

Abstract
Triphosphate tunnel metalloenzymes (TTMs) are a superfamily of phosphotransferases with a distinctive active site located within an eight-stranded β barrel. The best understood family members are the eukaryal RNA triphosphatases, which catalyze the initial step in mRNA capping. The RNA triphosphatases characteristically hydrolyze nucleoside 5′-triphosphates in the presence of manganese and are inept at cleaving inorganic tripolyphosphate. We recently identified a TTM protein from the bacterium Clostridium thermocellum (CthTTM) with the opposite substrate preference. Here we report that CthTTM catalyzes hydrolysis of guanosine 5′-tetraphosphate to yield GTP and Pi (Km = 70 μm, kcat = 170 s-1) much more effectively than it converts GTP to GDP and Pi (Km = 70 μm, kcat = 0.3 s-1), implying that a nucleoside interferes when positioned too close to the tunnel entrance. CthTTM is capable of quantitatively cleaving diadenosine hexaphosphate but has feeble activity with shorter derivatives diadenosine tetraphosphate and diadenosine pentaphosphate. We propose that the tunnel opens to accommodate the dumbbell-shaped diadenosine hexaphosphate and then closes around it to perform catalysis. We find that CthTTM can exhaustively hydrolyze a long-chain inorganic polyphosphate, a molecule that plays important roles in bacterial physiology. CthTTM differs from other known polyphosphatases in that it yields a ∼2:1 mixture of Pi and PPi end products. Bacterial/archaeal TTMs have a C-terminal helix located near the tunnel entrance. Deletion of this helix from CthTTM exerts pleiotropic effects. (i) It suppresses hydrolysis of guanosine 5′-tetraphosphate and inorganic PPPi; (ii) it stimulates NTP hydrolysis; and (iii) it biases the outcome of the long-chain polyphosphatase reaction more strongly in favor of Pi production. We discuss models for substrate binding in the triphosphate tunnel.
The RNA triphosphatase components of the mRNA capping systems of fungi, protozoa, and several DNA viruses are the founding members of the triphosphate tunnel metalloenzyme (TTM)2 superfamily of phosphohydrolases (1-21). The signature biochemical property of this branch of the TTM superfamily is the ability to hydrolyze NTPs to NDPs and Pi in the presence of manganese. The crystal structure of the Saccharomyces cerevisiae RNA triphosphatase Cet1 bound to manganese and sulfate (a proposed mimetic of the product complex with phosphate) revealed what was then a novel tertiary structure in which the active site is situated within a topologically closed tunnel composed of eight antiparallel β strands (2). The Cet1-like TTM fold is conserved in the crystal structure of mimivirus RNA triphosphatase, which has an acetate anion bound in its tunnel (21). A plausible mechanism has been proposed whereby Cet1-like proteins catalyze direct attack of a nucleophilic water on the γ-phosphorus of NTP or pppRNA substrates (4). The putative water nucleophile is oriented by an essential glutamate contributed by the fifth β strand of the tunnel wall. A proposed pentacoordinate transition state is stabilized by interactions of the γ-phosphate oxygens with the divalent cation and a layer of essential lysine and arginine side chains that emanate from the third and sixth β strands of the tunnel walls. The octahedral metal coordination complex is formed by a set of essential conserved glutamates from strands β1 and β8 and an aspartate from β7 that are clustered on the floor of the tunnel.
The tertiary structure and active site architecture of yeast and mimivirus RNA triphosphatases are recapitulated in the crystal structures of several archaeal and bacterial proteins, of mostly unknown biochemical function, that have been solved as a part of structural genomics initiatives; these include proteins from Pyrococcus (Protein Data Bank codes 1YEM, 2DC4, and 2EEN), Vibrio (2ACA), Nitrosomonas (2FBL), and Yersinia (2FJT) (3, 22). Primary structure similarity searches against these proteins readily identify a large bacterial/archaeal TTM clade widely distributed among taxa (23). Given that bacterial and archaeal RNAs do not have 5′ caps, it is reasonable to think that members of this clade might play novel and diverse roles in prokaryotic physiology, to which insights might be gained by examining the activity and substrate specificity of exemplary family members. This theme is underscored by the identification and structural characterization of a mammalian enzyme, thiamine triphosphatase, as a TTM protein with extreme specificity for metal-dependent hydrolysis of thiamine triphosphate to thiamine diphosphate and Pi (Protein Data Bank codes 3BHD and 2JMU) (24, 25).
Here we focus our attention on Clostridium thermocellum TTM (CthTTM), the first bacterial TTM-type phosphohydrolase to be subjected to detailed biochemical and structure-function analysis (26). Purified recombinant CthTTM is a vigorous inorganic tripolyphosphatase that converts PPPi to PPi plus Pi in the presence of manganese or magnesium. In fact, CthTTM is 2 orders of magnitude more active in cleaving PPPi than it is in hydrolyzing the β-γ phosphoanhydride of ATP (26). By contrast, the Cet1-like RNA triphosphatases are much more active with ATP than PPPi. Indeed, PPPi is a potent competitive inhibitor of their ATPase activity (11, 14). We are interested in understanding the basis for the distinctive substrate preference of CthTTM and have been guided in this effort by crystal structures of homologous TTM proteins.
The primary structure of the 156-amino acid CthTTM protein is
aligned in Fig. 1A to
two related prokaryotic TTM proteins, from Nitrosomonas europea
(NeuTTM; 151 amino acids) and Pyrococcus horikoshii
(PhoTTM; 165 amino acids), for which crystal structures have been
solved (Fig. 1B),
albeit without any functional characterization. As expected, the positions of
side chain identity/similarity in the three proteins (indicated by
dots) are located within the eight β strands that comprise the
triphosphate tunnel of PhoTTM
(Fig. 1A). The
NeuTTM fold is distinguished by the fact that its antiparallel β
barrel is not a closed tunnel, as in PhoTTM, but rather an open
C-shaped “cup” that is prevented from closing by virtue of the
insertion of a C-terminal helix into one end of the tunnel
(Fig. 1B). In the open
state, the essential amino acid side chains that comprise the active site are
displaced from the catalytically active arrangement seen in the
Cet1-Mn2+/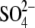 complex
(2). We speculated previously
that binding of the polyanionic substrate to the open conformation of the
bacterial/archaeal-type TTM apoenzyme might trigger closure of the tunnel and
the formation of a proper Michaelis complex
(26). Similar inferences
regarding substrate-induced conformational changes have been drawn for
mammalian thiamine triphosphatase
(25), based on documentation
of alternative atomic structures analogous to the open and closed states
depicted in Fig. 1B.
By contrast, Cet1 has a topologically closed “O-shaped” tunnel
that cannot open, because the crossover strands of the Cet1 tunnel penetrate
into a globular pedestal domain upon which the tunnel rests
(2). Unfortunately, because no
TTM protein structure has been solved with a true substrate (NTP, RNA, or
thiamine triphosphate) bound at the active site, we cannot confidently know
the orientation of the substrate and the site of asymmetric hydrolysis of the
phosphoanhydride linkage. Nevertheless, many TTM structures have been solved
with anionic ligands in the tunnel (e.g. sulfate, phosphate, citrate,
acetate, or chloride).
complex
(2). We speculated previously
that binding of the polyanionic substrate to the open conformation of the
bacterial/archaeal-type TTM apoenzyme might trigger closure of the tunnel and
the formation of a proper Michaelis complex
(26). Similar inferences
regarding substrate-induced conformational changes have been drawn for
mammalian thiamine triphosphatase
(25), based on documentation
of alternative atomic structures analogous to the open and closed states
depicted in Fig. 1B.
By contrast, Cet1 has a topologically closed “O-shaped” tunnel
that cannot open, because the crossover strands of the Cet1 tunnel penetrate
into a globular pedestal domain upon which the tunnel rests
(2). Unfortunately, because no
TTM protein structure has been solved with a true substrate (NTP, RNA, or
thiamine triphosphate) bound at the active site, we cannot confidently know
the orientation of the substrate and the site of asymmetric hydrolysis of the
phosphoanhydride linkage. Nevertheless, many TTM structures have been solved
with anionic ligands in the tunnel (e.g. sulfate, phosphate, citrate,
acetate, or chloride).
FIGURE 1.

Structural similarities between CthTTM, NeuTTM, and PhoTTM. A, the amino acid sequence of CthTTM is aligned to the homologous polypeptides encoded by N. europaea (Neu) and P. horikoshii (Pho). Gaps in the alignment are indicated by dashes. Positions of amino acid side chain identity or similarity in all three proteins are indicated by dots. The site of the C-terminal truncation of CthTTM is denoted by the bent arrow. The peptides comprising the eightβ strands of the PhoTTM tunnel are depicted as thick arrows below the sequence. B, the tertiary structures of NeuTMM (left) and PhoTTM (right) are shown with β strands colored magenta and helices colored cyan. The amino (N) and carboxyl (C) termini are indicated. Three chloride anions in the PhoTTM tunnel are depicted as spheres. C, stereo view of the closed tunnel of PhoTTM. The conserved side chains are shown in a stick representation; the side chain name and number are given for CthTTM. Waters and chlorides are depicted as red and green spheres, respectively.
In this respect, it is striking that the closed tunnel conformation in the PhoTTM structure correlates with the presence of a linear array of three chloride anions in the tunnel cavity (Fig. 1B), which are putative mimetics of three phosphates of the enzyme's substrate. A detailed stereo view of the PhoTTM tunnel is shown in Fig. 1C (albeit with the conserved amino acids of the tunnel numbered according to their positions in CthTTM). The tunnel is oriented in Fig. 1 so that the chloride anion closest to the viewer corresponds to the sulfate in Cet1; thus, we regard this atom as a potential analog of the γ-phosphate and, by extension, the two other chlorides serially deeper in the tunnel as rough analogs of the β- and α-phosphates, respectively. Many of the conserved polar side chains surrounding the chlorides have been shown by mutational analysis to be essential for CthTTM triphosphatase activity (26).
It is critical to note that the C-terminal α helix of PhoTTM sterically occludes the back opening of the tunnel even after closure has occurred (Fig. 1, B and C). This C-terminal “plug” helix is a conserved feature of all of the structurally characterized bacterial and archaeal TTM proteins as well as mammalian thiamine triphosphatase. By contrast, a plug helix is conspicuously absent from yeast and mimivirus RNA triphosphatases, which have unobstructed “entrance” and “exit” sides to their tunnels (2, 21). The plug helix in PhoTTM blocks the side corresponding to the putative entrance to the Cet1 tunnel; the open side closer to the viewer in Fig. 1C corresponds to the putative exit side of the Cet1 tunnel. We envision that the plug helix must move away from the tunnel entrance in order for an NTP to bind productively and for efficient hydrolysis and product release. This idea was supported by the initial finding that an alanine mutation of CthTTM residue Lys8, which in Pho-TTM tethers the plug helix to the tunnel opening via a hydrogen bond to the main chain of the plug helix (Fig. 1C), stimulated the ATPase activity of CthTTM (26).
Here we delve further into the catalytic repertoire of CthTTM by studying its ability to cleave longer nucleotide 5′-polyphosphate and inorganic polyphosphate substrates. Our results suggest that opening of the tunnel is relevant to productive substrate binding, and they underscore the role of the C-terminal helix as a substrate filter.
EXPERIMENTAL PROCEDURES
Materials—Guanosine 5′-tetraphosphate (Gpppp), P1,P4-di(adenosine-5′) tetraphosphate (AppppA), P1,P5-di(adenosine-5′) pentaphosphate (ApppppA), P1,P6-di(adenosine-5′) hexaphosphate (AppppppA), and Crotalus adamanteus nucleotide pyrophosphatase were purchased from Sigma. [γ-32P]ATP (3000 Ci/mmol) was obtained from PerkinElmer Life Sciences. Calf intestine phosphatase (CIP) was purchased from Roche Applied Science. Ni2+-nitrilotriacetic acid-agarose was purchased from Qiagen. Malachite green reagent was purchased from BIOMOL Research Laboratories. Polyethyleneimine (PEI)-cellulose TLC plates were from Baker.
Recombinant CthTTM—Full-length CthTTM and the C-terminal truncation mutant CthTTM-(1-137) were produced in Escherichia coli BL21(DE3) as N-terminal His10-tagged fusions and purified from soluble bacterial extracts by sequential Ni2+-agarose and DEAE-Sephacel column chromatography steps, as described previously (26). The protein concentrations were determined by using the Bio-Rad dye reagent with bovine serum albumin as the standard. The yields of CthTTM and CthTTM-(1-137) from a 250-ml bacterial culture were 11 and 9 mg, respectively. The protein preparations were stored at -80 °C.
Recombinant Polyphosphate Kinase (PPK)—The E. coli ppk open reading frame was amplified by PCR from genomic DNA with primers that introduced an NdeI site at the start codon and a BamHI site immediately after the stop codon. The ppk gene was inserted into pET16B so as to fuse the coding sequence to a 5′ leader encoding a His10 tag. The pET-PPK plasmid was transformed into E. coli BL21(DE3). A 500-ml culture derived from a single transformant was grown at 37 °C in Luria-Bertani medium containing 0.1 mg/ml ampicillin until the A600 reached ∼0.6, at which time the culture was adjusted to 2% ethanol and 0.2 mm isopropyl-β-d-thiogalactopyranoside. The culture was then incubated at 17 °C for 16 h with continuous shaking. Cells were harvested by centrifugation, and the pellet was stored at -80 °C. All subsequent procedures were performed at 4 °C. Thawed bacteria were resuspended in 50 ml of buffer C (50 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 10% sucrose). Lysozyme, phenylmethylsulfonyl fluoride, and Triton X-100 were added to final concentrations of 1 mg/ml, 0.2 mm, and 0.1%, respectively. The lysate was sonicated to reduce viscosity, and the insoluble material was removed by centrifugation. The soluble extract was applied to a 5-ml column of Ni2+-nitrilotriacetic acid-agarose (Qiagen) that had been equilibrated with buffer A (50 mm Tris-HCl, pH 8.0, 0.25 m NaCl, 10% glycerol, 0.05% Triton X-100). The column was washed with 15 ml of the same buffer and then eluted stepwise with 2-ml aliquots of 50, 100, 200, and 500 mm imidazole in buffer A. The polypeptide compositions of the column fractions were monitored by SDS-PAGE. The recombinant His10-PPK polypeptide was recovered predominantly in the 500 mm imidazole fraction, which was then dialyzed against 200 mm NaCl in buffer B (50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1 mm dithiothreitol, 10% glycerol, 0.05% Triton X-100). The yield was 0.2 mg of PPK from a 500-ml bacterial culture. The PPK preparation was stored at -80 °C.
Preparation of [32P]Polyphosphate—A reaction mixture (0.5 ml) containing 50 mm Tris-HCl (pH 7.5), 40 mm (NH4)2SO4, 10 mm MgCl2, 4 mm ATP, 100 μCi of [γ-32P]ATP, and 18 pmol of E. coli PPK was incubated at 37 °C for 45 min. The reaction was quenched by adding 50 μl of 0.5 m EDTA. Conversion of ATP to polyphosphate was gauged by PEI-cellulose TLC analysis of aliquots of the reaction mixture with or without PPK. Ascending TLC with 0.75 m LiCl, 1 m HCOOH separated the free radiolabeled ATP from the polymeric PPK reaction product that remained at the origin. Approximately 20% of input [γ-32P]ATP was converted to polyphosphate via this procedure. The [32P]polyphosphate was purified as described by Keasling et al. (27), with some modifications. The quenched reaction mixture was extracted once with an equal volume of phenol-CHCl3 and then twice with CHCl3. Isopropyl alcohol (0.7 volume) was then added to the aqueous phase, and the solution was left at room temperature for 1 h. The mixture was microcentrifuged at 15,000 rpm, and the supernatant was discarded. Cold 100% ethanol was added to the polyphosphate-containing precipitate, and the tube was kept on dry ice for 30 min. This was followed by centrifugation at 4 °C at 15,000 rpm. The supernatant was discarded again, and the precipitate was rinsed with cold 70% ethanol. After repeat centrifugation and removal of the ethanol supernatant, the precipitated [32P]polyphosphate was dried and then dissolved in water. PEI-cellulose TLC confirmed that all free radiolabeled ATP had been removed. The total phosphate concentration of the polyphosphate preparation was determined by digestion with CIP, which quantitatively liberated the radiolabel as 32Pi, as verified by TLC. The phosphate content of the CIP hydrolysate was determined colorimetrically with the malachite green reagent. The concentrated [32P]polyphosphate preparation (11.7 mm phosphate) was stored at -20 °C and diluted prior to use in polyphosphatase assays.
RESULTS
Nucleoside Tetraphosphatase Activity of CthTTM—Prompted by our initial finding that CthTTM was ∼150-fold more active with PPPi than ATP (26), we speculated that the nucleoside might sterically hinder productive substrate binding in the active site. This hypothesis engenders a prediction that CthTTM should regain activity if the interfering nucleoside moiety were shifted away from the terminal phosphoanhydride linkage that undergoes enzymatic hydrolysis. Thus, we queried whether CthTTM could catalyze phosphate release from a nucleoside 5′-tetraphosphate substrate, Gpppp. We found that CthTTM did cleave 100 μm Gpppp in the presence of 200 μm manganese and that the extent of Pi release as a function of input enzyme was acutely biphasic (Fig. 2A). Pi release was steeply proportional to the amount of enzyme added up to 1 ng of CthTTM, at which point the molar yield of phosphate was equal to the initial amount of Gpppp substrate. Further phosphate release displayed a much shallower dependence on increasing CthTTM from 50 to 400 ng. The specific activities calculated from the slope of the steep and shallow phases corresponded to turnover numbers of 150 and 0.07 s-1, respectively. We ascribe the initial steep phase to efficient and quantitative conversion of Gpppp to GTP and Pi at low enzyme concentrations, followed by a shallow phase of inefficient conversion of GTP to GDP and Pi at higher enzyme concentrations. Indeed, the enzyme dependence of Pi release from 100 μm GTP substrate under the same reaction conditions was 0.06 s-1 (Fig. 2A), a value virtually identical to that of the shallow phase of the Gpppp titration profile. Thus, CthTTM is 2100-fold more active on a nucleoside 5′-tetraphosphate than on a standard NTP under these conditions.
FIGURE 2.

Nucleoside tetraphosphatase activity of CthTTM. A, reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 8.5), 200 μm MnCl2, 100 μm Gpppp (•), or 100 μm GTP (○) and CthTTM as specified were incubated for 30 min at 37 °C. The reaction was quenched by adding 1 ml of malachite green reagent. Phosphate release was determined by measuring A620 and interpolating the value to a phosphate standard curve. B, reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 8.5), 20 mm MgCl2, 100 μm Gpppp (▪), or 100 μm Gppp (□) and CthTTM as specified were incubated for 30 min at 37 °C. C, reaction mixtures (400 μl) containing 50 mm Tris-HCl (pH 8.5), 200 μm MnCl2, 100 μm Gpppp, and 2 ng (○), 4 ng (gray circles), or 8 ng (•) of CthTTM were incubated at 37 °C. Aliquots (50 μl) were withdrawn at the times specified and quenched immediately. D, reaction mixtures (400 μl) containing 50 mm Tris-HCl (pH 8.5), 20 mm MgCl2, 100 μm Gpppp, and 4 ng (□), 12 ng (gray squares), or 24 ng (▪) of CthTTM were incubated at 37 °C. Aliquots (50 μl) were withdrawn at the times specified and quenched immediately.
A different profile of enzyme dependence was observed when CthTTM was reacted with 100 μm Gpppp in the presence of magnesium (Fig. 2B). Here, Pi release was monophasic and proportional to enzyme (estimated turnover number 70 s-1). The reaction attained an insurmountable plateau when 1 mol of phosphate was released/mol of input Gpppp substrate, presumably corresponding to quantitative conversion of Gpppp to a GTP product. To verify this interpretation, the fate of the Gpppp nucleotide moiety in the presence of CthTTM and magnesium was monitored by PEI-cellulose TLC in buffer containing 1.25 m LiCl, 1 m HCOOH. This procedure revealed complete conversion of Gpppp to GTP, with no detectable GDP formation (not shown). Moreover, a parallel titration experiment with GTP as the substrate verified that the enzyme was unable to catalyze release of Pi from GTP in the presence of magnesium (Fig. 2B). These results agree with previous findings (26) that CthTTM performs ATP hydrolysis with manganese but not magnesium.
Further characterization of the tetraphosphatase showed that Pi accumulated steadily with time and that the rate of release of Pi from Gpppp was proportional to enzyme concentration with either manganese or magnesium as the cofactor (Fig. 2, C and D). Hydrolysis of Gpppp by CthTTM required divalent metal ion and was optimal in Tris buffer at pH 8.5 with either manganese or magnesium (not shown). Various divalent metal ions were tested at concentrations ranging from 0.1 to 20 mm. As noted above, manganese supported the highest specific activity (150 s-1) at its optimal concentration of 200 μm, whereas magnesium was half as effective (70 s-1) at its optimum concentration of 20 mm. Cobalt supported much lower activity (5 s-1) at its optimum of 200 μm. We detected no tetraphosphatase activity with calcium, cadmium, copper, nickel, or zinc.
The extent of Pi release in the presence of 0.2 mm manganese displayed a hyperbolic dependence on Gpppp concentration (not shown). From a double-reciprocal plot of the data, we calculated a Km of 70 μm Gpppp and a kcat of 170 s-1. The steadystate kinetic parameters for hydrolysis of GTP to GDP and Pi under optimal reaction conditions (pH 8.0; 2 mm MnCl2) were as follows: Km = 70 μm GTP and kcat = 0.3 s-1. Thus, CthTTM discriminated between the nucleoside tetraphosphate and triphosphate by a factor of 570 at the level of reaction chemistry rather than initial substrate binding.
The kinetic parameters for Gpppp hydrolysis in 20 mm magnesium were as follows: Km = 86 μm Gpppp and kcat = 96 s-1. The turnover number for magnesium-dependent Gpppp hydrolysis was similar to the value of 63 s-1 reported for magnesium-dependent hydrolysis of PPPi (26). The failure of CthTTM to hydrolyze GTP in the presence of magnesium was at least partly reflective of weak substrate binding, insofar as the addition of excess GTP had only a weak inhibitory effect on magnesium-dependent hydrolysis of Gpppp. Specifically, the extents of Pi release from 0.2 mm Gpppp in the presence of 0.4, 0.6, 0.8, and 1 mm GTP were 98, 90, 85, and 78% of the control reaction lacking GTP (not shown).
Effects of Alanine Mutations on CthTTM Tetraphosphatase Activity—We surveyed a collection of alanine mutants of CthTTM (26) for their ability to hydrolyze Gpppp (Fig. 3). Mutants E6A, R39A, R41A, K52A, E62A, K87A, R89A, and E117A were shown previously to have <1% of the specific activity of wild-type CthTTM in manganese-dependent ATP hydrolysis; mutants E4A and E115A were 3% as active as wild-type CthTTM in ATP hydrolysis (26). Here we find that each of these changes, involving amino acids in the tunnel active site (Fig. 1C), had similarly severe effects on the hydrolysis of Gpppp, no matter what the divalent cation cofactor (Fig. 3). E64A, shown previously to have 3% of wild-type ATPase activity and 7% of wild-type tripolyphosphatase activity in the presence of manganese (26), was found here to be 12% (Mn2+) or 17% (Mg2+) as active as wild-type CthTTM in hydrolyzing guanosine tetraphosphate (Fig. 3). E100A, which was 22 and 40% as active as wild-type CthTTM in manganese-dependent cleavage of ATP and PPPi, respectively (26), was seen here to have 20% of wild-type activity in hydrolyzing Gpppp in the presence of manganese (Fig. 3). These concordant results suggest that hydrolysis of nucleoside tri- and tetraphosphates and tripolyphosphate requires the same core constellation of active site functional groups.
FIGURE 3.

Effects of alanine mutations on CthTTM nucleoside tetraphosphatase activity. A, reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 8.5), 200 μm MnCl2, 100 μm Gpppp, and 0.3 ng of wild-type (WT) or mutant CthTTM, as indicated, were incubated for 30 min at 37 °C. B, reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 8.5), 20 mm MgCl2, 100 μm Gpppp, and 0.7 ng of wild-type or mutant CthTTM, as specified, were incubated for 30 min at 37 °C. Each datum is the average of three separate experiments. Bars, S.E.
The K8A mutant, which was reported previously to have lower tripolyphosphatase activity in the presence of manganese (14% of wild type) and magnesium (<1%), was found here to have reduced activity in hydrolyzing Gpppp, again with the activity decrement in manganese (40% of wild type) being less than that in magnesium (9% of wild type) (Fig. 3). The concordant trend of K8A toward lower activity with Gpppp and PPPi contrasts with the 21-fold increase in kcat for ATP hydrolysis by K8A compared with wild-type CthTTM (26). This disparity underscores that Lys8 is a determinant of substrate specificity, simultaneously making positive contributions to Gpppp and PPPi cleavage while suppressing nucleoside triphosphate hydrolysis. Reference to the PhoTTM structure shows that the equivalent of Lys8 makes two potentially significant contacts: (i) a hydrogen bond to the main chain of the plug helix and (ii) a contact to the chloride anion proposed to mimic the α-phosphate of ATP (Fig. 1C). Additional experiments will be presented that address the contributions of the plug helix to the disparate effects of K8A on activity with different substrates and different divalent cations.
Steric Restrictions on CthTTM Hydrolysis of Dinucleoside Polyphosphates—The vigorous activity of CthTTM with tripolyphosphate and a nucleoside tetraphosphate, but not nucleoside triphosphates, highlights the hindrance of catalysis by a nucleoside positioned too close to the scissile phosphoanhydride. In order to probe whether catalysis can occur when both ends of the phosphoanhydride bridge are “blocked,” we reacted CthTTM with a series of dinucleoside 5′,5′-polyphosphates: AppppA, ApppppA, and AppppppA. These dinucleotides yielded no inorganic phosphate when treated with CIP, an enzyme that requires a “free” (R-O-PO3) phosphate terminus (Fig. 4, CIP). However, treatment with snake venom nucleotide pyrophosphatase, which cleaves within the polyphosphate bridge, generates adenylates with free R-O-PO3 ends that are sensitive to CIP digestion, so that the adenylate 5′-phosphates are converted nearly quantitatively to Pi by treatment with nucleotide pyrophosphatase plus CIP (Fig. 4). No phosphate was released by nucleotide pyrophosphatase per se (Fig. 4).
FIGURE 4.

Dinucleoside polyphosphatase activity of CthTTM. Reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.5); 5 mm MnCl2; 250 μm AppppA (A), ApppppA (B), or AppppppA (C); and CthTTM (1.6 μg; 8 μm enzyme) with or without CIP (2 units), as specified, were incubated at 37 °C for 60 min. CIP-alone control reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.9); 10 mm MnCl2; 100 mm NaCl; 1 mm dithiothreitol; 250 μm AppppA (A), ApppppA (B), or AppppppA (C); and 2 units of CIP were incubated at 37 °C for 60 min. Nucleotide pyrophosphatase control reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.9); 10 mm MgCl2; 100 mm NaCl; 1 mm dithiothreitol; 250 μm AppppA (A), ApppppA (B), or AppppppA (C); and nucleotide pyrophosphatase (0.2 units) with or without CIP (2 units) were incubated at 37 °C for 60 min. The reactions were quenched by adding 1 ml of malachite green reagent. Phosphate release was determined by measuring A620 and interpolating the value to a phosphate standard curve. The molar amounts of Pi release from the diadenosine polyphosphate substrates by combined treatment with nucleotide pyrophosphatase plus CIP corresponded closely to the total phosphate residues in the reaction mixtures, which were 10, 12.5, and 15 nmol for AppppA, ApppppA, and AppppppA, respectively.
The instructive findings were that CthTTM (added at 8 μm concentration) failed to hydrolyze the tetraphosphate bridge of 250 μm AppppA to form a CIP-sensitive product (Fig. 4A), and it displayed feeble activity in cleaving within the pentaphosphate bridge of ApppppA, such that only 10% of the available nucleotide phosphate was released as Pi in the presence of CIP (Fig. 4B). By contrast, CthTTM did hydrolyze the hexaphosphate linker of AppppppA, resulting in the release of 89% of the available nucleotide phosphate groups as Pi in the presence of CIP (Fig. 4C). Omission of CIP treatment reduced the extent of CthTTM-catalyzed Pi release from AppppppA to a level corresponding to 1.9 nmol of Pi/nmol of input AppppppA (Fig. 4C), indicating that one or more of the adenylate nucleotides formed by incision of the hexaphosphate bridge was itself a substrate for the CthTTM phosphohydrolase. These results highlight that the capacity of CthTTM to cleave a polyphosphate chain with nucleosideblocked ends is strictly limited by the length of the phosphate linker. Whereas CthTTM cleaved AppppppA in the presence of 5 mm manganese, we detected no activity with this substrate in the presence of 5 mm magnesium or cobalt (not shown). The E6A mutant of CthTTM catalyzed no detectable AppppppA cleavage (not shown).
Tracking the Outcome of AppppppA Cleavage—The fate of the 250 μm AppppppA nucleotide during its reaction with 8 μm CthTTM in the presence of manganese was monitored by withdrawing aliquots at various times, quenching the reaction with EDTA, binding the nucleotides to an anion exchange column, and then following their elution profile (by UV absorbance) as the column was developed with a linear salt gradient (Fig. 5). Absent CthTTM, the substrate consisted of a nearly homogeneous UV-absorbing species eluting at high salt that corresponds to diadenosine hexaphosphate; trace amounts of AMP were seen eluting at lower salt concentration (Fig. 5; 0 min). The amount of AppppppA decayed steadily with time, concomitant with the appearance of a novel UV-absorbing product eluting at lower salt that corresponded to ADP. The reaction attained an end point at 75 min, at which time all of the AppppppA had been converted to ADP; there was no change in the amount of the minor AMP contaminant at any point during the reaction. Low levels of ATP (eluting between ADP and AppppppA) were detected at 5, 15, and 30 min (Fig. 5). The ATP level declined at 50 min and then disappeared at the 75 min reaction end point. This kinetic profile suggested that ATP might be an intermediate in the conversion of AppppppA to ADP. In the simplest scheme, AppppppA is cleaved symmetrically by CthTTM to yield two molecules of ATP, which are then hydrolyzed to the ADP end product. The low abundance of the putative ATP intermediate implies that the rate of AppppppA hydrolysis must be significantly slower than the rate of ATP hydrolysis. By integrating the areas under the UV absorbance curves, we calculated a turnover number for the consumption of AppppppA of 0.68 min-1, a value that is indeed much less than the kcat of 25 min-1 for ATP hydrolysis by CthTTM in the presence of manganese (26). Although we did not detect any UV-absorbing material eluting between ATP and AppppppA that could correspond to Apppp (Fig. 5), it is possible that the reaction proceeds via an alternative scheme in which AppppppA is cleaved asymmetrically to yield ADP and Apppp, followed by very rapid conversion of Apppp to ATP and finally slower conversion of ATP to ADP.
FIGURE 5.

Kinetics of CthTTM reaction with diadenosine hexaphosphate. A reaction mixture (400 μl) containing 50 mm Tris-HCl (pH 7.5), 5 mm MnCl2, 250 μm AppppppA, and 64 μg of CthTTM (8 μm enzyme) was incubated at 37 °C. Aliquots (50 μl) were withdrawn at the times specified and quenched immediately with 5 μl of 100 mm EDTA. The time 0 sample was taken prior to adding CthTTM. The nucleotides were analyzed by anion exchange chromatography. Each sample was applied to a MonoQ column (5.5 × 0.5 cm) equilibrated in 20 mm Tris-HCl (pH 7.5), 10 mm EDTA. The nucleotides were eluted with a 120-ml linear gradient of 0-1 m NaCl in 20 mm Tris-HCl (pH 7.5), 10 mm EDTA. UV absorbance (A254) was monitored during the gradient elution. The elution profiles are stacked from top to bottom as a function of reaction time. The MonoQ column was standardized by parallel analysis of a mixture of AppppppA, ATP, ADP, and AMP nucleotides (not shown).
It is striking that the observed rate of AppppppA cleavage is 4 orders of magnitude slower that the rate of hydrolysis of a Gpppp or tripolyphosphate. Although steric hindrance by the second nucleoside in AppppppA could account for this disparity, another contributing factor might be differential reactivity of CthTTM at a free terminal phosphate R-O-PO3 (present in Gpppp and PPPi) versus an internal phosphoanhydride linkage R-O-(PO2)-O-R′ (present in AppppppA). Note that CthTTM activity with longer dinucleoside polyphosphate substrates could not be assessed readily, because these molecules are not available from commercial sources.
Inorganic Polyphosphatase Activity of CthTTM—Inorganic polyphosphate is a ubiquitous intracellular macromolecule with an important role in the response of many bacterial species to environmental stress (28, 29). Therefore, we considered the prospect that long-chain polyphosphates might be a physiological substrate for CthTTM. We prepared [32P]polyphosphate via enzymatic synthesis from [γ32P]ATP by E. coli PPK (27). We found that reaction of CthTTM with [32P]polyphosphate in the presence of 10 mm manganese resulted in the release of 32Pi and 32PPi products that could be resolved from the [32P]polyphosphate substrate by PEI-cellulose TLC (Fig. 6A). The identity of the slower migrating product as PPi was verified by treating the reaction products with yeast inorganic pyrophosphatase, which completely depleted the slower migrating species and elicited a concomitant increase in the level of 32Pi (not shown).
FIGURE 6.

Inorganic polyphosphatase activity of CthTTM. A, a reaction mixture (10 μl) containing 50 mm Tris-HCl (pH 7.5), 10 mm MnCl2, [32P]polyphosphate (59 μm phosphate groups), and CthTTM (1.6 μg; 8 μm enzyme) was incubated at 37 °C for 60 min (lane +). CthTTM was omitted from a control reaction mixture (lane -). Aliquots (1 μl) were applied to a PEI-cellulose TLC plate, which was developed in 0.75 m LiCl, 1 m HCOOH. An autoradiograph of the TLC plate is shown. The species corresponding to Pi,PPi, and polyphosphate (polyP) are indicated on the right. B, a reaction mixture (80 μl) containing 50 mm Tris-HCl (pH 7.5), 10 mm MnCl2,[32P]polyphosphate (59 μm phosphate groups; ∼85 nm polyphosphate, assuming an average chain length of 700 phosphate groups), and CthTTM (3.2 μg; 2 μm enzyme) was incubated at 37 °C. Aliquots (10 μl) were withdrawn at the times specified and quenched immediately with 5 μl of loading buffer (270 mm Tris borate (pH 8.3), 26 mm EDTA, 30% sucrose). The time 0 sample was taken prior to adding CthTTM. The samples were analyzed by electrophoresis through a 40-cm 6% polyacrylamide gel containing 7 m urea in 90 mm Tris borate, 2.7 mm EDTA. The gel was prerun at 750 V for 30 min before loading the samples in the wells. A tracking dye mix containing bromphenol blue, xylene cyanol, and an aliquot of 32Pi was loaded in a separate well. Electrophoresis was continued at 750 V until the bromphenol blue had migrated ∼14 cm into the gel. Radiolabeled material was visualized by autoradiography.
We also monitored the progress of the polyphosphatase reaction by gel electrophoresis (Fig. 6B). Absent CthTTM, the [32P]polyphosphate substrate migrated as a heterogeneous high molecular weight polymer near the top of the gel. Although we do not have polyphosphate standards to accurately calibrate polymer size, previous studies established that the product of the in vitro synthesis of [32P]polyphosphate by PPK has a chain length of ∼700 phosphate groups (30). CthTTM catalyzed a time-dependent decrease in the size of the radiolabeled polyphosphate (reflected in its increased electrophoretic mobility), which was eventually converted into a rapidly migrating end product that corresponded to the mixture of 32PPi and 32Pi observed in the TLC analysis. There is an apparent area of exclusion of the labeled polyphosphate about two-thirds of the way down the gel, with label compression above and below the excluded area. This electrophoretic anomaly for polyphosphates was seen previously by Wurst and Kornberg (31), who indicated that the excluded area corresponds to polyP4-15.
To verify that the inorganic polyphosphatase activity is mediated by CthTTM, we subjected the enzyme to zonal velocity sedimentation through a 15-30% glycerol gradient (Fig. 7A). Marker proteins catalase (248 kDa), bovine serum albumin (66 kDa), and cytochrome c (13 kDa) were included as internal standards and were resolved accordingly, as gauged by SDS-PAGE analysis of the gradient fractions. The peak positions for the markers are denoted in Fig. 7A. Aliquots of even-numbered gradient fractions were assayed for hydrolysis of [32P]polyphosphate via the TLC method; the yields of 32Pi and 32PPi products are plotted separately in Fig. 7A. The activity profiles for formation of the two products were identical, peaking in fractions 16-18, and they coincided with the sedimentation profile of the CthTTM polypeptide (not shown). We conclude that CthTTM has an inherent inorganic polyphosphatase activity.
FIGURE 7.

Characterization of the polyphosphatase activity of CthTTM. A, velocity sedimentation. A mixture of CthTTM (100 μg) and internal standards catalase (50 μg), bovine serum albumin (50 μg), and cytochrome c (50 μg) was applied to a 4.8-ml 15-30% glycerol gradient containing 50 mm Tris-HCl (pH 7.5), 0.2 m NaCl, 1 mm EDTA, 1 mm dithiothreitol, and 0.05% Triton X-100. The gradient was centrifuged for 17 h at 4 °C at 50,000 rpm in a Beckman SW55Ti rotor. Fractions (∼0.2 ml) were collected from the bottom of the tube. Aliquots of even-numbered fractions were analyzed by SDS-PAGE and assayed for polyphosphatase activity. Reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.5), 10 mm MnCl2,[32P]polyphosphate (59 μm phosphate groups), and 2 μl of the glycerol gradient fractions were incubated at 37 °C for 60 min. The reaction products were analyzed by TLC as described in the legend to Fig. 6A. The 32P-labeled products were quantified by scanning the TLC plate with a FUJIX BAS2500 imager. The extents of release of 32PPi (•) and 32Pi (○) are shown. B, reaction mixtures (80 μl) containing 50 mm Tris-HCl (pH 7.5), [32P]polyphosphate (59 μm phosphate groups) were incubated for the specified time intervals at 37 °C with 10 mm MnCl2 and 6.4 μg of CthTTM. Aliquots (10 μl) were withdrawn at the times specified and quenched immediately with EDTA. The amounts of 32PPPi (gray circles), 32PPi (•), and 32Pi (○) formed (per 10 μl of reaction mixture) were determined by PEI-cellulose TLC in 1.25 m LiCl, 1 m HCOOH. C, reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.5); 10 mm MnCl2; either 59 μm (⋄), 240 μm (♦), 940 μm (□), or 3800 μm (▪)[32P]polyphosphate (as total phosphate concentration; corresponding to ∼0.085, 0.34, 1.3, and 5.4 μm polyphosphate chains); and the specified amounts of CthTTM were incubated at 37 °C for 60 min. The extents of product formation (32PPi plus 32Pi) were plotted as a function of input CthTTM for each polyphosphate substrate concentration.
Characterization of the CthTTM Polyphosphatase Reaction—Polyphosphatase activity was optimal in Tris-HCl buffer at pH 7.5-8.0. No activity was detectable in the absence of a divalent cation. Manganese supported optimal activity at 10-20 mm concentration; product formation at 2 and 5 mm manganese was 39 and 57% of the value at 10 mm (not shown). Cadmium (10 mm) was 40% as effective as manganese, whereas 10 mm magnesium, cobalt, copper, and nickel were ineffective (not shown). It was apparent that the molar yield of 32Pi was consistently about twice that of the 32PPi product (Fig. 7A and other data not shown). To better evaluate this phenomenon, we examined product release from [32P]polyphosphate as a function of time, using a different TLC buffer that allowed us to separate and quantify the 32PPPi, 32PPi, and 32Pi species, whereas larger polyphosphates remained at the origin. This experiment revealed simultaneous accumulation of 32Pi and 32PPi products at rates of 0.27 and 0.14 min-1, respectively (Fig. 7B). Both products plateaued at 60-90 min, at which time the 32Pi/32PPi ratio was 1.7 (Fig. 7B). This kinetic profile is not consistent with a precursor-product relationship of 32PPi to 32Pi. Indeed, we showed previously that CthTTM has no detectable inorganic pyrophosphatase activity (26). A key finding here was that only trace levels of 32PPPi were detected during the CthTTM polyphosphatase reaction (Fig. 7B). These results suggest that (i) CthTTM can directly liberate either PPi or Pi from the end of the polyphosphate chain and that it prefers the option of Pi release to PPi release by about a factor of 2 (possible explanations for this preference will be discussed) and/or (ii) any 32PPPi that was liberated from the end of the polyphosphate chain was rapidly hydrolyzed to 32PPi plus 32Pi.
The standard polyphosphate assays in the preceding experiments were performed at a relatively low concentration of [32P]polyphosphate (59 μm total phosphate groups; ∼85 nm polyphosphate chains, assuming an average chain length of 700 phosphate groups). Whereas the combined yield of the two reaction products (32Pi plus 32PPi) from this amount of [32P]polyphosphate was proportional to the amount of CthTTM added to the reaction (Fig. 7C), the calculated specific activity was only 6.6 pmol of product released per pmol of enzyme during the 60-min reaction. A series of enzyme titrations at increasing concentrations of [32P]polyphosphate over a 64-fold range (from 0.085 to 5.4 μm polyphosphate chains) revealed a steady increase in specific activity, up to 90 pmol of product released per pmol of enzyme at the highest level tested (Fig. 7C).
Effect of Deleting the Plug Helix—We produced and purified a truncated protein, CthTTM-(1-137), that lacks the plug helix. CthTTM-(1-137) was then tested for its activity with a variety of substrates utilized by wild-type CthTTM. Fig. 8A shows manganese-dependent hydrolysis of Gpppp and GTP as a function of input CthTTM-(1-137) protein. The relatively smooth shape of the titration curve for release of Pi from Gpppp contrasted with the sharply biphasic pattern seen with wild-type CthTTM (Fig. 2A). The specific activity for Gpppp hydrolysis by CthTTM-(1-137) calculated from the slope of the curve at lower protein levels in Fig. 8A was 0.8 s-1; this value is 190-fold lower than the guanosine tetraphosphatase activity of wild-type CthTTM under the same assay conditions. By contrast, the manganese-GTPase specific activity of CthTTM-(1-137) calculated from the data in Fig. 8A (0.23 s-1) was 4-fold greater than the GTPase activity of wild-type CthTTM (Fig. 2A). The opposing effects of deleting the plug helix on Gpppp hydrolysis versus GTPase activity were qualitatively similar to the consequences of loosening the tether of the plug helix to the tunnel by virtue of the K8A mutation. We detected no hydrolysis of Gpppp by CthTTM-(1-137) in the presence of magnesium (data not shown).
FIGURE 8.

Phosphohydrolase activities of CthTTM-(1-137). A, hydrolysis of Gpppp and GTP. Reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 8.5), 200 μm MnCl2, 100 μm Gpppp (•), or 100 μm GTP (○) and CthTTM-(1-137) as specified were incubated for 30 min at 37 °C. B, hydrolysis of tripolyphosphate. Reaction mixtures (50 μl) containing 50 mm Tris-HCl (pH 9.0), 0.5 mm MnCl2, 100 μm PPPi, and wild-type CthTTM or CthTTM-(1-137), as specified, were incubated for 30 min at 37 °C. Pi release was determined using the malachite green method and is plotted as a function of input enzyme. C and D, reaction mixtures (10 μl) containing 50 mm Tris-HCl (pH 7.5), 10 mm MnCl2, [32P]polyphosphate (59 μm phosphate), and either wild-type CthTTM (C) or CthTTM-(1-137) (D), as specified, were incubated for 60 min at 37 °C. The extents of release of 32PPPi, 32PPi, and 32Pi were determined by TLC and are plotted as a function of input enzyme.
Deletion of the plug helix reduced manganese-dependent inorganic tripolyphosphatase activity by 16-fold compared with wild-type CthTTM (Fig. 8B), again reprising the effects of the K8A change noted previously (26). We detected no hydrolysis of PPPi by CthTTM-(1-137) in the presence of magnesium (not shown). The specific activity of CthTTM-(1-137) in manganese-dependent cleavage of AppppppA was 15-fold lower than that of wild-type CthTTM (not shown).
A comparison of the hydrolysis of inorganic polyphosphate by wild-type CthTTM (Fig. 8C) versus CthTTM-(1-137) (Fig. 8D) revealed little difference in total product formation (32PPi plus 32Pi) as a function of input enzyme. However, the distribution of products was quite different. Whereas wild-type CthTTM liberated a 1.9-fold molar excess of 32Pi over 32PPi, the truncated enzyme displayed a stronger bias toward 32Pi, which was produced in 4.1-fold excess over 32PPi. A control experiment showed that CthTTM-(1-137) catalyzed no detectable conversion of PPi to Pi under the same reaction conditions (not shown). Thus, we surmise that deleting the plug helix directly affects the outcome of the polyphosphate hydrolysis reaction, by favoring cleavage at the terminal phosphate.
DISCUSSION
By surveying a variety of nucleotidyl and inorganic polyphosphates as substrates for CthTTM, we gain a new appreciation of its catalytic potential and the constraints to substrate utilization imposed by steric hindrance and the choice of metal cofactor. Initial studies showed that CthTTM hydrolyzed NTPs to NDPs and Pi, exclusively with manganese, without apparent preference for any of the standard purine or pyrimidine bases or deoxy versus ribo sugars. However, NTP hydrolysis was conspicuously feeble compared with hydrolysis of tripolyphosphate, which was supported by manganese and magnesium (26). Here we find that a nucleoside 5′-tetraphosphate is a far better substrate for CthTTM than the cognate NTP and that the reaction occurs with either manganese or magnesium. These findings engender a model for productive substrate binding whereby the nucleoside hinders the formation of a proper Michaelis complex when it is located too close to the terminal phosphate (Fig. 9).
FIGURE 9.

Inferred substrate binding modes in the CthTTM tunnel. The CthTTM tunnel is depicted as a magenta barrel (“entrance” side on the left) with three phosphate-binding sites (e.g. for the α-, β-, and γ-phosphates of a nucleoside triphosphate or PPPi). The divalent metal (cyan sphere) is located at a fixed site on the tunnel floor that dictates hydrolysis (↓) of the overlying phosphoanhydride bond. The inferred binding modes for the nucleotide polyphosphate substrates GTP, Gpppp, and AppppppA are shown at the top, and the reaction products are indicated at the right. The binding modes and reaction outcomes for hydrolysis of inorganic polyphosphate substrates are illustrated at the bottom.
Based on the structures of Cet1 and PhoTTM, we envision that three phosphates of the substrate bind within the tunnel so that the terminal scissile phosphoanhydride is poised over the catalytic metal. This establishes an asymmetry to the active site, even when the substrate is chemically symmetrical, as in the case of tripolyphosphate. We and others (25) have proposed that NTPs and thiamine triphosphate, which are asymmetric chemically, bind the tunnel so that the NDP or ThDP leaving group is located on the “entrance” side of the tunnel. In the case of the bacterial/archaeal TTMs and mammalian thiamine triphosphatase, this corresponds to the face of the tunnel that is occupied by the plug helix (which apparently must be displaced to some degree when substrate binds productively). The sharp increase in activity with nucleoside tetraphosphate versus NTP would thereby reflect the movement of the nucleoside further from the tunnel entrance when the β, γ, and δ phosphates of Gpppp occupy the active site so as to result in hydrolysis of the γ-δ phosphoanhydride (Fig. 9). The effects of deleting the C-terminal plug helix underscore its role as a substrate filter, which minimizes reactivity with NTPs. How the plug helix exerts opposite effects on hydrolysis of NTP versus PPPi and Gpppp substrates is not clear at present.
CthTTM acts solely as an “exophosphatase” in releasing Pi from nucleoside 5′-triphosphate, nucleoside 5′-tetraphosphate, and inorganic tripolyphosphate substrates. In this mode, the TTM enzymes are proposed to catalyze in-line attack by water on a free R-O-PO3 end, which is nominally a dianion at the mild alkaline pH used for the enzyme assays. The situation is quite different when CthTTM is presented with a dinucleoside polyphosphate, where it must act as an “endophosphatase” on an internal phosphoanhydride R-O-(PO2)-O-R′ that poses distinctive steric and electrostatic problems. First, the scissile phosphate is an obligate monoanion. Second, the R′ group, if bulky and anionic in its own right (as in the case of a nucleotide), might interfere with the proper orientation of the water nucleophile. Given these differences, it is remarkable that CthTTM can catalyze hydrolysis of diadenosine hexaphosphate via the endo mechanism. As noted above, we cannot clearly assign the slow rate of AppppppA cleavage (compared with Gpppp with a free end) to the endo versus exo mechanism, steric hindrance by a second nucleoside, or a combination of both factors.
In addition to demonstrating that the endophosphatase mode is feasible for CthTTM, our studies of dinucleoside polyphosphate cleavage provide evidence that tunnel opening precedes productive substrate binding. The dinucleoside hexaphosphate cleaved by CthTTM contains two bulky terminal groups, analogous to a dumbbell, that constitute a major impediment to penetration of the polyphosphate bridge into the triphosphate site of the tunnel, because the aperture is too narrow when the tunnel is in the closed conformation (Fig. 1C). Therefore, we invoke initial engagement of the dinucleoside hexaphosphate by CthTTM in the open conformation (as in Fig. 1B), followed by closure around the polyphosphate bridge. In light of the results for Gpppp versus GTP, we speculate that AppppppA binds such that the β-, γ-, and δ-phosphates occupy the triphosphate site in the tunnel (Fig. 9), which results in cleavage of the γ-δ phosphoanhydride to form two molecules of ATP. We attribute the inability of CthTTM to effectively hydrolyze AppppA and its weak activity with ApppppA to steric hindrance of tunnel closure by the more closely spaced nucleosides. It is not yet clear whether tunnel opening, which must occur to accommodate AppppppA, is an obligate step when CthTTM acts on PPPi or Gpppp. Because these substrates have a free end, we see no inherent obstacle to their binding directly to the closed tunnel via insinuation of the free triphosphate end into the tunnel entrance (as posited for the Cet1 clade of RNA triphosphatases). The greater leeway of PPPi or Gpppp binding to the closed tunnel conformation could be another factor in their superior activity if CthTTM is in equilibrium between open and closed tunnel conformations prior to substrate binding.
The ability of CthTTM to degrade a long-chain inorganic polyphosphate is intriguing in light of the multiple roles imputed to polyphosphate in bacterial physiology (28, 29). Although evaluation of CthTTM in polyphosphate biology ultimately hinges on a genetic analysis via targeted gene disruption (to our knowledge, not currently practiced in C. thermocellum), the properties of the inorganic polyphosphatase activity reported here are quite interesting and distinct from other known polyphosphate catabolizing enzymes: Ppx1, PPX, and Ppn1. Yeast Ppx1 and E. coli PPX are metal-dependent exopolyphosphatases that cleave the terminal phosphoanhydride linkage to yield Pi as the sole product of each catalytic cycle (31, 32). The structures of yeast Ppx1 (33) and E. coli PPX (34) have been solved; they are unrelated to each other and bear no resemblance to the TTM fold. Yeast Ppn1 is an endopolyphosphatase that cleaves long polyphosphate chains internally to yield shorter polyphosphates, ultimately forming a mixture of Pi and PPPi as the end products (35).
By contrast, CthTTM cleaves polyphosphate to yield a mixture of Pi and PPi, with Pi being formed in ∼2-fold molar excess over PPi, with no significant accumulation of PPPi during the reaction. This product distribution implies that CthTTM hydrolyzes polyphosphate in both exo mode when forming Pi and endo mode when generating PPi. Several plausible substrate-binding scenarios are depicted in Fig. 9 to account for this pattern of product formation. In the exo mode, we propose that the polymer accesses the tunnel on the entrance side so that the terminal phosphate is over the metal and hydrolysis liberates Pi. The endo mode of PPi hydrolysis could entail (i) polymer access through the tunnel entrance, albeit with the penultimate n-1 phosphate over the metal and the terminal phosphate protruding from the tunnel exit or (ii) polymer entry through the exit side of the tunnel, with the three terminal phosphates bound so that the cleavage occurs between the n-1 and n-2 phosphates. The balance between exo and endo modes is apparently flexible, because we find that deletion of the plug helix shifts the reaction outcome toward generation of Pi.
Finally, our studies of CthTTM emphasize the need for skepticism regarding the data base annotations of dozens of uncharacterized bacterial/archaeal TTMs as “adenylate cyclases.” CthTTM has no such activity (26). As shown here and previously, it exhibits the signature manganese-dependent phosphohydrolase function of TTM proteins (1) but has its own distinctive substrate specificity and reaction outcomes. Surveying the properties of additional bacterial and archaeal TTMs, especially those for which crystal structures are in hand, should provide important clues to the function and evolution of this enzyme superfamily.
This work was supported, in whole or in part, by National Institutes of Health Grant GM52470. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TTM, triphosphate tunnel metalloenzyme; CthTTM, C. thermocellum TTM; NeuTTM, N. europea TTM; PhoTTM, P. horikoshii TTM; Gpppp, guanosine 5′-tetraphosphate; AppppA, P1,P4-di(adenosine-5′) tetraphosphate; ApppppA, P1,P5-di(adenosine-5′) pentaphosphate; AppppppA, P1,P6-di(adenosine-5′) hexaphosphate; CIP, calf intestine phosphatase; PPK, polyphosphate kinase; PEI, polyethyleneimine.
References
- 1.Ho, C. K., Pei, Y., and Shuman, S. (1998) J. Biol. Chem. 273 34151-34156 [DOI] [PubMed] [Google Scholar]
- 2.Lima, C. D., Wang, L. K., and Shuman, S. (1999) Cell 99 533-543 [DOI] [PubMed] [Google Scholar]
- 3.Gong, C., Smith, P., and Shuman, S. (2006) RNA 12 1468-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bisaillon, M., and Shuman, S. (2001) J. Biol. Chem. 276 17261-17266 [DOI] [PubMed] [Google Scholar]
- 5.Bisaillon, M., and Bougie, I. (2003) J. Biol. Chem. 278 33963-33971 [DOI] [PubMed] [Google Scholar]
- 6.Pei, Y., Schwer, B., Hausmann, S., and Shuman, S. (2001) Nucleic Acids Res. 29 387-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pei, Y., Lehman, K., Tian, L., and Shuman, S. (2000) Nucleic Acids Res. 28 1885-1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho, C. K., and Shuman, S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 3050-3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takagi, T., and Buratowski, S. (2001) Mol. Biochem. Parasitol. 114 239-244 [DOI] [PubMed] [Google Scholar]
- 10.Ho, C. K., and Shuman, S. (2001) J. Biol. Chem. 276 46182-46186 [DOI] [PubMed] [Google Scholar]
- 11.Gong, C., Martins, A., and Shuman, S. (2003) J. Biol. Chem. 278 50843-50852 [DOI] [PubMed] [Google Scholar]
- 12.Hausmann, S., Vivarès, C. P., and Shuman, S. (2002) J. Biol. Chem. 277 96-103 [DOI] [PubMed] [Google Scholar]
- 13.Hausmann, S., Altura, M. A., Witmer, M., Singer, S. M., Elmendorf, H. G., and Shuman, S. (2005) J. Biol. Chem. 280 12077-12086 [DOI] [PubMed] [Google Scholar]
- 14.Gong, C., and Shuman, S. (2002) J. Biol. Chem. 277 15317-15324 [DOI] [PubMed] [Google Scholar]
- 15.Myette, J., and Niles, E. G. (1996) J. Biol. Chem. 271 11945-11952 [DOI] [PubMed] [Google Scholar]
- 16.Gong, C., and Shuman, S. (2003) Virology 309 125-134 [DOI] [PubMed] [Google Scholar]
- 17.Jin, J., Dong, W., and Guarino, L. A. (1998) J. Virol. 72 10011-10019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins, A., and Shuman, S. (2001) J. Biol. Chem. 276 45522-45529 [DOI] [PubMed] [Google Scholar]
- 19.Martins, A., and Shuman, S. (2003) Nucleic Acids Res. 31 1455-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Souliere, M. F., Perreault, J. P., and Bisaillon, M. (2008) Nucleic Acids Res. 36 451-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benarroch, D., Smith, P., and Shuman, S. (2008) Structure 16 501-512 [DOI] [PubMed] [Google Scholar]
- 22.Gallagher, D. T., Smith, N. N., Kim, S. K., Heroux, A., Robinson, H., and Reddy, P. T. (2006) J. Mol. Biol. 362 114-122 [DOI] [PubMed] [Google Scholar]
- 23.Iyer, L. M., and Aravind, L. (2002) BMC Genomics 3 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakaye, B., Makarchikov, A. F., Antunes, A. F., Zorzi, W. Coumans, B., DePauw, E., Wins, P., Grisar, T., and Bettendorf, L. (2002) J. Biol. Chem. 277 13771-13777 [DOI] [PubMed] [Google Scholar]
- 25.Song, J., Bettendorf, L., Tonelli, M., and Markley, J. L. (2008) J. Biol. Chem. 283 10939-10948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keppetipola, N., Jain, R., and Shuman, S. (2007) J. Biol. Chem. 282 11941-11949 [DOI] [PubMed] [Google Scholar]
- 27.Keasling, J. D., Bertsch, L., and Kornberg, A. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 7029-7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown, M. R. W., and Kornberg, A. (2008) Trends Biochem. Sci. 33 284-290 [DOI] [PubMed] [Google Scholar]
- 29.Sureka, K., Dey, S., Datta, P., Singh, A. K., Dasgupta, A., Rodrigue, S., Basu, J., and Kundu, M. (2007) Mol. Microbiol. 65 261-276 [DOI] [PubMed] [Google Scholar]
- 30.Tzeng, C. M., and Kornberg, A. (2000) J. Biol. Chem. 275 3977-3983 [DOI] [PubMed] [Google Scholar]
- 31.Wurst, H., and Kornberg, A. (1994) J. Biol. Chem. 269 10996-11001 [PubMed] [Google Scholar]
- 32.Akiyama, M., Crooke, E., and Kornberg, A. (1993) J. Biol. Chem. 268 633-639 [PubMed] [Google Scholar]
- 33.Ugochukwu, E., Lovering, A. L., Mather, O. C., Young, T. W., and White, S. A. (2007) J. Mol. Biol. 371 1007-10021 [DOI] [PubMed] [Google Scholar]
- 34.Rangarajan, E. S., Nadeau, G., Li, Y., Wagner, J., Hung, M., Schrag, J. D., Cygler, M., and Matte, A. (2006) J. Mol. Biol. 359 1249-1260 [DOI] [PubMed] [Google Scholar]
- 35.Shi, X., and Kornberg, A. (2005) FEBS Lett. 597 2014-2018 [DOI] [PubMed] [Google Scholar]