

Abstract
A central aim of neuroscience is to map neural circuits, in order to learn how they account for mental activities and behaviours and how alterations in them lead to neurological and psychiatric disorders. However, the methods that are currently available for visualizing circuits have severe limitations that make it extremely difficult to extract precise wiring diagrams from histological images. Here we review recent advances in this area, along with some of the opportunities that these advances present and the obstacles that remain.
Understanding how the precise interconnections of neurons account for brain functions has been a preoccupation of neuroscientists for over a century. To accomplish this aim, neuroscientists will need various types of data. First, we will need a physical map of the neurons that comprise a circuit and the sites at which they form synapses with each other. Second, we will need to understand how electrical signals flow through the circuit when a stimulus is perceived, a decision is made or an action is taken. Third, if the circuitry undergoes modifications, we will need information on how circuits and their signals change over time. Finally, and arguably most demanding, we will need ways to transform the raw data into useful forms so that we can extract meaning from the physical maps. Neuroscientists are hard at work developing the tools that are needed to accomplish each of these; here we discuss several technological advances that might provide solutions to the first problem: obtaining a complete physical map of the nervous system.
Circuit-mapping strategies
Over the past century, neuroscientists have used three main sets of anatomical approaches to study neural connectivity: single-cell impregnation, optically based tract-tracing and electron microscopy (FIG. 1). In this section, we briefly discuss these methods and their limitations.
Figure 1. Circuit-mapping strategies.
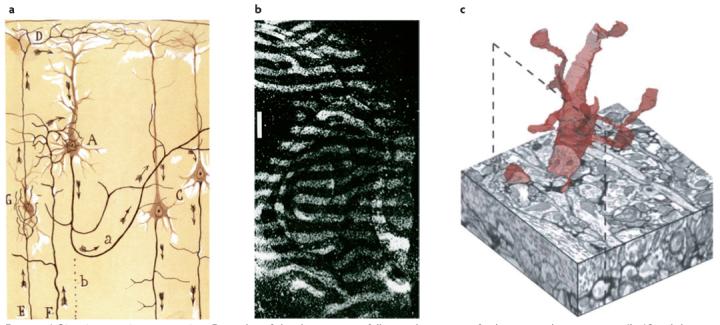
Examples of the three main approaches that have been used to study the connectivity of neurons. a | Single-cell staining by dye impregnation. The best-known of these techniques is the Golgi staining method, which was used by Ramón y Cajal to describe the principles of neuronal circuit organization39. b | The introduction of diffusible or transportable labelling agents to discrete areas. In some cases the label crosses a synapse to reach a second-order target. A classical example is the identification of ocular dominance columns in the visual cortex following the injection of radioactive proline into one eye40. c | Serial electron microscopy can be used to reconstruct neurons and their processes with the best attainable resolution. Shown here is a reconstruction from the adult rat barrel cortex that was segmented from a three-dimensional image stack obtained by serial block-face imaging16. Part a reproduced, with permission, from REF. 39 © (1914) Herederos de santiago Ramón y Cajal. Part b reproduced, with permission, from REF. 40 © (1977) royal society of London. Part c reproduced, with permission, from REF. 16 © (2006) Elsevier Sciences.
Single-cell staining by dye impregnation
This was the first and most influential approach. Single-cell impregnation is rooted in the extraordinary work of Santiago Ramón y Cajal (1852-1934), who used Golgi’s ‘black reaction’ — a silver-staining technique that labels neurons with a dark precipitate. This method stains the entirety of just a few individual nerve cells and all their processes (axons and dendrites), thereby making evident shapes that would otherwise be indiscernible in the dense meshwork of the neuropil1. Using the Golgi stain, Cajal was able to not only identify many different neuronal types in the brain, but also describe their pattern of interconnections and discover principles of neuronal circuit organization (FIG. 1a).
Despite their power, the Golgi method and other single-cell labelling techniques — for example, intracellular injection of dyes — have significant limitations. First, because the chemicals with which the neurons are impregnated must be applied to relatively small blocks of tissue, they cannot be used to trace long-distance connections. Second, because of diffraction, neuronal processes that come closer to each other than approximately 0.25 μm are not resolvable in the light microscope. It is therefore difficult to trace slender axons and dendrites without there being any ambiguities. Third, if all or even many of the elements in a circuit are stained, they become impossible to distinguish from one another. Therefore, circuit descriptions are almost always based on a statistical analysis of small amounts of data from multiple individuals, rather than on full analyses of any actual circuit. Indeed, these problems are so severe that, with the exception of Cajal, few scientists have been able to draw general conclusions from such material that have stood the test of time2.
Diffusion or transport of labelling agents placed at discrete positions
The problem of visualizing long-distance connectivity can be addressed, at least in part, by circuit-tracing techniques, which involve marking tracts that project from one brain region to another. Tracts are generally labelled by mechanically introducing a specific labelling agent — for instance, through a surgical procedure. The first tract-tracing methods to be applied, however, took advantage of the fact that axons degenerate following injury and leave behind debris that persists for days to weeks before it is cleared. This debris could be stained with silver salts. Thus, following local damage to a neuronal population or tract, the silver stain marked sites to which neurons had projected3.
By the 1970s, degeneration methods had largely been replaced by a suite of techniques in which labels were placed into circumscribed regions of the central or peripheral nervous systems and then diffused or actively transported along axons to other sites (FIG. 1b). A wide range of labels proved useful for this application: radioactive amino acids or sugars, proteins such as the cholera toxin B subunit, enzymes such as horseradish peroxidase, and fluorescent organic molecules such as DiI, DiO and Fluorogold4. Most recently, genetically encoded tracers, including fluorescent proteins (see below), have been expressed under the control of subtype- or region-specific regulatory elements or from viral vectors5-9. These methods have several advantages over methods that use chemical labels, including the possibility of indelible marking and, for the transgenic approaches, avoidance of the need for surgery. An additional exciting development is the ability to introduce genetically encoded trans-synaptic tracers that can cross from a neuron to its synaptic partners8-12, thus revealing (with some caveats; see REF. 9) connectivity.
Despite the encouraging progress that there has been along these lines, difficulties remain. First, tracing from a set of somata to a terminal field, or vice versa, will in most cases leave local interneurons unlabelled or impossible to resolve. Second, because these methods typically label groups of neurons rather than individual cells, tract-tracing methods can seldom be used to trace the highly complex branching patterns of the thousands of interdigitated axons and dendrites in each cubic μm of brain.
Electron microscopy
Serial section electron microscopy has been the method of choice for overcoming the limited resolution of light microscopy and the insufficiencies of tract-tracing. In pioneering work, Levinthal, Lopresti and Macagno used this method to map connections in the optic lobe of the small crustacean Daphnia13, and White et al. used it to provide a nearly complete map of connectivity in the roundworm, Caenorhabditis elegans14. Serial electron microscopy is so laborious, however, that its application has been infrequent. Fortunately, this situation is now changing thanks to recent developments in the automation of electron microscopy15-17. One promising approach is to image the thin surface layer (tens of nanometers) of a block of plastic-embedded and heavy-metal-stained brain tissue by looking at backscattered electrons with a scanning electron microscope15. After taking the image, the top section of the block is removed with a diamond knife and the block face is reimaged. This procedure can now be iterated thousands of times to give perfectly aligned stacks of digital electron micrographs from which neuronal processes can be traced (FIG. 1c). Other approaches use new microtome designs involving a rotary lathe and a tape transport system to automate the cutting and collection of serial thin sections. Along with new methods in computational segmentation that improve tracing and three-dimensional reconstruction (see below), these innovations are likely to restore electron microscopy to a position of prominence in circuit analysis. Nonetheless, large-scale reconstruction, especially over long distances, remains a distant hope. For example, reconstructing paths from the retina to the thalamus or from the thalamus to the cortex would require hundreds of thousands or even millions of sections to be acquired and analysed, and even then there would be no obvious way to verify the accuracy of the tracing.
The multicolour solution
Introducing a marker into the reconstructed neuron allows its identity to be verified in each section. However, the need for sparse labelling when using monochromatic labels remained an intractable limitation until recently. The obvious solution is to use multiple, distinct labels so that nearby processes can be distinguished from each other. This approach is exemplified by the use of ‘rainbow’ cables in electronic devices: the various colours of the wires help people keep track of them. Several methods designed along this principle have been applied to the task of neuronal circuit visualization. One, called Diolistics, involves shooting tissues with biolistic metal beads that have been coated with various combinations of different coloured lipophilic dyes, such as DiI, DiO, and DiD, with the result that impregnated cells are labelled with different colours18. However, Diolistic labelling is best done ex vivo on slices, so it is poorly suited for long-range reconstruction.
Another approach is based on the green fluorescent protein (GFP) revolution19, which has led to a technical renaissance in imaging. Over the past several years, dozens of spectral and photophysical fluorescent protein variants (XFPs) have been discovered or engineered20, and many of these have been introduced as intrinsic neuronal labels in transgenic mice. In some of the mice that produce these XFPs, the expression is confined to small, quasi-random subsets of cells, creating the equivalent of a vital Golgi stain21. By crossing two such lines of mice expressing spectrally distinct XFPs, one can obtain ‘bicolour’ or even ‘tricolour’ animals in which the interactions between two neurons can be visualized21-24. Another method for expressing two XFPs in a mosaic manner is the ingenious mosaic analysis with double markers (MADM) strategy25. In this method, the XFP genes are artificially split by a synthetic intron that contains a loxP site (Box 1). Reciprocally chimeric genes made by pairing the exons that encode the two XFPs are inserted at identical positions in a pair of homologous chromosomes. These elements remain non-functional until Cre recombinase (Box 1) triggers interchromosomal recombination to reconstitute active XFP-encoding genes. Subsequent mitoses segregate the alleles such that different cells inherit different products of recombination and thus express distinct colours.
Box 1. Brainbow strategies.

Brainbow transgenes drive the combinatorial expression of several fluorescent proteins (XFPs) in neurons, resulting in the colour-tagging of individual cells. Here we outline the methodology that we used to generate these constructs.
The strategy makes use of the Cre/lox recombination system. Cre recombinase specifically recognizes and catalyses recombination between a pair of 34-nucleotide sequences called loxP. This reaction leads to the excision of a DNA segment that is flanked by two loxP sites of the same orientation, and to the inversion of a DNA segment that is flanked by loxP sites of opposite orientation (see figure, part c).
One strategy, Brainbow-1, makes use of mutant lox sites that can recombine among themselves but which bear mutations that render them incompatible with the canonical loxP site7; see figure, part a). Alternating canonical (wild-type) loxP sites with variant sites creates mutually exclusive excision possibilities (see figure, part b). Implementation of this strategy with three lox variants creates four possible outcomes following excision by Cre recombinase: three different products of recombination plus the unrecombined initial state. In all cases, only the XFP directly following the promoter is expressed. A second strategy, Brainbow-2, uses Cre-recombinase-mediated DNA inversion. The DNA segment can invert repeatedly while Cre recombinase is present but will stabilize in a random orientation when Cre recombinase stops acting, offering two possibilities of expression. Moreover, when two such invertible segments are arranged in tandem, the number of possibilities is increased: additional inversion and excision possibilities create a total of four expression possibilities (see figure, part d).
Both of these strategies are in essence stochastic: the Cre recombinase enzyme is given a choice and the outcome is unpredictable. This rolling of the ‘molecular dice’ allows each cell to independently make a random choice from several XFPs. In the transgenes, a choice is initially given between several recombination possibilities, each leading to a distinct final configuration (that is, the different sides of the molecular dice) corresponding to the expression of a particular gene.
For the generation of the first-generation Brainbow mice26, transgenes were placed under the control of Thy1 genomic elements to obtain strong neuronal expression; these animals were mated to other transgenic mice that expressed Cre recombinanse or a ligand-activated variant, CreER. CFP, cyan fluorescent protein; GFP, green fluorescent protein; OFP, orange fluorescent protein; RFP, red fluorescent protein; YFP, yellow fluorescent protein. Figure modified, with permission, from REF. 26 © (2007) Macmillan Publishers Ltd.
Despite their utility, bi- and tricolour mice have far too few labels to disambiguate one neural process from the thousands of axons and dendrites that it will closely approach as it courses through the nervous system. Recently, we developed a combinatorial colour method, called Brainbow26, that addresses this problem by marking individual neurons in one of >100 colours rather than one of 2-3 colours. The method is based on combinatorial and stochastic expression of multiple XFPs from a single transgene. In principle, combinatorial expression of three different XFPs could colour neurons in one of ten hues (FIG. 2). However, three XFPs can produce a much larger number of hues if they are expressed in different amounts in different neurons. A good analogy is a TV monitor, which combines different intensities of three channels (red, green and blue) to generate almost the entire colour spectrum that the human visual system can perceive (FIG. 2b). Our thought was to use such colour combinations to distinguish neurons in a circuit by generating a random mixture of three fluorescent proteins in each neuron. This stochastic colour expression was accomplished in two steps. First, we designed genetic constructs that could be recombined in several mutually exclusive ways to give any one of three or four colours (see Box 1 for a detailed description of the method). Second, we inserted multiple copies of the transgene into the genome. Independent expression from each copy allows a range of colours to result from combinatorial expression of the XFPs (FIG. 2c). As a result, individual neurons express one of many distinguishable hues (∼100), enabling the spectral differentiation of individual neurons and their processes (FIG. 3).
Figure 2. Combinatorial expression of three distinct fluorescent proteins can generate a large spectrum of colours.

a | several spectrally distinct fluorescent proteins (XFPs) are now available, including ones that emit in red (RFP), green (YFP) and blue (CFP) frequencies. b | The combinatorial expression of red, green and blue XFPs at various levels is sufficient to encode a colour space analogous to the one that is generated by an RGB video monitor. c | An example showing how ten distinct colours can be generated by expressing a trimeric combination of three different XFPs. In Brainbow mice, this outcome would result if three copies of a trichromatic transgene (illustrated at the top of the panel; see Box 1 for details) each recombined independently (Box 1). Triangles represent lox sites (see Box 1 for details). CFP, cyan fluorescent protein; P, promoter; RFP, red fluorescent protein; YFP, yellow fluorescent protein.
Figure 3. Multicolour neuronal labelling in Brainbow transgenic mice.
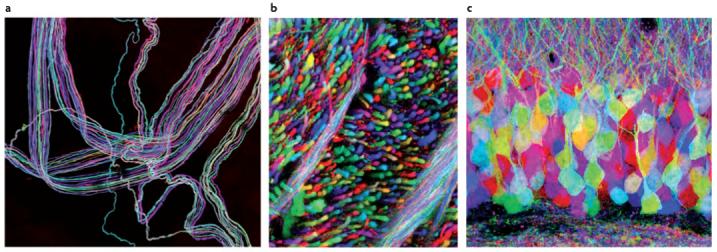
a | A motor nerve innervating ear muscle. b | An axon tract in the brainstem. c | The hippocampal dentate gyrus. In the Brainbow mice from which these images were taken, up to ∼160 colours were observed as a result of the co-integration of several tandem copies of the transgene into the mouse genome and the independent recombination of each by Cre recombinase (see FIG. 2c). The images were obtained by the superposition of separate red, green and blue channels. The image in part a is courtesy of ryan Draft.
The role of Brainbow in connectomics
Brainbow transgenes have generated striking images and excited considerable interest, but how can they actually be expected to contribute to the mapping of neuronal circuits? Their potential is illustrated by our recent experience with perhaps the simplest connectome: the innervation of skeletal muscles by motor axons. A few years ago, before Brainbow lines were available, we began by using a mouse line in which all motor axons are labelled the same colour21 to reveal the entire connectional map in a single muscle. We worked on a single small muscle that was innervated by only 15 or so motor axons (J. Lu, personal communication). A complete map requires that all axons be labelled, and this is one of the limitations of the now-existing ‘first generation’ Brainbow mice (for a detailed discussion of these limitations see below). Our thought was that, with only 15 axons innervating ∼200 muscle fibres, such a mapping would not be difficult. we were wrong. In order to get sufficient resolution to track all of the axons and at the same time image the whole innervation field, we had to assemble tens of thousands of high magnification confocal images into hundreds of stacks and then stitch the stacks together to generate a montage of the whole muscle. Use of a motorized computer-automated stage made it possible to collect the images, which together comprised hundreds of gigabytes of data. Then, using computer-assisted tracing, we tracked the entire branching tree of each motor axon and its synaptic contacts (a motor unit) to reconstruct the entire ‘connectome’ of the muscle. This enterprise required several months of work for each individual muscle, highlighting the scale of the technical hurdles that would have to be surmounted to accomplish the same task for any region of the CNS. By contrast, as we have begun to reconstruct neuromuscular connectomes with Brainbow lines, we have found that the whole process can be accomplished in an order of magnitude less time. Indeed, in some cases it is possible to identify the destination of an axon without having to trace its path, as its unique colour remains recognizable along its length.
Based on these initial encouraging results, we are hopeful that Brainbow technology can also be applied to model nervous systems with limited numbers of axons in order to further deduce principles of nervous system connectivity. Brainbow transgenes are already being used for multicolour labelling of the zebrafish sensory nervous system (Y.A. Pan and A. Schier, personal communication), and attempts are underway to generate Brainbow Drosophila.
A much more ambitious goal is to use Brainbow methods in the mammalian CNS. Here the challenges include the sheer density of the wiring, the very fine calibre of much of the neuropil, the enormous diversity of neuronal types and the interesting fact that many axons travel very long distances to innervate neurons throughout the brain. These obstacles make it unlikely that complete connectomes of large brain regions will be obtainable in the next few years. Working towards that end seems a valuable goal, however, and even partial connectomes — for example, of the retinal inner plexiform layer or of a cortical column — would provide considerable insight into the design principles of the brain.
Moreover, such partial connectomes might be useful in studying mouse models of psychiatric disorders. There is a growing suspicion that defects in the pattern, number or proportions of connections might underlie behavioural disorders with a developmental component, such as schizophrenia and autism. The primary causes of such disorders might include various genetic variants or environmental insults that might affect various molecular cascades or developmental events, but the final common pathway could be a quantitative or qualitative defect in circuitry. That is, some disorders of this sort might be ‘connectopathies’. Because Brainbow labelling facilitates the surveying of quantitative and qualitative aspects of circuitry in diverse brain regions, it might enable this hypothesis to be tested.
It will also be valuable to use Brainbow transgenes to learn how connectional patterns change over the lifespan of an animal. For example, the neurobiological underpinnings of healthy aging remain a mystery. Solving this mystery is not only important in its own right, but might also be required if we are to understand the pathological alterations to which the aging nervous system is especially vulnerable. Likewise, circuit modifications that underlie the critical period in early postnatal life are incompletely understood, as are the compensatory changes that make the young nervous system so resilient following injury.
In studying these issues, the microscopic analysis that we have used so far will be useful. Greater insights might eventually come from time-lapse imaging of circuit alterations as they occur. XFPs make ideal labels for this purpose, and time-lapse methods have now been applied to many parts of the peripheral and central nervous systems of one- and two-colour XFP mice27-30. Further improvements in spectral separation, acquisition rate and detector sensitivity should make it possible to expand the method to Brainbow mice.
So far, we have only discussed connections among the minority cellular population of the brain: neurons. Brainbow methods can also be used to map connections among the majority population, glia, or between neurons and glia. Indeed, some Brainbow lines already label some glial subsets, including astrocytes, Schwann cells and Bergman glia; it should be straightforward to generate others.
Finally, Brainbow can be used to highlight connections of another sort: the lineage relations among cell populations. If recombination is induced in neural progenitors with a ligand-activated Cre recombinase (for example, Tamoxifen-dependent CreER) (Box 1), all offspring of a particular blast cell should be labelled with the same colour. This approach might allow the analysis of the interactions of many clonal sets of cells (each with their own colour) in the same piece of tissue.
Technological challenges
For more complex circuits, the diffraction-limited resolution of the light microscope is a serious limitation. Fortunately, several solutions to this problem are already emerging. One is to cut extremely thin sections of labelled tissue31,32. By cutting sections that are several-fold thinner than the optical section thickness, the superimposition of multiple neurites in the same focal thickness is avoided. In addition, the thin samples scatter light much less, and this improves the contrast and clarity of the labelled material.
To make such thin sections, the neural tissue must be embedded in a hard resin. This, in turn, requires the XFPs to retain their fluorescence following the dehydration and embedding steps. To some extent this might be possible; however, Brainbow transgenes could alternatively be redesigned to include epitope tags that would be recognized by antibodies33. This would allow traditional staining methods to be used after the tissue has been cut and would therefore allow the labelling by the endogenous fluorescent protein to be augmented or replaced. The seemingly simple solution of using antibodies to detect the XFPs themselves is unfortunately not yet feasible, because several of the XFPs (for example, GFP, cyan fluorescent protein and yellow fluorescent protein) differ by only a few amino acids and are, in effect, antigenically identical. If immunohistochemical detection is used, one can also localize sites of synaptic contacts on or between marked cells, using antibodies to pre- or postsynaptic components. Methods such as array tomography, in which thin sections of brain material are assayed with an assemblage of antibody probes, provide an elegant protocol for detecting many antigens (or epitope tags) in single thin sections31.
A more radical solution to the resolution problem might be provided by the new and rapidly growing field of nanoscopy, which overcomes the resolution limitations of light diffraction34-38. These new imaging modalities provide resolutions of tens of nm which, alone or with the addition of spectral information, might be sufficient to trace the finest neuropil.
In parallel, it will be important to design new Brainbow transgenes to increase the range of neuronal types and developmental stages to which the method can be applied. There are several major limitations of the first-generation Brainbow lines. The regulatory elements that are used (from the Thy1 gene) direct high levels of expression in many projection neurons, but most interneurons are poorly marked21. Also, in most lines, transgene expression is low in embryos and during the first postnatal week, so these lines are unsuitable for developmental studies. Furthermore, some colours are dim (especially red shades), primarily because the proteins are insufficiently photostable. Additionally, several bright XFPs are difficult to distinguish from each other (for example, yellow versus green or orange versus red) because their spectra are insufficiently distinct. Moreover, although the number of colours is presently large (∼100), it is nonetheless insufficient for some purposes. There are few conceptual difficulties in designing ‘second-generation’ Brainbow transgenes that circumvent these limitations but, as with the first generation, large numbers of alternatives might need to be assayed before success is achieved.
It also remains to be seen whether multicolour labelling can generate complex connectomes once these technical problems have been solved. If it cannot, or even if it can, the rapid advances in high-throughput electron microscopy might mean this technique emerges as a viable alternative16. Finally, given that all of these methods are evolving in parallel, it might be that the optimal solution to connectomics will be a hybrid of techniques that includes not only Brainbow but also electron microscopy, array tomography and advanced pathwaytracing methods.
Conclusion
we call wiring diagrams connectomes to highlight the analogy to genomes. Obtaining the sequence of the human genome was difficult and expensive. The quest for connectomic information will probably be even more difficult. Like genomics, connectomics requires the development of new techniques, the comparison of alternatives and ways to make mundane and automatic what is initially difficult and labour-intensive. Unlike genomics, however, connectomics might require the parallel application of multiple approaches for labelling, imaging, reconstructing and analysing neurons. Fortunately, the field is now moving at an astonishing pace, and the approaches we have outlined here represent only a subset of those that are being developed independently and energetically to overcome the challenges that lie ahead.
References
- 1.Sotelo C. Viewing the brain through the master hand of Ramon y Cajal. Nature Rev. Neurosci. 2003;4:71–77. doi: 10.1038/nrn1010. [DOI] [PubMed] [Google Scholar]
- 2.Callaway EM, Sanes JR. New technologies. Curr. Opin. Neurobiol. 2006;16:540–542. doi: 10.1016/j.conb.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 3.Nauta WJ. Some early travails of tracing axonal pathways in the brain. J. Neurosci. 1993;13:1337–1345. doi: 10.1523/JNEUROSCI.13-04-01337.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callahan CA, Yoshikawa S, Thomas JB. Tracing axons. Curr. Opin. Neurobiol. 1998;8:582–586. doi: 10.1016/s0959-4388(98)80084-6. [DOI] [PubMed] [Google Scholar]
- 5.Feinstein P, Bozza T, Rodriguez I, Vassalli A, Mombaerts P. Axon guidance of mouse olfactory sensory neurons by odorant receptors and the β2 adrenergic receptor. Cell. 2004;117:833–846. doi: 10.1016/j.cell.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nature Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- 7.Dymecki SM, Kim JC. Molecular neuroanatomy’s “Three Gs”: a primer. Neuron. 2007;54:17–34. doi: 10.1016/j.neuron.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song CK, Enquist LW, Bartness TJ. New developments in tracing neural circuits with herpesviruses. Virus Res. 2005;111:235–249. doi: 10.1016/j.virusres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horowitz LF, Montmayeur JP, Echelard Y, Buck LB. A genetic approach to trace neural circuits. Proc. Natl Acad. Sci. USA. 1999;96:3194–3199. doi: 10.1073/pnas.96.6.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshihara Y, et al. A genetic approach to visualization of multisynaptic neural pathways using plant lectin transgene. Neuron. 1999;22:33–41. doi: 10.1016/s0896-6273(00)80676-5. [DOI] [PubMed] [Google Scholar]
- 12.Wickersham IR, et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macagno ER, Levinthal C, Sobel I. Three-dimensional computer reconstruction of neurons and neuronal assemblies. Annu. Rev. Biophys. Bioeng. 1979;8:323–351. doi: 10.1146/annurev.bb.08.060179.001543. [DOI] [PubMed] [Google Scholar]
- 14.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 15.Denk W, Horstmann H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Briggman KL, Denk W. Towards neural circuit reconstruction with volume electron microscopy techniques. Curr. Opin. Neurobiol. 2006;16:562–570. doi: 10.1016/j.conb.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Hayworth K, Kasthuri N, Schalek R, Lichtman JW. Automating the collection of ultrathin serial sections for large volume TEM reconstructions. Microsc. Microanal. 2006;12:2. [Google Scholar]
- 18.Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- 19.Tsien RY. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 20.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nature Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 21.Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 22.Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- 23.Kasthuri N, Lichtman JW. The role of neuronal identity in synaptic competition. Nature. 2003;424:426–430. doi: 10.1038/nature01836. [DOI] [PubMed] [Google Scholar]
- 24.Lichtman JW, Sanes JR. Watching the neuromuscular junction. J. Neurocytol. 2003;32:767–775. doi: 10.1023/B:NEUR.0000020622.58471.37. [DOI] [PubMed] [Google Scholar]
- 25.Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 27.Gan WB, Kwon E, Feng G, Sanes JR, Lichtman JW. Synaptic dynamism measured over minutes to months: age-dependent decline in an autonomic ganglion. Nature Neurosci. 2003;6:956–960. doi: 10.1038/nn1115. [DOI] [PubMed] [Google Scholar]
- 28.Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen QT, Sanes JR, Lichtman JW. Pre-existing pathways promote precise projection patterns. Nature Neurosci. 2002;5:861–867. doi: 10.1038/nn905. [DOI] [PubMed] [Google Scholar]
- 30.Trachtenberg JT, et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- 31.Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters MF, et al. Differential membrane localization and intermolecular associations of α-dystrobrevin isoforms in skeletal muscle. J. Cell Biol. 1998;142:1269–1278. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fritze CE, Anderson TR. Epitope tagging: general method for tracking recombinant proteins. Methods Enzymol. 2000;327:3–16. doi: 10.1016/s0076-6879(00)27263-7. [DOI] [PubMed] [Google Scholar]
- 34.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heintzmann R, Ficz G. Breaking the resolution limit in light microscopy. Methods Cell Biol. 2007;81:561–580. doi: 10.1016/S0091-679X(06)81026-5. [DOI] [PubMed] [Google Scholar]
- 36.Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 37.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shroff H, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl Acad. Sci. USA. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramón y Cajal S. Estudios Sobre la Degeneración y Regneración del Sistema Nervioso. Moya; Madrid: 19131914. reprinted and edited with additional translations by DeFelipe, J. & Jones, E. G. Cajal’s Degeneration and Regeneration of the Nervous System (Oxford Univ. Press, New York, 1991) [Google Scholar]
- 40.Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1977;278:377–409. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]