

Abstract
Brain tumours are the commonest solid tumours of childhood, and pilocytic astrocytomas (PAs) are the most common central nervous system tumour in 5-19 year-olds. Little is known about the genetic alterations underlying their development. Here we describe a tandem duplication of ∼2Mb at 7q34 occurring in 66% of pilocytic astrocytomas. This rearrangement, which was not observed in a series of 244 higher-grade astrocytomas, results in an in-frame fusion gene incorporating the kinase domain of the BRAF oncogene. We further show that the resulting fusion protein has constitutive BRAF kinase activity, and is able to transform NIH3T3 cells. This is the first report of BRAF activation through rearrangement as a frequent feature in a sporadic tumor. The frequency and specificity of this change underline its potential both as a therapeutic target and a diagnostic tool.
Keywords: Pilocytic astrocytoma, BRAF, fusion, tandem duplication
Introduction
Pilocytic astrocytomas are the most common pediatric brain tumor, with peak incidence in the first decade of life (1). They are not usually infiltrating, with malignancy grade I in the World Health Organisation classification (2), and progression to higher grades is extremely rare. While gross total resection may result in a cure, recurrence is seen in up to 19% of cases (3) and both the initial tumor and subsequent treatment are associated with considerable morbidity.
Classic histology includes bipolar tumour cells, eosinophilic Rosenthal fibres and granular bodies alternating with microcystic areas comprising loosely arranged astrocyte-like cells. However, PAs can show a wide morphological spectrum, with areas resembling oligodendroglioma or higher-grade astrocytoma. Necrosis, vascular proliferation and mitotic figures do not have the same significance as in other gliomas (2), and definitive histological diagnosis can be challenging. This is particularly important when considering the treatment modalities employed to combat PAs (usually surgery with careful follow-up) versus most other gliomas (generally surgery followed by radio- and chemotherapy).
Little is known about the molecular mechanisms involved in the tumorigenesis of pilocytic astrocytomas. Cytogenetic and array-based studies have indicated grossly normal genetic complements in most cases, with whole chromosomal gains, particularly of chromosomes 5 and 7, observed in some (e.g. (4, 5)). Two recent studies have reported recurrent gains at 7q34 in PAs. The first concludes that this leads to gain of BRAF, with subsequent BRAF overexpression (6). The second reports HIPK2 as the target for amplification and overexpression (7). However, our data demonstrate that tandem duplication at this locus produces a novel oncogenic fusion gene incorporating a constitutively active BRAF kinase domain. This is the first report of a common, specific genetic alteration underlying the formation of pilocytic astrocytomas. The frequency and specificity of this change underline its potential both as a therapeutic target and a diagnostic marker.
Materials and Methods
Patients, DNA, RNA and microarray analysis
Clinicopathological data are given in Supplementary Table S1. Nucleic acid extraction and the whole-genome microarray have been described previously (5), and the array data have been deposited in GEO, accession no. GSE11263. A tiling-path array with overlapping clones covering >92% of chromosome 7 was constructed as per the whole-genome array. Clones were obtained from the Wellcome Trust Sanger Institute, Hinxton, UK. These data have also been deposited with GEO, accession no. GSE11265. An oligonucleotide array covering the ends of the duplicated region was constructed by Oxford Gene Technologies Ltd, Oxford, UK. Hybridizations and data extraction for this array were performed by the manufacturer.
5′ rapid amplification of cDNA ends, mutation screening, polymerase chain reaction and sequencing
5′ RACE was carried out with an RNA ligase-mediated kit (Invitrogen, Carlsbad, CA) as per manufacturer's instructions, with nested reverse primers (PC4588 and PC4590) towards the start of the known reference sequence. Primer sequences and conditions for polymerase chain reactions are described in Supplementary Table S2. Cycle sequencing was performed with BigDye v3.1 chemistry and run on a 3100-Avant Genetic Analyser (Applied Biosystems, Foster City, CA).
Cloning and expression constructs
Full open reading frames for long- and short-form KIAA1549:BRAF transcripts (KEx16BEx9) as well as wild-type and V600E-mutant BRAF were cloned into a pCR4 vector using a TA-cloning kit (Invitrogen). These constructs were used as templates for the addition of C-terminal HA-tags and subsequent cloning into expression vectors. A pcDNA3.1-based vector (Invitrogen) was used for the kinase assay. A pBABE-puro vector (a kind gift from Dr S Turner, University of Cambridge) was used for stable transductions.
Cell culture
Cos-7 (CRUK Cell Services, London, UK) and Phoenix cells (a kind gift from Dr S Turner) were grown in 5% CO2 in high-glucose DMEM (PAA Laboratories Ltd, Somerset, UK) supplemented with 10% FBS (PAA). For NIH3T3 cells (a kind gift from Professor Y Yuasa, Tokyo Medical and Dental University), 10% calf serum (PAA) was used in place of FBS. Cells were grown at 37°C unless otherwise stated.
Kinase assay
HA-tagged l-KIAA1549:BRAF, s-KIAA1549:BRAF, BRAFWT and BRAFV600E were transfected into Cos-7 cells using Lipofectamine 2000 (Invitrogen). Cells were harvested in RIPA buffer supplemented with Complete Protease Inhibitor Cocktail (Roche Applied Science, West Sussex, UK). Tagged protein was purified with an anti-HA immunoprecipitation kit (Sigma-Aldrich, St. Louis, MO). BRAF kinase activity was determined with a chemiluminescent kinase assay kit (Millipore (UK) Ltd, Hertfordshire, UK). Kinase activity was normalised for input protein quantity as assessed by blotting with an antibody against BRAF C-terminus (C-19, Santa Cruz Biotechnology, Santa Cruz, CA). Western blots were visualised with ECL+ reagent (GE Healthcare, Buckinghamshire, UK) using a Fujifilm LAS-4000 imager and quantified with AIDA analysis software (Fujifilm UK Ltd, Bedfordshire, UK).
Soft agarose assay
A viral packaging line (Phoenix) was transfected with s-KIAA1549:BRAF, BRAFWT, BRAFV600E, RasV12 and empty pBABE-puro vector using FuGene 6 reagent (Roche) with 4μg plasmid and 2μg helper phage per 10cm dish. At 24h post-transfection, cells were transferred to 32°C. Forty-eight hours post-transfection, supernatant was collected and filtered through a 45μm filter before being overlayed onto NIH3T3 cells. Twenty-four hours post-infection, medium was changed and cells moved back to 37°C. Mass selection with 2μg/ml puromycin (Sigma-Aldrich) was applied at 48hrs post-infection. Resistant cells were harvested, and 1×104 were mixed with 2ml 0.35% agarose in complete medium and overlayed onto 2ml 0.6% agarose in a 6-well plate. A further overlay of 2ml complete medium was changed every 72hrs. Cell growth was examined at 11 days post-plating.
Statistical analysis
Equality of survival distributions by fusion gene status was assessed by a log-rank test using SPSS v15 (SPSS Inc, Chicago, IL).
Results
Microarray copy number analysis reveals a recurrent region of gain at 7q34 in PAs
A series of forty-four well-documented pilocytic astrocytomas (no pilomyxoid variants) were investigated using large-insert clone microarrays. Clinicopathological details are given in Supplementary Table S1. Large-scale changes observed on the 1Mb whole-genome array have been described previously (5). A chromosome 7 tiling-path array was used to validate a recurrent change observed on the 1Mb array (Fig. 1A). Our analysis revealed a region of gain of ∼2Mb within 7q34, between clones RP11-355D18 and RP11-543P6, in 29/44 cases (66%, Fig. 1B). Analysis of a subset of matched blood samples with the same platform eliminated the possibility of either a copy number polymorphism or a germline alteration (data not shown). The tumour microarray data have been deposited in NCBIs Gene Expression Omnibus (GEO 1, accession numbers GSE11263 and GSE11265).
Figure 1. Identification of copy number gain at 7q34 in pilocytic astrocytomas.

A, A representative 1Mb array plot from PA2 showing gain of clone RP5-886O8 at 7q34. B, A chromosome 7 tiling-path array plot from the same tumor, showing gain of approximately 2Mb spanning 22 clones between RP11-355D18 and RP11-543P6. C & D, A custom oligonucleotide array covering the ends of the region of gain, showing a change in copy number within the KIAA1549 and BRAF genes.
Tandem duplication at 7q34 produces a novel fusion gene
The location of the gain as a tandem duplication was demonstrated by interphase fluorescence in-situ hybridization (i-FISH, Supplementary Fig. S1). To determine the ends of the duplication, a custom oligonucleotide array covering the breakpoints was constructed. This indicated a break in intron 16 of the uncharacterised gene KIAA1549, and intron 8 of BRAF (Fig. 1C,D). KIAA1549 exons are numbered according to accession no. AM989467 in the EMBL Nucleotide Sequence Database 2. The rearrangement is shown schematically in Fig. 2A. RT-PCR with primers in exon 16 of KIAA1549 (PC4578) and exon 9 of BRAF (PC4579) gave the predicted product in many tumors, but not in all cases showing gain at 7q34 by microarray analysis. Primers in more distal exons (PC4644 and PC4645) revealed three mRNA breakpoints occurring with varying frequency (Fig. 2B,C). RT-PCR confirmed the presence of a fusion transcript in all cases where 7q34 duplication was indicated by the microarrays. The most common fusion was between KIAA1549 exon 16 and BRAF exon 9 (KEx16BEx9, 20 cases), followed by KIAA1549 exon 15 - BRAF exon 9 (KEx15BEx9, 7 cases) and KIAA1549 exon 16 - BRAF exon 11 (KEx16BEx11, 2 cases). RT-PCR products were sequenced for confirmation, and verified retention of an open reading frame in all fusion transcripts (Fig. 2D, and data not shown).
Figure 2. Tandem duplication at 7q34 produces a fusion gene between KIAA1549 and BRAF.

A, A schematic diagram of the tandem duplication event observed at 7q34. Clones flanking the region of gain and the 1Mb clone from the region are indicated. Fluorescence in-situ hybridization analysis confirmed a tandem duplication (See Supplementary Fig. S1). B, RT-PCR analysis with primers in KIAA1549 exon 15 (PC4645) and BRAF exon 11 (PC4644) showing the three different mRNA fusion junctions observed (top) and a control locus in wild-type BRAF exons (bottom, expected product 214bp). PA25 does not harbour the 7q34 gain. PCR products were electrophoresed on a 1.5% agarose gel and visualised on a UV-transilluminator (UVP Ltd, Cambridge, UK). L; 100bp DNA size ladder (Invitrogen), NB; Normal brain cDNA (Ambion, Austin, TX), −ve: no template control. C, A schematic of the mRNA/proteins formed by the three different fusion products, and their frequency in our series. All three retain intact open reading frames. KD; BRAF kinase domain. D, A sequence trace confirming a fusion between KIAA1549 exon 16 and BRAF exon 9.
Identification of full-length KIAA1549 and an internal promoter producing a shorter isoform
In order to clone the KIAA1549:BRAF fusion gene, it was necessary to determine the true 5′ end of KIAA1549, since previously documented sequences (e.g. Ensembl transcript ENST00000242365 3) suggested 5′ truncation. 5′ rapid amplification of cDNA ends (5′ RACE) analysis revealed an additional exon ∼60kb upstream of the previously documented KIAA1549 exon 1. This exon was located in an Ensembl-annotated CpG island with a transcriptional start site (TSS) predicted by the Eponine TSS finder (8), and contains two start AUG codons in-frame with exon 2. The new first intron has canonical GT/AG splice donor/acceptor sites.
In addition to this, GenBank sequence XM_935390 indicated the existence of a shorter KIAA1549 transcript originating from a promoter within intron 8, with a start codon in this intronic sequence. Shorter forms of both wild-type KIAA1549 (s-KIAA1549) and KIAA1549:BRAF fusions were confirmed by RT-PCR (data not shown). The presence of stop codons in all three reading frames in the 5′UTR of the short form excludes the possibility of it being a rare splice variant of the longer form. Furthermore, an alternative splice acceptor site in intron 19 gives two variants of KIAA1549 exon 20 (ex20b being 48bp longer than ex20a). Sequences of long- and short-form wild-type KIAA1549 variants and KIAA1549:BRAF fusion transcripts have been deposited in the EMBL database, as described in Supplementary Table S3.
The novel BRAF fusion gene shows constitutive kinase activity and transforms NIH3T3 cells
The full coding sequence of the long (l-) and short (s-) forms of the most common KIAA1549:BRAF fusion (KEx16BEx9), a constitutively active mutant BRAF (V600E) and wild-type BRAF were cloned and used to transfect Cos-7 cells. The common KEx16BEx9 fusion was taken to be representative of KIAA1549:BRAF fusion as a whole. The breakpoint variants are expected to be functionally similar, since they all possess an in-frame BRAF kinase domain with a substituted amino terminus lacking the BRAF autoregulatory domain (see Discussion). Both isoforms of the fusion protein showed constitutive kinase activity at levels similar to or higher than BRAFV600E (Fig. 3A). NIH3T3 cells were then stably transduced with either empty pBABE-puro vector, HRasV12, BRAFV600E or s-KEx16BEx9. Cells with the fusion construct displayed anchorage-independent growth in soft agarose (Fig. 3B), demonstrating its transforming potential in this model system.
Figure 3. The KIAA1549:BRAF fusion gene shows constitutive kinase activity and transforms NIH3T3 cells.
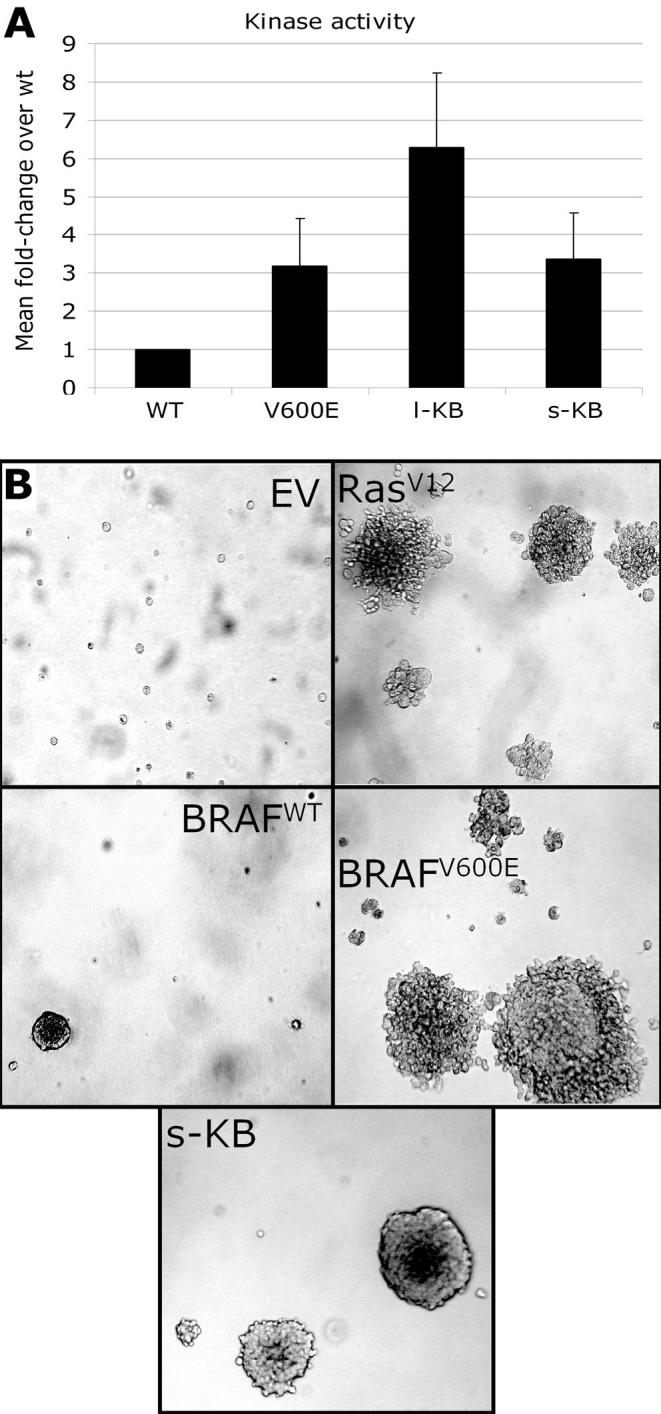
A, An in vitro BRAF kinase activity assay showing the fusion proteins to have constitutive kinase activity at a level similar to or higher than that of mutant BRAF. WT; wild-type BRAF, V600E; BRAFV600E, l-KB: long-form KIAA1549-BRAF (KEx16BEx9) fusion, s-KB: short-form KIAA1549-BRAF (KEx16BEx9) fusion. Bars show the fold increase in activity over wild-type, averaged over triplicate assays, with standard deviation indicated by error bars. Two independent transfections gave similar results. B, NIH3T3 cells transduced with pBABE-puro vector alone (EV), HRasV12 (RasV12), wild-type BRAF (WT), BRAFV600E, or short-form KIAA1549-BRAF (KEx16BEx9) fusion (s-KB) were grown in soft agarose. Active RAS, mutant BRAF and the fusion protein all display anchorage-independent growth. Photos shown are representative fields taken at 11 days after plating.
Gain at 7q34 is highly specific for pilocytic astrocytoma
This 7q34 duplication appears to be highly specific for pilocytic astrocytoma. Gain of clone RP5-886O8 on the 1Mb array was seen in 29/29 PAs known to harbour the ∼2Mb duplication (Fig. 1A), and was therefore considered an accurate marker of gain at this locus. RP5-886O8 gain was not observed in any of 60 anaplastic astrocytomas or 184 glioblastomas (data not shown). Screening of 22 grade II astrocytomas and 96 oligodendroglial or mixed astrocytic/oligodendroglial tumors showed six of these to have gain at 7q34, confirmed by tiling-path array analysis (data not shown). These cases are all children or young adults with posterior fossa lesions and extremely long survival (all greater than 12 years, see Supplementary Table S4). It is thus likely that these 6 tumors represent pilocytic astrocytomas that were originally diagnosed otherwise. However, even if these cases are not PAs, they are biologically benign gliomas, and the specificity of the change is still excellent (6 false-positives from 406 cases, specificity = 98.5%), highlighting its potential use as a molecular diagnostic marker.
The impact of KIAA1549:BRAF on survival
The effect of the fusion gene on survival was examined. At last follow-up (Sept. 2006), data was available on 37/44 cases and 35 were still alive (average follow-up 150 months; range 105 – 215 months). The two deaths occurred 5 months and 167 months post-operatively (cause unknown). The tumors from the deceased patients both showed 7q34 gain. However, there was no significant difference in survival at latest follow-up with respect to presence of the KIAA1549:BRAF fusion (OS = 91% v 100%, respectively, p=0.26, log-rank test).
Mutational analysis of BRAF and H-, K-, N-RAS
Three individual cases of pilocytic astrocytoma with a mutation in KRAS have previously been reported (9-11), and a recent report demonstrated BRAF mutation in 3/53 cases (6%, (6)). Tumors not showing 7q34 gain were screened for point mutations in BRAF (exons 11 & 15) and H-, K-, and N-RAS (exons 2 & 3) by PCR amplification and direct sequencing. Two cases (PA7 & PA25) showed a valine-to-glutamate substitution at codon 600 (BRAFV600E), a known mutational hotspot (data not shown). No mutations were observed in the RAS genes.
Discussion
Our results demonstrate a tandem duplication at 7q34 leading to a fusion between KIAA1549 and BRAF in 66% of pilocytic astrocytomas. Three different fusion breakpoints are observed, but all retain an open reading frame encoding the BRAF kinase domain, in a situation analogous to that of the BCR/ABL fusion. A recent report describes 11 tandem duplications in 2 cancer cell lines, suggesting that this class of rearrangement may be more common than previously thought (12). Our data indicate that re-evaluation of single-copy gains seen in other tumors may therefore be warranted, and further highlight the importance of activating gene rearrangements in solid tumors.
N-terminal truncation of BRAF through deletion or rearrangement leads to constitutive kinase activity (13), since the activity of wild-type BRAF is regulated by interaction of its autoregulatory N-terminal region, including the Ras-binding domain, with the kinase domain (14). Oncogenic activation of BRAF is well-documented in many tumors (15). However, this usually results from point mutation rather than gene rearrangement, with a hotspot at residue 600 (BRAFV600E; 4). An activating rearrangement of BRAF in a primary tumor was first reported in four papillary thyroid carcinomas, where an AKAP9:BRAF fusion was described in short-latency, post-Chernobyl tumors (13). Translocations involving BRAF have also been reported in two cases of large congenital melanocytic naevi (16). The present findings are thus unique in reporting BRAF activation through rearrangement as a common feature of a sporadic tumor.
Recent reports have shown that constitutive activation of BRAF can lead to oncogene-induced senescence (OIS) in benign tumours (17). Thus, OIS may contribute to the benign course and slow growth of pilocytic astrocytomas. The role of activated BRAF in regulating the mitotic spindle checkpoint is also of interest in light of reports of aneuploidy in PAs (5, 18).
The importance of RAS/RAF signalling in pilocytic astrocytomas is further supported by the increased incidence of PAs in Neurofibromatosis type 1 (NF1). Mutations of the NF1 gene in this syndrome lead to hyperactive RAS signalling and RAF activation (19). Three tumors in our series (PA19, PA41 and PA42) were diagnosed as having clinical features of NF1. None of these possessed the KIAA1549:BRAF fusion, suggesting that only one ‘hit’ on the MAPK pathway is required for PA development. The total number of cases with an identified alteration in this pathway in our series is 34/44 (77%, see Supplementary Table S1).
The identification of a recurrent, transforming genetic event in the majority of pilocytic astrocytomas represents a significant increase in our understanding of this entity. The KIAA1549:BRAF fusion is seen across all ages and in various locations in our series, including the cerebellum, ventricles, hypothalamus and optic nerve. This is in contrast with previous reports suggesting differences in genomic and/or expression signatures with respect to age and tumor location (5, 20), and may suggest a precursor cell common to most cases of PA that displays a degree of oncogene tropism.
In conclusion, due to the frequency and transforming activity of the recurrent fusion event presented here, we consider it likely that this is the initiating lesion in the majority of pilocytic astrocytomas. The prevalence and specificity of this change strongly indicate potential uses both as a diagnostic marker and in a targeted therapeutic setting.
Supplementary Material
Acknowledgements
We thank Dr M G McCabe, Dr M Dimitriadi, Mr R Chan, Miss A Goatly, Miss S Rigby, Miss F McDuff, Dr S Turner and Professor Y Yuasa for help and for reagents. We also thank the Mapping Core, Map Finishing and Microarray Facility groups of the Wellcome Trust Sanger Institute, Hinxton, UK and the Centre for Microarray resources in the Department of Pathology, University of Cambridge for assistance. This work was supported by grants from the Samantha Dickson Brain Tumour Trust, Cancer Research UK, the Jacqueline Seroussi Memorial Foundation for Cancer Research and CAMPOD.
Footnotes
References
- 1.Central Brain Tumor Registry of the United States . Statistical Report: Primary Brain Tumors in the United States, 1998-2002. CBTRUS; Chicago, IL: 2006. [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. IARC Press; Lyon, France: 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dirven CM, Mooij JJ, Molenaar WM. Cerebellar pilocytic astrocytoma: a treatment protocol based upon analysis of 73 cases and a review of the literature. Childs Nerv Syst. 1997;13:17–23. doi: 10.1007/s003810050033. [DOI] [PubMed] [Google Scholar]
- 4.Zattara-Cannoni H, Gambarelli D, Lena G, et al. Are juvenile pilocytic astrocytomas benign tumors? A cytogenetic study in 24 cases. Cancer Genet Cytogenet. 1998;104:157–60. doi: 10.1016/s0165-4608(97)00455-x. [DOI] [PubMed] [Google Scholar]
- 5.Jones DT, Ichimura K, Liu L, Pearson DM, Plant K, Collins VP. Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol. 2006;65:1049–58. doi: 10.1097/01.jnen.0000240465.33628.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008 doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshmukh H, Yeh TH, Yu J, et al. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene. 2008 doi: 10.1038/onc.2008.110. [DOI] [PubMed] [Google Scholar]
- 8.Down TA, Hubbard TJ. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002;12:458–61. doi: 10.1101/gr.216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janzarik WG, Kratz CP, Loges NT, et al. Further Evidence for a Somatic KRAS Mutation in a Pilocytic Astrocytoma. Neuropediatrics. 2007;38:61–3. doi: 10.1055/s-2007-984451. [DOI] [PubMed] [Google Scholar]
- 10.Maltzman TH, Mueller BA, Schroeder J, et al. Ras oncogene mutations in childhood brain tumors. Cancer Epidemiol Biomarkers Prev. 1997;6:239–43. [PubMed] [Google Scholar]
- 11.Sharma MK, Zehnbauer BA, Watson MA, Gutmann DH. RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology. 2005;65:1335–6. doi: 10.1212/01.wnl.0000180409.78098.d7. [DOI] [PubMed] [Google Scholar]
- 12.Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–9. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran NH, Wu X, Frost JA. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J Biol Chem. 2005;280:16244–53. doi: 10.1074/jbc.M501185200. [DOI] [PubMed] [Google Scholar]
- 15.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 16.Dessars B, De Raeve LE, El Housni H, et al. Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J Invest Dermatol. 2007;127:1468–70. doi: 10.1038/sj.jid.5700725. [DOI] [PubMed] [Google Scholar]
- 17.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 18.Cui Y, Guadagno TM. B-Raf(V600E) signaling deregulates the mitotic spindle checkpoint through stabilizing Mps1 levels in melanoma cells. Oncogene. 2008;27:3122–33. doi: 10.1038/sj.onc.1210972. [DOI] [PubMed] [Google Scholar]
- 19.Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–16. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma MK, Mansur DB, Reifenberger G, et al. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res. 2007;67:890–900. doi: 10.1158/0008-5472.CAN-06-0973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.