

Abstract
Based on the structures of several potent inhibitor molecules for γ-aminobutryric acid aminotransferase (GABA-AT) that were previously reported, six modified fluorine-containing conformationally-restricted analogues were designed, synthesized, and tested as GABA-AT inhibitors. The syntheses of all six molecules followed from a readily synthesized ketone intermediate. Three of the molecules were found to be irreversible inhibitors of GABA-AT with comparable or larger kinact/KI values than that of vigabatrin, a clinically used antiepilepsy drug, and the other three were reversible inhibitors. A possible mechanism for inactivation by one of the inactivators is proposed.
There are two principal neurotransmitters involved in the regulation of brain neuronal activity: γ-aminobutyric acid (GABA), one of the most widely distributed inhibitory neurotransmitters, and L-glutamic acid, an excitatory neurotransmitter.1 The GABA concentration is regulated by two pyridoxal 5′-phosphate (PLP)-dependent enzymes, L-glutamic acid decarboxylase (GAD), which catalyzes the conversion of L-glutamate to GABA, and GABA aminotransferase (GABA-AT), which degrades GABA to succinic semialdehyde (SSA) concomitant with the conversion of α-ketoglutarate to L-glutamate.2 When the concentration of GABA diminishes below a threshold level in the brain, convulsions result;3 raising the brain GABA levels terminates the seizure.4 A reduction in the concentrations of GABA and of the enzyme GAD has been implicated not only in the symptoms associated with epilepsy5,6 but also with several other neurological diseases such as Huntington's chorea, 7,8 Parkinson's disease,9,10 Alzheimer's disease,11 and tardive dyskinesia.12 GABA brain levels cannot be increased by administration of GABA because it does not cross the blood-brain barrier. An approach that has been successful to increase brain GABA levels is the use of a compound that crosses the blood-brain barrier and then inhibits or inactivates GABA-AT, thereby increasing the GABA concentration. Numerous competitive inhibitors of GABA-AT, particularly compounds having a similar backbone structure to GABA, 13 show anticonvulsant activity. A variety of mechanismbased inactivators14 of GABA-AT15 also have been shown to be effective anticonvulsant agents. The most effective of the mechanism-based inactivators as an anticonvulsant agent is 4-amino-5-hexenoic acid (1, Figure 1; γ-vinyl GABA),16 which has the commercial generic name vigabatrin. This compound, which has high potency,17 has been shown to be an effective treatment for epilepsies that are resistant to other anticonvulsant drugs18 and currently is prescribed in over 60 countries worldwide, but it was not approved in the U. S. It also was found that vigabatrin prevents cocaine addiction in rats and baboons.19 This activity was observed with other forms of addiction, including nicotine addiction20 and methamphetamine, alcohol, and heroin addictions.21 Self-administration of cocaine by rats decreased or was prevented by vigabatrin administration in a dose-dependent fashion, without affecting the craving for food.22 GABA was found to antagonize the extracellular dopamine levels responsible for drug addiction, and by positron emission tomography (PET) in primates, it was shown that vigabatrin, which causes a rise in GABA levels, inhibits these drug-induced dopamine increases.21 However, because vigabatrin is not approved in the U. S., new GABA-AT inhibitors and inactivators are needed.
Figure 1.

Vigabatrin (1) and conformationally-restricted analogues
Conformationally restricted analogues of GABA are of considerable interest among medicinal chemists.23 We previously reported the design and synthesis of a series of conformationally restricted analogues of GABA. Among them, (1R, 4S)-4-aminocyclopent-2-enecarboxylic acid (2) was shown to be a potent inhibitor and substrate of GABA-AT, but it was not a time-dependent inactivator.24 (1S, 3S)-3-Amino-4-methylenecyclopentane carboxylic acid (3) was found to inactivate GABA-AT, but only in the absence of 2-mercaptoethanol (indicating that a reactive species was produced that was released from the enzyme prior to inactivation), while (1S, 3S)-3-amino-4-difluoromethylenecyclopentanecarboxylic acid (4) is a much more potent time-dependent inhibitor of γ-aminobutyric acid aminotransferase, even in the presence of 2-mercaptoethanol (indicating that any reactive species generated are not released prior to inactivation).25 Compound 4 is 187 times more potent of an inactivator of GABA-AT than vigabatrin, and the corresponding E- and Z-monofluoro analogues of 3 also inactivate GABA-AT, but not with the same potency as 4. This suggests an important significance for fluorine in the molecules.
Fluorine and fluoroalkyl substitutents have an important electronic effect on the neighboring groups in a molecule and also mimic hydrogen and alkyl groups, respectively.26 Therefore, the introduction of one or more fluorine-containing groups into molecules alters their physical properties, as well as their biological activities.27 Fluorinated substrates also can be used as mechanistic probes and inhibitors for obtaining information about the catalytic mechanism of various enzymatic transformations.28 Consequently, it was thought that other fluorinated conformationally-restricted analogues of 2 and 3 should be evaluated for their inhibitory activities. Based on that hypothesis, we designed six new fluorine-containing conformationally-restricted vigabatrin analogues (5-10, Figure 2). Compounds 5, 6 and 7 are C-3 fluorine or fluoroalkyl substituted analogues of 2, while 8, 9 and 10 are trifluoromethylated analogues of 3 and 4. The presence of strongly electron-withdrawing fluorine and fluoroalkyl groups also may enhance binding and/or reactivity of these compounds during GABA-AT turnover based on a Michael addition mechanism.23a The results of this investigation are reported here.
Figure 2.

New fluorinated conformationally-restricted potential inhibitors and inactivators of GABA-AT
Results and Discussion
Chemistry
Key intermediate 16 for the synthesis of all of the analogues was synthesized according to a modification of a procedure we reported previously29 (Scheme 1). Protection of 11 was realized by treatment with p-methoxybenzyl chloride (PMBCl) in the presence of sodium hydride in DMF instead of LiHMDS in THF as reported. This revised procedure turned out to be much easier and gave higher yields of product.
Scheme 1.

Reagents and conditions: a, PMBCl, NaH, Bu4NI, DMF, 0 °C - r.t., 2h, 83%; b, DBDMH, HOAc, r.t., 20h, 97%; c, Bu3SnH, AIBN, PhH, reflux, 12h, 95%; d, K2CO3, MeOH, H2O, r.t., 2h, 87%; e, NMO, TPAP, DCM, r.t., 24h, 70%.
The synthesis of 5 was first attempted by a direct halogen exchange reaction of 17. However, treatment of 17 with (PhSO2)2NF/nBuLi resulted in the ent-12 reduction product as the major product. Conversion of 17 to organotin intermediate 18, followed by fluorine-metal exchange with XeF2 afforded 19 in a satisfactory yield (Scheme 2). Deprotection of 19 with ceric ammonium nitrate (CAN) followed by acidic hydrolysis gave 5.
Scheme 2.

Reagents and conditions: a, NH2NH2.H2O, Et3N, EtOH, reflux, 1 h; b, I2, Et3N, benzene, r.t., 2h; c, t-BuOK, ether, r.t., 20 h, 60% in three steps; d, Me3SnSnMe3, Pd(PPh3)4, PhMe, reflux, 30 min, 49%; e, XeF2, AgOTf, 2,6-bis(tert-butyl)-4-methylpyridine, DCM, r.t., 8 min, 30%; f, CAN, CH3CN, H2O, r.t., 2 h, 55%; g, 4N HCl (aq.), 70 °C, 0.5-1 h, 64%
Syntheses of target molecules 6 and 7 via elimination of an alcohol or the corresponding tosylated derivative were unsuccessful (Scheme 3). Treatment of 16 with TMSRf (Rf = CF3 or CF3CF2) in the presence of a catalytic amount of TBAF afforded tertiary alcohols 21a and 21b in excellent yields.30 However, direct dehydration of 21a or 21b with various dehydration conditions failed to produce the desired products 22a and 22b. Activation of 21a and 21b with TsCl in the presence of NaH successfully gave the tosylates 23a and 23b, respectively. Unfortunately, attempted elimination reactions of 23a and 23b with various bases always resulted in formation of 21a and 21b, respectively, with only a trace amount of the desired products detected.
Scheme 3.

Reagents and conditions: a, TMSRf, TBAF (cat.), THF, r.t., 1 h, 95%; b, TsCl, NaH, ether, 0 °C, 16 h, 79%.
An alternative synthesis of target molecule 6 (Scheme 4) started with iodination of 16 with hydrazine and iodine followed by elimination of one molecule of HI in the presence of potassium tert-butoxide to give 17. Direct trifluoromethylation of 17 with in situ generated CF3Cu from FSO2CF2CO2Me (methyl fluorosulfonyldifluoroacetate; MFSDA) and CuI31 successfully afforded 22a. Removal of the PMB group with CAN followed by acidic hydrolysis gave 6.
Scheme 4.

Reagents and conditions: a, MFSDA, CuI, DMF, HMPA, 20 h, 75%; b, CAN, MeCN/H2O, r.t., 3 h, 61%; c, 4N HCl (aq.), 70 °C, 0.5-1 h, 85%.
Compound 7 was synthesized from 17 using steps similar to those used to prepare 6 (Scheme 5). A first attempt at pentafluoroethylation of 17 with CF3CF2CO2Na/CuI at 140 °C only resulted in decomposition of the substrate. Treatment of 17 with CF3CF2SiMe3/KF/CuI, however, afforded 22b in good yields.
Scheme 5.

Reagents and conditions: a, CF3CF2SiMe3/KF/CuI, NMO/DMF(1/1), 75 °C, 24 h, 57%; b, CAN, CH3CN, H2O, r.t., 2 h, 76%; c, 4N HCl (aq.), 70 °C, 0.5-1 h, 82%.
A Wittig reaction of 16 with CHBr=PPh3, generated in situ from bromomethyltriphenylphosphonium bromide and tert-BuOK afforded an E/Z mixture of bromomethylenes 25a and 25b, which was easily separated by column chromatography on silica gel. The conformations of the double bonds in the two isomers were determined based on NOE experiments. Trifluoromethylation of 25a and 25b with CF3Cu under similar conditions used for 17 produced compounds 26a and 26b, respectively. Removal of the PMB protecting group with CAN followed by acidic hydrolysis gave 8 and 9 (Scheme 6).
Scheme 6.
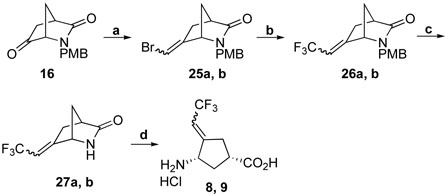
Reagents and conditions: a, BrCH2PPh3.Br, tert-BuOK, THF, -78 C, 20 h, 72% E/Z; b, MFSDA, CuI, DMF, HMPA, 75 °C, 20 h, 95%; c, CAN, CH3CN, H2O, r.t., 2 h, 76%; d, 4N HCl (aq.), 70 °C, 10-12 h, 79%.
Treatment of 16 with CBr4 and PPh3 in toluene afforded 28, which was double trifluoromethylated with in situ generated CF3Cu to give 29. Removal of the PMB group with CAN, followed by hydrolysis with 4 N HCl (aq.) at 75 °C gave 10 (Scheme 7).
Scheme 7.

Reagents and conditions: a, CBr4, PPh3, toluene, reflux, 22 h, 86%; b, MFSDA, CuI DMF, HMPA, 75 °C, 50 h, 82%; c, CAN, CH3CN, H2O, r.t., 1 h, 56%; d, 4N HCl (aq.), 70 °C, 10-12 h, 77%.
It is noteworthy that, compared to the compounds with an exocyclic double bond (27a, 27b, and 30), the hydrolysis of 20, 24a, and 24b, which have endocyclic double bonds, was found to be much easier. The reaction is usually completed in one hour when the compounds are treated with 4 N aq. HCl at 70 °C. Prolonged heating and stirring of these compounds under these conditions resulted in major side reactions. No such side reactions were observed from hydrolysis of 27a, 27b, and 30.
Enzyme inhibition results
Compounds 6, 8, and 9 showed concentration and time-dependent inhibition of pig brain GABA-AT in the presence of β-mercaptoethanol (Table 1). Compounds 5, 7, and 10 showed only weak reversible inhibition of GABA-AT in the presence of β-mercaptoethanol. None of the three reversible inhibitor target molecules was more potent than 2 or 4. However, the irreversible inhibitors were comparable to vigabatrin as inactivators of GABA-AT. It is interesting that although 6 was designed to be a reversible inhibitor of GABA-AT because a simple elimination of HF was not initially apparent, it was found to be an irreversible inhibitor. Compounds 5 and 7, which differ from 6 only by the length of the fluoroalkyl chain, are reversible inhibitors of GABA-AT. Although 8 and 9 are irreversible inhibitors of GABA-AT, as expected, introduction of a second trifluoromethyl group makes 10 a weak reversible inhibitor.
Table 1.
Kinetic constants for 5-10
| Compound | kinact (min−1) | KI (mM) | kinact/ KI (min−1mM−1) | Ki (mM) |
|---|---|---|---|---|
| 5 | 1.3 | |||
| 6 | 0.68 | 4.11 | 0.17 | |
| 7 | 2.8 | |||
| 8 | 6.96 | 39.23 | 0.18 | |
| 9 | 2.89 | 7.74 | 0.37 | |
| 10 | 4.2 | |||
| vigabatrina | 0.24 | 0.85 | 0.28 |
From reference 25
Exhaustive dialysis of GABA-AT that was inactivated by 6, 8, and 9 against potassium pyrophosphate buffer (pH 8.5) resulted in different extents of irreversibility of inhibition. Compared to the control, 48% and 13% of enzyme activity was recovered for 6 and 8, respectively, after 22 h of dialysis, while the percentage of recovered enzyme activity for 9 continued to increase with extended dialysis. These results suggest that at least part of the adducts formed between the enzyme and 6 and 8 are stable, while at least one with 9 can be slowly hydrolyzed upon dialysis. Incubation of 6, 8 and 9 with GABA-AT in the presence of GABA resulted in a significant increase in their t½ of inactivation (slower rate of inactivation), indicating active-site binding to GABA-AT by these compounds.32
A possible mechanism for inactivation of GABA-AT by 6 is shown in Scheme 8. Initial Schiff base formation with the PLP followed by tautomerization gives 31. Michael addition to this trifluoromethyl-activated Michael acceptor33 by Lys329 (X = Lys329) or hydroxide (X = HO−) results in 32, which can eliminate fluoride ion to give a highly reactive exocyclic difluoromethylene group32 conjugated to the PMP iminium (33). This should lead to rapid nucleophilic attack,32 either by the enzyme (pathway a) or by water (pathway b) to give 34 or 36, respectively. Fluoride ion elimination and hydrolysis would give 35 (inactivation) or 38 (turnover). Inactivation by 8 and 9 may be related to the mechanism of inactivation proposed for 4.25
Scheme 8.

To test this mechanism, fluoride ion release was monitored during inactivation. GABA-AT was incubated with all six compounds for two hours, and the fluoride ion concentration was monitored. With 6, 270 fluoride ions were released per enzyme dimer inactivated. This suggests that possibly 90 molecules of 6 are turned over for each inactivation event (partition ratio of 89). Possibly, the activated intermediate 33 undergoes reaction with water (pathway b) 89 times for each attack by an active site residue (pathway a). Depending on what enzyme residue reacts with 33, the carboxylic acid derivative product formed may or may not be stable to conditions of dialysis, leading to partial reactivation of the enzyme during dialysis. For 7, 109 fluoride ions were released, and for the other four compounds, less than 6 fluoride ions were released during inactivation.
Enzyme activity was monitored concomitantly with fluoride ion release during inactivation of GABA-AT by 6. A good correlation was observed (Figure 3), which suggests that inactivation occurs as a consequence of fluoride ion release. When GABA-AT was substituted by human albumin, no fluoride ion was released, indicating that the presence of a protein is not responsible for the fluoride ion release.
Figure 3.

Enzyme activity and fluoride ion release during inactivation of GABA-AT with 6 (2 mM) as a function of time. Fluoride ion concentration when GABA-AT was incubated with 6 (diamonds); fluoride ion concentration when human albumin was incubated with 6 (squares); remaining relative enzyme activity as compared to control when GABA-AT was incubated with 6 (triangles).
Figure 4 shows the time-dependent increase in fluoride ion concentration when GABA-AT was incubated with 7, a weak reversible inhibitor. The fluoride ion concentration reached it highest level in 2-3 hours. No fluoride ion was released when 7 was incubated with albumin, indicating a GABA-AT-dependent reaction. This suggests that the corresponding reactive intermediate (related to 33 in Scheme 8) only undergoes hydrolysis (possibly because of steric hindrance by the trifluoromethyl group of 7 that replaces the fluoride in 6).
Figure 4.

Enzyme activity and fluoride ion release during inactivation of GABA-AT with 7 (2 mM) as a function of time. Fluoride ion concentration when GABA-AT was incubated with 7 (diamonds); remaining relative enzyme activity as compared to control when GABA-AT was incubated with 7 (squares).
Conclusion
Two series of fluorinated compounds were designed as potential inhibitors and inactivators of GABA-AT based on the high potency of 4. The kinact/KI value for 8 (0.37 mM−1min−1) is greater than that for the epilepsy drug vigabatrin (0.28 mM−1min−1); that for 6 (0.16 mM−1min−1) and 9 (0.18 mM−1min−1) are slightly lower. None of these, however, is as efficient an inactivator as 4 (52 mM−1min−1). Compounds 5-7 were designed as reversible inhibitors, so it was unexpected that 6 was a potent time-dependent inactivator. It appears that there is more to the effective binding of compounds to GABA-AT than just the fluorine atoms. Other structures that more closely resemble 4 are being constructed.
4. Experimental
General Methods and Reagents
Optical spectra and GABA-AT assays were recorded on a Perkin-Elmer Lambda 10 UV/Vis spectrophotometer. 1H NMR spectra were recorded on a Varian Gemini 300 MHz NMR spectrometer. Chemical shifts are reported as δ values in parts per million downfield from TMS as the internal standard in CDCl3. For samples run in D2O, the HOD resonance was arbitrarily set at 4.60 ppm. CClF3 was selected as an external standard with δ 0.0 ppm for 19F NMR. Melting points were determined on a Fisher-Johns melting point apparatus and are uncorrected. Combustion analyses were performed by Oneida Research Laboratories, NY. High resolution mass spectra and accurate mass spectra were obtained on a VG-70-250SE high-resolution spectrometer and a Micromass Quattro II LC/MS spectrometer. An Orion Research Model 702A pH meter with a general combination electrode was used for pH measurements. Fluoride ion concentration measurements were obtained using an Orion Research model 702A pH meter with an Orion Research model 96-09 combination fluoride electrode. Flash column chromatography was carried with Merck silica gel 60 (230-400 mesh ASTM). TLC was run with EM Science silica gel 60 F254 precoated glass plates.
All reagents were purchased from Aldrich Chemical Co. and were used without further purification except anhydrous ether and tetrahydrofuran (THF), which were distilled from sodium metal under nitrogen and anhydrous dichloromethane, which was distilled from calcium hydride.
(1S,4R)-6-Iodo-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]hept-5-en-3-one (17)
Compound 16 (0.246 g, 1.0 mmol)29 was dissolved in anhydrous ethanol (5 mL) and was added via syringe to a solution of hydrazine hydrate (51% of hydrazine, 1.02 mL, 21.0 mmol) and Et3N (2.18 mL, 15.7 mmol) in anhydrous ethanol (5 mL) while stirring. The resulting colorless solution was heated to reflux and stirred under argon for 1 h. The reaction mixture was then evaporated under vacuum to give the crude hydrazone product as a colorless oil, which was used directly in the next step without further purification. I2 (0.508 g, 2.0 mmol) was dissolved in anhydrous benzene (6 mL) and was added dropwise into a solution of the above hydrazone and Et3N (1.1 mL, 8.0 mmol) in anhydrous benzene (5 mL) with stirring at room temperature. The resulting brown suspension was stirred at room temperature under argon for 1.5 h. Water (20 mL) was added, and the resulting two layers were separated; the aqueous layer was further extracted with ether (20 mL × 3). The combined organic layers were washed with aqueous HCl (0.5 N, 10 mL), water (10 mL), saturated aqueous NaHCO3 (20 mL), brine (20 mL × 2), and dried over MgSO4. The solvent was removed under vacuum, and the resulting crude product was purified via column chromatography on silica gel (hexanes/EtOAc 9:1) to give the diiodo intermediate as a light brown oil, which was dissolved in anhydrous ether and treated by dropwise addition of two equiv of tert-BuOK at room temperature; the resulting brown suspension was stirred overnight. Water was added, and the two layers were separated. The aqueous layer was further extracted with ether, and the combined ether solutions were washed with brine and dried over anhydrous MgSO4. Ether was removed under vacuum, and the resulting crude product was purified via column chromatography on silica gel (hexanes/EtOAc 9:1) to give 17 as a light green oil (213 mg, 60%). 1H NMR (400MHz, CDCl3) δ 7.19 (2H, d, J = 8.5 Hz), 6.96 (1H, s), 6.89 (2H, d, J = 8.5 Hz), 4.53 (1H, d, J = 14.5 Hz), 4.06 (1H, s), 4.01 (1H, d, J = 14.5 Hz), 3.82 (3H, s), 3.36 (1H, s), 2.31 (1H, d, J = 7.5 Hz), 2.22 (1H, d, J = 7.5 Hz). 13C NMR (100MHz, CDCl3) δ 178.50, 159.36, 144.04, 129.84, 128.73, 114.32, 98.91, 71.65, 58.13, 56.33, 55.38, 46.83. m/z (ESI, MeOH): 378 (M + Na)+. HRMS calcd. For C14H14O2NI: 355.0064. Found 355.0057.
(1S,4R)-2-(4-Methoxybenzyl)-6-trimethylstannanyl-2-azabicyclo[2.2.1]hept-5-en-3-one (18)
Compound 17 (41 mg, 0.115 mmol) was dissolved in anhydrous toluene (4 mL) under argon, followed by addition of Pd(PPh3)4 (20 mg, 0.017 mmol) and Me3SnSnMe3 (45 mg, 0.139 mmol). A light yellow solution formed, which was heated to reflux under argon and stirred for 1 h. Toluene was evaporated under vacuum, and the residue was purified via column chromatography on silica gel (hexanes/EtOAc 3:1) to give 18 (25 mg, 56%) as a colorless oil. 1H NMR (500MHz, CDCl3) δ 7.14 (2H, d, J = 8.5 Hz), 6.88 (2H, d, J = 8.5 Hz), 6.85 (1H, m), 4.67 (1H, d, J = 15.0 Hz), 4.13 (1H, s), 3.81 (3H, s), 3.47 (1H, s), 3.41 (1H, d, J = 15.0 Hz), 2.24 (1H, d, J = 7.5 Hz), 2.00 (1H, d, J = 8.0 Hz), 0.16 (9H, s). 13C NMR (CDCl3, 125.6 MHz) δ 179.83, 159.03, 153.07, 148.19, 129.26, 129.12, 114.12, 66.20, 58.27, 55.68, 55.34, 47.56, -9.28. m/z (EI, DCM): 389.0 (M)+, 374.0 (M – CH3)+, 211.0 (M – PMB – CONH – CH3)+, 121.0 (PMB)+. HRMS calcd. for C17H23O2N116Sn (M)+: 389.0741. Found 389.0749; calcd for C16H20O2N116Sn (M – CH3)+: 374.0506. Found 374.0504.
(1S,4R)-6-Fluoro-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]hept-5-en-3-one (19)
AgOTf (48 mg, 0.187 mmol) was dissolved in dry DCM under argon. Compound 18 (73.4 mg, 0.187 mmol) and 2,6-di-tert-butyl-4-methylpyridine (19 mg, 0.094 mmol) in dry DCM (1 mL) was added via syringe, followed by immediate addition of XeF2 (38 mg, 0.225 mmol) in DCM. The resulting reaction mixture was shielded from light and stirred at room temperature for 15 min. The reaction mixture was then partitioned between saturated aq. NaHCO3 and CHCl3, and the aqueous layer was further extracted with CHCl3 (20 mL). The combined CHCl3 solutions were washed with brine (20 mL) and dried over anhydrous MgSO4. The CHCl3 was removed under vacuum, and the residue was purified via column chromatography on silica gel (hexanes/EtOAc 3:1) to give 19 (20 mg, 30%) as a light yellow oil. 1H NMR (400MHz, CDCl3) δ 7.15 (2H, d, J = 8.8 Hz), 6.86 (2H, d, J = 8.4 Hz), 5.57 (1H, s), 4.48 (1H, d, J = 14.8 Hz), 4.08 (1H, d, J = 14.8 Hz), 3.89 (1H, s), 3.80 (1H, s), 3.24 (1H, s), 2.33 – 2.38 (2H, m). 19F NMR (376MHz, CDCl3) δ - 119.72 (1F, s). 13C NMR (CDCl3, 100.63 MHz) δ 177.01, 173.97, 159.20, 129.68, 128.27, 114.11, 105.37 (d, J = 7.3 Hz), 62.64, 57.16, 55.33, 49.62, 47.12. m/z (EI, DCM): 247.1 (M)+, 163.1, 121.1 (PMB)+. HRMS calcd. for C14H14O2NF (M)+: 247.1003. Found 247.1003.
(1S,4R)-6-Fluoro-2-azabicyclo[2.2.1]hept-5-en-3-one (20)
This compound was synthesized from 19 using general procedure II (see below) in a 55% yield. 1H NMR (400.168 MHz, CDCl3) δ 6.41 (1H, s), 5.59 (1H, s), 4.12 (1H, s), 3.10 (1H, s), 2.44 (2H, s). 19>F NMR (376.492 MHz, CDCl3) δ -120.59 (1F, s). 13C NMR (CDCl3, 125.614 MHz) δ 184.95, 175.63 (d, JFC = 304.9 Hz), 105.88, 59.52, 58.49, 49.45. m/z (CI, DCM): 128.1 (M+1)+, 85.3 (M-CON)+. HRMS calcd. For C6H7ONF: 128.0506. Found 128.0511.
(1R,4S)-4-Amino-3-fluoro-cyclopent-2-ene carboxylic acid (5)
This compound was synthesized from 20 using general procedure III in a yield of 64% as a light yellow solid, mp 201.0-202.5 °C; 1H NMR (D2O, 499.511 MHz) δ 5.68 (1H, s), 4.45 (1H, d, J = 2.9 Hz), 3.66 (1H, d, J = 4.0 Hz), 2.83 (1H, dt, J = 14.5 Hz, 9.0 Hz), 2.20 (1H, dt, J = 14.5 Hz, 4.5 Hz). 19F NMR (D2O, 376.493 MHz) δ -128.97 (1F, d, J = 4.5 Hz). 13C NMR (D2O, 125.614 MHz) δ 177.18, 157.61 (d, JFC = 280.2 Hz), 109.21 (d, JFC = 12.1 Hz), 51.37 (d, JFC = 22.2 Hz), 42.57 (d, JFC = 8.2 Hz), 29.32 (d, JFC = 4.0 Hz). m/z (EI, MeOH): 145.1 (M)+, 126.1, 100.1 (M-CO2H)+. HRMS calcd. For C6H8OFCNF: 145.0534. Found 145.0530. Anal. (C6H8O2NF.0.5H2O) C, H, N.
(1S,4S,6SR)-6-Hydroxy-6-trifluoromethyl-2-(4′-methoxybenzyl)-azabicyclo[2.2.1]heptan-3-one (21a)
To a solution of 16 (0.123 g, 0.5 mmol) in dry THF (4 mL) was added TMSCF3 (0.107 mg, 0.75 mmol). Tetrabutylammonium fluoride (TBAF) (1.0 M solution in THF, 50 μL, 0.05 mmol) was added dropwise. The resulting mixture was stirred under nitrogen for 2 h and was concentrated under vacuum. The residue was purified by column chromatography (hexanes: EtOAc 2:1 to 1:1), giving 21a (0.149 g, 94%) as a white solid. 1H NMR (400MHz, CDCl3) δ 7.19 (2H, d, J = 8.4 Hz), 6.87 (2H, d, J = 8.4 Hz), 5.00 (1H, d, J = 14.8 Hz), 4.56 (1H, br. s), 4.04 (1H, d, J = 14.4 Hz), 3.81 (3H, s), 3.74 (1H, s), 2.87 (1H, s), 2.33 (1H, dd, J = 14.0 Hz, 3.6 Hz), 1.80–1.91 (3H, m). 19F NMR (376MHz, CDCl3) δ -78.19 (3F, s). m/z (EI, DCM): 316.1 (M + 1)+, 315.1 (M)+, 121.1 (CH3OC6H4CH2)+. HRMS calcd. For C15H16O3NF3: 315.1070. Found 315.1077.
(1S,4S,6SR)-6-hydroxyl-6-pentafluoroethyl-2-(4′-methoxybenzyl)-azabicyclo[2.2.1]heptan-3-one (21b)
This compound was synthesized by the same method as for 21a. 1H NMR (400MHz, CDCl3) δ 7.17 (2H, d, J = 8.8 Hz), 6.87 (2H, d, J = 7.6 Hz), 4.98 (1H, d, J = 15.2 Hz), 4.97 (1H, s), 4.01 (1H, d, J = 14.8 Hz), 3.86 (1H, s), 3.80 (3H, s), 2.84 (1H, s), 2.42 (1H, d, J = 13.6 Hz), 1.78–1.89 (3H, m). 19F NMR (376MHz, CDCl3) δ -79.02 (3F, s), -120.08 (2F, ab, J = 277.5 Hz). m/z (EI, DCM): 366.1 (M + 1)+, 365.1 (M)+, 121.1 (CH3OC6H4CH2)+ HRMS calcd. For C16H16O3NF5: 365.1050. Found 365.1045.
(1S,4R,6S)-Toluene-4-sulfonic acid 2-(4-methoxybenzyl)-3-oxo-6-trifluoromethyl-2-azabicyclo[2.2.1]hept-6-yl ester (23a)
NaH in mineral oil (14 mg, 60%, 0.3 mmol) was washed with anhydrous pentane, suspended in dry ether (3 mL), and cooled to 0 °C in an ice bath. Compound 21a (52 mg, 0.16 mmol) in dry ether (1 mL) was then added dropwise via syringe. The resulting white suspension was stirred at 0 °C for 15 min under argon followed by addition of TsCl (64 mg, 0.3 mmol) in dry ether (1 mL). The resulting white suspension was allowed to slowly warm up to room temperature and was stirred under argon for 16 h. Water was added, and the two layers were separated. The aqueous layer was further extracted with ether (10 mL × 3), and the combined ether solutions were washed with brine (10 mL × 2) and concentrated under vacuum. The residue was separated by flash column chromatography on silica gel (hexanes/EtOAc 1:1) to give 23a (58 mg, 79%) as a white solid. 1H NMR (500MHz, CDCl3) δ 7.82 (2H, d, J = 8.0 Hz), 7.40 (2H, d, J = 6.0 Hz), 7.04 (2H, d, J = 6.4 Hz), 6.82 (2H, d, J = 6.4 Hz), 4.75 (1H, d, J = 12.0 Hz), 3.84 (1H, s), 3.78 (3H, s), 3.43 (1H, d, J = 12.0 Hz), 3.09 (1H, d, J = 12.0 Hz), 2.96 (1H, s), 2.47 (3H, s), 2.40 (1H, dd, J = 12.4 Hz, 2.8 Hz), 1.81 - 1.84 (2H, m). 19F NMR (376MHz, CDCl3) δ -74.41 (3F, s). 13C NMR (CDCl3, 125.6 MHz) δ 175.17, 159.24, 145.72, 134.22, 130.13, 129.54, 128.30, 127.76, 123.16 (q), 114.21, 63.32, 55.31, 45.83, 45.03, 39.13, 31.11, 21.80.
(1S,4R,6S)-Toluene-4-sulfonic acid 2-(4-methoxybenzyl)-3-oxo-6-pentafluoroethyl-2-azabicyclo [2.2.1] hept-6-yl ester (23b)
This compound was synthesized from 21b similar to the synthesis of 23a. 1H NMR (400MHz, CDCl3) δ 7.84 (2H, d, J = 8.0 Hz), 7.40 (2H, d, J = 8.8 Hz), 7.04 (2H, d, J = 8.4 Hz), 6.83 (2H, d, J = 8.0 Hz), 4.95 (1H, d, J = 15.6 Hz), 3.90 (1H, s), 3.78 (3H, s), 3.54 (1H, d, J = 14.8 Hz), 3.29 (1H, d, J = 15.6 Hz), 2.96 (1H, s), 2.48-2.51 (4H, m), 1.85 (1H, d, J = 11.2 Hz), 1.79 (1H, d, J = 10.4 Hz). 19F NMR (376MHz, CDCl3) δ -78.99 (3F, s), -116.41 (2F, ab). 13C NMR (CDCl3, 125.6 MHz) δ 174.98, 159.22, 145.86, 133.85, 130.04, 129.62, 127.98, 114.21, 63.21, 55.33, 46.13, 44.59, 30.77, 21.87. m/z (EI, MeOH): 519.0 (M)+, 520.0 (M + 1)+, 365 (M − CH3C6H4SO2)+, 121.0. HRMS calcd. For C23H22O5NF5S: 519.183. Found 519.1135.
(1S,4R)-6-Trifluoromethyl-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]hept-5-en-3-one (22a). General procedure I
Methyl fluorosulfonyldifluoroacetate (MFSDA; 48 mg, 0.25 mmol) in anhydrous DMF (1 mL) was added dropwise via syringe to a suspension of 17 (35.5 mg, 0.1 mmol) and CuI (23 mg, 0.12 mmol) in anhydrous DMF (2 mL) and HMPA (1 mL) at 75 °C under argon over a period of 1 h. The resulting suspension was stirred at 75 °C under argon for 72 h and evaporated under high vacuum. The residue was purified via column chromatography on silica gel (hexanes/EtOAc 3:1) to give 22a (22.1 mg, 75%) as a light brown oil. 1H NMR (500MHz, CDCl3) δ 7.18 (2H, d, J = 8.5 Hz), 7.11 (1H, s), 6.89 (2H, d, J = 8.5 Hz), 4.77 (1H, d, J = 15.0 Hz), 4.23 (1H, s), 3.82 (3H, s), 3.56 (1H, s), 3.54 (1H, d, J = 15.0 Hz), 2.40 (1H, d, J = 8.0 Hz), 2.32 (1H, d, J = 8.0 Hz). 19F NMR (376.49 MHz, CDCl3) δ -64.69 (3F, s). 13C NMR (CDCl3, 125.6 MHz) δ 178.04, 159.30, 143.97 (q, JCF = 36.6 Hz), 141.52 (q, JCF = 5.4 Hz), 129.54, 128.26, 122.44 (q, JCF = 267.8 Hz), 114.25, 61.35, 59.31, 55.35, 54.06, 46.81. m/z (EI, MeOH): 297.1 (M)+, 298.1 (M + 1)+, 163.1 (M – PMBN)+, 121.0 (PMB)+. HRMS calcd. For C15H14O2NF3: 297.0971. Found 297.0970.
(1S,4R)-6-Trifluoromethyl-2-azabicyclo[2.2.1]hept-5-en-3-one (24a). General procedure II
Ceric ammonium nitrate (CAN; 1.38 g, 2.525 mmol) in water (3 mL) was added to a solution of 22a (150 mg, 0.505 mmol) in acetonitrile (6 mL). The resulting red solution was stirred at room temperature for 1.5 h. The reaction mixture was diluted with ethyl acetate, washed with saturated aq. NaHCO3, then brine, and dried over anhydrous MgSO4. The solution was concentrated under vacuum, and the residue was purified via column chromatography on silica gel (hexanes/EtOAc 1:1) to give 24a (54 mg, 61%) as a white solid. 1H NMR (500MHz, CDCl3) δ 7.07 (1H, s), 6.15 (1H, br. s), 4.51 (1H, s), 3.42 (1H, s), 2.54 (1H, d, J = 8.0 Hz), 2.43 (1H, d, J = 8.0 Hz). 19F NMR (376MHz, CDCl3) δ -66.42 (3F, s). 13C NMR (CDCl3, 125.6 MHz) δ 182.3, 144.3 (m), 141.0 (q, JFC = 5.5 Hz), 122.4 (q, JFC = 268.2 Hz), 60.1, 58.6, 53.7. m/z (CI, MeOH): 178.3 (M + 1)+, 115.2. HRMS calcd. for C7H7ONF3 (M + H): 178.0474. Found 178.0481.
(1R,4S)-4-Amino-3-trifluoromethyl-cyclopent-2-ene carboxylic acid (6). General procedure III
Compound 24a (44 mg, 0.249 mmol) was dissolved in 4 N aq. HCl (5 mL), and the resulting clear solution was heated to 70 to 75 °C and stirred for 40 to 45 min. The resulting colorless clear solution was extracted with EtOAc (3 mL × 2) and evaporated to dryness under high vacuum to give the crude product, which was further purified via ion exchange chromatography (0.15 N aq. HCl as eluant) to give 6 as a white solid (49 mg, 85%), mp 195.0-196.0 °C; 1H NMR (D2O, 400.169 MHz) δ 6.96 (1H, s), 4.69 (1H, s), 3.92 (1H, s), 2.87-2.95 (1H, m), 2.29-2.35 (1H, m). 19F NMR (D2O, 376.493 MHz) δ -64.35 (3F, s). 13C NMR (D2O, 125.614 MHz) δ 175.12, 142.54 (q, JFC = 4.6 Hz), 130.82 (q, JFC = 33.7 Hz), 121.62 (q, JFC = 269.7 Hz), 53.64, 48.80, 32.11. m/z (EI, MeOH): 196.1 (M)+, 179.1 (M-OH)+, 159.1. HRMS calcd. For C7H9O2NF3: 196.0580. Found 196.0576. Anal. (C7H9O2NF3) C, H, N.
(1S,4R)-6-Pentafluoroethyl-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]hept-5-en-3-one (22b)
A 5 mL screw-capped Pyrex tube was charged with CuI (19 mg, 0.10 mmol), KF (14.5 mg, 0.25 mmol), and a stirring bar. The tube was dried with a heat gun under vacuum for 20 min and filled with argon. Compound 17 (18 mg, 0.05 mmol) in anhydrous NMP (1 mL) was added via syringe to the above reaction tube followed by TMSC2F5 (48 mg, 0.25 mmol) in anhydrous DMF (1 mL). The resulting suspension was heated to 80 °C and stirred vigorously for 48 h. The reaction mixture was then evaporated under high vacuum, and the residue was purified via column chromatography on silica gel (hexanes/EtOAc 3:1) to give 22b (10 mg, 57%) as a light yellow oil. 1H NMR (400.168 MHz, CDCl3) δ 7.17 – 7.19 (3H, m), 6.89 (2H, d, J = 8.8 Hz), 4.80 (1H, d, J = 15.2 Hz), 4.26 (1H, s), 3.82 (3H, s), 3.60 (1H, s), 3.52 (1H, d, J = 14.4 Hz), 2.40 (1H, d, J = 6.4 Hz), 2.32 (1H, d, J = 8.0 Hz). 19F NMR (376.49 MHz, CDCl3) δ -84.12 (3F, s), -113.67 (2F, ab, Jab = 279.4 Hz). 13C NMR (CDCl3, 125.6 MHz) δ 177.59, 159.30, 144.14 (t, JCF = 8.0 Hz), 143.03 (t, JCF = 26.0 Hz), 129.51, 128.21, 118.81 (dt, JCF = 285.4 Hz, 37.8 Hz), 114.24, 61.90, 59.79, 55.32, 54.54, 46.64. m/z (EI, DCM): 347.1 (M)+, 121.0 (PMB)+. HRMS calcd. For C16H14O2NF5: 347.0939. Found 347.0933.
(1S,4R)-6-Pentafluoroethyl-2-azabicyclo[2.2.1]hept-5-en-3-one (24b)
was synthesized from 22b in a yield of 74% using general procedure II. 1H NMR (500MHz, CDCl3) δ 7.14 (1H, s), 6.64 (1H, br. s), 4.52 (1H, s), 3.43 (1H, s), 2.52 (1H, d, J = 7.0 Hz), 2.41 (1H, d, J = 7.0 Hz). 19F NMR (376MHz, CDCl3) δ -84.45 (3F, s), -116.41 (2F, ab, Jab = 276.3 Hz, Δ v = 268.h Hz). 13C NMR (CDCl3, 125.6 MHz) δ 182.28, 143.40 – 143.78 (m, 2C), 111.80 – 122.15 (m, 2C, C2F5), 60.72, 59.23, 54.13. m/z (CI, DCM): 228.0 (M+1)+, 185.3 (M-CON)+, 165.3 (M-CONH-F)+. HRMS calcd. For C16H14O2NF5: 347.0939. Found 347.0933.
(1R,4S)-4-Amino-3-pentafluoroethyl-cyclopent-2-ene carboxylic acid (7)
was synthesized using general procedure III from 24b as a white solid in a yield of 82%, mp 187.0-188.5 °C; 1H NMR (D2O, 499.511 MHz) δ 7.00 (1H, s), 4.71 (1H, d, J = 3.5 Hz), 3.92 (1H, s), 2.85-2.91 (1H, m), 2.29-2.33 (1H, m). 19F NMR (D2O, 376.493 MHz) δ -84.60 (3F, t, J = 18.8 Hz), -113.07 (2F, ab, J = 283.9 Hz, Δ = 62.5 Hz). 13C NMR (D2O, 125.614 MHz) δ 174.89, 145.37 (t, JFC = 7.2 Hz), 129.63 (t, JFC = 25.0 Hz), 109.70-119.59 (2C, m), 54.61, 49.27, 32.10. m/z (EI, MeOH): 246.1 (M+1)+, 245.1 (M)+, 200.1 (M-CO2H)+, 176.1. HRMS calcd. For C8H8O2NF5: 245.0470. Found 245.0475. Anal. (C8H8O2NF5.1.5H2O) C, H, N.
(1S,4R)-6-Bromomethylene-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]heptan-3-one (25)
(Bromomethyl)triphenylphosphonium bromide (0.567 g, 1.3 mmol) was suspended in dry THF (4 mL), cooled to −78 °C, and then treated by addition of tert-BuOK (1.0 M in THF, 1.2 mL, 1.2 mmol). The resulting brown suspension was stirred at −78 °C for 1.5 h and then treated with 16 (0.246 g, 1.0 mmol) in dry THF (1 mL). The resulting light brown mixture was allowed to gradually warm up to room temperature and was stirred overnight. The reaction was quenched via addition of water, and the mixture was extracted with dichloromethane (10 mL × 3). The combined organic layers were washed with brine (20 mL × 2) and dried over anhydrous MgSO4. The solvent was removed under vacuum, and the residue was purified via column chromatography on silica gel (hexanes/EtOAc 1:1) to give 25a (Z-isomer, 100 mg, 31%) as a light yellow oil and 25b (E-isomer, 131 mg, 41%) as a white solid.
25a
1H NMR (500MHz, CDCl3) δ 7.26 (2H, d, J = 8.0 Hz), 6.88 (2H, d, J = 8.0 Hz), 6.01 (1H, s), 4.76 (1H, d, J = 15.0 Hz), 4.35 (1H, s), 3.81 (3H, s), 3.70 (1H, d, J = 15.0 Hz), 3.02 (1H, s), 2.47 (1H, d, J = 16.0 Hz), 2.31 (1H, d, J = 15.5 Hz), 1.97 (1H, d, J = 9.5 Hz), 1.54 (1H, d, J = 10.0 Hz). 13C NMR (126MHz, CDCl3) δ 177.51, 159.15, 143.88, 129.64, 128.94, 114.01, 97.82, 61.88, 55.31, 45.81, 44.36, 39.78, 32.77. m/z (ESI, MeOH): 344.6 (M + Na)+, 346.5 (M + 2 + Na)+. HRMS calcd. For C15H16O2NBr: 321.0359. Found 321.0359.
25b
1H NMR (500MHz, CDCl3) δ 7.14 (2H, d, J = 8.5 Hz), 6.86 (2H, d, J = 8.5 Hz), 6.10 (1H, s), 4.62 (1H, d, J = 15.0 Hz), 3.86 (1H, s), 3.80 (3H, s), 3.79 (1H, d, J = 15.0 Hz), 2.97 (1H, s), 2.43 (1H, dt, J = 16.5 Hz, 3.0 Hz), 2.26 (1H, d, J = 16.5 Hz), 2.06 (1H, d, J = 9.5 Hz), 1.60 (1H, d, J = 9.5 Hz). 13C NMR (126MHz, CDCl3) δ 177.79, 159.19, 144.28, 129.42, 128.53, 114.15, 100.00, 63.46, 55.33, 44.89, 43.95, 41.16, 33.49. m/z (ESI, MeOH): 344.6 (M + Na)+, 346.2 (M + 2 + Na)+. HRMS calcd. For C15H16O2NBr: 321.0359. Found 321.0359.
(Z)-(1S,4R)-2-(4-Methoxybenzyl)-6-(2,2,2-trifluoro-ethylidene)-2-azabicyclo[2.2.1]heptan-3-one (26a)
was synthesized in a 95% yield from 25a using general procedure I (reaction time 20 h). 1H NMR (400MHz, CDCl3) δ 7.18 (2H, d, J = 8.8 Hz), 6.88 (2H, d, J = 8.4 Hz), 5.68 (1H, q, J = 8.0 Hz), 4.81 (1H, d, J = 14.8 Hz), 4.44 (1H, s), 3.82 (1H, s), 3.58 (1H, d, J = 15.2 Hz), 2.95 (1H, s), 2.57 (1H, d, J = 17.2 Hz), 2.41 (1H, d, J = 17.2 Hz), 2.06 (1H, d, J = 8.8 Hz), 1.58 (1H, d, J = 10.4 Hz). 19F NMR (376.49 MHz, CDCl3) δ -58.02 (3F, m). m/z (EI, DCM): 311.1 (M)+, 312.1 (M + 1)+, 121.0 (PMB)+. HRMS calcd. For C16H16O2NF3: 311.1128. Found 311.1133.
(E)-(1S,4R)-2-(4-Methoxybenzyl)-6-(2,2,2-trifluoroethylidene)-2-azabicyclo[2.2.1]heptan-3-one (26b)
was synthesized in a 95% yield from 25b using general procedure I (reaction time 20 h). 1H NMR (400 MHz, CDCl3) δ 7.15 (2H, d, J = 8.8 Hz), 6.87 (2H, d, J = 8.4 Hz), 5.51 (1H, q, J = 7.2 Hz), 4.57 (1H, d, J = 14.4 Hz), 3.90 (1H, d, J = 15.2 Hz), 3.86 (1H, s), 3.80 (3H, s), 2.98 (1H, s), 2.62 (1H, dq, J = 17.2 Hz, 2.4 Hz), 2.48 (1H, dq, J = 17.2 Hz, 2.4 Hz), 2.05 (1H, d, J = 9.2 Hz), 1.56 (1H, d, J = 10.0 Hz). 19F NMR (376.49 MHz, CDCl3) δ -60.98 (3F, m). m/z (EI, DCM): 311.1 (M)+, 312.1 (M + 1)+, 121.0 (PMB)+. HRMS calcd. For C16H16O2NF3: 311.1128. Found 311.1133.
(Z)-(1S,4R)-6-(2,2,2-Trifluoroethylidene)-2-azabicyclo[2.2.1]heptan-3-one (27a)
was synthesized in a 74% yield from 26a using general procedure II. 1H NMR (400MHz, CDCl3) δ 6.30 (1H, s), 5.58 (1H, q, J = 8.0 Hz), 4.61 (1H, s), 2.83 (1H, s), 2.55 (1H, d, J = 17.2 Hz), 2.36 (1H, d, J = 16.4 Hz), 2.18 (1H, dd, J = 10.4 Hz, 1.2 Hz), 1.63 (1H, d, J = 9.6 Hz). 19F NMR (376MHz, CDCl3) δ -58.28 (3F, m). 13C NMR (CDCl3, 125.6 MHz) δ 179.92, 152.44 (q, JFC = 5.5 Hz), 122.93 (q, JFC = 270.1 Hz), 110.02 (q, JFC = 34.8 Hz), 56.67, 43.58, 41.49, 32.23. m/z (EI, DCM): 192 (M + 1)+, 191 (M)+, 79.1. HRMS calcd. for C8H8ONF3 : 191.0553. Found 191.0559.
(E)-(1S,4R)-6-(2,2,2-Trifluoroethylidene)-2-azabicyclo[2.2.1]heptan-3-one (27b)
was synthesized in a 74% yield from 26a using general procedure II. 1H NMR (400MHz, CDCl3) δ 6.57 (1H, s), 5.77 (1H, d, J = 7.6 Hz), 4.13 (1H, s), 2.87 (1H, s), 2.65 (1H, d, J = 16.0 Hz), 2.48 (1H, d, J = 16.8 Hz), 2.14 (1H, d, J = 9.6 Hz), 1.62 (1H, d, J = 9.6 Hz). 19F NMR (376MHz, CDCl3) δ -61.08 (3F, m). 13C NMR (CDCl3, 125.6 MHz) δ 180.27, 151.69 (q, JFC = 4.6 Hz), 123.20 (q, JFC = 270.1 Hz), 110.82 (q, JFC = 35.7 Hz), 60.10, 44.60, 41.52, 30.53. m/z (EI, DCM): 192 (M + 1)+, 191 (M)+, 163 (M-CO)+, 79. HRMS calcd. for C8H8ONF3: 191.0553. Found 191.0553.
(1S,3S)-(Z)-3-Amino-4-(2,2,2-trifluoroethylidene)cyclopentane carboxylic acid (8)
was synthesized in a 66% yield as a white solid from 27a using general procedure III; mp 212.0-214.0 °C; 1H NMR (D2O, 499.511 MHz) δ 5.97 (1H, q, J = 7.5 Hz), 4.37 (1H, s), 3.09-3.16 (2H, m), 2.92-2.95 (1H, m), 2.56-2.62 (1H, m), 1.98 (1H, q, J = 12.5 Hz). 19F NMR (D2O, 376.493 MHz) δ -60.78 (3F, d, JHF = 6.0 Hz). 13C NMR (D2O, 125.614 MHz) δ 177.93, 150.92 (q, JFC = 5.0 Hz), 123.11 (q, JFC = 270.2 Hz), 114.77 (q, JFC = 35.3 Hz), 53.76, 40.69, 32.95, 32.43. m/z (EI, MeOH): 209.1 (M)+, 208.1 (M-1)+, 189.1 (M-H2O-2)+, 164.1 (M-CO2H)+. HRMS calcd. For C8H10O2NF3: 209.0658. Found 277.0656. Anal. (C8H10O2NF3.0.25H2O) C, H, N.
(1S,3S)-(Z)-3-Amino-4-(2,2,2-trifluoroethylidene)cyclopentane carboxylic acid (9)
was synthesized from 27b in a 79% yield as a white solid using general procedure III; mp 206.0-207.5 °C; 1H NMR (D2O, 499.749 MHz) δ 6.09 (1H, qd, J = 9.0 Hz, 1.5 Hz), 4.65 (1H, s), 3.02-3.09 (1H, m), 2.91-2.92 (2H, m), 2.57-2.63 (1H, m), 2.13-2.18 (1H, m). 19F NMR (D2O, 376.493 MHz) δ -60.37 (3F, d, JHF = 7.9 Hz). 13C NMR (D2O, 125.673 MHz) δ 178.64, 149.42 (q, JFC = 5.4 Hz), 120.43 (q, JFC = 269.3 Hz), 116.73 (q, JFC = 34.3 Hz), 50.15, 40.74, 37.00, 34.44. m/z (EI, MeOH): 210.1 (M+1)+, 209.1 (M)+, 191.1 (M-H2O)+, 164.1 (M-CO2H)+. HRMS calcd. For C8H10O2NF3: 209.0664. Found 277.0675. Anal. (C8H10O2NF3.0.25H2O) C, H, N.
(1S,4R)-6-Dibromomethylene-2-(4-methoxybenzyl)-2-azabicyclo[2.2.1]heptan-3-one (28)
Compound 16 (0.246 g, 1.0 mmol), carbon tetrabromide (0.498 g, 1.5 mmol) and triphenylphosphine (0.787 g, 3.0 mmol) were dissolved in anhydrous toluene (10 mL), and the resulting light yellow suspension was stirred while refluxing under argon for 22 h. The reaction mixture was then filtered, washed with benzene, and evaporated to give the crude product, which was purified via column chromatography on silica gel (hexanes: EtOAc 1:1) to give 28 (377 mg, 94%) as a light yellow oil. 1H NMR (500MHz, CDCl3) δ 7.21 (2H, m), 6.85 (2H, m), 4.72 (1H, d, J = 12.5 Hz), 4.20 (1H, s), 3.73-3.78 (4H, m), 3.03 (1H, s), 2.45 (1H, d, J = 16.5 Hz), 2.29 (1H, d, J = 16.5 Hz), 2.02 (1H, s), 1.63 (1H, s). 13C NMR (126MHz, CDCl3) δ 177.41, 159.15, 145.83, 129.45, 128.51, 114.04, 81.58, 64.27, 55.28, 45.66, 44.49, 40.84, 36.61. m/z (ESI, MeOH): 399.9 (M)+, 401.9 (M + 2)+, 404 (M + 4)+, 322.0 (M - Br)+, 293.9 (M – CH3OC6H4)+. HRMS calcd. For C15H15O2NBr2: 398.9464. Found 398.9460.
(1S,4R)-2-(4-Methoxybenzyl)-6-(2,2,2-trifluoro-1-trifluoromethylethylidene)-2-azabicyclo[2.2.1]heptan-3-one (29)
was synthesized from 28 in an 82% yield using general procedure I (50 h). 1H NMR (400MHz, CDCl3) δ 7.15 (2H, d, J = 8.8 Hz), 6.88 (2H, d, J = 8.8 Hz), 4.82 (1H, d, J = 15.2 Hz), 4.62 (1H, s), 3.81 (3H, s), 3.65 (1H, d, J = 15.2 Hz), 3.03 (1H, s), 2.82 (1H, d, J = 18.4 Hz), 2.73 (1H, d, J = 18.4 Hz), 2.12 (1H, d, J = 10.4 Hz), 1.63 (1H, d, J = 10.4 Hz). 19F NMR (376.49 MHz, CDCl3) δ -57.12 - -57.03 (3F, m), -59.97 - -59.91 (3F, m). m/z (EI, DCM): 379.1 (M)+, 380.1 (M + 1)+, 136.1 (PMB + NH)+, 121.0 (PMB)+. HRMS calcd. For C16H16O2NF3: 379.1001. Found 379.1003.
(1S,4R)-6-(2,2,2-Trifluoro-1-trifluoromethylethylidene)-2-azabicyclo[2.2.1]heptan-3-one (30)
was synthesized from 29 in a 56% yield using general procedure II. 1H NMR (400MHz, CDCl3) δ 5.62 (1H, s), 4.78 (1H, s), 2.96 (1H, s), 2.83 (1H, s), 2.74 (1H, s), 2.29 (1H, s), 1.71 (1H, s). 19F NMR (376MHz, CDCl3) δ -57.32 (3F, s), -60.25 (3F, s). m/z (EI, DCM): 260.1 (M + 1)+, 259.1 (M)+, 189.1 (M – CF3)+. HRMS calcd. for C9H7ONF6 : 259.0426. Found 259.0434.
(1S,3S)-3-Amino-4-(2,2,2-trifluoro-1-trifluoromethylethylidene)cyclopentane carboxylic acid (10)
was synthesized from 30 in a 77% yield as a white solid using general procedure III; mp 198.0-199.5 °C; 1H NMR (D2O, 400.169 MHz) δ 4.92 (1H, s), 3.29-3.34 (1H, m), 3.16-3.20 (2H, m), 2.57-2.65 (1H, m), 2.20-2.27 (1H, m). 19F NMR (D2O, 376.493 MHz) δ -59.97 (3F, q, J = 9.0 Hz), -60.47 (3F, q, J = 9.0 Hz). 13C NMR (D2O, 100.749 MHz) δ 178.08, 158.14, 122.40, 119.16-119.67 (m), 52.56 (d, JFC = 2.3 Hz), 40.73, 36.31 (q, JFC = 3.8 Hz), 33.68. m/z (EI, MeOH): 278.1 (M+1)+, 277.1 (M)+, 259.1 (M-H2O)+, 232.0 (M-CO2H)+. HRMS calcd. For C9H9O2NF6: 277.0538. Found 277.0545. Anal. (C9H9O2NF6.H2O) C, H, N.
Enzyme and Assays
GABA aminotransferase was isolated from pig brains by a known procedure.34 Succinic semialdehyde dehydrogenase (SSDH) was isolated from GABAse, a commercially available mixture of SSDH and GABA aminotransferase, by inactivation of the GABA aminotransferase with gabaculine as described previously.35 GABA activity assays were carried out using a modification of the coupled assay developed by Scott and Jakoby.36 The assay solution contains 11 mM GABA, 5.3 mM α-ketoglutarate, 1.1 mM NADP+, and 5 mM β-mercaptoethanol in 100 mM potassium pyrophosphate, pH 8.5, and excess SSDH. Using this assay, the change in absorbance at 340 nm indicates production of NADPH, which is directly proportional to the activity of GABA aminotransferase.
Time-Dependent Inactivation of GABA Aminotransferase
Incubation solutions (200 μL) contained enzyme (30 μL), potassium pyrophosphate buffer (volume dependent on volume of inhibitor, 50 mM, pH 8.5), α-ketoglutarate (20 μL, 16 mM in 50 mM potassium pyrophosphate buffer, pH 8.5), 2-mercaptoethanol (2.0 mM), and various volumes of the inhibitor solutions (6, 8 and 9, 0.2M in doubly distilled H2O). The concentration of inhibitors was calculated based on a total volume of 200 μL. At time intervals, aliquots (27 μL) from the incubation solution were added to the assay solution (568 μL) with excess succinic semialdehyde dehydrogenase (SSDH). Rates were measured spectrophotometrically at 340 nm, and the logarithm of the remaining activity (percentage) was plotted against time for each concentration to determine the half-time. Then a secondary plot of half-time versus the reciprocal of the inhibitor concentration was obtained to determine the kinact and KI values.
Exhaustive Dialysis of 6, 8 and 9 Inactivated GABA-AT with Potassium Pyrophosphate Buffer
Incubation solutions (200 μL) contained enzyme (30 μL), potassium pyrophosphate buffer (147 μL, 50 mM, pH 8.5), α-ketoglutarate (20 μL, 16 mM in 50 mM potassium pyrophosphate buffer, pH 8.5), 2-mercaptoethanol (2.0 mM), and the inhibitor solutions (6, 8 and 9, 3.0 μL, 0.2 M in doubly distilled H2O). A control solution (200 μL) containing enzyme (30 μL), potassium pyrophosphate buffer (150 μL, 50 mM, pH 8.5), α-ketoglutarate (20 μL, 16 mM in 50 mM potassium pyrophosphate buffer, pH 8.5), 2-mercaptoethanol (2.0 mM), and no inhibitor solution was also prepared. The incubation solution was allowed to stand at room temperature for about 1 h to ensure most of the enzyme was inactivated. The incubation solutions and the control solution were injected into a Slide-A-Lyzer@ Dialysis Cassette (Extra Strength, 10000 MWCO, 0.5-3.0 mL capacity), respectively, and stirred in a 1 L beaker containing a stirring bar and 800 mL of 50 mM potassium pyrophosphate buffer (pH 8.5). The buffer was changed every 4 h, at which point, an aliquot was taken out to assay.
Substrate Protection against Inhibition of GABA-AT by Compounds 6, 8 and 9
Incubation solutions (200 μL) contained enzyme (30 μL), potassium pyrophosphate buffer (volumes dependent on the volume of substrate, 50 mM, pH 8.5), α-ketoglutarate (20 μL, 16 mM in 50 mM potassium pyrophosphate buffer, pH 8.5), 2-mercaptoethanol (2.0 mM), inhibitor solutions (2 μL or 0 μL in control, 6, 8 or 9, respectively in doubly distilled H2O, 0.2 M) and GABA solution [0 μL (control), 17.6 μL and 35.2 μL]. The concentrations of GABA in the incubation solutions were 0 mM (control), 3 mM, and 6 mM, respectively. At time intervals, aliquots (27 μL) from the incubation solution were added to the assay solution (568 μL) with excess succinic semialdehyde dehydrogenase (SSDH). Rates were measured spectrophotometrically at 340 nm, and the remaining activity (percentage) was determined against the control and plotted versus time to determine the t1/2.
Fluoride Ion Released During Inhibition of GABA-AT by 5-10
GABA-AT (25 μL) was incubated for 3 h with each of the six compounds 5-10 (0.8 mM) in 50 mM potassium pyrophosphate buffer, pH 8.5, containing 2.0 mM β-mercaptoethanol and 0.64 mM α-ketoglutarate in a total volume of 250 μL at 25 °C. A control containing no enzyme (25 μL of buffer was added instead) was also run. The fluoride ion concentration (200 μL) of each sample mixed with 1.00 mL of total ionic strength adjusting buffer (TISAB low level), 790 μL of potassium pyrophosphate buffer, and 10 μL of diluted standard fluoride solution (2.38 × 10−4 M), was measured with a fluoride ion electrode.
Fluoride Ion Release During Inhibition of GABA-AT by 6 and 7 with Time
GABA-AT (120 μL) was incubated with 6 or 7 (12 μL, 0.2 M in doubly distilled water) in 50 mM potassium pyrophosphate buffer, pH 8.5, containing 2.0 mM β-mercaptoethanol and 0.64 mM α-ketoglutarate in a total volume of 1.2 mL at 25 °C. Another two samples containing no GABA-AT but serum albumin or just potassium pyrophosphate buffer (control) were also run. At time intervals, 200 μL of each sample was removed and mixed with 1.00 mL of total ionic strength adjusting buffer (TISAB low level), 790 μL of potassium pyrophosphate buffer, and 10 μL of diluted standard fluoride solution (2.38 × 10−4 M). The fluoride ion concentration was measured with a fluoride ion electrode.
Supplementary Material
Acknowledgment
The authors are grateful to the National Institutes of Health (GM66132) for financial support of this research.
Abbreviations
- CAN
ceric ammonium nitrate
- DBDMH
1,3-dibromo-5,5-dimethylhydantoin
- GABA
γ-aminobutyric acid
- GABA-AT
γ-aminobutyric acid aminotransferase
- GAD
L-glutamic acid decarboxylase
- LiHMDS
lithium hexamethyldisilazide
- MFSDA
methyl fluorosulfonyldifluoroacetate
- PMBCl
p-methoxybenzyl chloride
- SSA
succinic semialdehyde dehydrogenase
- TBAF
tetrabutylammonium fluoride
- TPAP
tetra-n-propylammonium perruthenate
Footnotes
Supporting Information
Supporting Information Available: Combustion analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.McGeer EG, McGeer PL, Thompson S. GABA and Glutamate Enzymes. In: Hertz L, Kvamme E, McGeer EG, Schousboe A, editors. Glutamine, Glutamate, and GABA in the Central Nervous System. Liss; New York: 1983. pp. 3–17. [Google Scholar]
- 2.Baxter CF, Roberts E. The γ-Aminobutyric Acid-α-Ketoglutaric Acid Transaminase of Beef Brain. J. Biol. Chem. 1958;233:1135–1139. [PubMed] [Google Scholar]
- 3.Karlsson A, Fonnum F, Malthe-Sorrensen D, Storm-Mathisen J. Effect of the convulsive agent 3-mercaptopropionic acid on the levels of GABA, other amino acids and glutamate decarboxylase in different regions of the rat brain. Biochem. Pharmacol. 1974;23:3053–3061. doi: 10.1016/0006-2952(74)90281-0. [DOI] [PubMed] [Google Scholar]
- 4.Gale K. GABA in epilepsy: the pharmacologic basis. Epilepsia. 1989;30(Suppl 3):S1–S11. doi: 10.1111/j.1528-1157.1989.tb05825.x. [DOI] [PubMed] [Google Scholar]
- 5.Bakay RAE, Harris AB. Neurotransmitter, receptor and biochemical changes in mondey cortical epileptic foci. Brain Res. 1981;206:387–404. doi: 10.1016/0006-8993(81)90539-4. [DOI] [PubMed] [Google Scholar]
- 6.Loyd KG, Munari C, Bossi L, Stoeffels C, Talairach J, Morselli PL. Biochemical Evidence for the Alterations of GABA-Mediated Synaptic Transmission in Pathological Brain Tissue (Stereo EEG or Morphological Definition) from Epileptic Patients. In: Morselli PL, Loescher W, Loyd KG, editors. Neurotransmission, Seizures, Epilepsy. Raven Press; New York: 1981. pp. 325–338. [Google Scholar]
- 7.(a) Perry TL, Hansen S, Lesk D, Kloster M. Amino Acids in Plasma, Cerebrospinal Fluid, and Brain of Patients with Huntington's Chorea. Adv. Neurol. 1972;1:609–618. [Google Scholar]; (b) McGeer PL, McGeer EG. The GABA System and Function of the Basal Ganglia: Huntington's Disease. In: Roberts E, Chase TN, Tower DB, editors. GABA in Nervous System Function. Raven Press; New York: 1976. pp. 487–495. [Google Scholar]
- 8.(a) Butterworth J, Yates CM, Simpson J. Phosphate-activated glutaminase in relation to Huntington's disease and agonal state. J. Neurochem. 1983;41:440–447. doi: 10.1111/j.1471-4159.1983.tb04761.x. [DOI] [PubMed] [Google Scholar]; (b) Spokes EGS. Brain temperature after death. J. Neurosci. Meth. 1980;2:105–106. doi: 10.1016/0165-0270(80)90050-3. [DOI] [PubMed] [Google Scholar]; (c) Wu JY, Bird ED, Chen MS, Huang WM. Abnormalities of neurotransmitter enzymes in Huntington's chorea. Neurochem. Res. 1979;4:575–586. doi: 10.1007/BF00964435. [DOI] [PubMed] [Google Scholar]; (d) Iversen LL, Bird ED, Mackay AVP, Rayner CN. Analysis of glutamate decarboxylase in post-mortem brain tissue in Huntington's chorea. J. Psychiat. Res. 1974;11:255–256. doi: 10.1016/0022-3956(74)90099-5. [DOI] [PubMed] [Google Scholar]
- 9.Nishino N, Fujiwara H, Noguchi-Kuno S-A, Tanaka C. GABA receptor but not muscarinic receptor density was decreased in the brain of patients with Parkinson's disease. Jpn. J. Pharmacol. 1988;48:331–339. doi: 10.1254/jjp.48.331. [DOI] [PubMed] [Google Scholar]
- 10.(a) Maker HS, Weiss C, Weissbarth S, Silides DJ, Whetsell W. Regional activities of metabolic enzymes and glutamate decarboxylase in human brain. Ann. Neurol. 1981;10:377–383. doi: 10.1002/ana.410100410. [DOI] [PubMed] [Google Scholar]; (b) Rinne UK, Laaksonen H, Riekkinen P, Sonninen V. Brain glutamic acid decarboxylase activity in Parkinson's disease. Eur. Neurol. 1974;12:13–19. doi: 10.1159/000114599. [DOI] [PubMed] [Google Scholar]; (c) McGeer PL, McGeer EG, Wada JA, Jung E. Effects of globus pallidus lesions and Parkinson's disease on brain glutamic acid decarboxylase. Brain Res. 1971;32:425–431. doi: 10.1016/0006-8993(71)90334-9. [DOI] [PubMed] [Google Scholar]
- 11.(a) Aoyagi T, Wada T, Nagai M, Kojima F, Harada S, Takeuchi T, Takahashi H, Hirokawa K, Tsumita T. Increased γ-aminobutyrate aminotransferase activity in brain of patients with Alzheimer's disease. Chem. Pharm. Bull. 1990;38:1748–1749. doi: 10.1248/cpb.38.1748. [DOI] [PubMed] [Google Scholar]; (b) Davies P. Neurotransmitter-related enzymes in senile dementia of the Alzheimer type. Brain Res. 1979;171:319–327. doi: 10.1016/0006-8993(79)90336-6. [DOI] [PubMed] [Google Scholar]; (c) Perry EK, Gibson PH, Blessed G, Perry RH, Tomlinson BE. Neurotransmitter enzyme abnormalities in senile dementia. Choline acetyltransferase and glutamic acid decarboxylase activities in necripsy brain tissue. J. Neurol. Sci. 1977;34:247–265. doi: 10.1016/0022-510x(77)90073-9. [DOI] [PubMed] [Google Scholar]; (d) Bowen DM, White P, Flack RHA, Smith CB, Davison NA. Brain-decarboxylase activities as indices of pathological change in senile dementia. Lancet. 1974;1:1247–1249. doi: 10.1016/s0140-6736(74)90003-8. [DOI] [PubMed] [Google Scholar]; (e) Kodama K, Kaitani H, Nanba M, Kondo T, Mikame F, Yoshida H, Sato K, Yanaihara N. Neurotransmitter analogs in body fluids of patients with dementia. Shinkei Kagaku. 1981;20:496. [Google Scholar]
- 12.Gunne LM, Haeggstroem JE, Sjoequist B. Association with persistent neurolepticinduced dyskinesia of regional changes in brain GABA synthesis. Nature (London) 1984;309:347–349. doi: 10.1038/309347a0. [DOI] [PubMed] [Google Scholar]
- 13.(a) Johnston GAR, Curtis DR, Beart PM, Game CJA, McColloch RM, Twichin B. Cis- and trans-4-aminocrotonic acid as GABA analogues of restricted conformation. J. Neurochem. 1975;24:157–160. doi: 10.1111/j.1471-4159.1975.tb07642.x. [DOI] [PubMed] [Google Scholar]; (b) Schon F, Kelly JS. The Characterisation of [3H]GABA Uptake into the Satellite Glial Cells of Rat Sensory Ganglia. Brain Res. 1974;66:289–300. [Google Scholar]; (c) Johnston GAR, Stephanson AL, Twichin B. Piperazic acid and related compounds as inhibitors of GABA uptake in rat brain slices. J. Pharm. Pharmacol. 1977;29:240–241. doi: 10.1111/j.2042-7158.1977.tb11297.x. [DOI] [PubMed] [Google Scholar]; (d) Brehm L, Hjeds H, Krogsgarrd-Larsen P. The structure of muscimol, a GABA analogue of restricted conformation. Acta Chem. Scand. 1972;26:1298–1299. doi: 10.3891/acta.chem.scand.26-1298. [DOI] [PubMed] [Google Scholar]; (e) Krogsgaard-Larsen P, Johnston GAR, Lodge D, Curtis DR. A new class of GABA agonist. Nature (London) 1977;268:53–55. doi: 10.1038/268053a0. [DOI] [PubMed] [Google Scholar]; (f) Beart PM, Curtis DR, Johnston GAR. 4-aminotetrolic acid: new conformational-restricted analogue of γ-aminobutyric acid. Nature (New Biol.) 1971;234:80–81. doi: 10.1038/newbio234080a0. [DOI] [PubMed] [Google Scholar]; (g) Beart PM, Johnston GAR. GABA [γ-aminobutyric acid] uptake in rat brain slices. Inhibition by GABA analogs and by various drugs. Aust. J. Chem. 1972;25:1359–1361. [Google Scholar]
- 14.(a) Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. I and II CRC Press; Boca Raton, FL: 1988. [Google Scholar]; (b) Silverman RB. Mechanism-Based Enzyme Inactivators. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 15.Nanavati SM, Silverman RB. Design of potential anticonvulsant agents: mechanistic classification of GABA aminotransferase inactivators. J. Med. Chem. 1989;32:2413–2421. doi: 10.1021/jm00131a001. [DOI] [PubMed] [Google Scholar]
- 16.Lippert B, Metcalf BW, Jung MJ, Casara P. 4-amino-hex-5-enoic acid, a selective catalytic inhibitor of 4-aminobutyric-acid aminotransferase in mammalian brain. Eur. J. Biochem. 1977;74:441–445. doi: 10.1111/j.1432-1033.1977.tb11410.x. [DOI] [PubMed] [Google Scholar]
- 17.Loscher W. Comparative assay of anticonvulsant and toxic potencies of sixteen GABAmimetic drugs. Neuropharmacol. 1982;21:803–810. doi: 10.1016/0028-3908(82)90068-5. [DOI] [PubMed] [Google Scholar]
- 18.(a) Tassinari CA, Michelucci R, Ambrosetto G, Salvi F. Double-blind study of vigabatrin in the treatment of drug-resistant epilepsy. Arch. Neurol. 1987;44:907–910. doi: 10.1001/archneur.1987.00520210009010. [DOI] [PubMed] [Google Scholar]; (b) Browne TR, Mattson RJ, Penry JK, Smith DB, Treiman DM, Wilder BJ, Ben-Menachem E, Miketta RM, Sherry KM, Szabo GK. A multicentre study of vigabatrin for drug-resistant epilepsy. Br. J. Clin. Pharmacol. 1989;27:95S–100S. doi: 10.1111/j.1365-2125.1989.tb03468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sivenius MR, Ylinen A, Murros K, Matilainen R, Riekkinen P. Double-blind dose reduction study of vigabatrin in complex partial epilepsy. Epilepsia. 1987;28:688–692. doi: 10.1111/j.1528-1157.1987.tb03701.x. [DOI] [PubMed] [Google Scholar]
- 19.Dewey SL, Morgan AE, Ashby CR, Jr., Horan B, Kushner SA, Logan J, Volkow ND, Fowler JS, Gardner EL, Brodie JD. A novel strategy for the treatment of cocaine addiction. Synapse. 1998;30:119–129. doi: 10.1002/(SICI)1098-2396(199810)30:2<119::AID-SYN1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 20.Dewey SL, Brodie JD, Gerasimov M, Horan B, Gardner EL, Ashby CR., Jr. A pharmacologic strategy for the treatment of nicotine addiction. Synapse. 1999;31:76–86. doi: 10.1002/(SICI)1098-2396(199901)31:1<76::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Gerasimov MR, Ashby CR, Gardner EL, Mills MJ, Brodie JD, Dewey SL. Gamma-vinyl GABA inhibits methamphetamine, heroin, or ethanol-induced increases in nucleus accumbens dopamine. Synapse. 1999;34:11–19. doi: 10.1002/(SICI)1098-2396(199910)34:1<11::AID-SYN2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Kushner SA, Dewey SL, Kornetsky C. The irreversible γ-aminobutyric acid (GABA) transaminase inhibitor γ-vinyl-GABA blocks cocaine self-administration in rats. J. Pharmacol. Exp. Ther. 1999;290:797–802. [PubMed] [Google Scholar]
- 23.(a) Nanavati SM, Silverman RB. Mechanism of Inactivation of γ-Aminobutyric Acid Aminotransferase by the Antiepilepy Drug γ-Vinyl GABA (Vigabatrin) J. Am. Chem. Soc. 1991;113:9341–9349. [Google Scholar]; (b) Allan RD, Twitchin B. Synthesis of Analogues of GABA. IV Three Unsaturated Derivatives of 2-Aminocyclopentane-1-carboxylic Acid. Aust. J. Chem. 1980;33:599–604. [Google Scholar]; (c) Allan RD, Fong J. Synthesis of Analogues of GABA. XV Preparation and Resolution of Some Potent Cyclopentene and Cyclopentane Derivatives. Aust. J. Chem. 1986;39:855–864. [Google Scholar]; (d) Galeazzi R, Mobbili G, Orena M. Modeling and Synthesis of Conformationally Restricted Amino Acids. Current Organic Chemistry. 2004;8:1799–1929. [Google Scholar]
- 24.Qiu J, Pingsterhaus J, Silverman RB. Inhibition and Substrate Activity of Conformationally Rigid Vigabatrin Analogues with γ-Aminobutyric Acid Aminotransferase. J. Med. Chem. 1999;42:4725–4728. doi: 10.1021/jm990271o. [DOI] [PubMed] [Google Scholar]
- 25.Pan Y, Qiu J, Silverman RB. Design, Synthesis, and Biological Activity of a Difluoro- Substituted, conformationally Rigid Vigabatrin Analogue as a Potent γ-Aminobutyric Acid Aminotransferase Inhibitor. J. Med. Chem. 2003;46:5292–5293. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 26.(a) Park BK, Kitteringham NR, O'Neill PM. Metabolism of fluorine-containing drugs. Ann. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]; (b) Ismail FMD. Important fluorinated drugs in experimental and clinical use. J. Fluorine Chem. 2002;118:27–33. [Google Scholar]
- 27.Iseki K. Asymmetric Fluoroalkylation. In: Soloshonok VA, editor. Enantiocontrolled Synthesis of Fluoro-organic Compounds. John Wiley & Sons; New York: 2001. pp. 33–62. [Google Scholar]
- 28.Pongdee R, Liu H–W. Elucidation of Enzyme Mechanisms Using Fluorinated Substrate Analogues. Bioorganic Chemistry. 2004;32:393–437. doi: 10.1016/j.bioorg.2004.06.012. For a recent review, see: [DOI] [PubMed] [Google Scholar]
- 29.Qiu J, Silverman RB. A New Class of Conformationally Rigid Analogues of 4-Amino-5- halopentanoic Acids, Potent Inactivators of γ-Aminobutyric Acid Aminotransferase. J. Med. Chem. 2000;43:706–720. doi: 10.1021/jm9904755. [DOI] [PubMed] [Google Scholar]
- 30.Krishnamurti R, Bellew DR, Prakash GKS. Preparation of Trifluoromethyl and Other Perfluoroalkyl Compounds with (Perfluoroalkyl)trimethylsilanes. J. Org. Chem. 1991;56:984–989. [Google Scholar]
- 31.Chen Q, Wu S. Methyl (fluorosulfonyl)difluoroacetate; a new trifluoromethylating agent. J. Chem. Soc. Chem. Commun. 1989. pp. 705–706.
- 32.Natuzzi D, Daddabbo L, Stipani V, Cappello AR, Miniero DV, Capobianco L, Stipani I. Inactivation of the Reconstituted Oxoglutarate Carrier from Bovine Heart Mitochondria by Pyridoxal 5′-Phosphate. J. Bioenergetics & Biomembranes. 1999;31:535–541. doi: 10.1023/a:1026414826457. [DOI] [PubMed] [Google Scholar]
- 33.Yamazaki T, Hiraoka S, Kitazume T. Ready Carbon-Carbon Bond Formation of 2- (Trifluoromethy1)acrylate via Michael Addition Reactions. J. Org. Chem. 1994;59:5100–5103. [Google Scholar]
- 34.Koo YK, Nandi D, Silverman RB. Multiple Active Enzyme Species of γ-Aminobutyric Acid Aminotransferase are not Isozymes, but are Proteolytic Fragments. Arch. Biochem. Biophys. 2000;374:248–254. doi: 10.1006/abbi.1999.1623. [DOI] [PubMed] [Google Scholar]
- 35.Jeffery D, Weitzman PDJ, Lunt GG. An improved assay for 4-aminobutyrate:2- oxoglutarate aminotransferase. Insect Biochem. 1988;28:347–349. [Google Scholar]
- 36.Scott EM, Jakoby WB. Soluble γ-Aminobutyric-Glutamic Transaminase from Pseudomonas fluorescens. J. Biol. Chem. 1958;234:932–936. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.