

Abstract
S-nitrosylation of proteins serves an important role in regulating diverse cellular processes including signal transduction, DNA repair and neurotransmission. Identification of the S-nitrosylation sites is crucial for understanding the significance of this post-translational modification (PTM) in modulating the function of a protein. However, it is very challenging to identify S-nitrosylation sites directly by mass spectrometric (MS) methods due to the labile nature of the S-NO bond. Here we describe a strategy for direct identification of protein S-nitrosylation sites in an electrospray ionization (ESI) quadrupole time-of-flight (QTOF) mass spectrometer without prior chemical derivatization of S-nitrosylated peptides. Both sample buffer composition and MS hardware parameters were carefully adjusted to ensure that S-nitrosylated peptide ions could be analyzed by the QTOF MS with optimal signal/noise ratios. It was crucial that the proteins were preserved in a sample solution containing 1 mM EDTA and 0.1 mM neocuproine at neutral pH. Proteins dissolved in this solution are amenable to in-solution tryptic digestion, which is important for the analysis of biological samples. S-nitrosylated peptides were effectively analyzed by LC/MS/MS on QTOF MS, with an optimized cone voltage of 20 V and collision energy of 4 V. We have successfully applied this method to thioredoxin, a key antioxidant protein, and identified within it an S-nitrosylation site at Cys73.
Introduction
The covalent modification of a cysteine sulfhydryl group with nitric oxide (NO) is called S-nitrosylation or S-nitrosation [1]. An increasing number of proteins have been found to undergo S-nitrosylation in vivo [2-4]. For example, the inhibition of protein-tyrosine phosphatases by S-nitrosylation is critical in regulating cellular signal transduction pathways [5]; similarly, S-nitrosylation can also suppress C-Jun N-terminal kinase activation in intact cells [6]. On the other hand, S-nitrosylation of OxyR has been shown to activate OxyR-dependent transcriptional events [7] while S-nitrosylation of ryanodine receptor promotes its ability to release Ca2+ from the sarcoplasmic reticulum [8]. Therefore, S-nitrosylation may function as an important regulatory mechanism for fine-tuning protein activities within diverse cellular processes and biochemical pathways, including signal transduction, DNA repair, ion channel regulation and apoptosis [9-11]. Not all cysteines in a protein are equally susceptible to S-nitrosylation; cysteine thiols with an acidic pKa appear to be particularly susceptible [12]. Additional regulatory mechanisms governing protein S-nitrosylation may also include protein localization and protein-protein interactions [13,14]. Recent studies have also suggested that S-nitrosylation may play key roles in regulating human disease development. Abnormal increase of protein S-nitrosylation events have been reported in patients with diabetes, multiple sclerosis, tuberculosis and asthma [15]. Therefore, it is of great interest for biologists to identify protein S-nitrosylation sites and quantify their dynamic status in an effort to determine how S-nitrosylation and denitrosylation are regulated in biological systems.
Several methods have been developed to detect S-nitrosylated proteins. They include the use of a SNO-specific antibody to detect in situ protein S-nitrosylation by immunohistochemistry [16]; and the use of a biotin-switch method coupled with immunoblotting, 2D-gel electrophoresis, or fluorescence gel electrophoresis to detect changes in protein S-nitrosylation status under different biological conditions [17-19]. However, none of these methods identifies the specific S-nitrosylation sites within the proteins. To this end, MS can be used downstream of these methods. However, the labile nature of NO attachment to the cysteine thiol presents unique challenges for MS analysis, as evidenced by the fact that S-nitrosylated peptides are rarely observed in MALDI-TOF MS spectra. Given the laser energies needed for peptide protonation, NO usually falls off the precursor ions in-source during ionization. Under gentle ESI conditions, S-nitrosylated peptides have been observed as a +29 Da ion (+NO and −H) over the unmodified peptide ions for each S-nitrosylated cysteine [20]. In MS/MS analysis, the localization of the S-nitrosylated cysteines in peptides containing more than one cysteine is still problematic, given that S-NO bonds are usually more easily fragmented than the peptide backbone [21]. Recently, a biotin-switch method coupled with MS identification of protein S-nitrosylation sites was developed. In this method, nitrocysteines in peptides are converted into the more stable biotinylated cysteines, which can be analyzed by MS/MS methods, either with MALDI or ESI techniques [22,23]. However, this method may produce some false results stemming from the difficulties of precisely controlling the multiple chemical reaction steps in the biotin-switch protocol [24]. It is therefore desirable to develop a method that can directly detect protein S-nitrosylation sites without complicated derivatization steps.
Published reports suggest that fine-tuning of MS instrument parameters may enable the determination of peptide S-nitrosylation sites using ESI. Indeed, reports demonstrating the identification of peptide S-nitrosylation sites by LC/MS/MS have been published by Mirza et al., using a Finnigan-MAT TSQ-700 triple quadrupole instrument [25]; by Taldone et al., using a Bruker Daltonics Esquire 3000 quadrupole ion trap instrument [26]; by Chen et al. using an ABI QSTAR-XL hybrid QTOF instrument [12]; and by Hao et al., using an Agilent Technologies MSD Ultra ion trap [27] and Micromass Quatro II triple quadrupole instruments [28]. Since each type of instrument is configured differently, MS parameters should be optimized for each S-nitrosylation study. Here we describe a strategy for the direct identification of protein S-nitrosylation sites using a Waters QTOF mass spectrometer. The effectiveness of this strategy requires first, careful adjustment of the buffer composition and pH in order to minimize solution-phase denitrosylation; and second, fine-tuning of both cone and collision energy voltages for optimal ionization of S-nitrosylated peptides. This may be achieved at the expense of reduced ionization efficiency of non-nitrosylated peptides. We have successfully utilized this method to identify a site on thioredoxin that undergoes S-nitrosylation in vitro.
Experimental
Materials
All peptides including insulin B (9-23) (SHLVEALYLVCGERG, MH+ 1645.8, Catalog # 61532), beta-amyloid peptide (CFRHDSGY, MH+ 984.4, Catalog # 61979,), PKC zeta 410 (GDTTSTFCGTPN MH+ 1200.5, Catalog # 62025) and Caspase 3 (163-175) (CRGTELDCGIETD, MH+ 1411.6, Catalog # 60792) were purchased from AnaSpec (San Jose, CA). S-nitrosoglutathione (GSNO), neocuproine and recombinant human thioredoxin (Trx1) were purchased from Sigma (St. Louis, MO, USA). Acetonitrile (ACN) and water were obtained from Mallinckrodt Baker (Phillipsburg, NJ, USA). Formic acid (FA) was purchased from EMD Chemicals (Merck KGaA Darmstadt, Germany). EDTA solution (0.5 M, pH 8.0) was obtained from Cellgro (Mediatech Inc., Herndon, VA). Sequencing-grade trypsin was purchased from Promega (Madison, WI). All other chemicals and reagents were purchased from Sigma.
S-nitrosylation of synthetic peptides and Trx1
One nmol of each peptide was mixed with a 10-fold molar excess of GSNO in an aqueous solution containing 1 mM EDTA and 0.1 mM neocuproine (pH 6.8). The reaction was carried out at 37° C for 30 min in the dark. The resulting peptide solutions were analyzed directly on the QTOF MS. For the protein S-nitrosylation reaction, 25 μg of Trx1 was dissolved in 90 μL of 10% ACN containing 1 mM EDTA and 0.1 mM neocuproine (pH 6.8) and mixed with a 25-fold molar excess of GSNO. The reaction was carried out at 37° C for 30 min in the dark, followed by acetone precipitation to remove excess GSNO. Ninety microliters of Trx1 was precipitated by incubating samples with 360 μL of cold acetone at -20 °C for 1 h. After 10 min centrifugation at 8,000 g, the supernatant was removed and the protein pellet was resuspended in 30 μL of 10% ACN containing 1 mM EDTA and 0.1 mM neocuproine (pH 6.8). Then, 10 μL of this protein solution were directly infused into the QTOF for MS analysis. The remaining 20 μL of the protein solution was mixed with an NH4HCO3 buffer solution to a final concentration of 5 mM (pH 8.0). The protein was digested by trypsin (at a trypsin:Trx1 ratio of 1:5) at 37° C overnight in the dark. The resulting peptides were desalted via a PepClean™ C18 spin column (Pierce, Rockford, IL, USA) prior to LC/MS/MS analysis on the QTOF MS.
Reversed-phase nano LC
Reversed-phase nano LC separation of Trx1 tryptic peptides was carried out on an Ultimate 3000 nano LC system (Dionex Corporation, Sunnyvale, CA). The peptides were first captured on a reversed phase C18 trapping column (0.3 mm × 5 mm) and then resolved on a C18 capillary PepMap100 column (75 μm × 150 mm, 3 μm, 100 Å, C18, Dionex) with a 65-min gradient of Solvent A (2% ACN and 0.1% formic acid, FA) and Solvent B (98% ACN and 0.1% FA): at 0-8.5 min, from 3 to 6% B; at 35 min, to 18% B; at 57 min, to 50% B and at 65 min, to 95% B. The LC eluent was directly introduced into the nano-ESI source, fitted with a PicoTip™ Emitter (New Objective, Woburn, MA) for MS and MS/MS analysis on the QTOF MS.
Mass spectrometry
Peptide analyses were carried out on an API-US QTOF tandem MS system equipped with a nano-ESI source (Waters Corporation, Milford, MA). The ESI capillary voltage was set at 3,000 V. Both cone and collision energy voltages were optimized to achieve maximal ion intensities of the S-nitrosylated peptide ions in the MS mode (see Results and Discussion). The MS spectra (m/z 400 – 1900) were acquired in the positive ion mode. For the synthetic peptides, MS analyses were performed with direct infusion. The MS/MS spectra of selected precursors were acquired at a collision energy of 37 V. Argon was used as the collision gas. For the peptides derived from Trx1 tryptic digestion, LC/MS/MS analysis was performed. The collision energy was set within a range of 17 to 55 V, depending on the charge states and the m/z values. MS/MS spectra were acquired in Data Dependent Analysis mode, in which the top three most abundant precursors with two to five charges from each MS survey scan were selected for fragmentation.
Results and Discussion
Effects of pH and metal ion chelators on S-nitrosylation
Due to the chemical instability of the S-NO bond, it is crucial to optimize the buffer composition for direct identification of peptide S-nitrosylation sites using the ESI-QTOF MS approach. Two peptides, beta-amyloid peptide and insulin B (9-23) were incubated with 10-fold molar excess of GSNO in different sample solutions (Table 1). The MS spectra of the resulting peptides were acquired on a QTOF MS via direct infusion. The relative percentages of S-nitrosylated peptides among all variants of the same peptide in the MS spectra are plotted in Fig. 1. Under experimental conditions I and II, both sample solutions contained 1 mM EDTA and 0.1 mM neocuproine (a copper chelator) at pH 6.8. However, the pH of the sample solution was adjusted to 3.0 by adding FA prior to infusion analysis on the QTOF MS under condition II, while the peptides were analyzed without pH adjustments in condition I. Three major peptide fractions were observed in the MS spectra of both peptides: unmodified, S-nitrosylated and glutathiolated, distributed among different charge states. The relative percentages of S-nitrosylated peptide ions under experimental conditions I and II were 53% and 55% for beta-amyloid peptide, and 40% and 42% for insulin B (9-23), respectively (Fig. 1). These surprising results indicated that acidification from pH 6.8 to pH 3.0 after the S-nitrosylation reaction had little effect on the relative percentages of S-nitrosylated peptides ions observed in MS. By contrast, under experimental condition IV, in which both S-nitrosylation and ESI solutions were maintained at pH 3.0, the relative percentages of S-nitrosylated peptides were dramatically decreased to less than 5% (Fig. 1), indicating that the peptide S-nitrosylation efficiency was reduced in the acidic environment. Since thiol S-nitrosylation reactivity depends on its pKa, acidic pH decreases thiolate nucleophilicity, which may in turn reduce the yield of S-nitrosylated peptides.
Table 1. Peptide S-nitrosylation conditions tested.
| Experiment | S-nitrosylation reaction solution | MS analysis solution |
|---|---|---|
| I | 1 mM EDTA and 0.1 mM neocuproine (pH 6.8) | 1 mM EDTA and 0.1 mM neocuproine (pH 6.8) |
| II | 1 mM EDTA and 0.1 mM neocuproine, (pH 6.8) | 1 mM EDTA, 0.1 mM neocuproine and 0.1% formic acid, (pH 3.0) |
| III | H2O, (pH 6.8) | H2O, (pH 6.8) |
| IV | 1 mM EDTA, 0.1 mM neocuproine and 0.1% formic acid, (pH 3.0) | 1 mM EDTA, 0.1 mM neocuproine and 0.1% formic acid, (pH 3.0) |
Figure 1. Effects of pH and metal chelators on MS sensitivities for detecting S-nitrosylated peptide ions.
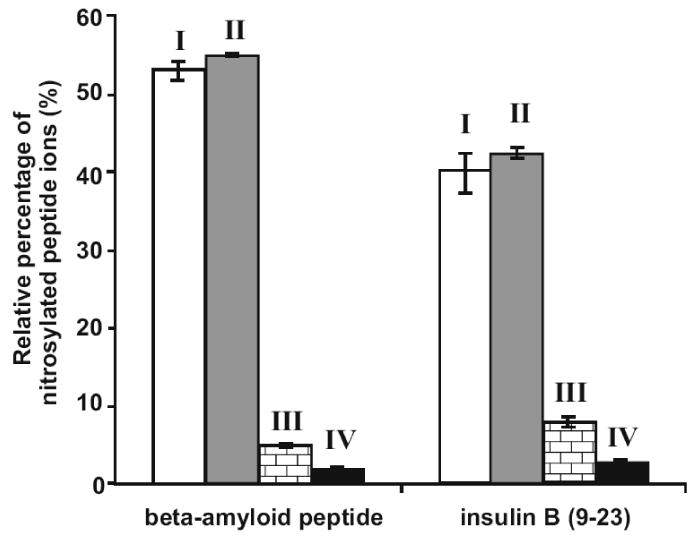
One nmol of synthetic peptides beta-amyloid peptide and insulin B (9-23) were treated with 10-fold molar excess of GSNO and analyzed on a QTOF MS under the different experimental conditions outlined in Table 1. Relative percentages of the S-nitrosylated peptide ion signals were calculated as the percentage ion intensities of the S-nitrosylated peptide divided by the sum of the ion intensities of all peptide species (peptide + peptide-GS + peptide-NO) derived from the same precursor. The MaxEnt3 module in the Masslynx software was used to convert all ion charge states into centroid singly-charged ions prior to the calculations.
EDTA and neocuproine were used as chelating agents to sequester metal ions during S-nitrosylation reactions. It has been reported that S-NO is very sensitive to the presence of copper ions; even low concentrations induce S-NO decomposition [29]. S-NO bond breakage can be inhibited by the copper chelator neocuproine [29]. We confirmed the stabilizing effects of metal ion chelators on S-NO bonds, observing that the relative percentages of S-nitrosylated peptides were dramatically decreased in the absence of EDTA and neocuproine (Fig. 1, conditions I and III).
Optimization of QTOF MS parameters for direct analysis of S-nitrosylated peptides
In order to obtain maximal ion signals for S-nitrosylated peptides, several QTOF MS instrument parameters were evaluated; among these, changes in cone and collision energy voltages had the most dramatic effects. In general, most ion intensities increased with the elevation of the cone voltages from 15 to 30 V, except that S-nitrosylated peptide ion intensities decreased once the cone voltages exceeded 20 V (Fig. 2A). It is well known that the S-NO bond is fragile. For example, we found that the S-NO bonds were easily cleaved by the MALDI process. No S-nitrosylated peptide MS signal for the synthetic peptides used in this study could be observed in a ABI 4800 MALDI-TOF/TOF MS (data not shown). Although ESI is a softer ionization method, when the cone voltage was too high, the labile S-NO bond was easily fragmented during ion transmission. The effects of increasing cone voltages on S-nitrosylated peptides ion stability may also be dependent upon the peptide sequence and the position(s) of the nitrocysteine(s). Therefore the optimal cone voltage for any given peptide may need to be determined empirically. However, the recommended 20-V setting reported here appeared to be optimal for two additional peptides (Supplemental Fig. 1), suggesting that this condition is effective for analyzing S-nitrosylated peptides in Waters QTOF MS. Consequently, a cone voltage value of 20 V was deemed optimal for studying S-nitrosylated peptides on the QTOF MS.
Figure 2. Effects of QTOF MS cone and collision energy voltages on MS ion intensities of S-nitrosylated peptides.
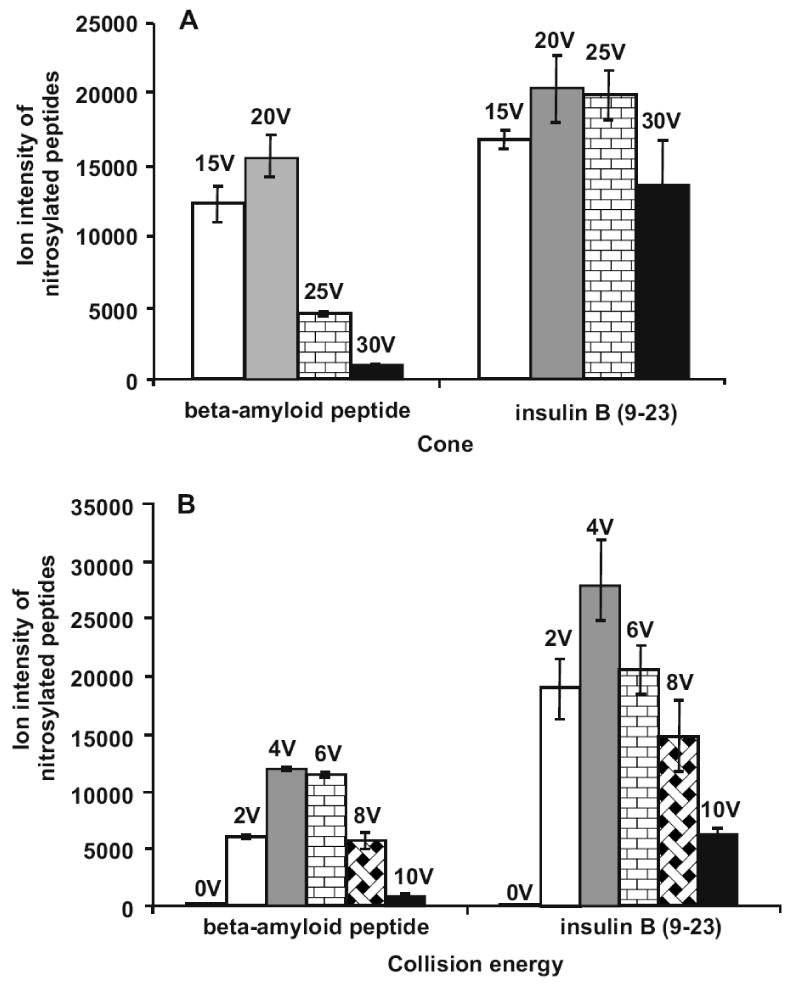
(A) With the collision energy set at 4 V, the effects of varied cone voltages on S-nitrosylated peptide ion signals were analyzed. (B) With the cone voltage set at 20 V, the effects of varied collision energy voltages were evaluated. The ion intensities were calculated using the MaxEnt3 module in the Masslynx software.
In the Waters Masslynx software, the cone voltage readback value in the tune page is the sum of cone, collision energy, extractor and RF lens voltages. Since the extractor and RF lens voltages made relatively small contributions to the readback value, the optimization of the collision energy became important for sensitive detection of S-nitrosylated peptide ions. As shown in Fig. 2B, when the collision energy was increased from 0 to 4 V, the ion intensities for S-nitrosylated peptides were significantly enhanced. Further increase of the collision energy resulted in the reduction of the ion intensities for S-nitrosylated peptides. Thus, the optimal collision energy for the analysis of S-nitrosylated peptides on the QTOF was set at 4 V.
The optimal cone voltage and collision energy determined in this study apply only to Waters QTOF instruments. For other instruments, the optimal parameters for analyzing S-nitrosylated peptides may vary due to the differences in hardware configuration and software design. In general, in order to preserve S-nitrosylated peptide ion stability, both cone and collision energy voltages should be set at much lower values than those used for conventional peptide detections. Typically the optimal cone voltage for detecting S-nitrosylated and non-nitrosylated peptides in our Waters QTOF MS is 20 V and 37 V, respectively. Similarly, the collision energy for analyzing S-nitrosylated peptide ions on QTOF MS is 4 V compared to 8 V for non-nitrosylated ions. Similar degrees of differences may apply to quadrupole time-of-flight instruments from other vendors. Obviously, the optimal conditions have to be determined under specific experimental conditions.
Identification of S-nitrosylation sites in model peptides
The peptides were S-nitrosylated using GSNO under the optimized experimental conditions II described above. The MS spectrum for insulin B (9-23) was acquired with a cone voltage of 20 V and collision energy voltage of 4 V. Prior to S-nitrosylation, doubly- (m/z 823.38) and triply-charged (m/z 549.25) ions were predominantly observed (Fig. 3A). After the S-nitrosylation reaction, the resulting singly- (m/z 1674.80), doubly- (m/z 837.89) and triply-charged (m/z 558.93) S-nitrosylated peptide ions were observed (Fig. 3B), indicating the formation of S-nitrosylated insulin B (9-23). Besides S-nitrosylated peptides, we also detected the doubly- (m/z 976.43) and triply-charged (m/z 651.28) ions corresponding to S-glutathiolated insulin B (9-23), which was an alternative reaction product of the GSNO treatment (Fig. 3B). After converting all the ion charge states into singly-charged species with the MaxEnt3 module in the Masslynx software, we found that ∼40% of insulin B (9-23) peptides were S-nitrosylated, 26% were S-glutathiolated and 34% were unmodified. We then performed an MS/MS analysis of the doubly-charged S-nitrosylated insulin B (9-23) ion (m/z 837.89) at a collision energy of 37 V. Using this high collision energy, the S-NO bond was fragmented prior to fragmentation of the peptide backbone, resulting in the neutral loss of 29 Da in many y-series ions. The S-nitrosylation of Cys19 was clearly identified in the MS/MS spectrum (Fig. 3C). Similar neutral loss of NO was also observed in the MS/MS spectrum of an amyloid peptide (Supplemental Fig. 2).
Figure 3. MS and MS/MS analysis of S-nitrosylated insulin B (9-23) on QTOF MS.
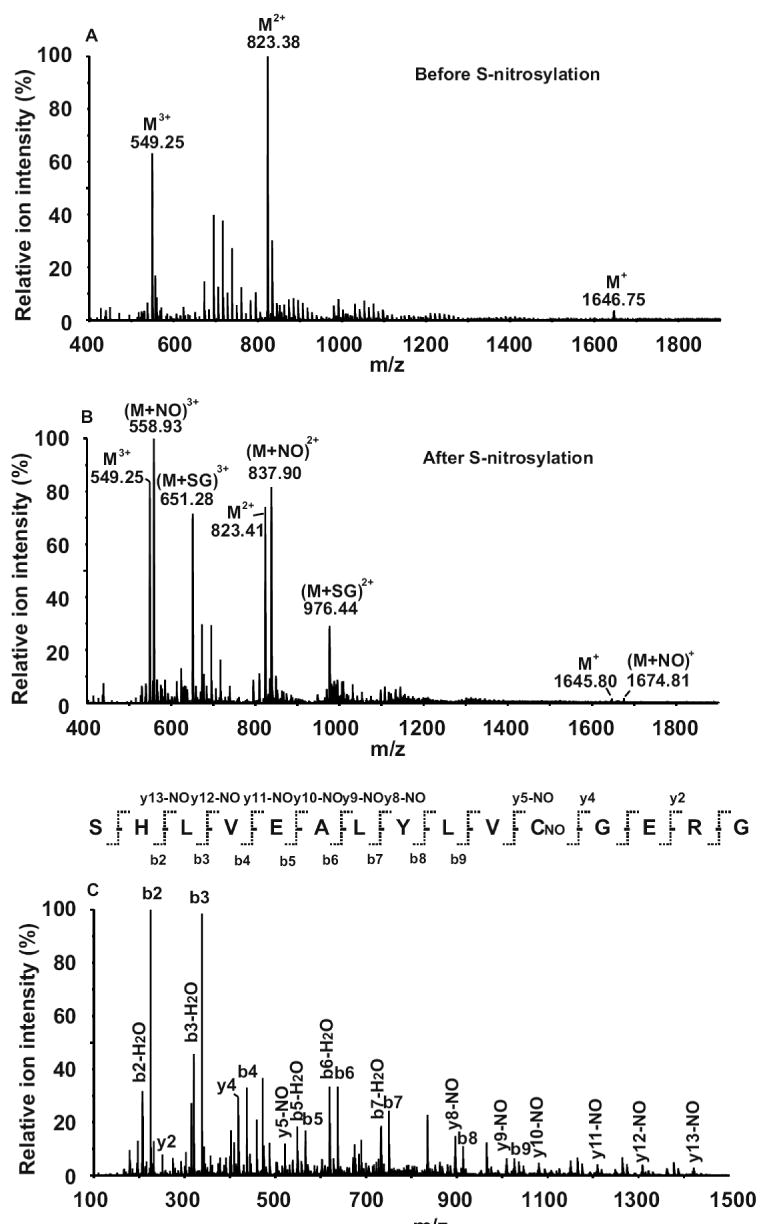
(A) MS spectrum of unmodified peptide. Triply- (m/z 549.25), doubly- (m/z 823.38) and singly-charged (m/z 1645.76) ions were observed. (B) MS spectrum of the peptide after GSNO treatment. The triply- (m/z 558.93), doubly- (m/z 837.89) and singly-charged (m/z 1674.80) ions corresponding to S-nitrosylated peptide (M+NO) were observed; while the triply- (m/z 651.28) and doubly-charged (m/z 976.43) ions corresponding to S-glutathiolated peptide (M+SG) were also detected. (C) MS/MS spectrum of the doubly-charged S-nitrosylated peptide ion of m/z 837.89. The S-nitrosylation site was located on Cys11. All the y-series ions containing the S-nitrosylated cysteine were found to have a neutral loss of 29 Da. The cone voltage was 20 V and the collision energy was 37 V for the MS/MS analyses.
Identification of an S-nitrosylation site in thioredoxin
Trypsin digestion is usually performed in preparing peptides for MS analysis. In order to determine (1) whether the digestion buffer affects protein S-nitrosylation status, and (2) whether trypsin activity is inhibited by the metal chelators present in the S-nitrosylation solution, we digested the S-nitrosylated insulin B (9-23) peptide with trypsin at 37° C overnight in the dark. From QTOF MS analysis, both triply- (m/z 539.99) and doubly-charged (m/z 809.49) ions corresponding to the tryptic fragment SHLVEALYLCNOGER were observed (Fig. 4A). It appeared that the S-NO bond was stable following tryptic digestion. As only about 60% of the S-nitrosylated peptide was digested (Fig. 4A), the proteolytic activity of trypsin was likely reduced by the metal chelators in the S-nitrosylation buffer; it is known that Ca2+ is important for maintaining trypsin activity. Consequently, for biological protein extraction and preparation, metal chelators should be included to preserve protein S-nitrosylation status; however, higher trypsin-to-protein ratios may be needed to ensure efficient protein digestion.
Figure 4. Identification of Trx1 S-nitrosylation site.
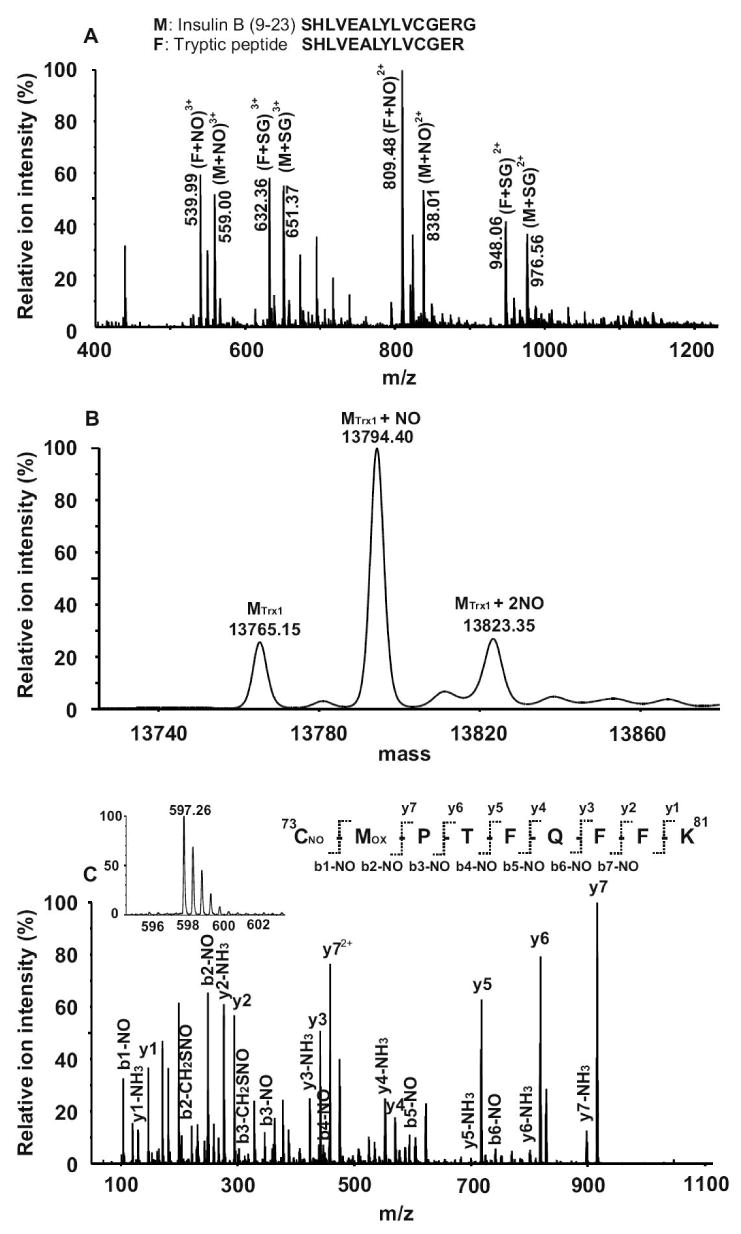
(A) Evaluation of tryptic digestion conditions on S-nitrosylation. One nmol of insulin B (9-23) peptide was nitrosylated by GSNO. The resulting peptides solution was mixed with an NH4HCO3 buffer solution to a final concentration of 5 mM (pH 8.0) and digested by trypsin (at a trypsin: peptide ratio of 1:50) at 37 °C overnight in the dark, followed by MS analysis on QTOF MS. The triply- (m/z 539.99) and doubly-charged (m/z 809.49) ions corresponding to the S-nitrosylated tryptic peptide fragment SHLVEALYLCGER were observed. Tryptic digestion was incomplete, as a substantial amount of undigested and S-nitrosylated insulin B (9-23) (m/z 559.00 and m/z 838.01) was also observed. (B) The deconvoluted spectrum of S-nitrosylated Trx1 protein after GSNO treatment. The unmodified (13765.15 Da), singly- (13794.40 Da) and doubly-S-nitrosylated (13823.35) Trx1 species were observed. (C) MS and MS/MS spectra of a S-nitrosylated tryptic Trx1 peptide. The doubly-charged ion (m/z 597.26, see insert) in the MS spectrum corresponded to S-nitrosylated 73CMPTFQFFK81, with methionine oxidized. In the MS/MS spectrum, both y-series and b-series ions confirmed the peptide sequence as CMPTFQFFK, with its S-nitrosylation site located at Cys73.
Thioredoxin-1 (Trx1) is a 12-kDa multifunctional protein involved in cell growth, death, protein reduction, and tissue development [30]. It has two cysteines located in its catalytic center, and it functions as a redox regulator in cells [31,32]. Earlier studies have reported that human Trx1 is readily S-nitrosylated both in vitro and in vivo, and the NO group in S-nitrosylated Trx1 can be transferred to other proteins, such as caspase-3 (at Cys163) [33] via a process called trans-nitrosylation. We used a 25-fold molar excess of GSNO to S-nitrosylate recombinant human Trx1. One-third of the resulting protein solution was directly infused into the QTOF for MS analysis. The MaxEnt1 module in the Masslynx software was used to deconvolute the raw MS spectrum to obtain the molecular weight of the S-nitrosylated Trx1. After the S-nitrosylation reaction, 66% of Trx1 was found to contain one additional NO group (29 Da), at a mass of 13794.40 Da, compared to the unmodified Trx1, at a mass of 13765.15 Da. In addition, we also observed that 17% of Trx1 contained two NO groups (13823.35 Da) (Fig. 4B). In order to identify the S-nitrosylation sites, Trx1 was digested and the resulting peptides were analyzed by LC/MS/MS. Since the Trx1 catalytic domain sequence is homologous to bovine pancreatic trypsin inhibitor, Trx1 may be able to inhibit trypsin activity [34]. Indeed, during the digestion, we had to increase the trypsin:Trx1 molar ratio to 1:5 in order to achieve a thorough digestion of Trx1. There are five cysteines in human Trx1; all cysteine-containing peptides were observed in our LC/MS/MS analysis. A doubly-charged ion (m/z 597.26) corresponding to 73CMPTFQFFK81 with methionine oxidized was found to be substantially S-nitrosylated at Cys73 (Fig. 4C). We also detected a doubly-charged ion (m/z 811.89) corresponding to 22LVVVDFSATWCGPCK36 with the Trx1 active site disulfide bond formed between Cys32 and Cys35 (Supplemental Fig. 3A). In addition, a triply-charged ion (m/z 906.72) corresponding to 49YSNVIFLEVDVDDCQDVASECEVK72 with both Cys62 and Cys69 in the reduced states was also observed (Supplemental Fig. 3B). We did not find detectable amounts of S-nitrosylated peptides containing Cys62 and Cys69 in this study, although Cys69 has been previously reported to be S-nitrosylated [4,30]. This discrepancy might be due to the different in vitro S-nitrosylation conditions used, including different GSNO concentrations and the duration of the reaction. At higher concentrations of GSNO or longer incubation times, we observed Trx1 with up to three NO groups added (data not shown). Mitchell et al. reported that Cys73 was S-nitrosylated after GSNO treatment [33]. Based on the crystal structure of Trx1, Cys73 is the most surface-accessible of Trx1 cysteines, and it is located close to the N-terminus of the protein, whereas Cys69 is only minimally exposed [33]. These results corroborate the findings reported by Mitchell et al. [33]. In this study, 66% of Trx1 was found to contain one NO modification and 17% contained two NO group additions. We could only find one tryptic peptide containing S-nitrosylated Cys73. Our inability to detect the second S-nitrosylation site may be due to the substantially lowered concentrations of the peptide(s) containing the second S-nitrosylation site; alternatively this S-nitrosylated peptide may be too hydrophilic and was lost during the desalting steps prior to LC/MS analysis.
Summary
We developed an effective LC/MS/MS strategy that enables us to directly identify S-nitrosylation sites in proteins using ESI-QTOF MS. Considering the labile nature of the S-NO bond, the sample solution should include both EDTA and neocuproine, and be maintained at neutral pH to prevent protein denitrosylation during protein extraction, digestion and analysis. Both cone and collision energy voltages in QTOF MS should also be fine-tuned to preserve the S-NO bonds. Using this method, we found that Cys73 in human Trx1 was specifically S-nitrosylated by GSNO treatment. A similar approach could be adapted for the identification of S-nitrosylation sites in biological samples.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Stroissnigg H, Trancikova A, Descovich L, Fuhrmann J, Kutschera W, Kostan J, Meixner A, Nothias F, Propst F. S-nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat Cell Biol. 2007;9:1035–1045. doi: 10.1038/ncb1625. [DOI] [PubMed] [Google Scholar]
- 3.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 4.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 5.Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280:14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
- 6.Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci U S A. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hausladen A, Privalle CT, Keng T, DeAngelo J, Stamler JS. Nitrosative stress: activation of the transcription factor OxyR. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 8.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 9.Derakhshan B, Hao G, Gross SS. Balancing reactivity against selectivity: the evolution of protein S-nitrosylation as an effector of cell signaling by nitric oxide. Cardiovasc Res. 2007;75:210–219. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broillet MC. S-nitrosylation of proteins. Cell Mol Life Sci. 1999;55:1036–1042. doi: 10.1007/s000180050354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benhar M, Stamler JS. A central role for S-nitrosylation in apoptosis. Nat Cell Biol. 2005;7:645–646. doi: 10.1038/ncb0705-645. [DOI] [PubMed] [Google Scholar]
- 12.Chen YY, Huang YF, Khoo KH, Meng TC. Mass spectrometry-based analyses for identifying and characterizing S-nitrosylation of protein tyrosine phosphatases. Methods. 2007;42:243–249. doi: 10.1016/j.ymeth.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 13.O'Brian CA, Chu F. Post-translational disulfide modifications in cell signaling--role of inter-protein, intra-protein, S-glutathionyl, and S-cysteaminyl disulfide modifications in signal transmission. Free Radic Res. 2005;39:471–480. doi: 10.1080/10715760500073931. [DOI] [PubMed] [Google Scholar]
- 14.Salanova M, Schiffl G, Rittweger J, Felsenberg D, Blottner D. Ryanodine receptor type-1 (RyR1) expression and protein S-nitrosylation pattern in human soleus myofibres following bed rest and exercise countermeasure. Histochem Cell Biol. 2008 doi: 10.1007/s00418-008-0399-6. [DOI] [PubMed] [Google Scholar]
- 15.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 16.Gow AJ, Davis CW, Munson D, Ischiropoulos H. Immunohistochemical detection of S-nitrosylated proteins. Methods Mol Biol. 2004;279:167–172. doi: 10.1385/1-59259-807-2:167. [DOI] [PubMed] [Google Scholar]
- 17.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 18.Kuncewicz T, Sheta EA, Goldknopf IL, Kone BC. Proteomic analysis reveals novel protein targets of S-nitrosylation in mesangial cells. Contrib Nephrol. 2004;141:221–230. doi: 10.1159/000074600. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 20.Lee SJ, Lee JR, Kim YH, Park YS, Park SI, Park HS, Kim KP. Investigation of tyrosine nitration and nitrosylation of angiotensin II and bovine serum albumin with electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:2797–2804. doi: 10.1002/rcm.3145. [DOI] [PubMed] [Google Scholar]
- 21.Knipp M, Braun O, Gehrig PM, Sack R, Vasak M. Zn(II)-free dimethylargininase-1 (DDAH-1) is inhibited upon specific Cys-S-nitrosylation. J Biol Chem. 2003;278:3410–3416. doi: 10.1074/jbc.M209088200. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell DA, Erwin PA, Michel T, Marletta MA. S-Nitrosation and regulation of inducible nitric oxide synthase. Biochemistry. 2005;44:4636–4647. doi: 10.1021/bi0474463. [DOI] [PubMed] [Google Scholar]
- 23.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 25.Mirza UA, Chait BT, Lander HM. Monitoring reactions of nitric oxide with peptides and proteins by electrospray ionization-mass spectrometry. J Biol Chem. 1995;270:17185–17188. doi: 10.1074/jbc.270.29.17185. [DOI] [PubMed] [Google Scholar]
- 26.Taldone FS, Tummala M, Goldstein EJ, Ryzhov V, Ravi K, Black SM. Studying the S-nitrosylation of model peptides and eNOS protein by mass spectrometry. Nitric Oxide. 2005;13:176–187. doi: 10.1016/j.niox.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Hao G, Gross SS. Electrospray tandem mass spectrometry analysis of S- and N-nitrosopeptides: facile loss of NO and radical-induced fragmentation. J Am Soc Mass Spectrom. 2006;17:1725–1730. doi: 10.1016/j.jasms.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 28.Hao G, Xie L, Gross SS. Argininosuccinate synthetase is reversibly inactivated by S-nitrosylation in vitro and in vivo. J Biol Chem. 2004;279:36192–36200. doi: 10.1074/jbc.M404866200. [DOI] [PubMed] [Google Scholar]
- 29.Gorren AC, Schrammel A, Schmidt K, Mayer B. Decomposition of S-nitrosoglutathione in the presence of copper ions and glutathione. Arch Biochem Biophys. 1996;330:219–228. doi: 10.1006/abbi.1996.0247. [DOI] [PubMed] [Google Scholar]
- 30.Weichsel A, Brailey JL, Montfort WR. Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Biochemistry. 2007;46:1219–1227. doi: 10.1021/bi061878r. [DOI] [PubMed] [Google Scholar]
- 31.Bai J, Nakamura H, Kwon YW, Hattori I, Yamaguchi Y, Kim YC, Kondo N, Oka S, Ueda S, Masutani H, Yodoi J. Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J Neurosci. 2003;23:503–509. doi: 10.1523/JNEUROSCI.23-02-00503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ago T, Sadoshima J. Thioredoxin and ventricular remodeling. J Mol Cell Cardiol. 2006;41:762–773. doi: 10.1016/j.yjmcc.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 34.Lunn CA, Pak P, Van Savage J, Pigiet V. The catalytic active site of thioredoxin: conformation and homology with bovine pancreatic trypsin inhibitor. Biochim Biophys Acta. 1986;871:257–267. doi: 10.1016/0167-4838(86)90207-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.