
liver and certain adult liver diseases, including hepatocellular carcinoma,
α1 subunit expression is diminished and replaced by α2, along with
the induction of MAT-IIβ expression
(27,
28).
MAT-α subunits are highly conserved across many species (22); by contrast MAT-IIβ is only found in mammals, associated with MAT-IIα2. In several of our previous studies, we showed that MAT-IIβ plays a crucial physiological role by lowering the Km of MAT-II for l-Met from 55–65 μm down to 3.5–20 μm (26, 29, 50). Inasmuch as the physiologic extrahepatic concentration of l-Met are 5–10-fold lower than that in the liver (30), we believe that the introduction of MAT-IIβ to lower the Km of the extrahepatic enzyme may have been an essential evolutionary event that allowed MAT-II to function in blood and other extrahepatic mammalian tissues, where l-Met levels are ∼10–25 μm (31–33).
We had reported that MAT-II expression and SAMe metabolism are considerably
different in normal and malignant lymphocytes
(4,
5,
8). MAT-IIβ expression in
established and primary human lymphocytic leukemia cells is significantly
higher than in quiescent or activated lymphocytes
(4). MAT-II activity, SAMe
utilization rate, and SAMe pool size are, respectively, 20-, 60-, and
60–100-fold higher in lymphocytic leukemia, than in normal lymphocytes
(4). Based on these previous
studies (4,
21,
26,
29), we predicted that if we
specifically ablated MAT-IIβ expression, we would shift MAT-II
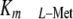 by at least 10-fold above physiologic
l-Met levels, and that this would consequently reduce SAMe pool
size and selectively diminish the growth of leukemic cells in physiological
fluids and extrahepatic tissues. We report that MAT-IIβ subunit specific
shRNA successfully silenced the expression of the MAT-IIβ regulatory
subunit in the Jurkat leukemic T cell line and increased the enzyme
by at least 10-fold above physiologic
l-Met levels, and that this would consequently reduce SAMe pool
size and selectively diminish the growth of leukemic cells in physiological
fluids and extrahepatic tissues. We report that MAT-IIβ subunit specific
shRNA successfully silenced the expression of the MAT-IIβ regulatory
subunit in the Jurkat leukemic T cell line and increased the enzyme
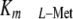 by 10–15-fold, consequently
depleting SAMe pools, inducing excessive apoptosis, and diminishing the growth
of these leukemic cells in physiologic l-Met concentrations, both
in vitro and in vivo in a humanized NOD/Scid
IL-2Rγnull mouse model of leukemia.
by 10–15-fold, consequently
depleting SAMe pools, inducing excessive apoptosis, and diminishing the growth
of these leukemic cells in physiologic l-Met concentrations, both
in vitro and in vivo in a humanized NOD/Scid
IL-2Rγnull mouse model of leukemia.
EXPERIMENTAL PROCEDURES
Leukemic Cells—Jurkat T cells (E6–1; ATCC, Manassas, VA) were maintained in RPMI 1640 medium, supplemented with either 10% fetal bovine serum or 1% HL-1 supplement (±l-Met), 2 mm l-glutamine, 50 μg/ml streptomycin, and 50 units/ml of penicillin.
Generation of pHSPG-shRNA Retrovirus—We designed several shRNA sequences to target MAT-IIβ expression and BLAST-searched these sequences against the human genome data base to ensure their specificity for our target gene. The pHSPG plasmid (Dr. Su, University of North Carolina) has a constitutively active polymerase III promoter in front of the multiple cloning site. We modified pHSPG by including a Histone 1 promoter needed for shRNA formation (34), followed by shRNA constructs of interest between the EcoRV and XbaI sites, in the multiple cloning site. Downstream of this construct is a PGK-GFP cassette whose expression is driven by the mouse mammary tumor virus promoter. The HSPG virus was chosen for its high transduction efficiency in hematopoetic cells (36, 37). The design of different MAT-IIβ-specific pHSPG-shRNA driven by the Histone 1 promoter is shown in supplemental Fig. S1.
The effect of several control and MAT-IIβ-specific pHSGP-shRNA constructs on MAT-IIβ expression were initially tested in transiently transfected Cos-1 cells. These studies showed that plasmid pHSGP-shRNA-1110 had the highest gene silencing activity and was not toxic to the Cos-1 cells, and thus we packaged it into HSPG-V1110 viral particles using HEK-293T cells and the CaCl2 transfection method (35). We also generated a control, empty virus without shRNA (V1302) and V1324 that encodes an shRNA for an irrelevant, mouse plexin A1 gene (37).
Transduction with HSPG-shRNA Retrovirus—We transduced Jurkat cells (106 cells) by adding 8 mg/ml polybrene plus 700 ml of viral supernatants and incubating for 20 min at room temperature. The cells were spun at 2000 rpm for 3 h and then resuspended in fresh 1.5 ml of RPMI 1640 complete medium. We cultured the cells in 12-well tissue culture plates, repeated the transduction after 24 and/or 48 h, and then assessed transduction efficiency by flow cytometry (FACSCalibur) to determine the percentage of GFP-positive cells. This procedure typically yielded >90% GFP+ cells, which were then sorted using FACSAria (BD Biosciences) to obtain a ≥98% transduced cell population.
MAT-II Subunits Expression Analysis—We lysed GFP+ sorted cells by three cycles of freeze-thawing in extraction buffer (50 mm Tris, pH 7.4, 50 mm NaCl, 5 mm MgCl2, 4 mm dithiothreitol), containing a mixture of proteolytic inhibitors (Roche Applied Science) as described (5). Protein concentration was determined in the cleared lysates by the bicinchoninic acid method (38). Equal amounts of protein extracts were separated on 10% SDS-PAGE and then transblotted onto nitrocellulose papers. Expression of α2 and β subunits was determined by Western blots and probed with antibodies to MAT-IIα and MAT-IIβ proteins (4, 39).
We also used quantitative real time PCR to assess MAT-IIα and MAT-IIβ mRNA expression. We constructed cRNA standards for each subunit and generated standard curves for each run to quantify mRNA copy number/2 μg of total RNA. Briefly, we transformed Escherichia coli strain JM109 with pTargeT/MAT-IIα subunit or pTargeT/MAT-IIβ subunit (26) and purified those plasmids using the Wizard PureFection DNA purification system (Promega). Correct pTargeT/MAT-IIα and pTargeT/MAT-IIβ plasmids were verified by sequencing, using a T7 promoter primer. T7 Ribomax large scale RNA production system (Promega) was used to generate MAT-IIα or MAT-IIβ cRNA. The number of cRNA molecules were calculated as follows: N(molecules/μl) = [C*182·5 × 1013]/K, where C = cRNA μg/μl and K = bp (40). For MAT-IIα cRNA, K = 1188 bp and C = 0.658 μg/μl; for MAT-IIβ cRNA, K = 1050 bp and C = 0.632.
To test the efficiency of MAT-IIβ silencing, we extracted RNA from 106 untransduced or transduced cells using RNA-STAT 60 (Tel-Test), removed residual contaminating genomic DNA by DNase I treatment (Qiagen), purified the RNA using Qiagen RNeasy kit, and then converted 2 μg of the purified RNA to cDNA using avian myeloblastosis virus reverse transcriptase, 10× random hexamers, and 10 mm dNTP in the presence of RNase inhibitor (Promega). Quantitative real time PCRs were run using the fluorogenic SYBR Green quantitative real time PCR system and the ABI prism 7900 Sequence Bio-Detector (PE Biosystems). In each run, serial dilutions of MAT-IIα or MAT-IIβ cRNA (104–1010 cRNA molecules/μl) were also converted to cDNA and used to generate the standard curve for each MAT-II subunit. The PCR mixture contained 25 μl of SYBR Green PCR Master mix (Applied Biosystems), 2.5 μl containing 1.5–12 pmol of each primer, and 5 μl of the template cDNA in a final volume of 50 μl. The sequence of the MAT-IIβ specific primers were (forward, 5′-CACCTTACAGAGAGGAAGA-3′ and reverse, 5′-CAGTCACAGCACTTTCTTC-3′); and for MAT-IIα2 specific primers (forward, 5′-AAAGTGGTTCGTGAAGCTGTTAAA-3′ and reverse, 5′-CCAAGGCTACCAGCACGTTAC-3′). An 18 S RNA primer mix (Qiagen) was used for normalization. To calculate N for each test sample, we first normalized the cycle threshold (CT) to the internal normalizer (18 S) and then determined N from the standard curve using the equation Y = [MX + C].
Assay for MAT Activity—We assayed MAT activity in cell extracts as described previously (21). For kinetic analyses, we used different l-Met concentrations (1.25–80 μm), using [14C]l-Met (57.9 mCi/mmol) and supplementing with cold l-Met. Reaction velocity is expressed as units/mg protein, where 1 unit = 1 nmol of adenosylmethionine/h (21). We calculated Km and Vmax using the GraphPad Prism program.
Cell Growth at Different l-Met Concentrations—We weaned the cells to grow in serum-free RPMI 1640 medium containing 1% HL-1 supplement (Cambrex, NJ) plus 2 mm l-glutamine, 50 μg/ml streptomycin, and 50 units/ml of penicillin (RPMI-HL1s medium). We seeded weaned cells (106/ml) in RPMI-HL1s medium at l-Met 5–100 μm, replaced the medium every 2 days, recorded numbers of viable cells, and then replated the cells at 106/ml for further analyses. Cell necrosis and apoptosis were determined by a flow cytometric quantification of propidium iodide and annexin-V (BD Pharmingen) stained cells, respectively. In some studies, the cells were cultured at 10 μm l-Met for 3d, harvested, counted, and replated at 106 cells/ml in fresh RPMI-HL1s medium with 10 μm l-Met. On day 6, the cells were harvested, washed, and replated in fresh medium containing 20 μm or 50 μm l-Met. The cell growth/death were determined every 3 days thereafter, replating 106 viable cells/ml each time.
Measurement of SAMe Levels—We quantified SAMe levels in a neutralized 2 n perchloric acid-soluble extract by HPLC as previously detailed (4). We calculated the SAMe concentration (pmol/106 cells) from a standard curve using different concentrations of SAMe standard (USB Corp.) and then converted the values to μm based on cell volume for Jurkat cells 0.76 ml/109 cells (4).
In Vivo Animal Studies—All of the animals were treated according to Institutional Animal Care and Use Committee regulations (protocol 1587). We irradiated highly immune deficient NOD/ScidIL-2Rγnull mice with 3.5GY, 24 h prior to transplanting them via intraperitoneal injection of 15 × 106 V1110 or V1302 Jurkat cells. We obtained sera from mice 12 days post-injection and then once a week thereafter to quantify levels of the surrogate tumor marker, soluble β2 microglobulin, using an enzyme-linked immunosorbent assay β2 microglobulin kit (R & D). At 4 and 5 weeks post-transplant, we sacrificed the transplanted mice (supplemental Table S1) and used fluorescence reflectance imaging to image GFP fluorescence in spleen prior to extracting splenocytes and bone marrow cells to assess tumor growth and engraftment by counting % human CD3/GFP-positive cells in each compartment by flow cytometry as detailed above.
Statistical Methods—We calculated statistical significance with one-way analysis of variance and Newman-Keuls Multiple Comparison test for all the experiments except for the in vivo experiment where we used a Mann-Whitney test using the GraphPad Prism program.
RESULTS
Successful Ablation of MAT-IIβ Expression—We designed several shRNA to ablate MAT-IIβ expression, cloned them into the retroviral plasmid pHSPG, which also expresses GFP, and screened their gene silencing efficiency and nonspecific toxicity (supplemental Fig. S1). All MAT-IIβ-specific shRNA constructs were designed to target the two splice variants of MAT-IIβ (28). Initial screening was done in COS-1 cells, and constructs with efficient gene silencing activity were then screened in Jurkat cells (supplemental Fig. S1). Construct V1110 had the highest MAT-IIβ silencing activity (supplemental Fig. S1) and least toxicity and was thus packaged into infectious HSPG-GFP virus (36).
We transduced Jurkat cells with the various recombinant HSPG-shRNA viruses and assessed MAT-IIβ ablation at the RNA and protein levels (supplemental Fig. S1). Controls included cells infected with an empty virus (V1302) or with a virus encoding shRNA construct (V1324) directed to the mouse plexin A1 gene (36). Although transduction efficiencies for all cells tested were ≥90%, significant reduction or complete ablation of MAT-IIβ RNA and protein expression was only seen in V1110-transduced cells (Fig. 1). By contrast, expression of MAT-IIα2 was essentially unaffected, thus indicating the MAT-IIβ specificity of V1110. In some cases, expression of MAT-IIα2 was insignificantly elevated, perhaps reflecting an attempt to increase enzyme synthesis to compensate for the lack of β expression and increase SAMe synthesis.
FIGURE 1.

Successful ablation of MAT-IIβ subunit using viral encoded shRNA. MAT-II α2 and β protein expression (60 μg of protein/well) and mRNA levels were analyzed in untransduced or ≥98% transduced cells as detailed under “Experimental Procedures.” Quantitative real time PCR was conducted using RNA extracted from cells maintained in 100 μm l-Met RPMI medium, and RNA copy number was calculated as detailed under “Experimental Procedures.” The results (means ± S.E.) of two combined experiments, each done in triplicate, are shown. Statistical differences were determined as described under “Experimental Procedures.” MAT-IIβ expression was significantly ablated in V1110 cells. ***, p ≤ 0.001. MAT-IIα2 was slightly elevated or unaffected (p > 0.05).
Effect of MAT-IIβ Silencing on MAT-II Activity, Kinetics,
SAMe Pool Size, and Cellular Growth—We next tested whether ablation
of MAT-IIβ expression will alter MAT-II
 and reduce SAMe pools in Jurkat
cells. As predicted, the
and reduce SAMe pools in Jurkat
cells. As predicted, the 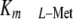 increased
from 3.5–6 μm in untransduced and control virus-transduced
Jurkat cells, up to 56–62 μm in V1110 cells
(Fig. 2A). Further, at
the high end of physiological l-Met levels (20 μm),
MAT-II specific activity was 4–5-fold lower in V1110 cells than in
untransduced or control virus-transduced Jurkat cells expressing MAT-IIβ
(Fig. 2B).
increased
from 3.5–6 μm in untransduced and control virus-transduced
Jurkat cells, up to 56–62 μm in V1110 cells
(Fig. 2A). Further, at
the high end of physiological l-Met levels (20 μm),
MAT-II specific activity was 4–5-fold lower in V1110 cells than in
untransduced or control virus-transduced Jurkat cells expressing MAT-IIβ
(Fig. 2B).
FIGURE 2.

MAT-IIβ ablation modulates MAT-II kinetics, reduces its specific activity, and diminishes intracellular SAMe levels. A, MAT-II Lineweaver-Burk kinetic plots of 1/V, where V is units of enzyme activity/mg of protein (units/mg; 1 unit = 1 nmol of adenosylmethionine/h). MAT assays were performed using extracts from untransduced, V110- or V1302-transduced cells in the presence of 2.5–80 μm l-Met, as detailed under “Experimental Procedures.” The data for each cell extract represents the means ± S.D. of three separate experiments, each assayed in triplicate. B, MAT specific activity (units/mg of protein) at 20 μm l-Met. C, SAMe levels (pmol/106 cells) as determined by HPLC in neutralized perchloric acid extracts from cells that were maintained at 50 or 20 μm l-Met. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Importantly, in the absence of MAT-IIβ, SAMe was undetectable in V1110 cells cultured at 10 μm l-Met (not shown), and even at 20 μm l-Met SAMe pool size was ∼85% less than in untransduced or control-transduced V1302 cells (9.5 ± 0.5 μm versus 62 ± 4 μm; p ≤ 0.05) (Fig. 2C). Significant 45–50%, reductions in SAMe pool size (p ≤ 0.01) were also found even when V1110 cells were maintained at 50 μm l-Met, which is ∼2.5-fold higher than the upper range of physiological l-Met levels.
The sharp reduction in SAMe pool size, consequent to MAT-IIβ ablation,
diminished the growth and viability of V1110 cells. At ≤20 μm
l-Met, V1110 cells ceased to grow, and at 50 μm their
growth was reduced by 33–36% compared with control-transduced V1302
cells (p < 0.01) (Fig.
3). By contrast, at 100 μm l-Met, V1110 cells grew
at essentially the same rates and for the same duration in culture as the
untransduced or control-transduced Jurkat cells (p > 0.05)
(Fig. 3). Thus, at
unphysiologically high l-Met levels approaching MAT-IIα2
Km without β (i.e. Km =
50–60 μm), V1110 leukemic cells appear to have satisfied
their SAMe needs and grew normally. These results indicate that diminished
V1110 cell growth at physiologic l-Met levels was a direct
consequence of specific MAT-IIβ silencing, which caused an upper shift in
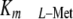 of MAT-II and a significant reduction
of SAMe synthesis and pools.
of MAT-II and a significant reduction
of SAMe synthesis and pools.
FIGURE 3.

MAT-IIβ ablation reduces leukemic cell growth in physiologic l-Met levels. We seeded 5 × 106 of indicated cells at 5–100 μm l-Met and monitored their growth every 2 days using the trypan blue exclusion method, adding fresh medium each time. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Cell Death and Apoptosis of Leukemic Cells Lacking MAT-IIβ—Significant reduction in growth rates of V1110 cells at physiological l-Met levels was associated with a significant increase in both cell necrosis and apoptosis (Fig. 4). At physiologic l-Met levels (5, 10, and 20 μm), the extent of necrosis in the V1110 cells was significantly higher (20–55%) than in untransduced or control V1302 cells (p < 0.001). Minimal cell death was seen at 50 μm l-Met; however, V1110 cells exhibited ∼70–80% higher levels of apoptosis than the control leukemic cells even at this unphysiological l-Met concentration (p < 0.01) (Fig. 4). This is likely attributed to the 50% reduction in SAMe pool size even at 50 μm l-Met (Fig. 2). Taken together, these findings indicate that the growth and viability of V1110 cells lacking MAT-IIβ expression remained at a selective disadvantage, even at more than twice the higher end of physiological l-Met levels.
FIGURE 4.

MAT-IIβ ablation induces high levels of apoptosis and necrosis of leukemic cells. A, propidium iodide-stained necrotic cells (%) at 20 μm l-Met. B, propidium iodide-stained necrotic cells (%) at 50 μm l-Met. C, necrotic cells (%) at 6 days in 5–50 μm l-Met. D, annexin-V-stained apoptotic cells (%) at 20 μm l-Met. D, annexin-V stained apoptotic cells (%) at 50 μm l-Met. F, apoptotic cells (%) at 6 days in 5–50 μm l-Met. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Attempts to Rescue Cells by Incrementally Elevating l-Met Levels—Following up on selective pressure imposed on the viability of MAT-IIβ-deficient Jurkat cells in physiologic l-Met conditions, we next tested whether V1110 cells can be rescued by incremental elevation of l-Met in culture. All of the cells grew at lower rates at the low end (5–10 μm) of physiologic l-Met levels compared with ≥20 μm. We therefore maintained V1110 and control Jurkat cells at 10 μm l-Met for 6 days in culture and then increased l-Met levels to either 20 or 50 μm to determine whether these cells can be rescued (Fig. 5). The growth of control untransduced and control V1302 Jurkat cells increased by almost 80 and 90% when l-Met levels were raised to 20 or 50 μm, respectively. By contrast, under the same conditions, V1110 cells could not be rescued with 20 μm l-Met (p < 0.01) and exhibited diminished growth even at 50 μm l-Met (p < 0.05) (Fig. 5). These data suggest that the initial insult to the V1110 cells, caused by low SAMe levels due to the absence of MAT-IIβ, is irreversible at the high end of physiologic l-Met levels (20 μm) and minimally reversible at 50 μm l-Met. Failure to rescue V1110 cells is consistent with the above observed increased apoptosis of these cells, even at 50 μm l-Met (Fig. 4).
FIGURE 5.

Differential rescue of Jurkat cells ± MAT-IIβ expression by incrementally increasing l-Met levels in culture. Untransduced and V1302- and V1110-transduced Jurkat cells (5 × 106) were cultured at 10 μm l-Met for 6 days, and then l-Met was raised to either 20 μm (A) or 50 μm (B). Viable cells were counted over 15 days, with the addition of fresh medium every 3 days. V1110 cells ceased to grow when l-Met was raised to 20 μm and exhibited diminished growth even at 50 μm.*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
MAT-IIβ Ablation Diminishes Leukemic Cell Growth in an NOD/Scid IL-2Rγnull Mouse Leukemia Model—To rule out possible in vitro culture artifacts, we investigated whether Jurkat cells that do not express MAT-IIβ would survive and multiply in vivo in our severely immunodeficient NOD/Scid IL-2Rγnull mouse model (41). These mice allow heightened engraftment with xenogeneic cells because, besides lacking T, B, and NK cells, they are also deficient in complement and macrophage function (41). After mildly irradiating these mice, we transplanted them intraperitoneally with 15 × 106 V1110 or control V1302 cells (n = 15 mice/group). Irradiated, noninjected mice served as additional controls. We monitored tumor engraftment and growth weekly for 5 weeks post-transplant by determining the percentage of human CD3 and GFP expression in bone marrow (BM)- and spleen-derived cells and by measuring serum levels of the surrogate tumor marker, β2-microglobulin (β2μ) (42). We also imaged GFP expression in whole spleens of sacrificed mice for a semi-quantitative measure of tumor burden (Fig. 6).
FIGURE 6.

MAT-IIβ ablation diminishes leukemic cell growth in vivo. Hyperimmune-deficient NOD/Scid IL-2Rγnull mice were irradiated with 3.5GY 24 h prior to intraperitoneal transplant with 15 × 106 of the indicated cells. Controls included irradiated but not injected mice and V1302 transplanted mice. Tumor engraftment and growth were monitored by measuring % human CD3+/GFP+ cells in mice spleens and bone marrow at 4 and 5 weeks post-transplant with either control V1302 or V1110 cells. Flow cytometry histograms of CD3+/GFP+ in spleen (A) or bone marrow (B) 5 weeks post-transplant. C, GFP expression in whole spleens of same mice prior to processing their splenocytes. D, number of mice engrafted with V1302 or V1110 leukemic cells at 4 and 5 weeks post-transplant. E and F show % CD3+/GFP+ cells in spleens (E) or bone marrow (F) of mice transplanted with either V1302 or V1110 leukemic cells, 5 weeks post-transplant. The statistical differences were calculated using a Mann-Whitney test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
During the first 3 weeks, none of the transplanted mice showed significant engraftment, but after 4 weeks, 57% of mice transplanted with control V1302 cells began to show variable levels of engraftment in spleen (3–44% human CD3+/GFP+ cells; mean 16 ± 7.6%) and in BM (3–12% human CD3+/GFP+ cells; mean 4 ± 1.6%). Engraftment was much less evident in spleens of mice transplanted 4 weeks earlier with V1110 cells (0.96 ± 0.32% CD3+/GFP+ cells; p = 0.004). This trend continued through 5 weeks post-transplant, with significantly higher human CD3+/GFP+ cells in spleen and BM of mice injected with V1302 versus V1110 cells (supplemental Table 1S and Fig. 6). At 5 weeks, 25% of V1110 cells transplanted mice started to show low levels of CD3+/GFP+ engraftment in spleen (1.1 ± 0.4%) and BM (2.4 ± 1%). In stark contrast, 75% of mice transplanted with V1302 cells showed heightened engraftment in spleen (30 ± 10%; p = 0.02) and BM (11 ± 4%; p = 0.04) (Fig. 6). Additionally, levels of the surrogate tumor marker β2μ were 2-fold higher in mice transplanted with control V1302 leukemic cells than those transplanted with V1110 cells (supplemental Fig. S2).
DISCUSSION
In this study, we showed that by silencing the expression of the regulatory
β subunit of MAT-II, we exerted significant growth disadvantage upon
human Jurkat leukemic T cells under physiologic l-Met levels in
vitro and in vivo. In the liver, l-Met levels are
80–100 μm, but in other tissues they range from 10 to 25
μm. Thus, the high Km hepatic isozyme,
MAT-III, can be fully functional in that organ without MAT-IIβ. By
contrast, MAT-II, the only SAMe-synthesizing enzyme in extrahepatic tissue can
only function when associated with its MAT-IIβ regulatory subunit, which
plays an important role in lowering MAT-II
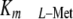 to values close to physiological
l-Met levels outside the liver
(26,
29,
43,
50). Without the β
subunit, MAT-II would function at minimal capacity and would not be able to
provide normal extrahepatic cells with adequate SAMe levels needed for their
growth and survival. In resting human T cells, MAT-IIβ is expressed at
low levels, and mitogenic activation of these cells does not induce further
expression of this subunit; by contrast, MAT-IIβ is constitutively
expressed at a much higher level in established and primary leukemic T cells
(4,
5). Induction of MAT-IIβ
expression has been reported in several types of cancer and has been shown to
confer a proliferative advantage to human hepatomas
(44).
to values close to physiological
l-Met levels outside the liver
(26,
29,
43,
50). Without the β
subunit, MAT-II would function at minimal capacity and would not be able to
provide normal extrahepatic cells with adequate SAMe levels needed for their
growth and survival. In resting human T cells, MAT-IIβ is expressed at
low levels, and mitogenic activation of these cells does not induce further
expression of this subunit; by contrast, MAT-IIβ is constitutively
expressed at a much higher level in established and primary leukemic T cells
(4,
5). Induction of MAT-IIβ
expression has been reported in several types of cancer and has been shown to
confer a proliferative advantage to human hepatomas
(44).
The role of MAT-IIβ in leukemic cells is even more crucial because leukemic T cells, both freshly explanted ALL-2 cells and the Jurkat cell line, utilize SAMe at significantly higher rates than normal resting or activated T cells (4, 5). The constitutive high expression of MAT-IIβ in leukemic cells allows high levels of SAMe synthesis needed to meet the growth requirements of malignant cells. In our quest to diminish SAMe in leukemic cells, it was unreasonable to target the MAT-IIα2 catalytic subunit, because that would be quite detrimental and toxic to normal cells that do not express the hepatic MAT-I/III isozyme, i.e. the majority of extrahepatic cells. However, targeting MAT-IIβ expression seemed much more appropriate and practical, because this would not block SAMe synthesis completely but would reduce it to where the rapid growth and proliferation of malignant lymphocytes and possibly other cancerous cells can no longer be sustained.
We succeeded in silencing the MAT-IIβ regulatory subunit without significantly affecting the expression of the MAT-IIα2 subunit. Under these conditions, our in vitro and in vivo studies provided strong evidence that MAT-IIβ ablation is detrimental to the growth of malignant T cells and that this is directly due to drastic reductions in SAMe levels needed to support their rapid growth. Inasmuch as it is quite feasible to modulate l-Met levels in vivo through dietary control (45), we believe these results are of particular interest, especially in the context of our quest to eventually translate these research findings into clinical applications. Indeed, there is a large body of evidence that the growth of several types of tumors is dependent on high l-Met and that l-Met deprivation causes cell cycle arrest in the G2 phase (46, 47). Several clinical studies demonstrated that diminishing l-Met in cancer patients by maintaining them on a Met- or choline-free diet and/or administering methioninase exerts selective growth disadvantage pressure on cancer cells in vitro and in vivo, inducing mitotic and cell cycle arrest, apoptotic death, and widespread necrosis in tumors (48). Induction of Jurkat cells apoptosis and death consequent to silencing MAT-IIβ is also in agreement with previous studies showing increased apoptosis and necrosis of tumor cells cultured in the absence of l-Met (1, 49). We observed significant apoptosis in Jurkat cells lacking MAT-IIβ at physiologic l-Met levels, and this indicates that direct depletion of SAMe may be more effective than l-Met depletion in inducing tumor cell apoptosis in vivo. We believe that targeting MAT-IIβ expression in combination with restricting dietary l-Met alone or in conjunction with chemotherapeutic agents may prove to be a powerful approach in the treatment of certain blood malignancies, and we are currently exploring these possibilities.
The growth of Jurkat cells with and without MAT-IIβ expression was reduced at 10 μm compared with ≥20 μm l-Met; however, when we attempted to rescue these cells by incrementally increasing l-Met levels in culture, we found that the growth of cells expressing MAT-IIβ was fully restored at 20 μm, whereas cells lacking MAT-IIβ could not be rescued at this concentration. Even at unphysiologically high 50 μm l-Met we still observed significant apoptosis in cells lacking MAT-IIβ. We therefore predict that it may be possible to adjust l-Met levels in vivo in a way that would rescue normal cells while still inducing death and apoptosis of leukemic cells that do not express this important regulatory subunit.
We ruled out the possibility that our in vitro observations are due to culture artifacts, because when we compared the ability of Jurkat cells that do and do not express MAT-IIβ to engraft and survive in an in vivo environment of physiologic l-Met levels in our hyperimmune deficient NOD/ScidIL-2Rγnull mouse model, we found significant differences in the in vivo tumor growth and engraftment, even in the absence of host immune rejection inasmuch as these mice were not reconstituted with a human immune system (41). Control transduced Jurkat cells grew well in this mouse model, whereas cells lacking MAT-IIβ expression showed significant diminution in engraftment for up to 5 weeks post-transplant. Although these results are very promising, we are currently designing studies to treat leukemic cells in vivo, after they have engrafted.
In summary, we had predicted that an approach that targets SAMe metabolism would provide a good adjunctive therapeutic tool for the treatment of leukemia because of the their excessive requirement for this central metabolic compound. Our in vivo data suggest that this approach, by itself, may have a more drastic effect on diminishing leukemic cell growth than we had originally anticipated. The stark difference in the in vivo survival and engraftment between the V1110- and V1302-transduced cells in mice lacking proper immune defenses lead us to be even more optimistic that it will be possible to generate targeted therapeutics and dietary protocols to selectively diminish the growth of malignant lymphocytes with little or no effect on normal cells.
Supplementary Material
Acknowledgments
We greatly appreciate the help of Dr. Waleed Gaber and Gogula Bharathi (University of Tennessee Health Science Center Biomedical Engineering and Imaging Department) with animal imaging.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA108792. This work was also supported by a Veterans Affairs Merit Review Award and a Senior Research Career Scientist Award (to M. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Table S1.
References
- 1.Kokkinakis, D. M., Brickner, A. G., Kirkwood, J. M., Liu, X., Goldwasser, J. E., Kastrama, A., Sander, C., Bocangel, D., and Chada, S. (2006) Mol. Cancer Res. 4 575-589 [DOI] [PubMed] [Google Scholar]
- 2.Pui, C. H., and Jeha, S. (2007) Nat. Rev. Drug. Discov. 6 149-165 [DOI] [PubMed] [Google Scholar]
- 3.Cunningham, L., and Aplenc, R. (2007) Expert Opin. Pharmacother. 8 2519-2531 [DOI] [PubMed] [Google Scholar]
- 4.De La Rosa, J., Geller, A. M., LeGros, H. L., Jr., and Kotb, M. (1992) Cancer Res. 52 3361-3366 [PubMed] [Google Scholar]
- 5.De La Rosa, J., LeGros, H. L., Jr., Geller, A. M., and Kotb, M. (1992) J. Biol. Chem. 267 10699-10704 [PubMed] [Google Scholar]
- 6.Cantoni, G. L. (1953) J. Biol. Chem. 204 403-416 [PubMed] [Google Scholar]
- 7.Tabor, C. W., and Tabor, H. (1984) Adv. Enzymol. Relat. Areas Mol. Biol. 56 251-282 [DOI] [PubMed] [Google Scholar]
- 8.Kotb, M., and Geller, A. M. (1993) Pharmacol. Ther. 59 125-143 [DOI] [PubMed] [Google Scholar]
- 9.Chiang, P. K., Gordon, R. K., Tal, J., Zeng, G. C., Doctor, B. P., Pardhasaradhi, K., and McCann, P. P. (1996) FASEB J. 10 471-480 [PubMed] [Google Scholar]
- 10.Mato, J. M., Alvarez, L., Ortiz, P., and Pajares, M. A. (1997) Pharmacol. Ther. 73 265-280 [DOI] [PubMed] [Google Scholar]
- 11.Loenen, W. A. (2006) Biochem. Soc. Trans. 34 330-333 [DOI] [PubMed] [Google Scholar]
- 12.Tabor, C. W., and Tabor, H. (1984) Annu. Rev. Biochem. 53 749-790 [DOI] [PubMed] [Google Scholar]
- 13.Coulter, A., Lombardini, J. B., Sufrin, J., and Talalay, P. (1974) Mol. Pharmacol. 10 319-334 [PubMed] [Google Scholar]
- 14.Sufrin, J., Coulter, A., and Talalay, P. (1979) Mol. Pharmacol. 15 661-677 [PubMed] [Google Scholar]
- 15.Federici, M. M., and Lotspeich, F. J. (1979) Biochem. Pharmacol. 28 1689-1693 [DOI] [PubMed] [Google Scholar]
- 16.Kappler, F., Vrudhula, V. M., and Hampton, A. (1988) J. Med. Chem. 31 384-389 [DOI] [PubMed] [Google Scholar]
- 17.Lavrador, K., Allart, B., Guillerm, D., and Guillerm, G. (1998) J. Enzyme Inhib. 13 361-367 [DOI] [PubMed] [Google Scholar]
- 18.Lombardini, J. B., and Sufrin, J. R. (1983) Biochem. Pharmacol. 32 489-495 [DOI] [PubMed] [Google Scholar]
- 19.Vrudhula, V., Kappler, F., Afshar, C., Ginell, S., Lessinger, L., and Hampton, A. (1989) J. Med. Chem. 32 885-890 [DOI] [PubMed] [Google Scholar]
- 20.Sufrin, J. R., Lombardini, J. B., and Alks, V. (1993) Biochim. Biophys. Acta 1202 87-91 [DOI] [PubMed] [Google Scholar]
- 21.Kotb, M., and Kredich, N. M. (1985) J. Biol. Chem. 260 3923-3930 [PubMed] [Google Scholar]
- 22.Kotb, M., Mudd, S. H., Mato, J. M., Geller, A. M., Kredich, N. M., Chou, J. Y., and Cantoni, G. L. (1997) Trends Genet. 13 51-52 [DOI] [PubMed] [Google Scholar]
- 23.Hoffman, J. L. (1983) Methods Enzymol. 94 223-228 [DOI] [PubMed] [Google Scholar]
- 24.Mato, J. M., Corrales, F. J., Lu, S. C., and Avila, M. A. (2002) FASEB J. 16 15-26 [DOI] [PubMed] [Google Scholar]
- 25.Suma, Y., Shimizu, K., and Tsukada, K. (1986) J. Biochem. (Tokyo) 100 67-75 [DOI] [PubMed] [Google Scholar]
- 26.Halim, A. B., LeGros, L., Geller, A., and Kotb, M. (1999) J. Biol. Chem. 274 29720-29725 [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Trevijano, E. R., Latasa, M. U., Carretero, M. V., Berasain, C., Mato, J. M., and Avila, M. A. (2000) FASEB J. 14 2511-2518 [DOI] [PubMed] [Google Scholar]
- 28.Yang, H., Ara, A. I., Magilnick, N., Xia, M., Ramani, K., Chen, H., Lee, T. D., Mato, J. M., and Lu, S. C. (2008) Gastroenterology 134 281-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeGros, H. L., Jr., Halim, A. B., Geller, A. M., and Kotb, M. (2000) J. Biol. Chem. 275 2359-2366 [DOI] [PubMed] [Google Scholar]
- 30.Finkelstein, J. D. (1990) J. Nutr. Biochem. 1 228-237 [DOI] [PubMed] [Google Scholar]
- 31.Finkelstein, J. D. (2006) J. Nutr. 136 (Suppl. 6) 1750S-1754S [DOI] [PubMed] [Google Scholar]
- 32.Loehrer, F. M., Schwab, R., Angst, C. P., Haefeli, W. E., and Fowler, B. (1997) J. Pharmacol. Exp. Ther. 282 845-850 [PubMed] [Google Scholar]
- 33.Barić, I., Fumić, K., Glenn, B., Ćuk, M., Schulze, A., Finkelstein, J. D., James, S. J., Mejasi-Bosnjak, V., Pazanin, L., Pogribny, I. P., Rados, M., Sarnavka, V., Scukanec-Spoljar, M., Allen, R. H., Stabler, S., Uzelac, L., Vugrek, O., Wagner, C., Zeisel, S., and Mudd, S. H. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4234-4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coffield, V. M., Helms, W. S., Jiang, Q., and Su, L. (2004) J. Exp. Med. 200 1315-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pear, W. S., Nolan, G. P., Scott, M. L., and Baltimore, D. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 8392-8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taxman, D. J., Livingstone, L. R., Zhang, J., Conti, B. J., Iocca, H. A., Williams, K. L., Lich, J. D., Ting, J. P., and Reed, W. (2006) BMC Biotechnol. 6 7-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong, A. W., Brickey, W. J., Taxman, D. J., van Deventer, H. W., Reed, W., Gao, J. X., Zheng, P., Liu, Y., Li, P., Blum, J. S., McKinnon, K. P., and Ting, J. P. (2003) Nat. Immunol. 4 891-898 [DOI] [PubMed] [Google Scholar]
- 38.Butron, K. (1956) Biochem. J. 62 315-31913293190 [Google Scholar]
- 39.Kotb, M., Geller, A. M., Markham, G. D., Kredich, N. M., De La Rosa, J., and Beachey, E. H. (1990) Biochim. Biophys. Acta 1040 137-144 [DOI] [PubMed] [Google Scholar]
- 40.Fronhoffs, S., Totzke, G., Stier, S., Wernert, N., Rothe, M., Bruning, T., Koch, B., Sachinidis, A., Vetter, H., and Ko, Y. (2002) Mol. Cell Probes 16 99-110 [DOI] [PubMed] [Google Scholar]
- 41.Shultz, L. D., Lyons, B. L., Burzenski, L. M., Gott, B., Chen, X., Chaleff, S., Kotb, M., Gillies, S. D., King, M., Mangada, J., Greiner, D. L., and Handgretinger, R. (2005) J. Immunol. 174 6477-6489 [DOI] [PubMed] [Google Scholar]
- 42.Zhang, M., Zhang, Z., Garmestani, K., Goldman, C. K., Ravetch, J. V., Brechbiel, M. W., Carrasquillo, J. A., and Waldmann, T. A. (2004) Cancer Res. 64 5825-5829 [DOI] [PubMed] [Google Scholar]
- 43.LeGros, H. L., Jr., Geller, A. M., and Kotb, M. (1997) J. Biol. Chem. 272 16040-16047 [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Chantar, M. L., Garcia-Trevijano, E. R., Latasa, M. U., Martin-Duce, A., Fortes, P., Caballeria, J., Avila, M. A., and Mato, J. M. (2003) Gastroenterology 124 940-948 [DOI] [PubMed] [Google Scholar]
- 45.Sanz, A., Caro, P., Ayala, V., Portero-Otin, M., Pamplona, R., and Barja, G. (2006) FASEB J. 20 1064-1073 [DOI] [PubMed] [Google Scholar]
- 46.Breillout, F., Antoine, E., and Poupon, M. F. (1990) J. Natl. Cancer Inst. 82 1628-1632 [DOI] [PubMed] [Google Scholar]
- 47.Guo, H., Lishko, V. K., Herrera, H., Groce, A., Kubota, T., and Hoffman, R. M. (1993) Cancer Res. 53 5676-5679 [PubMed] [Google Scholar]
- 48.Sun, X., Yang, Z., Li, S., Tan, Y., Zhang, N., Wang, X., Yagi, S., Yoshioka, T., Takimoto, A., Mitsushima, K., Suginaka, A., Frenkel, E, and Hoffman, R. M. (2003) Cancer Res. 63 8377-8383 [PubMed] [Google Scholar]
- 49.Goseki, N., Yamazaki, S., Shimojyu, K., Kando, F., Maruyama, M., Endo, M., Koike, M., and Takahashi, H. (1995) Jpn. J. Cancer Res. 86 484-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotb, M., Geller, A., and LeGros, L. (February 24, 2004) U. S. Patent 6,696,279
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.