

Abstract
Studies of adaptive mechanisms to hypoxia led to the discovery of the transcription factor called hypoxia inducible factor (HIF). HIF is a ubiquitously expressed, heterodimeric transcription factor that regulates a cassette of genes that can provide compensation for hypoxia, metabolic compromise, and oxidative stress including erythropoietin, vascular endothelial growth factor, or glycolytic enzymes. Diseases associated with oxygen deprivation and consequent metabolic compromise such as stroke or Alzheimer's disease may result from inadequate engagement of adaptive signaling pathways that culminate in HIF activation. The discovery that HIF stability and activation are governed by a family of dioxygenases called HIF prolyl 4 hydroxylases (PHDs) identified a new target to augment the transcriptional activity of HIF and thus the adaptive machinery that governs neuroprotection. PHDs lose activity when cells are deprived of oxygen, iron or 2-oxoglutarate. Inhibition of PHD activity triggers the cellular homeostatic response to oxygen and glucose deprivation by stabilizing HIF and other proteins. Herein, we discuss the possible role of PHDs in regulation of both HIF-dependent and -independent cell survival pathways in the nervous system with particular attention to the co-substrate requirements for these enzymes. The emergence of neuroprotective therapies that modulate genes capable of combating metabolic compromise is an affirmation of elegant studies done by John Blass and colleagues over the past five decades implicating altered metabolism in neurodegeneration.
Keywords: Hypoxia inducible factor, Prolyl 4-hydroxylase, Transcriptional regulation, Neuroprotection, Iron chelation
Introduction
‘Transcriptional regulation’ is a phenomena by which the amount and timing of appearance of the functional product of a gene is controlled. Processes critical to survival and death of cells such as metabolism are tightly regulated at the transcriptional level via intrinsic as well as extrinsic factors. The intrinsic transcription-machinery involves proteins called ‘transcription factors’ that bind to specific sites in DNA and recruit the basal transcriptional machinery, thereby regulating the process of gene expression. The regulation of activity of transcription factors is influenced by their protein levels or by post-translational modifications such as phosphorylation or acetylation. Signals that regulate transcription factor stability or activity are generated under physiological and pathological conditions. One of the key regulators of transcriptional activities of cells is oxygen.
Oxygen deprivation is a central feature of a number of neurological conditions including stroke, spinal cord injury and traumatic brain injury. Animals require oxygen to survive, and have evolved diverse mechanisms to sense and respond to low oxygen tensions. When faced with globally low blood oxygen levels (hypoxia), humans and other mammals reflexively engage a number of systemic physiological changes including an increase in the lung ventilation rate to restore normal oxygen tensions to vital organs. In addition to systemic homeostatic mechanisms to combat global hypoxia, organisms have also evolved cell intrinsic, homeostatic mechanisms that allow them to deal with both regional and global hypoxia [1, 2]. The ability of a single cell to gauge the oxygen concentrations in its microenvironment and to protect itself through internal regulation is a fundamental process in species of animals that depend critically on oxygen to maintain cellular function [3]. The adaptation to a sustained reduction in oxygen availability necessitates changes in gene expression, which would be predicted to lead to reduced oxygen consumption and increased oxygen delivery, and provide a means of counteracting the detrimental effects of hypoxia [3, 4]. This review will describe the cell intrinsic signaling pathways that are engaged to activate adaptive genetic responses to hypoxia in the brain and how these pathways may be manipulated for therapeutic advantage. Reduction or blockage of blood supply to the brain tissues, in conditions such as stroke, leads to a decrease in the supply of components such as glucose and oxygen necessary for the regular function of brain. After a few minutes without oxygen and glucose the cells of the brain begin to die. Among the cells that die are nerve cells. These nerve cells are responsible for controlling various parts and processes within the body. When these nerve cells cannot function properly, the body parts they are responsible for controlling also cannot function properly.
Unfortunately, dead brain cells are not spontaneously replenished and the loss of function caused by the death of these cells can be permanent. The severity and extent of the residual damage depends on the type of stroke, as well as the size and location. However there are circumstances when instead of being permanently damaged, the brain cells may be “in shock” or “bruised”. When this is the case they may start functioning properly again after they have recovered and some lost abilities may return. To facilitate this recovery the most challenging task for researchers is to identify the pathways and/or molecules that are involved in the process and regulate the recovery mechanisms and to design tools that will help manipulate the activity of these pathways in favor of recovery. One of the main hypothesis that is tested in our laboratory is that the therapeutic manipulation of endogenous transcriptional programs of neuroprotection and repair will facilitate recovery after stroke. Our studies have led us to focus on the adaptive responses to hypoxia and hypoxia-ischemia as a strategy for accomplishing this goal.
The identification of the HIF transcription system by Wang and Semenza [5] was a milestone in our understanding of oxygen physiology. Since then the HIF system has emerged as a key regulatory system of responses to hypoxia at both local and systemic levels. It is believed that approximately 1–1.5% of the genome is transcriptionally regulated by hypoxia and many of these genes are known to be regulated by HIF-1α. This remarkable protein or its homologs (e.g., HIF-2α, HIF-3α) appear to be synthesized constitutively in all of the cells of our body and constitutively degraded. Under conditions of hypoxia, hypoglycemia or hypoxia/hypoglycemia, an elegant series of molecular events are triggered that result in the stabilization of HIF proteins. The undegraded HIF-1α protein is free to dimerize with its constitutively expressed partner, HIF-1β Together HIF-1α and HIF-1β bind to specific hypoxia response elements in the promoters of a host of genes (e.g., erythropoietin, vascular endothelial growth factor, glycolytic enzymes) and activate expression of these genes (Fig. 1). These genes mediate biological changes (e.g., increased O2 delivery, increased angiogenesis, increased anaerobic glycolysis) that facilitate adaptation to hypoxia and associated metabolic compromise.
Fig. 1.
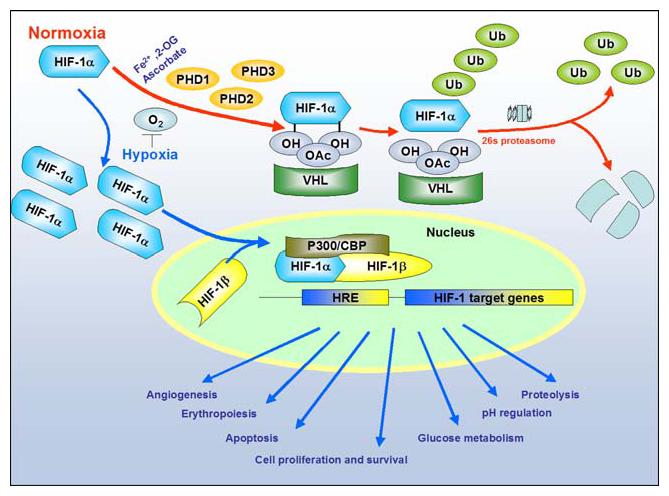
Cellular response to hypoxia: Levels of Hypoxia Inducible Factor-1 (HIF-1) are regulated by cellular oxygen by proline hydroxylation. The reaction is catalyzed by the enzymes prolyl 4-hydroxylases (PHD1:2 and 3). Under normoxia (red arrows), the intracellular level of HIF-1α is kept low by rapid ubiquitination and subsequent proteasomal degradation via recruitment of von Hippel-Lindau protein (pVHL), which depend on the hydroxylation of proline residues. In contrast, under hypoxia (blue arrows), both the intracellular level and the transcriptional activity of HIF-1α increase as a result of suppressed PHD activities. Consequently, HIF-1α forms a heterodimer with HIF-1β and changes the transcriptional rates of HIF-1-regulated genes under hypoxia (NOTE: For interpretation of the references to color in this figure legend, the reader is referred to the online version of this article)
In addition to its role in combating hypoxia, it is reasonable to propose that HIF-dependent gene expression can provide resistance to oxidative stress. This is because many of the genes regulated by HIF-1 or HIF-2 (e.g., erythropoietin, VEGF, MnSOD) can by themselves prevent oxidative stress-induced death [6-10]. Moreover, since activation of the HIF pathway is associated with a shift away from oxidative energy metabolism towards anaerobic energy metabolism, it is likely that this is accompanied by a decrease in mitochondrial free radical production [11, 12]. The putative ability of HIF directed transcription to fortify antioxidant defenses and to reduce oxidant load is intriguing given the compelling evidence for the role of oxidative stress in the destructive consequences of stroke. Oxidative stress is defined as “an imbalance between oxidants and antioxidants in favor of oxidants, potentially leading to damage” [13]. Recent evidence has pointed out that reactive oxygen species (ROS) generation occurs at multiple time points after stroke [14-16]. Oxidative stress in stroke develops as a result of excitotoxicity and inflammation leading to necrosis or apoptosis [17]. Pharmacological and genetic strategies that reduce oxidative injury also decrease brain damage. Accordingly, oxidative stress is an established target for drug development in stroke. The protection of many cellular components may be achieved by reducing oxidative damage by treating with antioxidants [18]. Activation of HIF by small molecule “drugs” potentially represents a distinct therapeutic strategy for treating oxidative stress. The advantage of this strategy as compared prior “antioxidants” is the ability of a single drug to activate more than seventy genes that could provide adaptation to ischemia and oxidative stress [19, 20]. However, the specificity of these pharmacological agents remains to be the main limitation factor in their use as therapeutic agents.
As mentioned above, one of the best-described pathways involved in adaptation to hypoxia, an important component of ischemia, includes the transcription factor, HIF-1. Studies on the expression of HIF-1 and its target genes in adult rat brain have shown that after focal cerebral ischemia, mRNAs encoding HIF-1α, glucose transporter-1 and several glycolytic enzymes including lactate dehydrogenase were up-regulated in the areas around the infarction [21]. HIF and its target genes are induced 7.5 h after the onset of ischemia and increased further at 19 and 24 h [22]. Similar findings were obtained in a rat neonatal stroke model [23]. These findings along with parallel studies in HIF-1α knockout animals indicate that HIF-1α is an essential component in changing the transcriptional repertoire of tissues as oxygen levels drop in animal models. Recent studies indicate that individuals with a certain HIF polymorphism are more likely to have a stroke [24]. These studies suggest that HIF-1 is an important target for stroke drug development in humans.
Regulation of hypoxic adaptation by HIF prolyl 4-hydroxylases
HIF is an oxygen-labile protein that becomes stabilized in neurons response to hypoxia, iron chelators and divalent cations such as cobalt chloride (CoCl2) [25]. The levels of HIF-1α are regulated by the activity of a group of enzymes called HIF-prolyl 4-hydroxylases (PHDs) (for general nomenclature, please refer to Table 1). These enzymes use iron, oxygen and 2-oxoglutarate to hydroxylate proline residues (pro-564 or pro-402) (amino acid positions designated in humans) in the HIF-1α molecule, allowing it to bind to the von Hippel-Lindau (pVHL)-E3 ubiquitin ligase complex, von Hippel-Lindau (pVHL) and undergo proteasomal degradation. So far three different iso-forms of PHDs have been identified, HIF-prolyl 4-hydroxylase-1:2 and 3 [27]. They share homology in the C-terminal catalytic domain but have significant difference in N-terminal sequences (discussed in detail in section 5).
Table 1.
Nomenclature of Prolyl 4-hydroxylase enzymes [26]
| Nomenclature 1 | Nomenclature 2 | Nomenclature 3 |
|---|---|---|
| PHD1 | HPH3 | EGLN2 |
| PHD2 | HPH2 | EGLN1 |
| PHD3 | HPH1 | EGLN3 |
The discovery of PHDs identified a new aspect of HIF regulation and indeed PHDs have become an attractive target for controlling HIF activity for therapeutic purposes. Pharmacological inhibition of these enzymes show that they can serve as a potential therapeutic target for the treatment of conditions that occur as a consequence of ischemia in a host of organs including the brain [27-34]. We previously used an in vitro model of oxidative stress to correlate the protective effects of iron chelators with their ability to activate HIF-1 [25]. In this scheme, iron chelators inhibit the activity of PHDs. Inhibition of PHDs prevents the attachment of an OH group (hydroxylation) to phylogenetically conserved proline residues at amino acid 402 and 564 in the protein HIF-1α [35]. Unhydroxylated HIF-1α does not bind the E3 ubiquitin ligase, von Hippel-Lindau protein [29, 30, 36]. HIF-1α is thus not ubiquitinated and degraded by the proteasome. Once stabilized, nuclear HIF-1α can heterodimerize with its partner, HIF-1β, bind to the pentanucleotide hypoxia response element (RCGTG) [37] in gene regulatory regions and increase the expression of established protective genes including vascular endothelial growth factor (VEGF) and erythropoietin (Epo) [2, 38]. According to this model, the protective effects of iron chelators may include inhibition of iron-dependent PHDs. In a more recent study conducted in our laboratory we have used small molecules and peptides that don't bind iron, but do inhibit the PHDs to show that PHD inhibition is sufficient to inhibit neuronal death due to oxidative stress in vitro and permanent focal ischemia in vivo [31].
Proline hydroxylation
Reaction mechanism
HIF Prolyl 4-hydroxylase enzymes (PHDs) belong to a superfamily of 2-oxoglutarate dependent hydroxylases, which employ non-heme iron in the catalytic moiety [39]. They require oxygen in the form of dioxygen, with one oxygen atom being incorporated in the prolyl residue, and the other into 2-oxoglutarate, yielding succinate and CO2 (Fig. 2). The 2-oxoglutarate is stoichiometrically decarboxylated during hydroxylation, with one atom of the O2 molecule being incorporated into the succinate and the other into the hydroxyl group formed on the proline residue. Past studies have made it possible to elucidate the reaction mechanism and structural features of the catalytic sites of dioxygenase enzymes [40-43]. The catalytic mechanism of these enzymes can be divided into two half-reactions: initial generation of the hydroxylating species, and its subsequent utilization for 4-hydroxyproline formation [40, 41]. Fe2+ is located in a pocket coordinated with the enzyme by three side-chains with two histidines and an aspartate forming the catalytic triad [41, 44, 45] (Fig. 3). Molecular oxygen is bound to the Fe2+ end-on in an axial position interacting with the dioxygen unit [41] (Fig. 3). One of the electron-rich orbitals of the dioxygen unit is directed to the electron-depleted orbital at the C2 of the 2-oxoglutarate bound to the iron (Fig. 3A), so that the C2 undergoes rehybridization and forms a covalent bond with the noncoordinated atom of the dioxygen unit. This weakens both the C–C bond in the 2-oxoglutarate and the O–O bond in the dioxygen [40, 41]. Decarboxylation then takes place simultaneously with the cleavage of the O–O bond leading to the formation of succinate. At the same time, a ferryl ion is formed, which hydroxylates a proline residue in the peptide substrate in the second half-reaction (Fig. 3B and C) [40, 41].
Fig. 2.
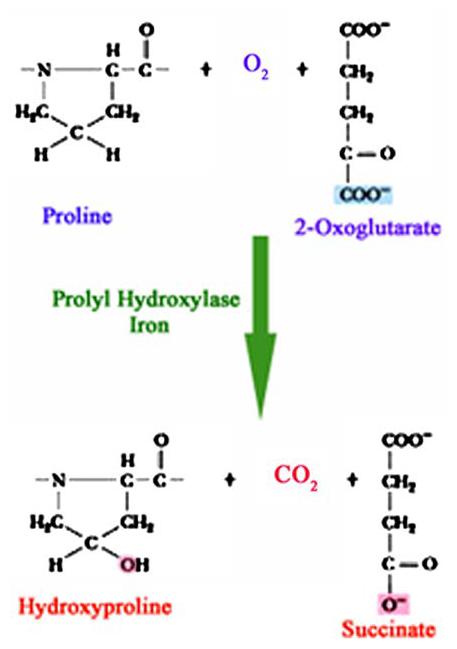
Schematic representation of the reaction catalyzed by prolyl 4-hydroxylases. The 2-oxoglutarate is stoichiometrically decarboxylated during the hydroxylation reaction in the presence of dioxygen and iron, resulting in the generation of carbon dioxide (CO2) and succinate
Fig. 3.
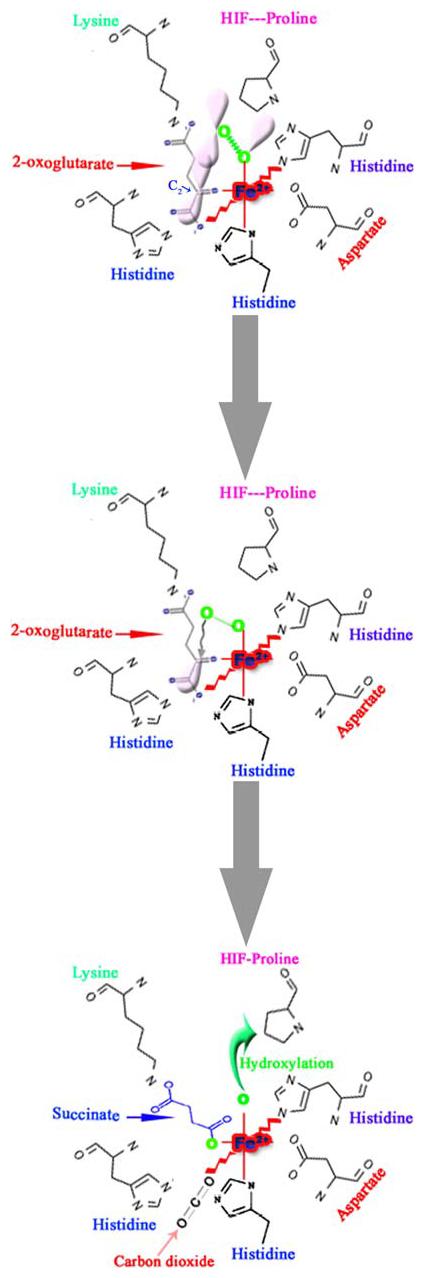
Schematic representation of the first half-reaction of prolyl 4-hydroxylase that takes place within the coordination sphere of the catalytic site iron (Fe2+) and the critical residues at the co-substrate binding sites. The Fe2+ is coordinated with the enzyme by two histidines (His) and an aspartate (Asp). The 2-oxoglutarate-binding site can be divided into two main subsites; subsite I consists of a Lysine residue (Lys), which ionically binds the C5 carboxyl group of 2-oxoglutarate, while subsite II consists of two cis-positioned equatorial coordination sites of enzyme-bound Fe2+ and is chelated by the C1 carboxyl and C2 oxo functions. Molecular oxygen is assumed to be bound end-on in an axial position, producing a dioxygen unit. (A) One of the electron-rich orbitals of the dioxygen is directed to the electron-depleted orbital at the C2 of the 2-oxoglutarate bound to the iron. (B) Nucleophilic attack on C2 generates a tetrahedral intermediate, with loss of the double bond in the dioxygen unit and of double bond characteristics in the oxo-acid moiety. (C) Elimination of CO2 coincides with the formation of succinate and a ferryl ion, which hydroxylates a proline residue in the peptide substrate in the second half-reaction. An additional amino acid in the vicinity of the catalytic triad is an important residue histidine, probably being involved in both coordination of the C1 carboxyl group of 2-oxoglutarate to Fe2+ and cleavage of the tetrahedral ferryl intermediate (Modified from Kivirikko et al. [43])
Although the PHD-mediated hydroxylation reaction is inherently dependent on ambient oxygen pressure, providing a molecular basis for the oxygen-sensing function of these enzymes [46], inhibition at other cofactor binding sites by 2-oxoglutarate analogs or iron chelators is equally capable of inhibiting the activity of the enzyme under normoxic conditions [31]. The sensitivity of the PHD activities in response to changing levels of fundamental molecules, like oxygen, 2-oxoglutarate and iron, is a very interesting and useful scheme for engaging cellular adaptive responses. A change in the cellular levels of oxygen, iron or 2-oxoglutarate could have deleterious consequences. The resulting interruption in the activity of prolyl 4-hydroxylases leads to the activation of rescue mechanisms that help the cell survive. This awards the PHD activity a potentially central role in the scheme of neuroprotection due to hypoxia, iron or 2-oxoglutarate deficiencies and makes them a specific controllable target to achieve this goal.
Oxygen
HIF Prolyl 4-hydroxylases appear to play a very important role in ensuring tight regulation of oxygen homeostasis in the brain to avoid metabolic compromise. The striking sensitivity of PHDs to graded levels of oxygen in vitro, mirrors the progressive increase in HIF-1 protein stability and transactivation activity observed when cells are subjected to graded hypoxia [27, 47]. The arterial pO2 has been reported to be around 90 mm Hg. PHDs have been found to have a striking low O2 affinity of 178 mm Hg- above the dissolved normobaric partial pressure of O2 in the air. The affinity of PHDs for O2 is much lower compared to collagen prolyl-4-hydroxylases that exhibit a Km of about 28 mm Hg [28]. A more recent study using different lengths of the HIF C- and N-terminal peptides shows about 40% change in the affinity of different isoforms of PHDs for oxygen [48]. This low affinity allows the PHDs to operate in a highly sensitive manner, in which small changes in oxygen concentration result in pronounced changes in enzymatic reaction velocity and thus HIF-1α turnover. Studies have shown that HIF-1α levels are generally low in rodent tissues under physiological conditions and are substantially increased in response to systemic hypoxia or tissue ischemia [49, 50]. Interestingly, HIF-1α levels remain low even in regions of these animals such as the renal medulla, which are characterized by low oxygen tensions. Furthermore, the extent and time course of induction as well as cell-type-specific expression of HIF-1α varies, suggesting that individual, cell-specific thresholds for activation of the response may exist. Disturbances in oxygen availability have been implicated in the central nervous system (CNS) pathology of a number of disorders including stroke, head trauma, neoplasia, vascular malformations and chronic neurodegenerative diseases. Specific redox sensitive transcription factor systems including HIF-1, nuclear factor kB (NF-kB), activator protein 1 (AP-1) and early growth response protein-1 (EGR-1), have been described that respond to changes in pO2 or an excess in reactive oxygen species (ROS) by activation of appropriate target gene expression [51, 52]. As mentioned above, the sensitivity of PHD to ambient oxygen levels in cells suggests a possible connection between these disorders and the activity of PHDs that needs to be explored.
Iron
In addition to pO2, PHD activity is regulated by the presence of ferrous iron (Fig. 3) [53]. Indeed, it had been known for some time that iron chelators or CoCl2 function as hypoxia “mimics”. Both agents stabilize HIF-1α and induce the expression of HIF target genes. Of note, they also can protect immature cortical neurons from oxidative stress [25, 31, 54]. The identification of PHDs as enzymes that contain a jellyroll motif with conserved iron and 2-oxoglutarate binding residues provided a ready explanation for the ability of iron chelators and CoCl2 to stabilize HIF. Treatment with CoCl2 also leads to stabilization of HIF-1α and a marked up-regulation of its target genes [25, 55]. Accordingly, this may be due to at least three mechanisms: partial inhibition of PHDs by depletion of ascorbate, which is required to maintain the PHDs in an active state, and/or direct binding of cobalt to HIF-1α, which may prevent its degradation by VHL-dependent and VHL-independent pathways. However, there have been recent reports that show that the enzyme retained a significant amount of activity after treatment with iron chelators such as Desferrioxamine (DFO). It was shown that only about 40–60% of the PHD activity was inhibited in vitro by DFO [55]. The studies show that the Km values for iron for PHDs are about 0.03–0.1 μM, significantly below the normal labile pool of iron (0.3–0.8 μM) [56]. Since there is a striking increase in not only HIF-1 protein stabilization but its target genes after DFO treatment, it is necessary to evaluate the decrease in the activity of these enzymes that is required to stabilize HIF-1 and downstream target genes in vivo. Using in vivo imaging system using animals expressing reporters coding for luminescent proteins such as luciferase will provide a better understanding of the effects of the pharmacological inhibitors of these enzymes in vivo [57].
Indeed HIF-1α is regulated in an iron- and redox-sensitive fashion by the HIF-prolyl 4-hydroxylases (PHDs). Oxidation of Fe2+ to Fe3+ may underlie the ability of ROS to promote HIF-1α buildup via inactivation of these enzymes under physiological conditions and in cancer [58]. Reports suggest that cysteine and histidine can also modulate iron redox status in a manner similar to ascorbate (Fig. 4) [60-62] and the relative cellular levels of these substances or of glutathione could thus play a significant role in determining the basal HIF-1α degradation rate of many cells. Deficiencies in iron reducing capacity may possibly produce greater PHD inactivation in neoplastic cells than in normal cells [63]. Treatment of embryonic cortical neurons with molecules that chelate iron such as DFO leads to inhibition of activity of the PHDs and HIF-1 upregulation [31] Fig. 4).
Fig. 4.
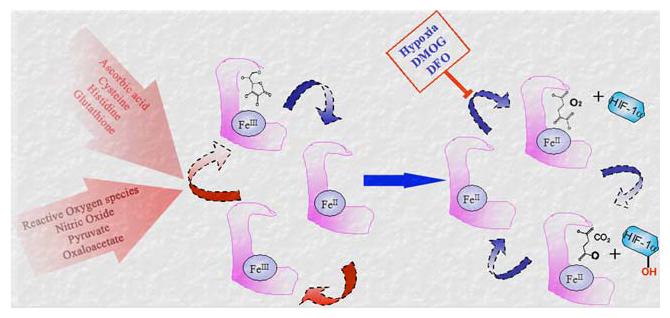
Regulation of redox state of iron at the catalytic site of the prolyl 4-hydroxylase. Ferrous (Fe2+) is required for the catalytic activity of the enzyme. The oxidation of Fe3+ to Fe2+ is facilitated by ascorbate, cysteine, Histidine or glutathione and is reversibly inhibited by reactive oxygen species, nitric oxide, pyruvate and oxaloacetate. Hypoxia, 2-oxoglutarate analogue dimethyl-oxalyl-glycine (DMOG) and iron chelator Desferrioxamine (DFO) inhibit the enzyme activity by reducing the availability of co-substrates at the catalytic site (Modified from Lu et al. [59])
Studies show that cancer cells deficient of JunD, a transcription factor involved in cell proliferation, generate excessive ROS and accumulate HIF-1 in an ascorbate- and cysteine-reversible manner [60]. A similar mechanism may underlie HIF-1 activation by ROS produced during cytokine signaling [64] or radiation therapy [65]. Similarly, redox regulatory systems such as Thioredoxin or Ref-1 have been shown to induce HIF-1 stabilization and transactivation function [66-70].
2-oxoglutarate
As shown in Fig. 3, the PHDs require a molecule of 2-oxoglutarate as a co-substrate to catalyze the transfer of a hydroxyl group at the proline 564 and 402 on HIF-1 protein. Structural analogue of 2-oxoglutarate such as dimethyl-oxalyl-glycine (DMOG) inhibit the activity of these enzymes. Normal levels of 2-oxoglutarate in various tissues range from 50 to 230 μM [71], reports suggest that similar to oxygen, the affinity of PHDs for 2-oxoglutarate is very low i.e., ∼60 μM as compared to that of collagen prolyl 4-hydroxylases that is 20 μM [55]. This low affinity provided high sensitivity towards changes in the levels of this co-substrate that might occur due to defects in other metabolic pathways such as Krebs cycle. Defects in the Krebs cycle are likely to influence 2-oxoglutarate levels compromising PHD function. Recent reports show that some other metabolic intermediates of TCA cycle such as pyruvate and oxaloacetate also effect PHD function in an isoform dependent manner [72]. Accordingly, pyruvate and oxaloacetate can activate HIF by binding reversibly to the 2-oxoglutarate site of two distinct isoforms of HIF-1 prolyl 4-hydroxylases, PHD2 and PHD3, at concentrations comparable to their normal physiological levels (Fig. 4). Both pyruvate and oxaloacetate can also induce expression of prolyl 4-hydroxylase isoforms 1 and 2 genes in human glioblastoma cells as a mechanism for limiting hypoxia-inducible gene expression and also for adapting to re-oxygenation. There are contradictory reports regarding the effects of other TCA cycle metabolites such as fumarate and succinate on HIF-1 activation [59, 73-75]. Little is known about the involvement of PHDs in pathological settings such as neoplasia or neurological disorders. However, reports suggest that PHD may be a target of growth factor signaling pathways and/or oncogenic transformation. It has been shown that certain oncogenes such as ras and src induce HIF-1 under normoxia by inhibiting prolyl hydroxylation on Pro 564 [76]. Recent findings also suggest a possible link between PHD function, tumorigenesis and neurodegeneration. Accordingly, the two ubiquitously expressed mitochondrial enzymes, succinate dehydrogenase (SDH) and fumarate hydratase (FH), catalyze sequential steps in the Krebs cycle. Interestingly, mutations in the autosomally encoded enzyme and enzyme subunits are associated with hereditary predispositions to various tumors [77-80]. Thus, both SDH and FH act as tumor suppressor genes. In contrast, homozygous germline mutations in SDH subunits cause mitochondriopathies such as Leigh's syndrome, which affect the central nervous system and skeletal muscles. The underlying mechanism through which loss of function leads to neoplasia or neurodegeneration remains unclear [81]. It is therefore likely that defects in the Krebs cycle influence 2-oxoglutarate levels or other metabolic intermediates compromising PHD function.
PHDs—beyond HIF-1
The above discussion suggests that it is reasonable to hypothesize that PHDs act as iron- and redoxsensors. It also strengthens the idea that PHDs are contributing towards the cell's fate via multiple mechanisms. There is indirect evidence that PHDs interact with molecules other than HIF-1 such as iron regulatory protein-2 (IRP2), RNA polymerase II (RNA pol II), mitogen-activated protein kinase organizer-1 (MORG-1) and other isoforms of HIF itself.
PHDs and other HIFs
HIF-2α and HIF-3α are the other isoforms of HIF-1α [82-84]. Like HIF-1α, HIF-2α (also known as endothelial PAS protein-1, EPAS-1) is also upregulated by hypoxia and activates transcription via binding of hypoxia response elements (HREs) and is regulated in the same way as HIF-1 [76, 83, 85-88]. The HIF-1α and HIF-2α proteins share only 48% identity, but they both contain the conserved oxygen degradation domain [89, 90], and are regulated by prolyl 4-hydroxylases. There is increasing evidence that HIF-1α and HIF-2α may have different functions [91-94]. For example, the targeted inactivation of HIF-1α and HIF-2α genes in embryonic stem cells results in different responses to various stimuli, such as hypoxia and hypoglycemia [95] and HIF-1α−/− and HIF-2α−/− knock out mouse embryos have different and variable phenotypes [96-98]. HIF-3α has high similarity with HIF-1α and HIF-2α in the regulatory oxygen degradation domains (ODD), but lacks structures for transactivation that are found in the C-terminus of HIF-1α and HIF-2α. The activity of HIF-3α is upregulated in low oxygen, it dimerises with HIF-β/ARNT and binds to the HRE core sequence, but interestingly, HIF-3α appears to suppress hypoxia-inducible HIF-mediated gene expression in the human kidney and could therefore be a negative regulator of hypoxia-inducible gene expression [99, 100]. These findings create a very interesting scenario with reference to the consequences of PHD activity. The differences in the functions of different isoforms of HIFα regulated by the prolyl 4-hydroxylases indicates that PHDs work via multiple molecules/pathways depending on the stimuli and physiological state of the cell that define the overall outcome of the event.
PHDs and IRP2
Oxidative stress is a hallmark of neurodegenerative disorders [101], such as Parkinson's and Alzheimer's diseases, where iron accumulates pathologically in the substantia nigra [102] or in senile plaques [103], respectively. Even though it is still not clear whether this is a primary cause or a secondary effect, iron is increasingly being considered as a pathogenic cofactor [104, 105]. IRP2 is regulated in response to the iron and oxygen supply. It is synthesized de novo under conditions of iron starvation, [106] remains stable in iron-starved or hypoxic cells, [107] and undergoes proteasomal degradation in iron-replete and normoxic cells [108]. Both iron and oxygen are required for the activity of PHDs. Pharmacological data suggest that a pathway for iron-mediated degradation of IRP2 requires the activity of PHDs [109-111], the co-substrate analogue dimethyl-oxalyl-glycine (DMOG) can efficiently inhibit the iron-dependent degradation of IRP2 in cells pretreated with DFO [109, 112]. The reports show that when human embryonic kidney cells are treated with an iron chelator, DFO, the IRP2 is stabilized, this effect is diminished by the addition of iron. However, addition of DMOG, a PHD inhibitor, diminishes the iron-induced degradation of IRP2. Accordingly, the pathways for iron- and oxygen-dependent degradation of IRP2 and HIF-1 share remarkable similarities [107]. However, DMOG fails to inhibit IRP2 degradation in cells not pretreated with DFO suggesting that this pathway operates in conjunction with the levels of the labile iron pool (LIP) and is activated only when previously iron-depleted cells are exposed to iron. These findings provide the groundwork for the identification of components of the pathway, including the 2-oxoglutarate-dependent enzymes in future.
PHDs and RNA Pol II
The von Hippel-Lindau (pVHL) protein regulates the accumulation of HIF-1 by acting as an E3 ubiquitin ligase complex, [29, 30, 36, 113], and thereby regulates the expression of hypoxia-inducible genes. The pVHL complex can function as an E3 ligase, which targets the hyperphosphorylated large subunit (Rpb1) of RNA polymerase II for ubiquitination and degradation [114]. Importantly, binding of pVHL to the full-size Rpb1 requires hydroxylation of proline-1465 within Rpb1 and phosphorylation of the carboxy terminal domain (CTD) [114-116]. Immobilized Rpb1 peptide (amino acids 1440–1475) containing the hydroxylated proline binds to pVHL, whereas the nonhydroxylated peptide does not bind pVHL as shown by the Kornberg laboratory [117]. This reaction occurs in a manner similar to HIF-1 regulation. However, it acquires pVHL-binding properties after incubation with PC12 cell extracts in the presence of Fe2+, ascorbic acid, and 2-oxoglutarate, cofactors required for PHD activity [114]. The hydroxylated, but not the nonhydroxylated, peptide competes with HIF for pVHL binding [114]. These data provide biochemical evidence that Rpb1 can be modified by proline hydroxylation. However, it is unclear whether proline hydroxylation requires CTD phosphorylation. Comparison of the sequence of the HIF-1α oxygen dependent degradation domain (ODDD) with representative libraries of protein structures revealed an identical region with similarities to a fragment of Rpb1 and the adjacent Rpb6 subunit. An almost 50-amino acid Rpb1 counterpart is 30% identical to HIF-1α and contains the L(XY)LAP motif, including proline at position 1465 similar to the proline residue at position 564 in HIF-1α. Computational models and experimental observations demonstrate significant structural similarity between the ODDD of HIF-1 and Rpb1/Rpb6. It is therefore reasonable to predict that the stability and thus function of RNA pol II in regulation of gene expression requires proline hydroxylation. The direct interaction between the PHDs and the Rbp1 and Rbp6 is thus an important piece of information that has yet to be provided by any group.
PHDs and MORG-1
The mitogen-activated protein kinases (MAPKs) extracellular signal-regulated kinase (ERK)-1 and ERK-2 are components of an evolutionarily conserved protein kinase cascade that participates in the regulation of various cellular processes, including gene expression, growth, differentiation, and apoptosis. A number of “scaffolding proteins”, both yeast and mammalian, were shown to associate with and enhance functional interaction of the components of MAPK pathways, although their physiological and molecular functions are incompletely understood. MAPK organizer (MORG)-1, which interacts with several components of the ERK cascade and functions as a scaffold protein linking ERK responses to specific agonists. Recent evidence shows that MORG-1 interacts with PHD3 and decreases HIF-1 mediated reporter gene induction [118]. It is interesting that PHD3 has been recently been implicated in the survival of sympathetic neurons following growth factor deprivation [119]. The physiological significance of MORG-1 interaction with PHD3 remains to be elucidated, however, as this report shows an interaction between the non-conserved region of PHD3 and a novel interactor MORG-1, it presents an important example that suggests yet another possible mechanism/means to modify the activity of PHDs in a very isoform specific manner since the three isoforms differ in their non-conserved regions. Such interactions also broaden the search for novel molecules that might be interacting with PHDs and effecting their activities, but do not contain potential hydroxylation sites in the primary structure. Detailed study of these novel interactions will not only identify novel pathways that are involved in the PHD regulated biology. The direct effect of these interaction on the overall well being and survival of neurons will not only uncover new pathways and targets for PHDs but will also help define the precise role of distinct PHD isoforms in neuroprotection.
PHDs and SIAH 2
Changes in oxygen affect PHD transcription and stability in addition to directly affecting PHD enzymatic activity [27, 120, 121]. Hypoxia also induces the accumulation of the ring finger protein seven in absentia (Drosophila) homolog 2 (SIAH2) [122] possibly in a HIF-independent manner [123]. SIAH2 is highly conserved mammalian homolog of drosophila seven in absentia (SIAH), which possesses potent RING finger E3 ubiquitin ligase activities. This activity is implicated in the degradation of diverse proteins including DCC, β-catenin, N-CoR, c-myb, Numb, and TRAF2 [124-129]. Analysis of steady-state levels of exogenously expressed PHD3 revealed almost undetectable expression in SIAH2+/+ cells, whereas it was clearly detected in SIAH2−/− cells [123], showing that SIAH2 negatively regulates PHD3 levels. The same report shows that SIAH2 poly ubiquitylates, and hence tags PHD1 and PHD3 for destruction [123]. The regulation of PHDs by the SIAH proteins indicates a feedback mechanism of PHD regulation that controls the activity of PHD3. A more recent study [130] shows that the isoform specific interaction of Siah2 with the PHD3 is due to the differences in the structure of the three molecules. The amino terminal domains of PHD1 and PHD2 limit their direct interaction with SIAH2 and that PHD3 can form complexes which include homo- and hetero-dimers/multimers, and that assembly of PHD3 into complexes affects its activity towards HIF-1α and susceptibility for degradation by SIAH2.
Isoforms of HIF-Prolyl 4-hydroxylase enzymes
To date three isozymes of the mammalian prolyl 4-hydroxylases specific to HIF-1α have been identified: PHD1, PHD2 and PHD3 [27] (corresponding to human HPH3, HPH2 and HPH1 [26] and C. elegans EGLN2, EGLN1, EGLN3 [131] (Table 1). All three orthologs appear to be a product of gene duplication as there is only one gene in C. elegans (EGL-9) [27] and D. melanogaster (CG1114) [132]. The three isoforms differ in expression regulation, tissue distribution, cellular localization, and ability to hydroxylate HIF-1α (Table 2) [38, 123]. They share homology in the C-terminal catalytic domain but have significant difference in N-terminal sequences [133-135].
Table 2.
Summary of the physiological properties of the three HIF prolyl 4-hydroxylases indicating their ability to modify Pro402/564 (designated position in human HIF), intracellular localization (N, nuclear; C, cytosolic), ability to mediate HIF-1 stability as determined by RNA interference, induction of hydroxylase mRNA expression in response to hypoxia, and tissue expression patterns [26]
| Gene | Size kDa | Substrate |
Localization | RNAi | Hypoxia induction |
Tissue distribution | |
|---|---|---|---|---|---|---|---|
| p402 | p564 | ||||||
| EGLN1/HPH-2/PHD2 | 46 | + | + | C/ > N | + + + | + | liver = heart > brain = kidney > lung |
| EGLN2/HPH-3/PHD1 | 43.6 | + | + | N | + | − | Testis > liver > kidney = heart = brain |
| EGLN3/HPH-1/PHD3 | 27.3 | − | + | C/N | − | + | Heart > liver > brain > kidney > lung |
PHD1 is a 43.6 kDa constitutively expressed protein that has no response to hypoxia or CoCl2 at mRNA level [136] but is induced by estrogen and stimulates cell proliferation [137]. PHD1 has high level of expression in the testis with low level expression in the kidney, liver and heart [138-140]. It was found that this protein localizes exclusively to the nucleus [134]. As mentioned before, there are two oxygen dependent domains (ODD) in HIF-1α. A C-terminal ODD (CODD) (around proline 402) and an N-terminal ODD (NODD) (around proline 564). PHD1 hydroxylates both N- and C-terminal ODD in HIF-1α. Two isoforms of PHD1 with molecular weights of 43 and 40 kDa, respectively are generated by alternative translational initiation [141]. Both PHD1 isoforms have similar HIF-1α prolyl 4-hydroxylase activity. Although it is commonly accepted that PHD1 is not transcriptionally upregulated by low oxygen tensions, a recent report shows upregulation of PHD1 mRNA in HT22 cells as well as in brains of hypoxic mice [142].
PHD2 is a 46 kDa protein whose expression can be regulated by hypoxia, DFO or CoCl2, all established inhibitors of prolyl 4-hydroxylase activity [134, 143]. Basal expression levels are high in heart and moderate in brain. The majority of PHD2 is localized to the cytoplasm with lower levels in the nucleus [134]. The dominant cytoplasmic localization of PHD2 in human osteosarcoma cells (U2OS) and human hepatoma cells (Hep3B) suggest about active exclusion of this isoform from nucleus [134]. Groulx and Lee provide evidence that the degradation of HIF-1α is initiated by strictly nuclear prolyl hydroxylation and is completed by the export of HIF-1α-pVHL complexes [144]. PHD2 hydroxylates both N-and C-terminal ODD in HIF-1α Studies have shown that although all three isoforms hydroxylate specific prolines in HIF subunits in a strongly conserved LXXLAP sequence motif (where X indicates any amino acid and P indicates the hydroxyl acceptor proline), mutation in the amino acid residues in the vicinity of these amino acids was least tolerated by PHD2 thus showing the highest specific activity toward the primary hydroxylation site of HIF-1α [27, 133, 134]. PHD2 has also been shown to have more influence over HIF-1α than HIF-2α [145]. Only inhibition of PHD2 but not PHD1 or PHD3 by siRNA is sufficient to upregulate HIF-1α in normoxia in various cell types such as HeLa (human cervical carcinoma cell line), CAL27 (human tongue squamous cell carcinoma cell line), HaCAT (Human keratinocyte cell line), HT29 (colon adenocarcinoma cell line), CAL51 (breast cancer cell line), RCC4/pVHL (RCC4 stably transfected with pcDNA3pVHL), WM9 (melanoma cell line) and FHN (primary fibroblasts) [146]. On the other hand, inhibition of HIF-1α by siRNA also leads to a loss of upregulation of PHD2 and PHD3 in hypoxia [147]. A hypoxia response element (the binding site for HIF-1) has been found in the human PHD2 gene. It is located at 412 bp upstream of start codon [148]. Analysis of PHD2 mRNA expression level in different organs of rat following hypoxia (8 h at 8% oxygen) showed its reduction in brain, whereas in heart and kidney there were no changes in the PHD2 mRNA levels. Liver on the other hand showed an increase in the mRNA levels of PHD2 [140]. Interestingly, the brain was the only organ where PHD2 mRNAs (and PHD3 mRNA in two out of three rats) were downregulated after 8 h systemic hypoxia. This does not appear to be a toxic effect since levels of the other genes analyzed were preserved or enhanced. The mechanism of this PHD downregulation is unknown, but it could potentially lead to enhanced HIF accumulation, mediating possible cerebral protective effects [140].
The third identified isoform, PHD3 is a 27.3 kDa protein. HRE consensus sequences in a region of 40 kb around the human EGLN3 gene have been identified [149]. PHD3 expression can be upregulated by hypoxia or the hypoxia mimetics, DFO or CoCl2. It is distributed evenly in nucleus and cytoplasm [134]. Despite it being an oxygen-dependent enzyme, PHD3 retains significant activity under conditions of hypoxia with greater activity towards HIF-2α [145] as opposed to HIF-1α, similar observation has been reported for PHD1. PHD3 preferentially hydroxylates CODD PHD3 hydroxylates the CODD but not NODD [27, 28, 145, 150]. In addition to full length PHD3 (27 kDa) at least two alternative slice forms have been found with predicted molecular weight 17 kDa and 24 kDa, respectively [151]. Expression patterns in the tissue show that the full length of PHD3 is detected as major product and the 17-kDa protein is expressed in the same tissue as a minor product. This small splice version has no prolyl 4-hydroxylase activity. In contrast the alternative splice variant that encodes a 24-kDa protein preserves prolyl 4-hydroxylase activity. It is interesting that the 24 kDa protein has been found only in primary cancer tissues [151]. Both active versions of PHD3 in Hep3B cells are localized to the nucleus. An interesting homolog for PHD3 reported in the literature is SM-20. SM-20 is a growth factor regulated gene in muscle cells that can promote cell death in neurons [131, 152, 153]. The extremely high degree of sequence identity between SM-20 and PHD3 indicates that they represent orthologous proteins in rats, mouse and humans [131].
Conclusion
The above discussion identifies some fundamental gaps in our knowledge of how PHD enzymes operate in brain: Are the neuroprotective effect of PHD inhibition observed after treatment with global inhibitors of PHDs mainly driven by inhibition of one isoform? If so which one? What is the subcellular localization of that isoform in brain? Is it localized in cellular compartments abundant in HIF-1? As discussed above, each one of these isoforms has distinct roles and specificity, depending upon their subcellular localization in different organs and cell types, however very little is known about these details in different regions and cell types in brain. Moreover the role of these enzymes in regulating mechanisms (other than HIF stabilization) that might be involved in the process of cell survival is also garnering support. Growing awareness of other substrates such as IRP2 [109] and RNA polymerase [114] or the RNA polymerase heavy subunit whose stability can be regulated by a hydroxylation reaction suggests that other plausible schemes in addition to, or exclusive of HIF-1 that may account for protection by HIF prolyl 4-hydroxylase inhibition. Identification of the isoform(s) of prolyl 4-hydroxylase enzymes that is neuroprotective and its novel target(s) to pinpoint the pathways that are regulated during this neuroprotection. This will allow researchers to design tools/molecules that will facilitate the manipulation of the activity and function of the relevant PHD isoform(s) as a therapeutic target in stroke and other diseases associated with metabolic compromise such as Alzheimer's disease. As metabolic compromise is firmly rooted in the pathophysiology of neurodegeneration, PHD inhibitors are poised to take center stage in CNS therapeutics. However, one important missing link for this approach is the risk associated with chronic administration of PHD inhibitors. For instance, transcriptional regulation of cell survival pathways need to be strictly controlled to eliminate risk of tumor growth. Further investigations are required to understand the risks as well as the possibilities to eliminate the risk by combinatorial therapy approaches.
Abbreviations
- AP-1
Activator protein 1
- CO2
Carbon dioxide
- CoCl2
Cobalt chloride
- CODD
Carboxy-terminal oxygen degradation domain
- DFO
Desferrioxamine
- DMOG
Dimethyl-oxalyl-glycine
- EGR-1
Early growth response protein-1
- ERK
Extracellular signal-regulated kinase
- Fe2+
Ferrous
- Fe3+
Ferric
- FH
Fumarate hydratase
- HIF
Hypoxia inducible factor
- HIF-1α
Hypoxia inducible factor-1 alpha
- HIF-1β
Hypoxia inducible factor-1 beta
- HRE
Hypoxia response elements
- IRP2
Iron regulatory protein-2
- LIP
Labile iron pool
- MnSOD
Manganese Superoxide Dismutase
- MORG-1
Mitogen-activated protein kinase organizer-1
- NF-kB
Nuclear factor kappa B
- NODD
N-terminal oxygen degradation domain
- O2
Oxygen
- ODD
Oxygen degradation domain
- ODDD
Oxygen dependent degradation domain
- PHD
prolyl 4-hydroxylase domain
- PVHL
von Hippel-Lindau protein
- RNA pol II
RNA polymerase II
- ROS
Reactive Oxygen Species
- SIAH2
Seven in absentia homolog 2
- SDH
Succinate dehydrogenase
- TCA
Tricarboxylic acid
- VEGF
Vascular endothelial growth factor
Footnotes
Special issue dedicated to John P. Blass.
References
- 1.Ikeda E. Cellular response to tissue hypoxia and its involvement in disease progression. Pathol Int. 2005;55:603–610. doi: 10.1111/j.1440-1827.2005.01877.x. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Chairman's summary: mechanisms of oxygen homeostasis, circa 1999. Adv Exp Med Biol. 2000;475:303–310. doi: 10.1007/0-306-46825-5_29. [DOI] [PubMed] [Google Scholar]
- 3.Peng YJ, Rennison J, Prabhakar NR. Intermittent hypoxia augments carotid body and ventilatory response to hypoxia in neonatal rat pups. J Appl Physiol. 2004;97:2020–2025. doi: 10.1152/japplphysiol.00876.2003. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 5.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 6.Josko J, Mazurek M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med Sci Monit. 2004;10:RA89–98. [PubMed] [Google Scholar]
- 7.Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207:3233–3242. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- 8.Nagy Z, Simon L, Bori Z. Regulatory mechanisms in focal cerebral ischemia. New possibilities in neuroprotective therapy. Ideggyogy Sz. 2002;55:73–85. [PubMed] [Google Scholar]
- 9.Scortegagna M, Ding K, Zhang Q, Oktay Y, Bennett MJ, Bennett M, Shelton JM, Richardson JA, Moe O, Garcia JA. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood. 2005;105:3133–3140. doi: 10.1182/blood-2004-05-1695. [DOI] [PubMed] [Google Scholar]
- 10.Zelko IN, Folz RJ. Extracellular superoxide dismutase functions as a major repressor of hypoxia-induced erythropoietin gene expression. Endocrinology. 2005;146:332–340. doi: 10.1210/en.2004-1007. [DOI] [PubMed] [Google Scholar]
- 11.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sies H, Cadenas E. Oxidative stress: damage to intact cells and organs. Philos Trans R Soc Lond B Biol Sci. 1985;311(1152):617–631. doi: 10.1098/rstb.1985.0168. [DOI] [PubMed] [Google Scholar]
- 14.Dawson SL, Blake MJ, Panerai RB, Potter JF. Dynamic but not static cerebral autoregulation is impaired in acute ischaemic stroke. Cerebrovasc Dis. 2000;10:126–132. doi: 10.1159/000016041. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36:1264–1269. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- 16.Mackensen GB, Patel M, Sheng H, Calvi CL, Batinic-Haberle I, Day BJ, Liang LP, Fridovich I, Crapo JD, Pearlstein RD, Warner DS. Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–4592. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrova ML, Bochev PG. Oxidative stress during the chronic phase after stroke. Free Radic Biol Med. 2005;39:297–316. doi: 10.1016/j.freeradbiomed.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 18.Koziol JA, Feng AC. On the analysis and interpretation of outcome measures in stroke clinical trials: lessons from the SAINT I study of NXY-059 for acute ischemic stroke. Stroke. 2006;37:2644–2647. doi: 10.1161/01.STR.0000241106.81293.2b. [DOI] [PubMed] [Google Scholar]
- 19.Ralph GS, Parham S, Lee SR, Beard GL, Craigon MH, Ward N, White JR, Barber RD, Rayner W, Kingsman SM, et al. Identification of potential stroke targets by lentiviral vector mediated overexpression of HIF-1 alpha and HIF-2 alpha in a primary neuronal model of hypoxia. J Cereb Blood Flow Metab. 2004;24:245–258. doi: 10.1097/01.WCB.0000110532.48786.46. [DOI] [PubMed] [Google Scholar]
- 20.Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, Gensert J, Chavez J. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35:2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 21.Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- 22.Sharp FR, Bergeron M, Bernaudin M. Hypoxia-inducible factor in brain. Adv Exp Med Biol. 2001;502:273–291. doi: 10.1007/978-1-4757-3401-0_18. [DOI] [PubMed] [Google Scholar]
- 23.Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis. 2003;14:524–534. doi: 10.1016/j.nbd.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Tupitsyna TV, Slominskii PA, et al. Association of the IVS9-675C > A polymorphism of the HIF-1alpha gene with acute ischemic stroke in the Moscow population. Genetika. 2006;42(6):858–861. [PubMed] [Google Scholar]
- 25.Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 27.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 28.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 29.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, Kaelin WG., Jr Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:13459–13464. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 31.Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elkins JM, Hewitson KS, et al. Structure of factor-inhibiting hypoxiainducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem. 2003;278(3):1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 33.Koivunen P, Hirsila M, et al. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279(11):9899–9904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 34.Schlemminger I, Mole DR, et al. Analogues of dealanylalahopcin are inhibitors of human HIF prolyl hydroxylases. Bioorg Med Chem Lett. 2003;13(8):1451–1454. doi: 10.1016/s0960-894x(03)00149-5. [DOI] [PubMed] [Google Scholar]
- 35.Willam C, Masson N, Tian YM, Mahmood SA, Wilson MI, Bicknell R, Eckardt KU, Maxwell PH, Ratcliffe PJ, Pugh CW. Peptide blockade of HIFalpha degradation modulates cellular metabolism and angiogenesis. Proc Natl Acad Sci USA. 2002;99:10423–10428. doi: 10.1073/pnas.162119399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001b;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 38.Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003;17:2614–2623. doi: 10.1101/gad.1145503. [DOI] [PubMed] [Google Scholar]
- 39.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 40.Hanauske-Abel HM. Prolyl 4-hydroxylase, a target enzyme for drug development. Design of suppressive agents and the in vitro effects of inhibitors and proinhibitors. J Hepatol. 1991;13(Suppl 3):S8–15. doi: 10.1016/0168-8278(91)90003-t. discussion S16. [DOI] [PubMed] [Google Scholar]
- 41.Hanauske-Abel HM, Gunzler V. A stereochemical concept for the catalytic mechanism of prolylhydroxylase: applicability to classification and design of inhibitors. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 42.Lamberg A, Pihlajaniemi T, Kivirikko KI. Site-directed mutagenesis of the alpha subunit of human prolyl 4-hydroxylase. Identification of three histidine residues critical for catalytic activity. J Biol Chem. 1995;270:9926–9931. doi: 10.1074/jbc.270.17.9926. [DOI] [PubMed] [Google Scholar]
- 43.Myllyharju J, Kivirikko KI. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. Embo J. 1997;16:1173–1180. doi: 10.1093/emboj/16.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE. Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature. 1995;375:700–704. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 45.Roach PL, Clifton IJ, Hensgens CM, Shibata N, Schofield CJ, Hajdu J, Baldwin JE. Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature. 1997;387:827–830. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- 46.Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207:3171–3188. doi: 10.1242/jeb.01075. [DOI] [PubMed] [Google Scholar]
- 47.Chavez JC, LaManna JC. Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J Neurosci. 2002;22:8922–8931. doi: 10.1523/JNEUROSCI.22-20-08922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem. 2006;281:28712–28720. doi: 10.1074/jbc.M604628200. [DOI] [PubMed] [Google Scholar]
- 49.Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. Faseb J. 2001;15:2445–2453. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 50.Wiesener MS, Maxwell PH. HIF and oxygen sensing; as important to life as the air we breathe? Ann Med. 2003;35:183–190. doi: 10.1080/0785389031000458233. [DOI] [PubMed] [Google Scholar]
- 51.Koong AC, Chen EY, Mivechi NF, Denko NC, Stambrook P, Giaccia AJ. Hypoxic activation of nuclear factor-kappa B is mediated by a Ras and Raf signaling pathway and does not involve MAP kinase (ERK1 or ERK2) Cancer Res. 1994;54:5273–5279. [PubMed] [Google Scholar]
- 52.Yan SF, Lu J, Zou YS, Soh-Won J, Cohen DM, Buttrick PM, Cooper DR, Steinberg SF, Mackman N, Pinsky DJ, Stern DM. Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- 53.Knowles HJ, Raval RR, et al. Effect of ascorbate on the activity of hypoxiainducible factor in cancer cells. Cancer Res. 2003;63(8):1764–1768. [PubMed] [Google Scholar]
- 54.Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 55.Hirsila M, Koivunen P, Xu L, Seeley T, Kivirikko KI, Myllyharju J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. Faseb J. 2005;19:1308–1310. doi: 10.1096/fj.04-3399fje. [DOI] [PubMed] [Google Scholar]
- 56.Esposito BP, Epsztejn S, Breuer W, Cabantchik ZI. A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal Biochem. 2002;304:1–18. doi: 10.1006/abio.2002.5611. [DOI] [PubMed] [Google Scholar]
- 57.Safran M, Kim WY, O'Connell F, Flippin L, Gunzler V, Horner JW, Depinho RA, Kaelin WG., Jr Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA. 2006;103:105–110. doi: 10.1073/pnas.0509459103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–41939. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 60.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 61.Myllyla R, Kuutti-Savolainen ER, Kivirikko KI. The role of ascorbate in the prolyl hydroxylase reaction. Biochem Biophys Res Commun. 1978;83:441–448. doi: 10.1016/0006-291x(78)91010-0. [DOI] [PubMed] [Google Scholar]
- 62.Winkler P, Schaur RJ, Schauenstein E. Selective promotion of ferrous ion-dependent lipid peroxidation in Ehrlich ascites tumor cells by histidine as compared with other amino acids. Biochim Biophys Acta. 1984;796:226–231. doi: 10.1016/0005-2760(84)90121-8. [DOI] [PubMed] [Google Scholar]
- 63.Shoichet SA, Baumer AT, Stamenkovic D, Sauer H, Pfeiffer AF, Kahn CR, Muller-Wieland D, Richter C, Ristow M. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum Mol Genet. 2002;11:815–821. doi: 10.1093/hmg/11.7.815. [DOI] [PubMed] [Google Scholar]
- 64.BelAiba RS, Djordjevic T, Bonello S, Flugel D, Hess J, Kietzmann T, Gorlach A. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem. 2004;385:249–257. doi: 10.1515/BC.2004.019. [DOI] [PubMed] [Google Scholar]
- 65.Moeller BJ, Dewhirst MW. Raising the bar: how HIF-1 helps determine tumor radiosensitivity. Cell Cycle. 2004;3:1107–1110. [PubMed] [Google Scholar]
- 66.Carrero P, Okamoto K, Coumailleau P, O'Brien S, Tanaka H, Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. Embo J. 1999;18:1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 69.Lando D, Pongratz I, Poellinger L, Whitelaw ML. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1alpha and the HIF-like factor. J Biol Chem. 2000;275:4618–4627. doi: 10.1074/jbc.275.7.4618. [DOI] [PubMed] [Google Scholar]
- 70.Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62:5089–5095. [PubMed] [Google Scholar]
- 71.Mans AM, DeJoseph MR, Hawkins RA. Metabolic abnormalities and grade of encephalopathy in acute hepatic failure. J Neurochem. 1994;63:1829–1838. doi: 10.1046/j.1471-4159.1994.63051829.x. [DOI] [PubMed] [Google Scholar]
- 72.Dalgard CL, Lu H, Mohyeldin A, Verma A. Endogenous 2-oxoacids differentially regulate expression of oxygen sensors. Biochem J. 2004;380:419–424. doi: 10.1042/BJ20031647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 74.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 75.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 76.Chan DA, Sutphin PD, Denko NC, Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. J Biol Chem. 2002;277:40112–40117. doi: 10.1074/jbc.M206922200. [DOI] [PubMed] [Google Scholar]
- 77.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 79.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 80.Pollard PJ, Wortham NC, Tomlinson IP. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann Med. 2003;35:632–639. doi: 10.1080/07853890310018458. [DOI] [PubMed] [Google Scholar]
- 81.Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- 82.Marti HH, Katschinski DM, Wagner KF, Schaffer L, Stier B, Wenger RH. Isoform-specific expression of hypoxia-inducible factor-1alpha during the late stages of mouse spermiogenesis. Mol Endocrinol. 2002;16:234–243. doi: 10.1210/mend.16.2.0786. [DOI] [PubMed] [Google Scholar]
- 83.Semenza GL. Perspectives on oxygen sensing. Cell. 1999;98:281–284. doi: 10.1016/s0092-8674(00)81957-1. [DOI] [PubMed] [Google Scholar]
- 84.Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. Faseb J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 85.Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755:107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Carroll VA, Ashcroft M. Targeting the molecular basis for tumour hypoxia. Expert Rev Mol Med. 2005;7:1–16. doi: 10.1017/S1462399405009117. [DOI] [PubMed] [Google Scholar]
- 87.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 88.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 89.Flamme I, Frohlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. doi: 10.1016/s0925-4773(97)00674-6. [DOI] [PubMed] [Google Scholar]
- 90.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 91.Akeno N, Robins J, Zhang M, Czyzyk-Krzeska MF, Clemens TL. Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2alpha. Endocrinology. 2002;143:420–425. doi: 10.1210/endo.143.2.8639. [DOI] [PubMed] [Google Scholar]
- 92.Jones A, Fujiyama C, Blanche C, Moore JW, Fuggle S, Cranston D, Bicknell R, Harris AL. Relation of vascular endothelial growth factor production to expression and regulation of hypoxia-inducible factor-1 alpha and hypoxia-inducible factor-2 alpha in human bladder tumors and cell lines. Clin Cancer Res. 2001;7:1263–1272. [PubMed] [Google Scholar]
- 93.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732. doi: 10.1097/01.asn.0000017223.49823.2a. [DOI] [PubMed] [Google Scholar]
- 94.Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63:6130–6134. [PubMed] [Google Scholar]
- 95.Brusselmans K, Bono F, Maxwell P, Dor Y, Dewerchin M, Collen D, Herbert JM, Carmeliet P. Hypoxia-inducible factor-2alpha (HIF-2alpha) is involved in the apoptotic response to hypoglycemia but not to hypoxia. J Biol Chem. 2001;276:39192–39196. doi: 10.1074/jbc.C100428200. [DOI] [PubMed] [Google Scholar]
- 96.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 97.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–267. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 98.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 99.Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun. 2001;287:808–813. doi: 10.1006/bbrc.2001.5659. [DOI] [PubMed] [Google Scholar]
- 100.Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 101.Sipe JC, Lee P, Beutler E. Brain iron metabolism and neurodegenerative disorders. Dev Neurosci. 2002;24:188–196. doi: 10.1159/000065701. [DOI] [PubMed] [Google Scholar]
- 102.Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, Becker G. Brain iron pathways and their relevance to Parkinson's disease. J Neurochem. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- 103.Bishop GM, Robinson SR, Liu Q, Perry G, Atwood CS, Smith MA. Iron: a pathological mediator of Alzheimer disease? Dev Neurosci. 2002;24:184–187. doi: 10.1159/000065696. [DOI] [PubMed] [Google Scholar]
- 104.Ke Y, Ming Qian Z. Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol. 2003;2:246–253. doi: 10.1016/s1474-4422(03)00353-3. [DOI] [PubMed] [Google Scholar]
- 105.Rouault TA. Systemic iron metabolism: a review and implications for brain iron metabolism. Pediatr Neurol. 2001;25:130–137. doi: 10.1016/s0887-8994(01)00260-0. [DOI] [PubMed] [Google Scholar]
- 106.Henderson BR, Kuhn LC. Differential modulation of the RNA-binding proteins IRP-1 and IRP-2 in response to iron. IRP-2 inactivation requires translation of another protein. J Biol Chem. 1995;270:20509–20515. doi: 10.1074/jbc.270.35.20509. [DOI] [PubMed] [Google Scholar]
- 107.Hanson ES, Foot LM, Leibold EA. Hypoxia post-translationally activates iron-regulatory protein 2. J Biol Chem. 1999;274:5047–5052. doi: 10.1074/jbc.274.8.5047. [DOI] [PubMed] [Google Scholar]
- 108.Guo B, Phillips JD, Yu Y, Leibold EA. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem. 1995;270:21645–21651. doi: 10.1074/jbc.270.37.21645. [DOI] [PubMed] [Google Scholar]
- 109.Hanson ES, Rawlins ML, Leibold EA. Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem. 2003;278:40337–40342. doi: 10.1074/jbc.M302798200. [DOI] [PubMed] [Google Scholar]
- 110.Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 111.Wang J, Chen G, Muckenthaler M, Galy B, Hentze MW, Pantopoulos K. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol Cell Biol. 2004;24:954–965. doi: 10.1128/MCB.24.3.954-965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang J, Pantopoulos K. The pathway for IRP2 degradation involving 2-oxoglutarate-dependent oxygenase(s) does not require the E3 ubiquitin ligase activity of pVHL. Biochim Biophys Acta. 2005;1743:79–85. doi: 10.1016/j.bbamcr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 113.Yu F, White SB, Zhao Q, Lee FS. Dynamic, site-specific interaction of hypoxia-inducible factor-1alpha with the von Hippel-Lindau tumor suppressor protein. Cancer Res. 2001a;61:4136–4142. [PubMed] [Google Scholar]
- 114.Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, Conaway JW, Conaway RC, Czyzyk-Krzeska MF. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci USA. 2003;100:2706–2711. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee KB, Wang D, Lippard SJ, Sharp PA. Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. Proc Natl Acad Sci USA. 2002;99:4239–4244. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mitsui A, Sharp PA. Ubiquitination of RNA polymerase II large subunit signaled by phosphorylation of carboxyl-terminal domain. Proc Natl Acad Sci USA. 1999;96:6054–6059. doi: 10.1073/pnas.96.11.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 118.Hopfer U, Hopfer H, Jablonski K, Stahl RA, Wolf G. The novel WD-repeat protein Morg1 acts as a molecular scaffold for hypoxia-inducible factor prolyl hydroxylase 3 (PHD3) J Biol Chem. 2006;281:8645–8655. doi: 10.1074/jbc.M513751200. [DOI] [PubMed] [Google Scholar]
- 119.Lee S, Nakamura E, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8(2):155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 120.D'Angelo G, Duplan E, Boyer N, Vigne P, Frelin C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem. 2003;278:38183–38187. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- 121.del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. The von Hippel-Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J Biol Chem. 2003;278:48690–48695. doi: 10.1074/jbc.M308862200. [DOI] [PubMed] [Google Scholar]
- 122.Frew IJ, Hammond VE, Dickins RA, Quinn JM, Walkley CR, Sims NA, Schnall R, Della NG, Holloway AJ, Digby MR, et al. Generation and analysis of Siah2 mutant mice. Mol Cell Biol. 2003;23:9150–9161. doi: 10.1128/MCB.23.24.9150-9161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 124.Hu G, Zhang S, Vidal M, Baer JL, Xu T, Fearon ER. Mammalian homologs of seven in absentia regulate DCC via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:2701–2714. doi: 10.1101/gad.11.20.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Matsuzawa S, Takayama S, Froesch BA, Zapata JM, Reed JC. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: suppression by BAG-1. Embo J. 1998;17:2736–2747. doi: 10.1093/emboj/17.10.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 127.Susini L, Passer BJ, Amzallag-Elbaz N, Juven-Gershon T, Prieur S, Privat N, Tuynder M, Gendron MC, Israel A, Amson R, et al. Siah-1 binds and regulates the function of Numb. Proc Natl Acad Sci USA. 2001;98:15067–15072. doi: 10.1073/pnas.261571998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tiedt R, Bartholdy BA, Matthias G, Newell JW, Matthias P. The RING finger protein Siah-1 regulates the level of the transcriptional coactivator OBF-1. Embo J. 2001;20:4143–4152. doi: 10.1093/emboj/20.15.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang J, Guenther MG, Carthew RW, Lazar MA. Proteasomal regulation of nuclear receptor corepressor-mediated repression. Genes Dev. 1998;12:1775–1780. doi: 10.1101/gad.12.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nakayama K, Gazdoiu S, et al. Hypoxia-induced assembly of prolyl-hydroxylase, PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem J. 2007;401(1):217–226. doi: 10.1042/BJ20061135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taylor MS. Characterization and comparative analysis of the EGLN gene family. Gene. 2001;275:125–132. doi: 10.1016/s0378-1119(01)00633-3. [DOI] [PubMed] [Google Scholar]
- 132.Lavista-Llanos S, Centanin L, Irisarri M, Russo DM, Gleadle JM, Bocca SN, Muzzopappa M, Ratcliffe PJ, Wappner P. Control of the hypoxic response in Drosophila melanogaster by the basic helix-loop-helix PAS protein similar. Mol Cell Biol. 2002;22:6842–6853. doi: 10.1128/MCB.22.19.6842-6853.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277:39792–39800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Burgel T, Jelkmann W, et al. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003;116:1319–1326. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 135.Ozer A, Wu LC, Bruick RK. The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF) Proc Natl Acad Sci USA. 2005;102:7481–7486. doi: 10.1073/pnas.0502716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cioffi CL, Liu XQ, Kosinski PA, Garay M, Bowen BR. Differential regulation of HIF-1 alpha prolyl-4-hydroxylase genes by hypoxia in human cardiovascular cells. Biochem Biophys Res Commun. 2003;303:947–953. doi: 10.1016/s0006-291x(03)00453-4. [DOI] [PubMed] [Google Scholar]
- 137.Seth P, Krop I, Porter D, Polyak K. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression) Oncogene. 2002;21:836–843. doi: 10.1038/sj.onc.1205113. [DOI] [PubMed] [Google Scholar]
- 138.Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–426. doi: 10.1139/o02-115. [DOI] [PubMed] [Google Scholar]
- 139.Oehme F, Ellinghaus P, Kolkhof P, Smith TJ, Ramakrishnan S, Hutter J, Schramm M, Flamme I. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochem Biophys Res Commun. 2002;296:343–349. doi: 10.1016/s0006-291x(02)00862-8. [DOI] [PubMed] [Google Scholar]
- 140.Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W, Wiesener M, Ratcliffe PJ, Eckardt KU, Pugh CW. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol. 2006;41(1):68–77. doi: 10.1016/j.yjmcc.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 141.Tian YM, Mole DR, Ratcliffe PJ, Gleadle JM. Characterization of different isoforms of the HIF prolyl hydroxylase PHD1 generated by alternative initiation. Biochem J. 2006;397:179–186. doi: 10.1042/BJ20051996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Khanna S, Roy S, Maurer M, Ratan RR, Sen CK. Oxygen-sensitive reset of hypoxia-inducible factor transactivation response: prolyl hydroxylases tune the biological normoxic set point. Free Radic Biol Med. 2006;40(12):2147–2154. doi: 10.1016/j.freeradbiomed.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aravind L, Koonin EV. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-research0007. RESEARCH0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Groulx I, Lee S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 2002;22:5319–5336. doi: 10.1128/MCB.22.15.5319-5336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 146.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. Embo J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381:761–767. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Metzen E, Stiehl DP, Doege K, Marxsen JH, Hellwig-Burgel T, Jelkmann W. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: identification of a functional hypoxia-responsive element. Biochem J. 2005;387:711–717. doi: 10.1042/BJ20041736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005;390:189–197. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cervera AM, Apostolova N, Luna-Crespo F, Sanjuan-Pla A, Garcia-Bou R, McCreath KJ. An alternatively spliced transcript of the PHD3 gene retains prolyl hydroxylase activity. Cancer Lett. 2006;233:131–138. doi: 10.1016/j.canlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 152.Freeman RS, Hasbani DM, Lipscomb EA, Straub JA, Xie L. SM-20, EGL-9, and the EGLN family of hypoxia-inducible factor prolyl hydroxylases. Mol Cells. 2003;16:1–12. [PubMed] [Google Scholar]
- 153.Wax SD, Rosenfield CL, Taubman MB. Identification of a novel growth factor-responsive gene in vascular smooth muscle cells. J Biol Chem. 1994;269:13041–13047. [PubMed] [Google Scholar]