

Abstract
Mitochondrial aldehyde dehydrogenase (ALDH2) may be involved in the biotransformation of glyceryl trinitrate (GTN), and the inactivation of ALDH2 by GTN may contribute to the phenomenon of nitrate tolerance. We studied the GTN-induced inactivation of ALDH2 by UV/visible absorption spectroscopy. Dehydrogenation of acetaldehyde and hydrolysis of p-nitrophenylacetate (p-NPA) were both inhibited by GTN. The rate of inhibition increased with the GTN concentration and decreased with the substrate concentration, indicative of competition between GTN and the substrates. Inactivation of p-NPA hydrolysis was greatly enhanced in the presence of NAD+, and, to a lesser extent, in the presence of NADH. In the presence of dithiothreitol (DTT) inactivation of ALDH2 was much slower. Dihydrolipoic acid (LPA-H2) was less effective than DTT, whereas glutathione, cysteine, and ascorbate did not protect against inactivation. When DTT was added after complete inactivation, dehydrogenase reactivation was quite modest (≤16%). The restored dehydrogenase activity correlated inversely with the GTN concentration but was hardly affected by the concentrations of acetaldehyde or DTT. Partial reactivation of dehydrogenation was also accomplished by LPA-H2 but not by GSH. We conclude that, in addition to the previously documented reversible inhibition by GTN that can be ascribed to the oxidation of the active site thiol, there is an irreversible component to ALDH inactivation. Importantly, ALDH2-catalyzed GTN reduction was partly inactivated by preincubation with GTN, suggesting that the inactivation of GTN reduction is also partly irreversible. These observations are consistent with a significant role for irreversible inactivation of ALDH2 in the development of nitrate tolerance.
Glyceryl trinitrate (nitroglycerin, GTN)2 has been used for the treatment of angina pectoris for well over a century (1–3). GTN is thought to cause vasodilatation mainly by activation of soluble guanylate cyclase (sGC), which is stimulated by the binding of NO to a regulatory heme group. Because GTN does not release NO spontaneously, it requires bioconversion. GTN activates sGC non-enzymatically in the presence of cysteine and a few structurally related thiols (4–6) via NO release (7). At sufficiently high concentrations, ascorbate can substitute for Cys in the activation of GTN (8). Although thiol- or ascorbate-supported decomposition of GTN might account for its biological effects (7, 8), bioconversion of GTN is probably largely enzymatic, and a number of mechanisms, featuring different enzymes, have been put forward (1, 5, 6). A recent proposal suggests a crucial role for the mitochondrial isoform of aldehyde dehydrogenase (ALDH2) in the transformation of GTN to 1,2-glyceryl dinitrate (1,2-GDN) and nitrite (3, 9–12). Because the active site thiol of ALDH2 is oxidized in the process, it must be regenerated by suitable reductants, which might explain the dependence of GTN bioconversion on the presence of thiols. One major unresolved question concerns the identity of the physiological reductant, because ALDH2-catalyzed GTN reduction is supported by the non-physiological thiol dithiothreitol (DTT) but not by glutathione (GSH), the most abundant physiological low-molecular weight thiol. A second problem adhering the hypothesis originates from the fact that the reaction produces nitrite rather than NO (9); it has been proposed that the ability of mitochondria to convert nitrite to nitric oxide or S-nitrosothiols provides the link between ALDH2-catalyzed GTN metabolism and vasodilatation (3, 11). Interestingly, activation of sGC by GTN has been accomplished in the presence of purified mitochondrial ALDH2, suggesting that the ALDH2-catalyzed decomposition of GTN yields NO (12); the physiological relevance of this observation is not clear yet.
Chronic application of GTN is severely hampered by the phenomenon of nitrate tolerance. Nitrate tolerance appears to be a multicause phenomenon, and numerous explanations for its occurrence have been proposed (3, 5, 6, 13–15). One attractive characteristic of the proposal that mitochondrial ALDH is responsible for GTN biotransformation is the fact that it can accommodate one of the more common explanations for nitrate tolerance in a slightly modified version. In view of the thiol dependence of GTN, it was suspected that thiol depletion might contribute to nitrate tolerance. The realization that each turnover with GTN requires regeneration of the active site thiol of ALDH2 by an exogenous reductant revitalized this hypothesis (9, 11). Accordingly, nitrate tolerance develops when regeneration of active ALDH2 fails, either because the unidentified cellular reductant runs low, or because a putative enzyme involved in the process is inactivated. In the present paper we investigated the inactivation of ALDH2 by GTN in the presence and absence of thiols to elucidate this issue.
EXPERIMENTAL PROCEDURES
Materials—Recombinant human ALDH2 was expressed in and purified from Escherichia coli (16). GTN (Nitropohl® ampoules; G. Pohl-Boskamp GmbH & Co, Hohenlockstedt Germany) was obtained from a local pharmacy as a 1 mg/ml aqueous isotonic solution, containing 49 mg/ml glucose monohydrate, and diluted in 50 mm triethanolamine/HCl (pH 7.4). [2-14C]GTN (50 mCi/mmol), manufactured by American Radiolabeled Chemicals, was supplied locally by Humos Diagnostica (Maria Enzersdorf, Austria). 1,2-GDN and 1,3-GDN, used as standards in radio-thin layer chromatography, were obtained from LGC Promochem (Wesel, Germany). 6-Methoxy-2-naphthaldehyde (Monal 62, 98%) was obtained from Sigma. Naphthoic acid was purchased from ABCR GmbH & Co KG (Karlsruhe, Germany). All other chemicals were from Sigma or Merck (Darmstadt, Germany).
Determination of the Inactivation Kinetics—To determine the rate of inactivation of ALDH2 by GTN, the optical absorption of a 1-cm quartz cuvette (type 105.200-QS, Hellma, Müllheim, Germany) containing substrate (acetaldehyde or p-NPA) and additional compounds like NAD+ or DTT as indicated in 50 mm potassium Pi (pH 7.4), was monitored at 340 (dehydrogenase) or 400 nm (esterase) in a Hewlett-Packard 8452A Diode Array spectrophotometer. After ∼1–2 min ALDH2 was added and the reaction was allowed to proceed for ∼2–3 min. Subsequently, GTN (or in some instances NAD+) was added to start the inactivation. Usually, when inactivation was complete, DTT, substrate, or both were added to reactivate the enzyme. The total volume of the samples after GTN addition was 250 μl. The total time over which the reaction was monitored was 20–30 min. Although new conditions were always explored with a single sample for optimal time resolution, concentration dependences or variations of cofactor or reductant (see “Results”) were usually studied with up to 6 samples per experiment with the 89073A multicell transport accessory. The temperature of the samples was kept at 20 °C by a refrigerating circulating water bath (Lauda RM6, Bartelt GmbH, Austria). The various segments of the time traces (initial activity, restored activities) were fitted linearly, except for the inactivation, which was fitted to a combination of a single-exponential and a straight line. The linear component yielded the residual activity; the exponential component yielded the apparent inactivation rate constant, as can be derived as follows: if the uninhibited activity is ve = ke·E, with ke and E representing the catalytic rate constant and the enzyme concentration, respectively, inactivation will lower the activity by decreasing the catalytically competent enzyme fraction in a mono-exponential fashion with Et = E0·e-kit, in which E0 is the total enzyme concentration and ki is the apparent inactivation rate constant. The activity will therefore change in time according to the equation ve(t) = ke · E0·e-kit = ve(0)·e·kit. Consequently, the absorbance will display simple monoexponential behavior during inactivation according,
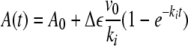 |
(Eq. 1) |
in which A0 and Δε correspond to the initial absorbance and the absorbance difference coefficient between product and substrate, respectively.
Inactivation of ALDH2 by Preincubation with GTN (“GTN Preconditioning”)—Purified ALDH2 (0.27 mg/ml) was preincubated with 1 μm GTN in 50 mm NaPi (pH 7.5) containing 0.2 mm MgCl2 and 2 mm NAD+ at 37 °C for 10 min. Dehydrogenase activity was determined as the formation of NADH from NAD+ in the presence of acetaldehyde by monitoring the increase in absorbance at 340 nm (ε340 = 6.22 mm-1 cm-1). Esterase activity was measured by monitoring the formation of p-nitrophenolate from p-nitrophenylacetate (p-NPA, ε400 = 16 mm-1 cm-1). The reaction mixture contained substrate (0.2 mm acetaldehyde or 0.1 mm p-NPA), 2 mm NAD+, 10 mm MgCl2, and 1.5 μg/ml ALDH2 in 50 mm NaPi (pH 7.5) at 25 °C.
In a second series of experiments, preincubation with 1 or 10 μm GTN was performed in 0.1 m MES (pH 6.5) containing 0.2 mm MgCl2 at 37 °C for 10 min, dehydrogenation was measured at pH 9.0 (50 mm NaPi), and esterase activity was determined in the absence of NAD+ (17). All other conditions were the same as in the first series.
Determination of 1,2-GDN Formation by Radio-thin Layer Chromatography—The rate of the conversion of GTN to 1,2- and 1,3-GDN was determined according to a described protocol (12). Purified ALDH2 (4 μg) was incubated with 14C-labeled GTN (2 μm; ∼50,000 dpm) at 37 °C for 10 min in a final volume of 100 μl of 50 mm potassium Pi (pH 7.4), containing 3 mm MgCl2, 1 mm NAD+, 1 mm EDTA, 1 mm EGTA, and 2 mm GSH with 2 mm DTT as indicated. After incubation, reaction mixtures were filtered over YM-10 columns (Millipore GmbH, Vienna, Austria) and washed twice with 60 μl of 50 mm potassium Pi (pH 7.4) to yield 200-μl filtrate and 20-μl residues containing the enzyme. The recovered protein was incubated again with 14C-labeled GTN (2 μm, ∼50,000 dpm) at 37 °C for 10 min in a final volume of 200 μl of potassium Pi buffer (50 mm, pH 7.4), containing 3 mm MgCl2, 1 mm NAD+, 1 mm EDTA, 1 mm EGTA, and 2 mm GSH with 2 mm DTT as indicated. Reaction products from the filtrate of the first incubation (pre-column) and from the reaction mixture of the second incubation (post-column) were extracted twice with 1 ml of diethyl ether, separated by thin layer chromatography, and quantified by liquid scintillation counting. Blank values, determined in the absence of protein under identical conditions, were subtracted. Results are expressed as nanomole of product per min and mg of protein. Mean values ± S.E. were calculated from reaction rates measured as duplicates in three independent experiments.
To evaluate the effect of the procedure on enzyme activity, dehydrogenase and esterase assays were performed before and after filtration with YM-10 columns. The filtered enzyme was washed twice with 60 μl of 50 mm NaPi (pH 9.0 or pH 7.5 for the dehydrogenase and esterase determination, respectively). Assay conditions were: 50 mm NaPi (pH 9.0), 0.2 mm acetaldehyde, 2 mm NAD+, 10 mm MgCl2, and 1.5 μg/ml ALDH2 for the dehydrogenase activity; 50 mm NaPi (pH 7.5), 0.1 mm p-NPA, 10 mm MgCl2, and 1.5 μg/ml ALDH2 for the esterase activity.
Inactivation by GTN and Reactivation by Thiols of ALDH in Mitochondria—Rat liver mitochondria were prepared according to a previously published method (18). Rat livers were homogenized in Tris buffer (250 mm sucrose, 10 mm Tris, 5 mm EGTA, 2 mm MgCl2, pH 7.4) and centrifuged at 600 × g (10 min at 4 °C). The supernatant was collected and centrifuged at 6,500 × g for 15 min. The pellet was resuspended in Tris buffer and centrifuged at 15,000 × g for 5 min. This step was repeated twice. The mitochondrial fraction (∼20–30 mg/ml protein) was frozen at -70 °C until use.
ALDH activity was determined by fluorescence spectroscopy (19). Mitochondria (0.8 mg/ml) were preincubated with GTN (100 μm) in Tris buffer for 10 min at 37 °C. After preincubation, the mixture was centrifuged (15,000 × g, 5 min at 4 °C) and washed three times with Tris buffer to remove GTN. The pellet was resuspended in Tris buffer at a final concentration of 8 μg/ml and mixed with 0.1 mm NAD+ in a cuvette at 37 °C. After 3 min, the dehydrogenase substrate Monal 62 (3 μm) was added and fluorescence was monitored (λex 310 nm, λem 360 nm) in a LS50B Luminescence Spectrometer (PerkinElmer Life Sciences). After another 3 min, thiol (DTT, β-mercaptoethanol, GSH, Cys, or LPA-H2) was added at the indicated concentrations and fluorescence was measured for 8 more min. At the end of the experiment, signal intensity was calibrated by comparison with the fluorescence of known concentrations of naphthoic acid. Mitochondria without preincubation with GTN served as controls.
RESULTS
Inhibition of Dehydrogenase Activity by GTN—To study the inactivation of ALDH2 by GTN in real time we monitored the formation of NADH from NAD+ spectrophotometrically at 340 nm and determined how the activity of ALDH2 was affected by the addition of GTN. Fig. 1A shows a typical trace recorded for the inhibition of dehydrogenase activity with acetaldehyde as the substrate. It illustrates how the linear increase of the NADH concentration is gradually reduced to a very low residual level. Fitting the curve corresponding to this process to a single exponential yielded a first-order rate constant that can be equated to the apparent inactivation rate constant under this particular set of conditions (see “Experimental Procedures”). Attempts to reactivate the enzyme by DTT resulted in modest restoration of activity. The magnitude of reactivation was not affected by the concentrations of DTT (between 0.2 and 2 mm) and acetaldehyde (up to 2 mm).
FIGURE 1.

Inhibition by GTN of ALDH2-catalyzed dehydrogenation of acetaldehyde. Panel A shows a time trace for the formation of NADH from NAD+, monitored at 340 nm. At t = 0 the cuvette contained 0.2 mm acetaldehyde and 0.2 mm NAD+ in 50 mm potassium Pi (pH 7.4). At t = 80 s, catalysis was initiated by the addition of 33 μg/ml ALDH2. Inactivation started at t = 480 s by the addition of 0.05 mm GTN. After inactivation of the enzyme, at t = 1160 s, an attempt was made to restore activity by the addition of 1 mm DTT. The dots represent the data points, whereas the continuous lines are best fits to the data. Linear fits were applied to the phases before (no catalysis (-0.10 ± 0.06) × 10-4 absorbance units (a.u.)/s), and after ALDH2 addition (initial activity, (8.03 ± 0.05) × 10-4 a.u./s), and to the activity after DTT admission (restored activity (1.378 ± 0.006) × 10-4 a.u./s); for the inactivation after GTN addition a combination of a single exponential and a linear fit was applied (apparent inactivation rate constant (7.77 ± 0.06) × 10-3 s-1; residual activity (0.133 ± 0.007) × 10-4 a.u./s). Panel B compares the residual and restored rates of acetaldehyde dehydrogenation after addition of GTN and DTT, respectively. Experimental conditions: 33 μg/ml ALDH2, 0.43 mm acetaldehyde, 0.4 mm NAD+, 0.4 mm DTT, and concentrations of GTN as indicated in 50 mm potassium Pi (pH 7.4). Initial dehydrogenase activities under the conditions applied here amounted to 289 ± 13 nmol min-1 mg-1, which corresponds to a turnover number of 69 ± 3 min-1.
The rate of inactivation increased when the GTN concentration was raised. For instance, at a fixed concentration of 0.43 mm acetaldehyde the inactivation rate constant increased from 2.96 ± 0.08 × 10-3 s-1 at 0.05 mm GTN to 21.1 ± 0.5 × 10-3 s-1 at 0.6 mm GTN. By contrast, inactivation slowed down at higher acetaldehyde concentrations: at a fixed concentration of 0.5 mm GTN the rate constant decreased from 23 ± 5 × 10-3 s-1 at 0.1 mm acetaldehyde to 2.9 ± 0.3 × 10-3 s-1 at 20 mm acetaldehyde. The latter observation is indicative of competition between acetaldehyde and GTN. A detailed description of the effects of substrate and inhibitor concentrations on the inactivation rate is given under the supplemental materials (Fig. S1, A and B, and accompanying text).
The residual activity and the extent of restoration in the presence of DTT diminished when the concentration of GTN was increased (Fig. 1B). Reactivation was not appreciably improved by extra DTT: at 0.032 mm acetaldehyde and 0.1 mm GTN, reactivation amounted to 8 ± 2 and 10 ± 1% with 0.2 and 1.0 mm DTT, respectively. Similarly, the substrate concentration had little effect on reactivation (supplementary Fig. S1C) and addition of extra substrate after inactivation did not affect the restored activity either.
As illustrated by Fig. 2A, LPA-H2 restored GTN-inactivated dehydrogenation to about the same level as DTT (from 6 ± 1 to 20 ± 3% of the initial activity with LPA-H2 compared with 26 ± 1% with DTT). GSH, on the other hand, had no effect (7 ± 2%). Although DTT could not completely restore activity after inhibition by GTN (Figs. 1B and 2A), it largely prevented inactivation if added before GTN (Fig. 2B). DTT slowed down inactivation by 0.2 mm GTN (7.4 ± 0.6)-fold3 with an EC50 of 10 ± 1 μm (supplementary Fig. S2). The results indicate that DTT strongly protects against inactivation, but cannot completely abolish it. Whereas GSH, Cys, and ascorbate did not prevent inactivation, LPA-H2 lowered the inactivation rate constant to a similar extent as DTT (results not shown).
FIGURE 2.

Effects of thiols on the inactivation of dehydrogenation by GTN. Panel A, restoration by different thiols of ALDH2-catalyzed dehydrogenation after inactivation by GTN. Plotted are the relative residual activities (vresidual/vinitial) after inactivation by 0.1 mm GTN and the relative restored activities (vrestored/vinitial) after addition of 1 mm DTT, LPA-H2, or GSH. Initial conditions: 25 μg/ml ALDH2, 0.45 mm acetaldehyde, and 0.4 mm NAD+ in 0.2 m potassium Pi (pH 7.4). Panel B, protection by DTT of dehydrogenase activity against GTN-mediated inactivation. The figure shows representative time traces of the ALDH2-catalyzed formation of NADH, as monitored at 340 nm before and after addition of GTN in the presence or absence of DTT. Experimental conditions: 33 μg/ml ALDH2, 0.45 mm acetaldehyde, 0.4 mm NAD+, and 0.1 mm GTN with or without 1 mm DTT in 50 mm potassium Pi (pH 7.4).
Inhibition of the Esterase Activity of ALDH2 by GTN—In addition to the NAD+-dependent dehydrogenation of aldehydes, ALDH is known to catalyze the hydrolysis of certain esters in a reaction that does not require NAD+ (20). When the effect of GTN on this activity was examined under standard conditions, i.e. in the absence of NAD+, GTN hardly affected the rate of hydrolysis of p-NPA; however, subsequent addition of NAD+ did cause inactivation (Fig. 3A). Esterase inactivation also occurred when GTN was added in the presence of NAD+ (Fig. 3B). The rate of inactivation was independent of the order in which GTN and NAD+ were added.
FIGURE 3.

Inhibition by GTN of ALDH2-catalyzed hydrolysis of p-nitrophenylacetate. The figure shows the formation of p-nitrophenol from p-NPA, as monitored at 400 nm. Panel A shows that, in the absence of NAD+, GTN did not significantly affect esterase activity. However, inactivation set in immediately after the addition of NAD+. Experimental conditions were: p-NPA (0.2 mm) in 50 mm potassium Pi (pH 7.4) was present at the start of the reaction; ALDH2 (33 μg/ml), GTN (0.1 mm), DTT (0.2 mm), and NAD+ (0.2 mm) were added as indicated. Curve fitting as described in the legend to Fig. 1 yielded (1.0 ± 0.5) × 10-5 absorbance units (a.u.)/s for the uncatalyzed reaction, (1.07 ± 0.02) × 10-3 a.u./s for the initial rate, (1.083 ± 0.008) × 10-3 a.u./s and (1.15 ± 0.01) × 10-3 a.u./s after addition of GTN and DDT, respectively, (1.051 ± 0.008) × 10-2 s-1 for the apparent inactivation rate constant after addition of NAD+, and (7.2 ± 0.1) × 10-5 a.u./s for the residual activity. Panel B demonstrates that GTN inactivates the enzyme in the presence of NAD+. Experimental conditions: p-NPA (0.2 mm) and NAD+ (0.2 mm) in 50 mm potassium Pi (pH 7.4) were present at the start of the reaction; ALDH2 (33 μg/ml) and GTN (0.1 mm) were added as indicated. Curve fitting yielded the following results: (0.048 ± 0.009) × 10-3 a.u./s for the uncatalyzed reaction, (3.65 ± 0.08) × 10-3 a.u./s for the initial activity, (2.6 ± 0.1) × 10-2 s-1 for the apparent inactivation rate constant, and (0.0567 ± 0.0008) × 10-3 a.u./s for the residual activity.
The rate of inactivation increased when the GTN concentration was raised, and decreased at higher concentrations of p-NPA. At a fixed concentration of 0.5 mm p-NPA the inactivation rate constant increased from 2.8 ± 0.3 × 10-3 s-1 at 0.05 mm GTN to 11.7 ± 0.8 × 10-3 s-1 at 1.0 mm GTN; at a concentration of 0.1 mm GTN the inactivation rate constant decreased from 12.6 ± 0.9 × 10-3 s-1 to 1.5 ± 0.5 × 10-3 s-1, when the p-NPA concentration was raised from 0.05 to 1.0 mm. More details are provided under supplemental materials (Fig. S3, and corresponding text).
The residual esterase activity after inactivation by GTN was affected by the concentration of p-NPA: the relative activity (vresidual/vinitial) increased from 0.024 ± 0.002 to 0.12 ± 0.03 when the substrate concentration was raised from 0.2 to 1.0 mm (supplementary Fig. S4A). On the other hand, the activity was not affected by the GTN concentration (supplementary Fig. S4B). Thiol-induced reactivation of p-NPA hydrolysis could not be determined, because of the ability of thiols (not just DTT but also GSH) to hydrolyze p-NPA in the absence of ALDH (results not shown).
In line with previous reports (21), NAD+ enhanced esterase activity, and an intermediate stimulation was observed in the presence of NADH (supplementary Fig. S5). Both with and without DTT, the apparent rate constant for GTN-induced inactivation increased by an order of magnitude in the presence of NAD+, and again the effect of NADH was intermediate (Fig. 4). Although the presence of NAD(H) is no prerequisite for the reduction of GTN by ALDH2, it is conceivable that the enzyme uses NADH as a facultative electron donor. However, we observed no absorbance changes at 340 nm upon incubating ALDH2 with GTN and NAD+ or NADH, which rules out the participation of either nucleotide as a redox agent in the decomposition of GTN.
FIGURE 4.

Effect of NAD+ and NADH on the rate of inactivation by GTN of ester hydrolysis. The figure shows the effects of NAD+ and NADH (0.2 mm) on the apparent inactivation rate constant in the absence and presence of 0.4 mm DTT. Results are presented as averages ± S.E. (n = 3). Further conditions: 33 μg/ml ALDH2, 0.2 mm p-NPA, and 0.1 mm GTN in 50 mm potassium Pi (pH 7.4).
Activities of ALDH2 after GTN Preconditioning—In the experiments described so far, we studied the inactivation of ALDH in real time by monitoring the dehydrogenase and esterase activities in the presence of GTN. Although this procedure enables the determination of the inactivation kinetics, the presence of GTN after inactivation may compromise subsequent attempts to restore enzyme activity. Moreover, because of the inevitable presence of the substrate during preincubation, fairly high GTN concentrations were required. Therefore, in a different procedure, referred to as GTN preconditioning, we preincubated the enzyme in the presence of GTN followed by determination of the activity (dehydrogenase or esterase) after ∼200-fold dilution of the preincubation mixture. In this way inactivation takes place in the absence of substrate, allowing application of much lower GTN concentrations and determination of activities in the virtual absence of GTN.
As is shown in Fig. 5, preincubation with 1 μm GTN at pH 7.5 resulted in complete inactivation of acetaldehyde dehydrogenation, whereas the enzyme remained active after preincubation in the absence of GTN. Part of the dehydrogenase activity that was lost after preincubation with GTN was recovered in the presence of DTT. Partial reactivation occurred in the presence of LPA-H2 as well, although to a smaller degree. Similar observations were made for the esterase reaction (results not shown). Comparable results were obtained when preincubation took place at pH 6.5 and when “standard” assay conditions were applied for both reactions (pH 9.0 with NAD+ for the dehydrogenase reaction, and pH 7.5 without NAD+ for the esterase reaction) (see supplemental Fig. S6 and accompanying text).
FIGURE 5.

Effect of preincubation of ALDH2 with GTN on acetaldehyde dehydrogenation. The figure shows the effects of preincubation of ALDH2 at pH 7.5 with 1 μm GTN on the subsequent determination of dehydrogenase activity in the presence of NAD+ at pH 7.4 in the absence and presence of 2 mm DTT and LPA-H2. Experimental conditions for preincubation were: 0.27 mg/ml ALDH2, 1 μm GTN, 0.2 mm MgCl2, 2 mm NAD+, 50 mm NaPi (pH 7.5) for 10 min at 37 °C. Assay conditions were: 1.5 μg/ml ALDH2, 2 mm NAD+, 10 mm MgCl2, and 0.2 mm acetaldehyde in 50 mm NaPi (pH 7.5) at 25 °C. See “Experimental Procedures” for further details.
Time Dependence of Irreversible ALDH2 Inactivation—To further elucidate the mechanism of irreversible inactivation, we performed a series of experiments of the type shown in Fig. 1A and varied the time of DTT addition, leaving all other conditions equal. The results, which are described in greater detail under supplemental materials (Fig. S7 and accompanying text), showed that most of the inactivation took place within the same time span in which NAD+ reduction was inhibited by GTN in the absence of DTT, although a slow additional inactivation was also apparent. The “rapid” irreversible inactivation was exacerbated when the GTN concentration was increased, whereas the slower process was not affected.
We also determined the effect of incubation time in experiments of the type described in Fig. 5, in which we preincubated the enzyme in the presence and absence of GTN (0.18 mm) and DTT (2 mm) for different lengths of time, and then measured the dehydrogenase activity after 15-fold dilution. The results, presented in Fig. 6, show that in the absence of DTT the enzyme tends to lose activity over time (34 ± 2% in 1 h). The same trend was observed in the presence of GTN, but in that case approximately half of the activity was additionally lost within the first 15 min. As a result, 13 ± 4% of the activity remained after 1 h of preincubation. DTT offered moderate protection against the gradual inactivation in the absence of GTN (20 ± 3% activity loss after 1 h). In the combined presence of DTT and GTN, virtually all activity was lost within 15 min.
FIGURE 6.

Effect of preincubation time on the dehydrogenase activity of ALDH2. The enzyme was preincubated for various times, after which dehydrogenase activities were determined by monitoring the linear absorbance increase at 340 nm over a period of 5 min. Preincubation was performed in the absence (open symbols) and presence (filled symbols) of DTT, and in the absence (circles) or presence (squares) of GTN. Preincubation conditions were: 0.48 mg/ml ALDH2, 1 mm NAD+, and 0.18 mm GTN and 0.2 mm DTT as indicated in 300 mm potassium Pi (pH 7.4). Assay conditions were: 19 μg/ml ALDH2, 1 mm acetaldehyde, 1 mm NAD+, and 2 mm DTT in 50 mm potassium Pi (pH 7.4). Both incubation and preincubation were performed at 20 °C.
Inactivation and Reactivation of ALDH2-catalyzed GTN Reduction—To assess how GTN affects its own reduction by mitochondrial ALDH we measured GTN reductase activity, removed low molecular weight ingredients from the assay mixture by gel filtration, and repeated the activity assay with the recovered enzyme. Before gel filtration, 1,2-GDN formation was always observed, but omission of DTT from the reaction mixture reduced pre-column activity by 85 ± 2% (Fig. 7). The activity in the absence of DTT corresponded to approximately one turnover (280 nm 1,2-GDN were formed by 332 nm ALDH), in line with the proposal that reduction of GTN results in oxidation of active site cysteine residues. After gel filtration, the recovered enzyme showed no activity in the absence of DTT, but produced a greater than stoichiometric amount of 1,2-GDN in its presence, in agreement with the proposed role of DTT in regeneration of active site thiols. Fig. 7 also shows that the recovered enzyme was significantly less active than before treatment, suggesting partially irreversible inactivation. In parallel experiments, the same procedure (enzymatic assay, enzyme recovery, repeated assay) did not affect dehydrogenase and esterase activities (dehydrogenation: 0.39 ± 0.05 and 0.37 ± 0.07 μmol/mg/min before and after enzyme recovery, respectively; ester hydrolysis: 0.219 ± 0.003 and 0.223 ± 0.004 nmol/mg/min before and after enzyme recovery, respectively), which rules out that partial inactivation of GTN reduction was an artifact.
FIGURE 7.

Effect of preincubation of ALDH2 with GTN on the ALDH2-catalyzed reduction of GTN. GTN reduction was determined in the absence or presence of DTT (pre-column activity). Subsequently, the enzyme was rapidly recovered from the assay mixture by YM-10 chromatography, and the GTN reductase activity was measured again in the absence or presence of thiols (post-column activity). Experimental conditions were: 4 μg of ALDH2, 2 μm [14C]GTN (50 000 dpm), 3 mm MgCl2, 1 mm NAD+, 1 mm EDTA, 1 mm EGTA, and 2 mm GSH with 2 mm DTT as indicated in 50 mm potassium Pi (pH 7.4) for 10 min at 37 °C. See “Experimental Procedures” for further details.
Inactivation by GTN and Reactivation by Thiols of ALDH in Intact Mitochondria—To probe the inactivation by GTN and reactivation by thiols in a physiologically more relevant setting, we investigated the inhibition of ALDH by GTN and its reactivation by thiols in isolated rat liver mitochondria. Dehydrogenase activity was determined as the conversion of the aldehyde Monal 62 to naphthoic acid by fluorescence spectroscopy in the presence of intact mitochondria preincubated with 100 μm GTN (37 °C, 10 min). As illustrated in Fig. 8, preincubation with GTN caused a loss of activity of 84 ± 2% that was partly (51 ± 5%) restored by 2 mm DTT. LPA-H2 (2 mm) and β-mercaptoethanol (β-ME) at the very high concentration of 50 mm reactivated the enzyme to comparable levels. At a concentration of 2 mm, β-ME, Cys, and GSH caused no or marginal reactivation; at a physiologically relevant concentration of 0.1 mm, LPA-H2 was ineffective as well (supplemental Fig. S8A). These results are in good agreement with the observations for the purified enzyme and with a previous report on ALDH inactivation in intact mitochondria (30). Raising the GTN concentration gradually increased the extent of inactivation from 20 ± 10% at 1 μm to 81 ± 4% at 100 μm GTN with an IC50 of 2.7 ± 0.5 μm (supplemental Fig. S8B). DTT (2 mm) and LPA-H2 (10 mm) restored activity to ∼80 and 60% of the original activity with 1 and 100 μm GTN, respectively. These results suggest that partially irreversible ALDH inactivation occurs in intact mitochondria as well.
FIGURE 8.

Inhibition by GTN and reactivation by thiols of ALDH in intact mitochondria. The figure depicts the effect of preincubation with GTN (0.1 mm, 10 min, 37 °C) on the dehydrogenation of Monal 62 in the absence and presence of various thiols. Experimental conditions were: 8.1 μg/ml rat liver mitochondria, 3 μm Monal 62, 0.1 mm NAD+, and DTT (2 mm), β-ME (50 mm), or LPA-H2 (0.1 mm) as indicated, in 10 mm Tris (pH 7.4), 250 mm sucrose, 5 mm EGTA, and 2 mm MgCl2.
DISCUSSION
Inactivation of Mitochondrial ALDH by GTN—In agreement with previous reports (10, 22, 23), we found that GTN caused time-dependent inactivation of both the dehydrogenase and esterase activities of ALDH2. Inactivation became faster with increasing GTN concentration and slower with increasing substrate concentration. The inactivation rates exhibited a tendency toward saturation at high GTN and low substrate concentrations, and we could fit the curves of kobs versus [GTN] and 1/[AcAld] or 1/[p-NPA] to hyperbolic functions. The data also show that any contribution from reversible competitive inhibition can be neglected (see supplemental materials for a discussion of this issue).
Rapid-equilibrium binding of substrate and inhibitor, followed by a slow inactivation step, is described by an equation of the form,
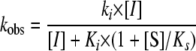 |
(Eq. 2) |
in which [I] and [S] are the concentrations of inhibitor and substrate, Ki and Ks are the respective dissociation constants, and ki is the inactivation rate constant. This model predicts the observed hyperbolic dependence of kobs on the GTN concentration and the reciprocal substrate concentration. However, our results suggest an apparent dissociation constant (Ks) for acetaldehyde some 2–3 orders of magnitude above the published Km. In fact, analysis of the data suggests values for the apparent dissociation constants for acetaldehyde, p-NPA, and GTN in the order of 10-4 m (see supplementary materials). Similar discrepancies have been reported for the mutual inhibition of esterase and dehydrogenase activities (21, 24). Interestingly, inhibition of GTN reduction by ALDH2 also required more than 1000-fold higher concentrations of acetaldehyde than might have been expected (9). This was ascribed to the absence of NAD+, which has been reported to increase the substrate affinity of mitochondrial ALDH (20). Because our results suggest that the phenomenon persists in the presence of NAD+, an alternative explanation seems to be required. It has been debated whether the dehydrogenase and esterase reactions take place at the same site. This matter appears settled now, with convincing evidence in favor of a single active site, although some of the earlier data that suggested a 2-site model remain unexplained (21, 24–28). To accommodate the disparate observations, we propose that all substrates react in the same active site pocket, but that the unnatural esterase substrates and GTN can occupy any of a number of poorly defined low-affinity positions, whereas the dehydrogenase substrates can and must be bound to a strictly defined high-affinity site to enable hydride transfer (supplementary materials Scheme S1).
Somewhat surprisingly, NAD+ profoundly affected the rate of inactivation of the esterase reaction by GTN. In the presence of DTT, inactivation was negligible unless NAD+ was present as well. A substantial decrease of the dissociation constant for GTN in the presence of NAD+ seems to be an implausible explanation.4 It is more likely that NAD+ binding exposes a target to GTN that is poorly accessible in the absence of the cofactor.
Protection and Restoration of the Dehydrogenase Reaction by Thiols—The inhibition of mitochondrial ALDH by isosorbide dinitrate is completely reversed by β-ME (23). Inhibition of yeast ALDH by NO was almost completely prevented and largely reversed by DTT, whereas β-ME and GSH were less efficient (29). Mitochondrial ALDH was partially protected against GTN-induced inactivation by DTT and LPA-H2, but not by GSH (30). It has been demonstrated that the conversion of GTN to 1,2-GDN by mitochondrial ALDH requires the presence of a reducing agent (3). The highest activity was observed in the presence of DTT, whereas LPA-H2 was about half as effective; activity was low in the presence of Cys and β-ME and virtually absent in the presence of GSH. In line with these reports, we found that DTT and LPA-H2, but not Cys or GSH, protected dehydrogenation against GTN-induced inhibition. Accordingly, inactivation of ALDH2 seems to be due to the oxidation and subsequent disulfide formation of an essential cysteine residue. The poorer performance of GSH compared with DTT has been ascribed to a combination of the lower redox potential of DTT and the presence of two thiol groups per molecule, which gives DTT a kinetic advantage over GSH (and β-ME) in achieving complete reduction of the disulfide bridge (29). The same arguments hold for LPA-H2 as well.
When DTT was added to the enzyme after complete inactivation by GTN, the thiol could only rescue a minor fraction of dehydrogenase activity, suggesting that at that time most of the enzyme was no longer present as a disulfide. Both residual and thiol-restored activities diminished when the GTN concentration was increased. This might suggest that the magnitude of residual and restored activity is determined by competition between GTN and substrate; however, the acetaldehyde concentration hardly affected either activity. Conceivably, the GTN concentration could also modulate restored activity by competition with DTT, because the GTN to DTT ratio might affect the steady-state levels of the inactive and active ALDH2 species in a reaction cycle involving oxidation/inactivation by GTN and reduction/reactivation by DTT. However, the DTT concentration also did not affect restored activity. We propose that, in addition to the DTT-reversible oxidation of the active site cysteine by GTN, the oxidized cysteinyl and/or some other active site residues may engage in a secondary reaction with GTN to yield an enzyme species that does not support dehydrogenation and is not reduced by DTT.
Scheme 1 shows a simple model for GTN-induced inactivation that accommodates most of the observations, particularly the observations that (i) irreversible inhibition in the absence of DTT is only partial and (ii) the extent of irreversible inhibition increases with the GTN concentration. Accordingly, partitioning between reversible and irreversible inhibition in the absence of a reductant will depend on the ratio of the rates of disulfide formation (krev) and irreversible inactivation (kirr·[GTN]), the latter of which depends on the GTN concentration. In the presence of a reductant (e.g. DTT), reversible inactivation will be abolished by regeneration of the active enzyme state (ESH) from the disulfide species (ES-S), and irreversible inhibition will be slowed down by the DTT-induced reduction of the oxidized thiol species (ES·). The model correctly predicts that the fraction of ALDH2 that is irreversibly inactivated by GTN in the absence of DTT will depend on the GTN concentration but not on the concentrations of DTT and acetaldehyde applied to reactivate the enzyme.
SCHEME 1.

Irreversible inactivation of ALDH-catalyzed GTN reduction. Under low turnover conditions ([GTN], [DTT] << KmGTN, KmDTT), the enzyme cycles between the reduced thiol/thiolate, oxidized thiyl, and oxidized disulfide states. At higher concentrations, GTN and the thiol reductant (here represented by DTT) compete for the oxidized thiyl state, resulting in direct regeneration of the reduced enzyme with DTT, or in irreversible inactivation with GTN (ESH, ES-, ES-S, and Ei represent the reduced (thiol/thiolate), oxidized thiyl, oxidized disulfide, and irreversibly inactivated enzyme states; kox, krev, kirr, kred, and kred′ stand for the rate constants for GTN-induced thiolate oxidation, DTT-reversible disulfide formation, GTN-induced DTT-irreversible inactivation, and DTT-exacted reduction of the ES-S and ES· enzyme states, respectively). Note that because of the branching of the reaction after thiol oxidation, irreversible inactivation in the absence of DTT will only be partial, with the extent depending on the concentration of GTN. Inactivation will be slower in the presence of DTT but, on account of the continuous recycling of the disulfide to the active reduced thiol, all of the enzyme should eventually be inactivated irreversibly.
As a rather trivial alternative explanation for the dependence of the extent of irreversible inactivation on the concentration of GTN, one could assume that GTN or one of its metabolites is inactivating the enzyme in a totally unrelated reaction. Although such a reaction would be expected to affect the rate rather than the extent of irreversible inactivation, it might explain the observations of Fig. 1, provided the reaction is slow and the time of GTN and DTT additions did not vary much between experiments. To investigate that possibility we performed the experiments illustrated by Figs. 6 and supplemental S7, in which we varied the time between GTN and DTT addition. The results indicated that most of the activity was lost within the same time frame in which GTN inhibited the enzyme (in the absence of DTT), in agreement with the mechanism-based model of inactivation presented in Scheme 1.
Although a slower reaction was also apparent, it did not depend on the GTN concentration and also occurred in the absence of GTN, suggesting that it represents unspecific deterioration of the enzyme upon prolonged incubation. In the preincubation experiments (Fig. 6) we made the observation that, although DTT protected the enzyme against inactivation in the absence of GTN, it exacerbated the inactivation in its presence. Although paradoxical at first sight, this observation is in line with expectations for the mechanism-based model of irreversible inactivation proposed by us. In the absence of DTT, the enzyme undergoes only one turnover in the presence of GTN, and mechanism-based inactivation will be halted after that. In the presence of DTT, multiple turnovers with GTN will occur, which will result in complete inactivation, even if DTT diminishes the fraction of irreversibly inactivated enzyme per turnover. Taken together, these results provide strong support for mechanism-based inactivation as the origin of irreversible inactivation.
The strictly kinetic nature of the present study precludes conclusions about the site or identity of the structural changes underlying the inactivation processes, although the literature may offer some clues. A secondary reaction, in which GTN or one of its metabolites reacts with an oxidized or nitrated thiol before generation of the disulfide can occur, might result in irreversible formation of thiol sulfinate and sulfonate products (6, 14). One candidate inactivating agent is nitroxyl (NO-), which has been proposed to be a product of GTN metabolism (31), and which was reported to cause partially irreversible inhibition of ALDH, presumably by the formation of sulfinamides (32). Another interesting possibility is that NO is produced in the inactivation process. GTN can stimulate sGC in the presence of purified ALDH2, and this stimulation is mediated by NO even though the proposed mechanism for ALDH-supported decomposition of GTN predicts nitrite as the only product (12). Formation of NO in the secondary reaction occurring at higher GTN concentrations might explain those observations. Additional structural studies will be necessary to resolve these issues. We are currently carrying out mass spectrometric studies with wild-type ALDH2 and several active site mutants to this end.
Physiological Implications—Nitrate tolerance is a complex and almost certainly multifactorial phenomenon, with potential contributions from impaired bioactivation, oxidative stress, sGC desensitization, and physiological counter-regulation (3, 6, 14, 15). The role of impaired GTN bioactivation depends on the physiologically relevant mechanism of GTN biotransformation. Because decomposition of GTN by ALDH2 results in oxidation of the active site thiol that must subsequently be reduced to sustain catalysis, the proposed involvement of ALDH2 in the bioactivation of GTN revived the early proposal that ascribed nitrate tolerance to thiol depletion (34). However, whereas the mechanism of bioactivation involves reversible thiol oxidation, an irreversible or slowly reversible process must underlie nitrate tolerance. The irreversible inhibition reported in the present study may, therefore, contribute to nitrate tolerance in vivo.
In the absence of DTT the rate of irreversible inactivation can be estimated from the observed inactivation rates (reversible + irreversible) and the relative activity after reactivation. Analysis of supplemental Fig. S1 yields an apparent inhibition rate constant of 3 × 102 m-1 s-1 with a limit of 0.04–0.06 s-1. According to Figs. 1 and 2 a large fraction of the inactivation (≥70%) will be irreversible, suggesting that the rate constant for irreversible inactivation is of similar magnitude. For GTN concentrations in the physiological range (0.01–0.1 μm) this would result in lifetimes for active ALDH2 between 6 and 60 h. For the inactivation in the presence of DTT a rate constant of ∼4 × 10-4 s-1 can be derived (Fig. 2), but this rate corresponds to saturating concentrations of GTN and DTT and cannot be easily extrapolated to physiologically relevant levels. Moreover, the present model does not account for the small but significant fraction of irreversible inhibition (∼7%) that seems to be refractory to DTT (Fig. S2).3
A crucial question regarding the physiological significance of irreversible ALDH2 inactivation concerns the applicability of the observations to GTN reduction. The results presented in Fig. 7 suggest that GTN reduction behaves like aldehyde dehydrogenation in this respect. In the absence of DTT the enzyme turned over only once and after recovery no more GTN was reduced, in line with the hypothesis that the reaction with GTN results in oxidation of the active site thiol. In the same time frame the enzyme underwent multiple turnovers (5.35 times) in the presence of DTT. However, activity diminished by 33 and 51% after preincubation with 2 μm GTN in the absence and presence of DTT, respectively. Considering that, during preincubation in the presence of DTT, ALDH2 underwent 5.35 turnovers on average, the 51% inhibition after recovery corresponds to a loss of active enzyme of 12.5% per turnover. Comparison with the 33% inhibition during one turnover in the absence of DTT indicates that DTT provides partial protection against irreversible inactivation by GTN. These results agree qualitatively with those for dehydrogenation.
The central observation of the present paper is that in addition to the previously noticed thiol-reversible inhibition, there is an irreversible aspect to the inactivation of ALDH2 by GTN. This irreversible component, which affects dehydrogenase activity as well as GTN reductase activity, may contribute to the phenomenon of nitrate tolerance. In line with this hypothesis, thiols are often unable to reverse nitrate tolerance completely (14, 30). In summary, the present study allows the following conclusions: (i) in the absence of DTT, GTN oxidizes the active site thiol of mitochondrial ALDH resulting in complete inactivation of dehydrogenation, ester hydrolysis, and GTN reduction; (ii) the rate of inactivation is affected by the concentrations of substrate and inhibitor, indicative of competitive binding, but the apparent dissociation constants (in the order of 10-4 m) are much higher than reported Km values for the respective reactions, which suggests that competition takes place outside of the high-affinity binding site; (iii) dehydrogenase activity is only restored to a modest extent by DTT, indicating that a large fraction of inactivation is irreversible; (iv) GTN reduction is irreversibly inactivated in a similar fashion as dehydrogenase activity, suggesting a role for irreversible ALDH2 inactivation in the development of nitrate tolerance.
Supplementary Material
This work was supported by the Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich (W901 DK Molecular Enzymology and P20669 to B. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Results, Figs. S1–S7, and Scheme S1.

Author's Choice—Final version full access.
Footnotes
The abbreviations used are: GTN, glyceryl trinitrate (nitroglycerin); 1,2-GDN and 1,3-GDN, 1,2- and 1,3-glyceryldinitrate; ALDH, aldehyde dehydrogenase; ALDH2, the main mitochondrial isoform of aldehyde dehydrogenase; DTT, dithiothreitol; LPA-H2, dihydrolipoic acid; β-ME, β-mercaptoethanol; Monal 62, 6-methoxy-2-naphthaldehyde; p-NPA, p-nitrophenylacetate; sGC, soluble guanylate cyclase; MES, 2-(N-morpholino)ethanesulfonic acid; NO, nitric oxide.
This underestimates the protection by DTT, because dehydrogenation already slowed down in the absence of GTN, resulting in an artifactual apparent inactivation rate constant of (4.4 ± 0.9) × 10-4 s-1. Correction of the observed apparent inactivation rate constant in the presence of DTT, (8.4 ± 1.6) × 10-4 s-1, yields a value of (4.0 ± 1.8) × 10-4 s-1, and a (15 ± 3)-fold retardation of inactivation by DTT.
Although Kd for GTN in the presence of NAD+ has not yet been reported, we obtained a Km of 7 ± 2 μm and a Vmax of 38 ± 4 nmol/mg/min in preliminary experiments. These values may be compared with the Km of 12 μm and Vmax of 30 nmol/mg/min, reported by Chen et al. (9) in the absence of NAD+.
References
- 1.Ignarro, L. J. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 7816-7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer, B. (2003) Angew. Chem. Int. Ed. 42 388-391 [DOI] [PubMed] [Google Scholar]
- 3.Chen, Z., and Stamler, J. S. (2006) Trends Cardiovasc. Med. 16 259-265 [DOI] [PubMed] [Google Scholar]
- 4.Feelisch, M. (1998) Naunyn-Schmiedeberg's Arch. Pharmacol. 358 113-122 [DOI] [PubMed] [Google Scholar]
- 5.Wang, P. G., Xian, M., Tang, X., Wu, X., Wen, Z., Cai, T., and Janczuk, A. J. (2002) Chem. Rev. 102 1091-1134 [DOI] [PubMed] [Google Scholar]
- 6.Thatcher, G. R. J., Nicolescu, A. C., Bennett, B. M., and Toader, V. (2004) Free Radic. Biol. Med. 37 1122-1143 [DOI] [PubMed] [Google Scholar]
- 7.Gorren, A. C. F., Russwurm, M., Kollau, A., Koesling, D., Schmidt, K., and Mayer, B. (2005) Biochem. J. 390 625-631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kollau, A., Beretta, M., Gorren, A. C. F., Russwurm, M., Koesling, D., Schmidt, K., and Mayer, B. (2007) Mol. Pharmacol. 72 191-196 [DOI] [PubMed] [Google Scholar]
- 9.Chen, Z., Zhang, J., and Stamler, J. S. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 8306-8311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daiber, A., Oelze, M., Coldewey, M., Bachschmid, M., Wenzel, P., Sydow, K., Wendt, M., Kleschyov, A. L., Stalleicken, D., Ullrich, V., Mülsch, A., and Münzel, T. (2004) Mol. Pharmacol. 66 1372-1382 [DOI] [PubMed] [Google Scholar]
- 11.Chen, Z., Foster, M. W., Zhang, J., Mao, L., Rockman, H. A., Kawamoto, T., Kitagawa, K., Nakayama, K. I., Hess, D. T., and Stamler, J. S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 12159-12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kollau, A., Hofer, A., Russwurm, M., Koesling, D., Keung, W. M., Schmidt, K., Brunner, F., and Mayer, B. (2005) Biochem. J. 385 769-777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sydow, K., Daiber, A., Oelze, M., Chen, Z., August, M., Wendt, M., Ullrich, V., Mülsch, A., Schulz, E., Keaney, J. F., Jr., Stamler, J. S., and Münzel, T. (2004) J. Clin. Investig. 113 482-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fung, H.-L. (2004) Annu. Rev. Pharmacol. Toxicol. 44 67-85 [DOI] [PubMed] [Google Scholar]
- 15.Münzel, T., Daiber, A., and Mülsch, A. (2005) Circ. Res. 97 618-628 [DOI] [PubMed] [Google Scholar]
- 16.Zheng, C. F., Wang, T. T., and Weiner, H. (1993) Alcohol. Clin. Exp. Res. 17 828-831 [DOI] [PubMed] [Google Scholar]
- 17.Klyosov, A. A., Rashkovetsky, L. G., Tahir, M. K., and Keung, W.-M. (1996) Biochemistry 35 4445-4456 [DOI] [PubMed] [Google Scholar]
- 18.Schenkman, J. B., and Jansson, I. (1999) in Current Protocols in Toxicology (Maines, M. D., Costa, L. G., Reed, D. J., Sassa, S., and Sipes, I. G., eds) pp. 4.1.9-4.1.11, John Wiley & Sons, Inc., New York
- 19.Wierzchowski, J., Wroczynski, P., Laszuk, K., and Interewicz, E. (1997) Anal. Biochem. 245 69-78 [DOI] [PubMed] [Google Scholar]
- 20.Feldman, R. I., and Weiner, H. (1972) J. Biol. Chem. 247 267-272 [PubMed] [Google Scholar]
- 21.Mukerjee, N., and Pietruszko, R. (1992) Arch. Biochem. Biophys. 299 23-29 [DOI] [PubMed] [Google Scholar]
- 22.Towell, J., Garthwaite, T., and Wang, R. (1985) Alcohol. Clin. Exp. Res. 9 438-442 [DOI] [PubMed] [Google Scholar]
- 23.Mukerjee, N., and Pietruszko, R. (1994) J. Biol. Chem. 269 21664-21669 [PubMed] [Google Scholar]
- 24.Tu, G.-C., and Weiner, H. (1988) J. Biol. Chem. 263 1218-1222 [PubMed] [Google Scholar]
- 25.Kitson, T. M. (1986) Biochemistry 25 4718-4724 [DOI] [PubMed] [Google Scholar]
- 26.Motion, R. L., Buckley, P. D., Bennett, A. F., and Blackwell, L. F. (1988) Biochem. J. 254 903-906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang, X., and Weiner, H. (1995) Biochemistry 34 237-243 [DOI] [PubMed] [Google Scholar]
- 28.Farrés, J., Wang, T. T. Y., Cunningham, S. J., and Weiner, H. (1995) Biochemistry 34 2592-2598 [DOI] [PubMed] [Google Scholar]
- 29.DeMaster, E. G., Redfern, B., Quast, B. J., Dahlseid, T., and Nagasawa, H. T. (1997) Alcohol 14 181-189 [DOI] [PubMed] [Google Scholar]
- 30.Wenzel, P., Hink, U., Oelze, M., Schuppan, S., Schaeuble, K., Schildknecht, S., Ho, K. K., Weiner, H., Bachschmid, M., Münzel, T., and Daiber, A. (2007) J. Biol. Chem. 282 792-799 [DOI] [PubMed] [Google Scholar]
- 31.Booth, B. P., Tabrizi-Fard, M. A., and Fung, H.-L. (2000) Biochem. Pharmacol. 59 1603-1609 [DOI] [PubMed] [Google Scholar]
- 32.DeMaster, E. G., Redfern, B., and Nagasawa, H. T. (1998) Biochem. Pharmacol. 55 2007-2015 [DOI] [PubMed] [Google Scholar]
- 33.Deleted in proof
- 34.Needleman, P., and Johnson, E. M., Jr. (1973) J. Pharmacol. Exp. Ther. 184 709-715 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.