

Abstract
LC-MS/MS has emerged as the method of choice for the identification and quantification of protein sample mixtures. For very complex samples such as complete proteomes, the most commonly used LC-MS/MS method, data-dependent acquisition (DDA) precursor selection, is of limited utility. The limited scan speed of current mass spectrometers along with the highly redundant selection of the most intense precursor ions generates a bias in the pool of identified proteins toward those of higher abundance. A directed LC-MS/MS approach that alleviates the limitations of DDA precursor ion selection by decoupling peak detection and sequencing of selected precursor ions is presented. In the first stage of the strategy, all detectable peptide ion signals are extracted from high resolution LC-MS feature maps or aligned sets of feature maps. The selected features or a subset thereof are subsequently sequenced in sequential, non-redundant directed LC-MS/MS experiments, and the MS/MS data are mapped back to the original LC-MS feature map in a fully automated manner. The strategy, implemented on an LTQ-FT MS platform, allowed the specific sequencing of 2,000 features per analysis and enabled the identification of more than 1,600 phosphorylation sites using a single reversed phase separation dimension without the need for time-consuming prefractionation steps. Compared with conventional DDA LC-MS/MS experiments, a substantially higher number of peptides could be identified from a sample, and this increase was more pronounced for low intensity precursor ions.
Over the past decade, MS has emerged as the method of choice for the identification and quantification of proteins in very complex biological samples (1). In the most widely used implementation, referred to as shotgun proteomics, protein samples are first digested, and the resulting peptide mixtures are then chromatographically separated and finally sequenced by automated MS/MS. Because of its conceptual and experimental simplicity, the shotgun approach has become a very popular method for the identification of proteins in a wide range of biological samples and, in combination with stable isotope labeling, for quantitative proteomics studies (2–4). Recent technical improvements in MS instrumentation, database searching, and result validation as well as advances in database annotation now make it possible to routinely identify hundreds to a few thousands of proteins in complex biological samples (5–8).
Despite this impressive progress, shotgun proteomics is not yet capable of characterizing whole proteomes and presents obvious biases, among them the discrimination against protein species of low abundance (5, 8). This is primarily a consequence of limited sequencing speed of current LC-ESI-MS/MS systems that are incapable of analyzing each precursor ion detected in complex samples together with the redundant selection of a subset of precursor ions even if precautions like dynamic exclusion are applied. Therefore, even in repeat analyses of the same sample exhaustive identification of the low intensity precursors is not achieved (9–11).
In contrast to these approaches based on data-dependent acquisition (DDA)1 precursor ion selection, directed peptide sequencing provides the advantage of focusing the MS/MS analysis on non-redundant and information-rich precursor ions, thereby better managing the analysis time and increasing the depth of analysis (12, 13). In this regard, a two-stage strategy by which all MS1 features that represent peptides are extracted from LC-MS maps and subsequently subjected to targeted sequencing, in principle, should lead to the identification of all detectable precursors (14). Because the acquisition of MS1 and MS2 spectra is naturally decoupled in MALDI-MS/MS, this platform is well suited for directed sequencing and has been applied to selectively analyze differential expression or modifications of proteins (15, 16).
The same principle is also applicable to ESI-MS, which has the potential to provide much higher sequencing speed in routine applications compared with MALDI-MS/MS. Because the peptides are not “immobilized” on the sample plate, repeat injections of the same sample are required: the first to detect the MS1 features and subsequent ones for directed sequencing. Naturally the decoupling of feature detection and sequencing demands highly reproducible elution times and high mass accuracy. A directed sequencing strategy has already been applied on high mass accuracy ESI instruments to specifically sequence peptides of single proteins in complex mixtures (17) and for the detection of low abundance peptide species (18). However, the number of targeted peptide ion masses was limited to a few hundred per run, a much lower number of sequencing attempts than modern ESI-MS/MS instruments are capable of performing in DDA mode. The number of targeted precursors could only be increased by sample consuming and time consuming multiple LC-MS/MS analyses of the same mixture.
In the present study, a high performance LTQ-FT-ICR mass spectrometer that allows segmentation of inclusion mass lists by LC elution time was used for directed sequencing. Each inclusion lists contained the m/z and elution time of the targeted precursors and was divided into segments of 3–5 min, thereby increasing the number of possible target masses to 3,000 in a 1-hour LC gradient. The strategy was supported by software tools to (i) automatically extract peptide features from MS1 maps and to align features over multiple LC-MS/MS patterns (13), (ii) generate inclusion lists from the identified features,2 (iii) control directed sequencing of the features on the inclusion list by sequential LC-MS/MS analyses of the same sample, and (iv) map the MS/MS data obtained back to the initial feature list. The potential of the directed MS/MS approach was evaluated by in-depth characterization of complex peptide and phosphopeptide mixtures obtained from Drosophila melanogaster lysates. The data demonstrated the high specificity and reproducibility of the method to identify a higher number of peptides from the same sample in a lower number of LC-MS/MS runs compared with standard DDA LC-MS/MS analysis, specifically in the class of low intensity precursor ions.
MATERIALS AND METHODS
Cell Culture and Phosphopeptide Enrichment—
All chemicals, if not otherwise mentioned, were bought at the highest available purity from Sigma-Aldrich.
Cell Culture, Lysis, and Protein Digestion—
D. melanogaster Kc167 cells were grown in Schneiders Drosophila medium (Invitrogen) supplemented with 10% fetal calf serum, 100 units of penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) in an incubator at 25 °C. To increase the degree of phosphorylation in the Drosophila proteins different batches of cells were pooled that were either growing in rich medium, growing in serum-starved medium, treated for 30 min with 100 nm rapamycin (LC Laboratories, Woburn, MA), treated for 30 min with 100 nm insulin, or treated for 30 min with 100 nm calyculin A. Then the cells were washed with ice-cold PBS and resuspended in ice-cold lysis buffer containing 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, and a protease inhibitor mixture (Roche Applied Science). To preserve protein phosphorylation, several phosphatase inhibitors were added to a final concentration of 20 nm calyculin A, 200 nm okadaic acid, 4.8 μm cypermethrin (all bought from Merck KGaA), 2 mm vanadate, 10 mm sodium pyrophosphate, 10 mm NaF, and 5 mm EDTA, respectively. After a 10-min incubation on ice, cells were lysed by homogenization in a Dounce homogenizer. Cell debris and nuclei were removed by centrifugation for 10 min at 4 °C at 5,500 × g. Then the cytoplasmic and membrane fractions were separated by ultracentrifugation at 100,000 × g for 60 min at 4 °C. The proteins of the cytosolic fraction (supernatant) were subjected to acetone precipitation. The protein pellets were resolubilized in 3 mm EDTA, 20 mm Tris-HCl, pH 8.3, and 8 m urea. The disulfide bonds of the proteins were reduced with tris(2-carboxyethyl)phosphine at a final concentration of 12.5 mm at 37 °C for 1 h. The produced free thiols were alkylated with 40 mm iodoacetamide at room temperature for 1 h. The solution was diluted with 20 mm Tris-HCl (pH 8.3) to a final concentration of 1.0 m urea and digested with sequencing grade modified trypsin (Promega, Madison, Wisconsin) at 20 μg/mg of protein overnight at 37 °C. Peptides were desalted on a C18 Sep-Pak cartridge (Waters) and dried in a SpeedVac. Finally 1 μg of peptide sample was utilized for each LC-MS/MS experiment.
Phosphopeptide Isolation—
Phosphopeptides were isolated using TiO2 affinity enrichment as described recently (20). 1 μg of the phosphopeptide sample was subjected to each LC-MS/MS analysis.
Reversed Phase HPLC—
Peptide samples were analyzed on an Agilent 1100 microflow system (Agilent Technologies) connected to a 7-tesla Finnigan LTQ-FT-ICR instrument (Thermo Electron, Bremen, Germany) equipped with a nanoelectrospray ion source (Thermo Electron). Peptides were separated on an RP HPLC column (150 μm × 15 cm) packed in house with C18 resin (Magic C18 AQ 5 μm; Michrom BioResources, Auburn, CA) using a linear gradient from 98% solvent A (0.15% formic acid) and 2% solvent B (98% acetonitrile, 2% water, and 0.15% formic acid) to 30% solvent B over 60 min (for cytosol digest) and 90 min (for phosphopeptide-enriched samples) at a flow rate of 1.2 μl/min.
Mass Spectrometry—
In DDA mode, each MS1 scan (acquired in the ICR cell) was followed by CID (acquired in the LTQ part) of the three (for feature extraction) and five (for comparison of DDA and directed LC-MS/MS) most abundant precursor ions with dynamic exclusion for 30 s. Only MS1 signals exceeding 150 counts were allowed to trigger MS2 scans with wide band activation enabled. Total cycle time was ∼1–1.5 s. For MS1, 106 ions were accumulated in the ICR cell over a maximum time of 500 ms and scanned at a resolution of 100,000 full-width at half-maximum (at 400 m/z). MS2 and MS3 spectra were acquired using the normal scan mode, a target setting of 104 ions, and an accumulation time of 250 ms. Singly charged ions and ions with unassigned charge state were excluded from triggering MS2 events. The normalized collision energy was set to 30%, and one microscan was acquired for each spectrum. For phosphopeptide analysis, the mass spectrometer automatically switched between MS, MS2, and neutral loss-dependent MS3 acquisition. Data-dependent settings were chosen to trigger an MS3 scan when a neutral loss of 97.97, 48.99, 32.66, 24.5, or 19.6 Da was detected among the 10 most intense fragment ions.
Peak Detection—
First the data of the initial three LC-MS (mapping) runs (raw format) was converted to the profile mzXML format (21). Then the in-house developed software system SuperHirn (22) was used for (i) detection, (ii) deisotoping, (iii) peak integration, and (iv) alignment of detected features over multiple LC-MS patterns. Peak intensities were measured by calculating peak areas from extracted ion chromatograms of each MS signal. Highly stringent criteria were applied to filter the detected peaks for peptide signals. Specifically the algorithm searches for peak patterns matching isotope distributions typical for peptides within the m/z value range under investigation. Peaks had to be detected in at least two subsequent MS1 scans and with a minimum of three isotopic peaks to be considered. Only peaks that could be found in at least two LC-MS runs were considered, and singly charged masses were excluded. Finally a list of the relevant features was generated and used to build mass inclusion lists for directed MS sequencing.
Generation of Inclusion Lists—
To make the generation of inclusion lists less time-consuming, less prone to human error, and easier to reproduce we developed the “Inclusion List Builder” software. The Inclusion List Builder has a rich graphical user interface and is implemented as a plug-in for the Prequips platform2 (Seattle Proteome Center).
The table containing all features extracted from initial LC-MS runs is imported through the Prequips data provider interface and converted into a so-called “master table” by the Inclusion List Builder. The master table contains the m/z ratios, retention times, averaged peak areas, and charge states for all features identified by the SuperHirn algorithm. Through interactive application of filters to feature attributes, inclusion lists are created as subsets of the master table and segmented by retention time. This is necessary because the number of features present in the table usually exceeds the number of possible sequencing cycles that the mass spectrometer can acquire in a single run. After segmentation the inclusion lists are exported as tables (.csv file format) that can be read by the MS instrument software.
In the study presented here, the following software settings were used for the directed analysis of the peptide mixtures. The 9,680 features extracted from the three map runs analyzing the Drosophila lysate digest were split by their intensity into five bins, each consisting of 2,000 masses (1,680 for the last bin containing the least intense features) using the following average peak area thresholds: very high, 2.6 × 109–3 × 107; high, 3.0-1.4 × 107; medium, 1.4 × 107–8.1 × 106; low, 8.1-4.9 × 106; and very low, 4.9 × 106–5.3 × 105. Each subset was further clustered into 3- or 5-min segments using the following elution time values: 0, 27.5, 32.5, 37.5, 40.5, 43.5, 46.5, 49.5, 52.5, 55.5, 58.5, 61.5, 66.5, 71.5, and 80 min. The start and stop time of each 5- (or 3)-min time segment was extended by 2.4 (1.4) and 2.5 (1.5) min, respectively, to compensate for variations in retention time. Because the instrument software requires a few seconds to load/delete the new/old masses in each time segment, a delay of 6 s was implemented for each start time. For directed analysis of the phosphopeptide-enriched sample (90-min gradient), the features were clustered using the following elution time bins: 0, 22.5, 27.5, 32.5, 37.5, 42.5, 47.5, 52.5, 57.5, 62.5, 67.5, 72.5, 77.5, 82.5, 87.5, 95, and 110 min. In summary, this setup allowed, assuming equal distribution, the inclusion of up to 3,000 features in a single directed LC-MS/MS analysis using a 60-min gradient. It is important to note that applying shorter time segments could further increase this number.
Directed MS Sequencing of Features—
The generated inclusion lists (.csv file format) were directly imported into the global mass list parent ion table of the MS operating software (Xcalibur 2.0 SR1, Thermo Electron) and activated. Basically the settings for targeted LC-MS/MS were similar to those described above with a few modifications. First the dynamic exclusion mass window that is also setting the m/z tolerance for the inclusion list masses was narrowed to ±10 ppm for all directed analyses with enabled monoisotopic precursor selection and to ±5 ppm when this option was turned off. Ion signals for which no charge could be assigned were also allowed to trigger MS2 scans. The dynamic exclusion time was reduced to 10 s to acquire multiple MS2 scans for each feature. For directed sequencing in preview off mode, this option was disabled, and the resolution of the MS1 scan in the ICR cell was reduced to 50,000. For sequencing features of low abundance, the monoisotopic precursor selection option was disabled, and to minimize unspecific sequencing, the threshold required for triggering MS2 events was raised from 150 to 3,000 counts.
Database Searching—
All acquired MS2 and MS3 spectra were searched against the Drosophila Flybase protein database (D. melanogaster, release 4.3; March 2006; 19,645 entries), which also contained the protein sequences of bovine trypsin and human keratins, using the Bioworks (Version 3.2) software (Thermo Electron, San Jose, CA). The search criteria were as follows: full tryptic specificity was required (cleavage after lysine or arginine residues unless followed by proline); two missed cleavages were allowed; carbamidomethylation (Cys) was set as fixed modification; oxidation (Met) and, if required, phosphorylation (Ser/Thr/Tyr) were applied as variable modifications; and mass tolerance of the precursor ion and the fragment ions was 10 ppm and 0.8 Da, respectively. In addition to this, each data set was searched against a decoy Flybase protein database (Version 4.3) as described previously to assess the number of false positive peptide identifications (23). Based on this approach, the error rate was set to a maximum of 1% (error rate = 2 × percentage of decoy hits) using the following thresholds: peptide probability <0.01 and final score >0.2 for unphosphorylated and >0.4 for phosphorylated peptides. For each peptide sequence identified, all matching gene numbers (Flybase gene ID (FBgn)) and protein accession entries (computed gene (CG)) were determined and displayed. In addition to this, Occam's razor logic as implemented in Protein Prophet (24) was applied to calculate the number of identified proteins (CG entries). In brief, redundant protein entries were removed by clustering peptides matching to multiple members of a protein family to a single protein group and considered as a single identification. Furthermore when multiple proteins shared a peptide sequence, it was only assigned to the protein identified with the highest number of peptide assignments. In-house software was used for the calculation of non-redundant phosphorylation sites present in the data sets obtained as recently reported (25).
For comprehensive intensity comparison of the peptides identified by DDA and directed LC-MS/MS analysis (see Fig. 3B), the MS1 signal intensities were determined using the Bioworks software to also include peptides exclusively identified in DDA mode. Therefore, the peak height of every single peptide precursor ion was determined by applying the following parameters: mass range of 0.03 Da, intensity threshold of minimal 1,000, and three smoothing points allowed. The intensity of the same peptide ions detected in multiple runs was averaged.
Fig. 3.

Comparison of data-dependent and directed LC-MS/MS analysis. A, unique peptide identifications of the cytosolic digest from the D. melanogaster Kc167 cell line obtained with multiple data-dependent LC-MS/MS experiments (DDA, blue line), the optimized directed approach described in Fig. 2 (INL, orange line), and a combination of both methods (Combined, green line). Here two DDA LC-MS/MS experiments were carried out, and only extracted features without MS2 data were subjected to two additional directed LC-MS/MS analyses. B, base peak intensity distribution of the peptide precursor ions identified using either DDA mode (DDA, blue line) or the combined approach shown in A (Combined, green line). The abundance profile of the peptides exclusively detected with the two directed LC-MS/MS runs in the combined approach is also indicated (INL (from combined), red line).
Assignment of Identified Peptides to Inclusion Lists—
An Excel table (in tab-delimited format) containing the search results from the directed LC-MS/MS runs was imported into the Prequips software. Spectral data in mzXML format was loaded to obtain access to the corresponding retention times. All identified peptides were mapped to one or more features in the master table through the MS2 scans associated with them.
For the mapping algorithm we define a scan as the triple (corrected retention time t′R, feature at mass m, and charge z) where the feature mass m is defined as follows.
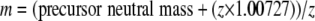 |
(Eq. 1) |
The corrected retention time t′R is computed as
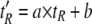 |
(Eq. 2) |
from the measured retention time tR. Because all LC-MS/MS experiments were performed using the same C18 column resulting in minimal shifts in feature elution times, no corrections of tR were required. Therefore, a was set to 1, and the value for b was set to 0. The following algorithm was applied for each identified peptide p.
For every scan s associated with p find all features f in the master table that fulfill each of the following conditions: t′R(s) − τ ≤ t′R(f) ≤ t′R(s) + τ, m(s) − μ ≤ m(f) ≤ m(s) + μ, and z(s) = z(f). The retention time tolerance τ (in min) and the mass tolerance μ (in ppm) define windows of 2τ and 2μ centered on t′R(s) and m(s), respectively.
Map p to all f fulfilling the conditions.
A single peptide can be mapped to the same feature multiple times if it is associated with more than one scan. Also more than one distinct peptide can be mapped to a feature. The peptide with the lowest retention time and mass difference is automatically selected as the best hit. The investigator can manually assign any other of the peptides mapped as the best hit for a given feature if there is supporting evidence.
For mapping identified features back to the initial inclusion list, the theoretical m/z values and the elution time of the identified peptides were matched with the precursor masses and elution times in the inclusion list. A mass tolerance of 0.01 Da and a time tolerance of 1 min were allowed. Because no precolumn was used during LC, very hydrophilic peptides, which were not or only partially retained by the C18 resin, showed very inconsistent retention times. Therefore, all peptides having LC retention times of less than 30 min were matched only by their m/z and charge values.
RESULTS
The Directed Precursor Ion Selection Work Flow—
The general work flow of the directed LC-MS/MS analysis described in this study is outlined in Fig. 1. It consists of the following steps.
Fig. 1.

Work flow of directed LC-MS/MS analysis. Aliquots of a complex peptide mixture are analyzed by LC-MS/MS, and all MS1 peaks are extracted. Subsequently subsets of features are selected and specifically sequenced by directed LC-MS/MS until MS/MS data are obtained for all detected features (see text for details).
Sample preparation. The proteins in a sample obtained from any source (cells, tissue, body fluids, etc.) are reduced, alkylated, and enzymatically cleaved with trypsin.
Feature detection. Sample aliquots are then analyzed by high performance LC-MS/MS whereby the MS1 features are recorded, and selected precursor ions are picked for CID by DDA. Usually we performed three independent LC-MS/MS runs, each consuming 1 μg of total peptide mass. The data obtained are subsequently analyzed off line by the in-house software tool SuperHirn (13) that automatically extracts all detectable MS1 signals from the individual LC-MS patterns by applying peak detection, noise reduction, deisotoping, and intensity determination algorithms to each detected peak. The detected ions are then aligned within the respective patterns.
Master table generation. The software overlays over all detected peaks an isotope distribution template that is typical for peptides, thereby discriminating between peptide and contaminant signals, and finally generates a master table of all masses that are very likely to be peptide ions. Each of these features is described by its m/z ratio, charge state, intensity, and chromatographic retention time. All features are then imported into a second in-house software tool, Prequips, that supports data filtering, generation of inclusion list files, and mapping MS2 data back to the respective MS1 features in an automated manner.2 The alignment of features over multiple patterns was a very effective method to reduce noise or signals from non-peptidic material if only MS1 signals that are found in at least two of the three replicate LC-MS runs were considered for further analysis. In the end, around 10,000 features are detected in a typical experiment.
Inclusion list creation. Because for most samples the number of features detected exceeded the number of available sequencing cycles, the feature list was further divided according to the sequencing speed of the MS instrument used. For the LTQ-FT-ICR instrument used in our study, a list of 10,000 features was divided into five LC-MS/MS inclusion lists of 2,000 features each. In general, the features were grouped into segments of 5 min (or 3 min for highly populated areas) and flanked by 2.5 (1.5) min extra time, respectively, resulting in segments of 10 (6) min, respectively. Hence each feature could tolerate a shift in retention time during LC of at least 2.5 (1.5) min. Initial tests showed that the current instrument control software (Xcalibur 2.0 SR1) limited the number of ions on an inclusion list to a maximum of 500. Thus the construction of the inclusion list required that the number of features from two overlapping segments did not exceed 500. In addition the lower number of features early and late in the gradient allowed the generation of longer segments with a higher time tolerance in those regions. The long time segment at the beginning is essential for targeting hydrophilic peptides because these molecules do not, or only weakly, bind to the RP LC column and consequently showed considerable shifts in elution times between different runs. For samples that showed a common feature distribution, a maximum of 3,000 features could be sequenced per hour (∼1 feature/s). It is worth mentioning that this number could be further increased by a further reduction of the time segments.
Feature identification. The MS2 data obtained from the directed LC-MS/MS analyses were searched against protein databases and, together with the corresponding mzXML files, mapped back to the master table using the Prequips software.
Reproducibility of LC-MS—
The success of an inclusion list experiment strongly depended on the reproducibility of both LC and MS performances. Supplemental Fig. S1 shows that variations in retention time were below 20 s, and mass accuracy was better than 2 ppm. These variations are well within the time and m/z tolerances of 2.5 min and 10 ppm, respectively, used for directed MS2. Naturally the time and m/z tolerances should be kept as small as possible to minimize random MS sequencing events.
Optimization of MS Parameters for Improved Identification of Features—
To optimize the number of features identified in a targeted precursor ion selection experiment we generated a peptide mixture from D. melanogaster Kc167 cells and subjected aliquots to repeat analyses in which relevant experimental parameters were varied.
Cells (108) were harvested and disrupted, and the cytosolic fraction was isolated. The proteins were alkylated and trypsinized, and three aliquots, each containing 1 μg of peptides, were analyzed as described above (Fig. 1). A total of 9,680 unique features could be detected (see supplemental Table S3) that were imported into the master table. The features were grouped by decreasing intensity into five inclusion lists, each containing around 2,000 features, and subjected to directed LC-MS/MS analysis.
Subsequently the influence of two important parameters for triggering MS2 in the LTQ-FT mass spectrometer used, the preview mode and the monoisotopic precursor selection (MPS), respectively, was evaluated. With preview mode on (default mode), the instrument acquires a low resolution MS1 spectrum (prescan) that takes around 20% of the total scan time and continues scanning the ions for high resolution while the LTQ is performing MS2 scans of selected ions in parallel (Fig. 2C). This significantly increases MS2 scan rates, but peptide ions for MS2 are selected from low resolution MS1 scans. By disabling the preview mode and reducing the resolution to 50,000 for MS1, the mass accuracy of the peptide ions selected on line for directed MS2 could be improved. Consequently 8% (13%) more features were identified (sequenced) from the Kc167 cell sample from the five inclusion lists with preview mode disabled (Fig. 2A and supplemental Tables S1, S4, and S5). Despite the slightly longer cycle times required (Fig. 2D), the speed of the MS instrument used was still sufficient to selectively sequence up to 2,000 features in a 60-min gradient (supplemental Table S1). It is important to note that the improvements were particularly pronounced for features with lower intensities. Additionally the MS2 selection efficiency of peptide ions of low abundance could be further improved by disabling MPS, which makes every single isotopic peak in the MS1 scans accessible for triggering MS2. As a consequence, the yield of identified low abundance peptide signals could be further improved from 33 to 40% and 26 to 35% for low and very low intensity features, respectively (Fig. 2A).
Fig. 2.

Evaluation of different MS conditions for improved feature sequencing. The 9,680 features extracted from the cytosolic digest from the D. melanogaster Kc167 cell line were split into five bins based on their intensity, each containing around 2,000 masses, and subjected to directed LC-MS/MS using an LTQ-FT mass spectrometer. The percentage of sequenced (A) and identified (B) features in each experiment using different MS parameters is shown. C, parallel acquisition of a high resolution MS1 scan in the ICR cell at a resolution of 100,000 and three MS2 spectra in the LTQ in preview on mode. D, sequential MS1 and MS2 data acquisition using a lower resolution in the ICR cell (50,000) in preview off mode. The time required for each MS cycle is indicated. Additional experiments for features with low and very low intensity were performed using the preview off mode and disabling monoisotopic precursor selection (Preview + MPS OFF).
It is worth mentioning that increasing the number of ions into the ICR cell for MS1 scans also slightly increased the numbers of identified low abundance ions, however, to a lower extent than by disabling MPS (data not shown). This is due to the fact that high ion numbers in the ICR cell lead to space charge effects that lower peptide mass accuracy (26, 27). Therefore, some features might fall out of the m/z tolerances applied and would not be sequenced if the ICR cell is filled with a high number of ions.
Using the optimized MS parameters, around 80% of all detected features were selected for sequencing, and 48% of those could be confidently identified, resulting in the identification of 3,931 unique peptides and 793 non-redundant gene products (supplemental Tables S1 and S6). In addition, the occurrence of random sequencing of MS signals not selected for directed MS2 was very low because only about 11.6% (516 peptides) of all identified peptides in the five inclusion list runs could not be mapped back to the master feature list. This demonstrates that the whole process starting with feature extraction from high resolution MS1 maps followed by directed sequencing using an inclusion list protocol and mapping the MS2 data back to the features is very effective and specific.
Comparison of Data-dependent and Directed LC-MS/MS—
To assess the performance of the directed LC-MS/MS approach in comparison with CID experiments using DDA, we compared the data obtained from applying the protocols optimized as described above with the results of five repeat LC-MS/MS runs using DDA. The sample was a tryptic digest of Kc167 cell cytosolic fraction, and in each analysis 1 μg of total peptide mass was injected. To increase the number of MS2 spectra acquired, the instrument was programmed to randomly select for CID the five most intense precursor ion signals detected in a survey scan in the LC-MS/MS experiment with DDA, and the peptides identified by either strategy were compared.
As shown in Fig. 3A (blue line), 2,493 unique peptides were identified in the first DDA LC-MS/MS run, but then the contribution of additional LC-MS/MS runs of the same sample to the overall number of peptides decreased rapidly, eventually identifying only 1,083 (43.4%) additional peptides in the four subsequent LC-MS/MS runs (supplemental Tables S2 and S7). This effect was even more apparent at the protein level where only 111 (21.0%) new gene products could be discovered by the additional four LC-MS/MS experiments. Notably the number of identified peptides over all five DDA runs was highly consistent, ranging from 2,461 to 2,493 (supplemental Tables S2 and S7). This clearly demonstrates a strong sequencing redundancy in shotgun LC-MS/MS in agreement with observations obtained from other recent studies (9–11).
In contrast, the directed approach presented here allowed the non-redundant sequencing of every single feature in only one LC-MS/MS run, and therefore, the degree of novel peptide identifications in each analysis was much higher, resulting in a much steeper curve (Fig. 3A, orange line). Here using the optimized MS parameters shown above (preview mode and MPS disabled; Fig. 2), 2,746 (237%) additional peptides could be determined after the first directed LC-MS/MS run, resulting in a total of 3,931 unique peptide identifications. Using non-redundant sequencing also allowed the identification of a significant number of additional proteins in each directed run compared with DDA LC-MS/MS (supplemental Table S1). Eventually a higher number of peptides and proteins were identified in five directed runs than in five DDA LC-MS/MS analyses despite the much lower number of sequencing events, clearly indicating the higher information content of the ions extracted off line over DDA LC-MS/MS (supplemental Tables S1 and S2). Interestingly the INL curve (Fig. 3A) is only slightly flattening with decreased feature signal intensity, which suggests that potentially even more peptides can be identified by the directed approach if less stringent peak picking criteria were applied and additional inclusion list runs were carried out on the same sample.
It is important to point out that current mass spectrometers offer possibilities to sequence additional low abundance peptides in DDA mode (28). However, most of them drastically reduce the number of MS1 scans acquired and therefore lower reliable quantification of complex peptide mixtures. Conversely directed LC-MS/MS enables the identification of low abundance peptides without affecting MS1 performance and thus quantification accuracy.
Combination of DDA and Directed LC-MS/MS—
As shown in Fig. 3A, three inclusion lists were necessary to identify as many peptides/proteins as in a single DDA LC-MS/MS experiment. Therefore, excluding already selected features from the initial LC-MS run for the generation of MS1 maps and specifically targeting the remaining features would be expected to reduce the number of directed LC-MS/MS runs required to sequence every detectable feature. We applied a combined strategy of two initial DDA runs with subsequent directed runs to the Kc167 cell sample described above. As shown in Fig. 3A, the results of more than 5,000 sequencing attempts of the first two DDA LC-MS/MS analyses runs could be mapped back to the feature list, thereby reducing the number of features from 9,680 to 4,323 that were specifically targeted in two additional LC-MS/MS experiments. The first directed run already identified as many novel peptides as the three additional DDA runs together, and eventually 4,273 unique peptides corresponding to 804 different proteins could be identified after only four LC-MS/MS runs (Fig. 3A, green line, and supplemental Table S8). In total, a higher number of peptides could be identified with less analytical effort and time by the combined approach than by the DDA or directed approach alone. Notably the high performance hybrid LTQ-FT mass spectrometer used for our experiments is very well suited for the combined approach because the ICR cell enables the acquisition of high resolution MS1 data while, in parallel, the LTQ part can collect MS2 data without sacrificing time, resolution, or sensitivity for MS1 data acquisition (Fig. 2C).
To evaluate the intensity distribution of the peptides identified by both DDA alone (Fig. 3A, blue line) and in combination with directed (Fig. 3A, green line) LC-MS/MS, the precursor ion abundances of these peptides were calculated and compared. To obtain an unbiased intensity determination of all identified precursor ions, their base peak intensities were calculated using the Bioworks software tool. As shown in Fig. 3B, most of the peptide ions identified in DDA mode had intensities between 106 and 2 × 106 counts (Fig. 3B, blue line). By contrast, the peak maximum of the distribution of peptides exclusively identified by the two inclusion list runs is clearly shifted toward precursor ions with lower abundance. The apex of the distribution of the peptides determined by the inclusion list in the combined approach was around 2 × 105–4 × 105 and thus about 5 times less intense than that of the DDA alone strategy (Fig. 3B, red line). In combination with the data obtained from the first two DDA runs, the intensity of peptides determined by the combined approach was much less biased toward high abundance signals and more equally distributed (Fig. 3B, green line). This clearly indicates that in complex peptide mixtures a higher number of peptide ions with low intensities could be identified by directed than by DDA LC-MS/MS approaches.
Reproducibility of Directed LC-MS/MS Sequencing—
To evaluate the reproducibility of the directed approach for both feature selection and identification, 200 identified features from each of the five intensity groups (very high to very low) were combined and reanalyzed five times. Overall the selected features spanned more than 3 orders of magnitude in signal intensity. The data are shown in Table I and supplemental Fig. S2. Of the 1,000 features targeted, more than 85% did trigger sequence attempts, and around 70% could be confidently identified in all five LC-MS/MS runs. Furthermore of the 1,000 features on the respective inclusion list, 984 did trigger an MS2 scan event, and 945 could be assigned to the correct peptide sequence in at least one run. Because the directed approach requires each feature to be detected on line, the selection of features for sequencing strongly depends on their signal intensity. Whereas all of the 200 features in the group with the highest signal intensities were selected for MS2 in all five repeat LC experiments, that number decreased to 148 (74%) in the group with the lowest signal intensities (supplemental Fig. S2B). A similar trend was observable for the number of correctly identified peptides albeit with a steeper decline at lower feature intensities. However, the MS2 spectra obtained can be useful for side-by-side comparison of correctly assigned spectra to confirm the feature identity (29) even if the spectral quality is not sufficient for the assignment of a peptide sequence by a database search engine.
Table I.
Reproducibility of directed LC-MS/MS
| No. of observations | Feature intensitya | No. of features targetedb | No. of features sequenced | No. of features identified |
|---|---|---|---|---|
| 1 | All | 1,000 | 984 | 945 |
| 2 | All | 1,000 | 972 | 906 |
| 3 | All | 1,000 | 953 | 863 |
| 4 | All | 1,000 | 923 | 803 |
| 5 | All | 1,000 | 865 | 695 |
| 5 | Very high | 200 | 200 | 192 |
| 5 | High | 200 | 186 | 164 |
| 5 | Medium | 200 | 170 | 129 |
| 5 | Low | 200 | 161 | 117 |
| 5 | Very low | 200 | 148 | 93 |
Feature intensity bins based on feature area determined by SuperHirn (very high, 2.6 × 109–3 × 107; high, 3.0-1.4 × 107; medium, 1.4 × 107–8.1 × 106; low, 8.1-4.9 × 106; and very low, 4.9 × 106–5.3 × 105).
The same 1,000 features were targeted in each run, including 200 features of each intensity bin.
Directed Analysis of a Complex Phosphopeptide Mixture—
Phosphopeptides, specifically those phosphorylated at serine or threonine residues, are notoriously difficult to identify because of their specific fragmentation patterns in ion trap instruments.
To evaluate the performance of directed sequencing for the identification of phosphopeptides we applied the combined directed approach described above (Fig. 3A, green line) to a sample consisting of phosphopeptides isolated by TiO2 affinity chromatography (20, 30) from a cytosolic fraction of Kc167 cells. The sample was separated using an RP LC gradient that was extended by 30 min compared with the one used for non-phosphorylated peptides to compensate for the longer MS acquisition times applied for phosphopeptide analysis (MS2 followed by MS3 of the neutral loss peak of −98 Da corresponding to the loss of phosphate). To maximize the number of identified features for a specific number of LC-MS runs, the experiment was designed such that in every run a different set of peptides was selected. Specifically the following protocol was applied. First the sample was analyzed by DDA LC-MS/MS, selecting precursor ions at [M + 2H]2+ in the first run and ions at charge states higher than [M + 2H]2+ in the second run, respectively. Then all MS1 peaks were extracted from the high resolution MS1 maps obtained in those first two runs and filtered as described above. A total of more than 7,776 features that aligned over both runs were detected (supplemental Table S9) about half of which were already sequenced by the two initial LC-MS/MS runs and excluded from the following directed LC-MS/MS. After generating a series of inclusion lists, the remaining 4,000 features were subjected to directed sequencing in two additional LC-MS/MS experiments. To further increase the number of detected phosphopeptides, the 2,000 most abundant features showing a neutral loss peak but that could not be confidently assigned to a peptide sequence were reanalyzed in one additional LC-MS/MS run using a different MS sequencing method especially designed for phosphopeptide analysis (31).
Every directed LC-MS/MS run identified a considerable number of previously unidentified peptides. In total, after five LC-MS/MS runs, more than 1,600 phosphorylation sites could be identified (Table II and supplemental Table S10 and Fig. S3). Interestingly 1,500 (87.2%) of the 1,721 identified peptides carried at least one phosphate group confirming a high specificity for phosphopeptides of the TiO2 affinity enrichment. Of the 1,628 phosphorylation events detected, the exact site of phosphorylation of 1,204 sites could be determined with a probability of more than 90% (ΔCN > 0.1) (32). The distribution of phosphorylated amino acids was similar to that in other studies (20, 33) with most sites found on serine (82.7%) followed by threonine (15.4%) and tyrosine (1.9%) residues. In addition to this, the majority of phosphopeptides identified contained one phosphogroup (92.1%), whereas only 110 (7.3%) and nine (0.6%) were phosphorylated on two or three different residues, respectively. More importantly, compared with the DDA runs, 65% additional protein phosphorylation sites were identified by the three directed LC-MS/MS runs (runs 3–5) of which 107 were identified by reanalyzing features that showed a neutral loss of phosphate (−98 Da) during CID (run 5). Compared with a single DDA LC-MS/MS analysis of this sample that detected a total of 720 phosphorylation sites (supplemental Table S11), the directed strategy used here enabled the identification of a 2.3 times higher number of phosphosites after five LC-MS/MS runs. Annotated spectra of all phosphopeptides identified by the directed approach and by the single DDA LC-MS/MS run are shown in supplemental Figs. S4 and S5, respectively.
Table II.
Identified phosphopeptides using directed LC-MS/MS
| LC-MS/MS runa | No. of peptides identifiedb | No. of phosphopeptides identifiedb | No. of phosphorylation sites identifiedb | Percentage of phosphopeptides identifiedb |
|---|---|---|---|---|
| 1 (DDA 2+) | 742 | 655 | 678 | 88.27 |
| 2 (DDA >2+) | 1,085 | 949 | 997 | 87.47 |
| 3 (INL-1) | 1,439 | 1,254 | 1,347 | 87.14 |
| 4 (INL-2) | 1,621 | 1,405 | 1,521 | 86.67 |
| 5 (INL-NL) | 1,721 | 1,500 | 1,628 | 87.16 |
Runs 1 (DDA 2+) and 2 (DDA >2+), only doubly or higher charged peptide ions, respectively, were allowed to trigger CID by DDA. Runs 3 (INL-1) and 4 (INL-2), directed LC-MS/MS of unsequenced features extracted from runs 1 and 2. Run 5 (INL-NL), directed LC-MS/MS of unassigned neutral loss-showing precursors.
Numbers calculated from Bioworks 3.2 search results using an in-house software tool (25).
The value of the additional information obtained with directed LC-MS/MS can be demonstrated by the increased protein phosphorylation coverage of the Wnt signaling pathway, which is implicated in the genesis of cancer (34, 35). Table III shows all eight identified phosphoproteins and their 13 phosphorylation sites that could be assigned to this pathway (36). Whereas five phosphoproteins and seven phosphorylation sites could be detected by the two initial DDA LC-MS/MS analyses (runs 1 and 2), the three following directed LC-MS/MS runs (runs 3–5) exclusively detected three phosphoproteins and six phosphorylation sites that increased the overall coverage of the pathway by 60 and 85.7%, respectively. For example, phosphorylation of ATP-dependent helicase SWR1 (gene name CG5899) at serines 169, 172, and 841 could be determined with high confidence only by additional directed LC-MS/MS. Although the precursor ion signal intensities for fragment ion spectra acquired by directed sequencing were generally lower than those of the DDA runs, their quality was high. For instance, the complete y-ion series, with the exception of y1 and y2, of the doubly phosphorylated peptide “K↓DQVYDpSDDpSDSEMSTK↓M” (where pS is phosphoserine) could be assigned from its fragment spectrum (Fig. 4B). Interestingly the precursor ion was not picked for fragmentation in the first two runs, although it is only 5 times less abundant than the highest peak present in the corresponding MS1 spectrum (Fig. 4A). Obviously the sequencing speed of the mass spectrometer was not sufficient to acquire MS2 spectra from the high number of co-eluting peptide ions present in the MS1 spectrum. This agrees very well with our results obtained from the sample consisting of non-phosphorylated peptides (Fig. 3B).
Table III.
Identified phosphorylation sites involved in the Wnt signaling pathway
| Accession numbera | Gene name | LC-MS/MS run detectedb | Precursor mass | Charge state | Peptidec | Probabilityd | Final scored | Xcorrd | ΔCNd |
|---|---|---|---|---|---|---|---|---|---|
| (Da) | |||||||||
| FBgn0001139 | gro | 1 (DDA 2+) | 1,182.51506 | 2 | R↓NSVS*PADREK↓Y | 1.83 × 10−3 | 0.85 | 3.26 | 0.31 |
| FBgn0001139 | gro | 1 (DDA 2+) | 1,356.67538 | 2 | R↓TRS*PLDIENDSK↓R | 1.52 × 10−6 | 0.82 | 3.28 | 0.43 |
| FBgn0002783 | mor | 1 (DDA 2+) | 1,520.71046 | 2 | K↓ANAALQSTAS*PAPGGK↓S | 1.03 × 10−3 | 0.70 | 3.34 | 0.05 |
| FBgn0015805 | Rpd3 | 1 (DDA 2+) | 2,141.75841 | 2 | R↓IVPENEYS*DS*EDEGEGGR↓R | 2.53 × 10−5 | 0.95 | 6.44 | 0.09 |
| FBgn0015805 | Rpd3 | 1 (DDA 2+) | 2,297.85952 | 2 | R↓IVPENEYS*DS*EDEGEGGRR↓D | 2.41 × 10−4 | 0.56 | 4.65 | 0.14 |
| FBgn0022131 | aPKC | 2 (DDA >2+) | 2,845.32267 | 3 | K↓EGIRPGDTTS*TFCGTPNYIAPEILR↓G | 1.20 × 10−3 | 0.53 | 4.04 | 0.30 |
| FBgn0003371 | sgg | 2 (DDA >2+) | 1,852.84120 | 3 | K↓QLLHGEPNVSY*ICSR↓Y | 2.23 × 10−3 | 0.84 | 4.09 | 0.08 |
| FBgn0003371 | sgg | 3 (INL-1) | 1,020.40338 | 2 | R↓TSS*FAEGNK↓Q | 8.72 × 10−3 | 0.55 | 1.92 | 0.29 |
| FBgn0032157 | CG5899 | 3 (INL-1) | 3,177.56749 | 3 | K↓LLTTALGLDKDQEEQLNNSLNNSIAS*PAK | 2.19 × 10−5 | 0.93 | 6.92 | 0.17 |
| FBgn0032157 | CG5899 | 4 (INL-2) | 1,981.62937 | 2 | K↓DQVYDS*DDS*DSEMSTK↓M | 2.29 × 10−4 | 0.95 | 5.60 | 0.09 |
| FBgn0015589 | Apc | 4 (INL-2) | 2,738.39852 | 3 | R↓RNS*VAGSGQNVDSPPVVIPASLQPLR↓S | 2.30 × 10−4 | 0.53 | 3.82 | 0.09 |
| FBgn0027492 | wdb | 5 (INL-NL) | 2,007.98993 | 2 | R↓YRNS*QDVELQQLPPLK↓A | 4.46 × 10−3 | 0.90 | 4.45 | 0.15 |
Accession number as indicated in Flybase 4.3.
Runs 1 (DDA 2+) and 2 (DDA >2+), only doubly or higher charged peptide ions, respectively, were allowed to trigger CID by DDA. Runs 3 (INL-1) and 4 (INL-2), directed LC-MS/MS of unsequenced features extracted from runs 1 and 2. Run 5 (INL-NL), directed LC-MS/MS of unassigned neutral loss-showing precursors.
Phosphorylation sites are italic and followed by an asterisk.
Search result values obtained from Bioworks 3.2.
Fig. 4.

Determination of two phosphorylation sites in ATP-dependent helicase SWR1 (gene name CG5899) by directed LC-MS/MS. A, MS spectrum showing the feature selected for sequencing during directed LC-MS/MS data acquisition. B, fragmentation pattern (MS/MS spectrum) obtained by collision-induced dissociation of the phosphopeptide precursor indicated in A. The sequence of the identified phosphopeptide, including the two phosphorylated serine residues (pS), is shown. Fragmentation of the peptide backbone during tandem mass analysis would result in the characteristic b- and y-ions displayed. All detected fragment ions (bold) including loss of phosphoric acid (*) are indicated.
DISCUSSION
This study describes a reproducible, sensitive, and integrated computational and mass spectrometric method for directed sequencing of a high number of peptide ions within complex mixtures. In contrast to concomitant MS1 and MS2 data acquisition during DDA ESI-LC-MS/MS, the directed approach described decouples MS1 and MS2 spectra collection. In a first step, potential peptide ion signals are extracted off line from the pattern generated in initial LC-MS/MS runs. For this task we developed the software tools SuperHirn and Prequips. In a second step, such features were subjected to directed sequencing in subsequent LC-MS/MS runs, and the identified features were automatically mapped back to the list of previously detected features. As we make the software tools developed for this method publicly available and because it uses generic data formats (21, 24, 37) for which converters for various MS instruments are available (Seattle Proteome Center), the presented approach is applicable to any high performance MS platform that allows direct sequencing by inclusion list. It is important to point out that high resolution scans are required only for MS1 data acquisition and not for MS2 sequencing. Notably hybrid MS instruments, like the LTQ-FT instrument used in this study, are preferred because they provide the unique advantage of parallel MS1 and MS2 spectra acquisition. Therefore, MS2 data can already be obtained in the initial LC-MS/MS runs with minimal impairment of the speed and sensitivity for acquiring high quality MS1 maps. Compared with current DDA LC-MS/MS, the described directed strategy provides several advantages.
First, the off-line feature extraction facilitates the identification of peptides with low intensity precursor ion signals. Whereas peak selection in DDA LC-MS/MS analysis is based on one single MS1 scan, off-line peptide ion detection allows summing up all isotopic signals of an eluting peptide ion. This simplifies the selection of potential peptide ions based on the distribution of isotopic clusters as well as the determination of charge state and monoisotopic precursor mass. Moreover the alignment of detected peptide ions over multiple runs helps to distinguish between peptide-derived signals and chemical and electronic noise. This significantly improved the generation of a feature list with a high content of peptide ions. As shown above, the directed sequencing of these information-rich features resulted in a higher number of unique peptides identified compared with on-line CID by DDA. It is worth mentioning that the number of MS2 scans acquired by the directed approach was 3 times lower than by DDA LC-MS/MS, considerably reducing the efforts for subsequent data analysis.
Second, in directed LC-MS/MS, the CID parameters can be adjusted and optimized according to the feature properties. For instance, a higher number of very intense features could be identified in one LC-MS/MS experiment by applying shorter ion accumulation and scan times, whereas longer gating and scan times were used for low abundance peptide ions. It is important to point out that the directed approach offers the opportunity of reanalyzing all features that could not be assigned to a peptide sequence in the first analysis using optimized MS parameters. As shown by the results obtained in this study, more than 100 additional phosphorylation sites were determined by iterative analysis of phosphopeptides that showed a neutral loss peak but could not be confidently identified on the basis of their MS2 or MS3 spectra using the multistage activation mode for MS/MS data acquisition (31). The application of optimized CID parameters for each feature not only allows for the identification of more peptides but can also be used to confirm peptide ions with questionable identity.
Third, the directed approach offers the possibility of selecting for CID precursor ions with specific properties. These include the charge state of a peptide that is most likely to yield an informative fragment ion spectrum, particular patterns after stable isotope labeling (13, 15, 16, 38), characteristic isotope distributions generated by tagging selected functional groups with suitable reagents (39), both isotopic versions of cross-linked peptides (40), and redundant identification of peptides of interest over multiple samples (e.g. time course experiments) (13).
For analyzing complex mixtures, the most important improvement of directed versus DDA LC-MS/MS results from the fact that the directed approach copes with the problem of “undersampling” during LC-MS/MS analysis meaning that not all precursor ions present in the MS1 scans can be sequenced using DDA-based LC-MS/MS (8–11). This results in the preferred identification of peptides of high abundance (Fig. 3A). Conversely using the directed approach, the sequencing speed of the MS instrument used is no longer a limiting factor with respect to the undersampling problem virtually allowing any detectable peptide ion in a sample to be MS-sequenced. Indeed a higher number of peptides were identified by using the inclusion list-based approach with most having a precursor intensity 5 times lower than that of the majority of peptides determined by the DDA strategy. The exclusive identification of low intensity peptide ions by directed LC-MS/MS was particularly pronounced for MS1 scans containing large numbers of peptide ions, clearly indicating incomplete sequencing by on-line DDA LC-MS/MS because of the limited MS sequencing speed and a more thorough sequencing of precursor ions by the directed approach.
Nonetheless the number of peptides identified by directed LC-MS/MS decreased rapidly with lower MS1 signal intensities. This can be ascribed to the fact that very low intensity peaks do not reach the detection limit in all replicate runs, show higher mass tolerances, and are consequently difficult to detect and to align across multiple maps. A higher dynamic range in the MS1 scans would be desirable; however, this is limited by the loading capacity of the ICR cell (26, 27). Certainly the dynamic intensity range of detected peptide ions can be extended by additional time-consuming sample fractionation steps (5, 19), which can be used in combination with the directed approach. More importantly, each feature needs to be detected on line by the MS instrument to trigger MS2. As shown above, changing the MS detection parameters increased this number, but for a considerable fraction of low abundance features detected off line, no MS2 scan was acquired. Therefore, further improvements in on-line monoisotopic peak detection of peptide-derived precursor ions could definitely increase the number of CIDs for low abundance peptide ions.
In conclusion, directed sequencing of non-redundant, high quality features enables the identification of a higher number of peptides with less analytical effort than current DDA LC-MS/MS-based methods. For instance, more than 1,600 phosphorylation sites could be identified using a single dimension reversed phase separation without the need for time-consuming sample prefractionation steps. The implemented software tools used are freely available and compatible with generic, publicly accessible data formats and therefore applicable to most high performance LC-MS (MS1 level) platforms. With the increasing availability of reproducible high performance LC-MS/MS systems and its capability to identify features of interest specifically and redundantly over a wide intensity range, the directed approach presented here is well suited for in-depth and high throughput characterization of complex protein samples and will find wide application in future LC-MS/MS-based proteome studies.
Acknowledgments
We thank Ralph Schiess and Reto Ossola for helpful discussions.
Footnotes
Published, MCP Papers in Press, May 29, 2008, DOI 10.1074/mcp.M700498-MCP200
The abbreviations used are: DDA, data-dependent acquisition; MPS, monoisotopic precursor selection; INL, inclusion list; RP, reversed phase; FBgn, Flybase gene ID; CG, computed gene.
N. Gehlenborg, W. Yan, H. Yoo, I. Lee, K. Nieselt, D. Hwang, R. Aebersold, and L. Hood, manuscript in preparation.
This work was supported, in whole or in part, by National Institutes of Health Grant N01-HV-28179 from the NHLBI. This work was also supported by F. Hoffmann La Roche Ltd. as well as the Swiss National Science Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Aebersold, R., and Mann, M. ( 2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2.Gygi, S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H., and Aebersold, R. ( 1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 3.Ong, S. E., Kratchmarova, I., and Mann, M. ( 2003) Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC). J. Proteome Res. 2, 173–181 [DOI] [PubMed] [Google Scholar]
- 4.Schmidt, A., Kellermann, J., and Lottspeich, F. ( 2005) A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics 5, 4–15 [DOI] [PubMed] [Google Scholar]
- 5.Brunner, E., Ahrens, C. H., Mohanty, S., Baetschmann, H., Loevenich, S., Potthast, F., Deutsch, E. W., Panse, C., de Lichtenberg, U., Rinner, O., Lee, H., Pedrioli, P. G., Malmstrom, J., Koehler, K., Schrimpf, S., Krijgsveld, J., Kregenow, F., Heck, A. J., Hafen, E., Schlapbach, R., and Aebersold, R. ( 2007) A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25, 576–583 [DOI] [PubMed] [Google Scholar]
- 6.Kislinger, T., Cox, B., Kannan, A., Chung, C., Hu, P., Ignatchenko, A., Scott, M. S., Gramolini, A. O., Morris, Q., Hallett, M. T., Rossant, J., Hughes, T. R., Frey, B., and Emili, A. ( 2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 [DOI] [PubMed] [Google Scholar]
- 7.Adachi, J., Kumar, C., Zhang, Y., Olsen, J. V., and Mann, M. ( 2006) The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 7, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Godoy, L. M., Olsen, J. V., de Souza, G. A., Li, G., Mortensen, P., and Mann, M. ( 2006) Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol. 7, R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuster, B., Schirle, M., Mallick, P., and Aebersold, R. ( 2005) Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 6, 577–583 [DOI] [PubMed] [Google Scholar]
- 10.Kristensen, D. B., Brond, J. C., Nielsen, P. A., Andersen, J. R., Sorensen, O. T., Jorgensen, V., Budin, K., Matthiesen, J., Veno, P., Jespersen, H. M., Ahrens, C. H., Schandorff, S., Ruhoff, P. T., Wisniewski, J. R., Bennett, K. L., and Podtelejnikov, A. V. ( 2004) Experimental Peptide Identification Repository (EPIR): an integrated peptide-centric platform for validation and mining of tandem mass spectrometry data. Mol. Cell. Proteomics 3, 1023–1038 [DOI] [PubMed] [Google Scholar]
- 11.Liu, H., Sadygov, R. G., and Yates, J. R., III ( 2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 12.Domon, B., and Aebersold, R. ( 2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 13.Rinner, O., Mueller, L. N., Hubalek, M., Muller, M., Gstaiger, M., and Aebersold, R. ( 2007) An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 25, 345–352 [DOI] [PubMed] [Google Scholar]
- 14.Domon, B., and Broder, S. ( 2004) Implications of new proteomics strategies for biology and medicine. J. Proteome Res. 3, 253–260 [DOI] [PubMed] [Google Scholar]
- 15.Bisle, B., Schmidt, A., Scheibe, B., Klein, C., Tebbe, A., Kellermann, J., Siedler, F., Pfeiffer, F., Lottspeich, F., and Oesterhelt, D. ( 2006) Quantitative profiling of the membrane proteome in a halophilic archaeon. Mol. Cell. Proteomics 5, 1543–1558 [DOI] [PubMed] [Google Scholar]
- 16.Griffin, T. J., Lock, C. M., Li, X. J., Patel, A., Chervetsova, I., Lee, H., Wright, M. E., Ranish, J. A., Chen, S. S., and Aebersold, R. ( 2003) Abundance ratio-dependent proteomic analysis by mass spectrometry. Anal. Chem. 75, 867–874 [DOI] [PubMed] [Google Scholar]
- 17.Calvo, S., Jain, M., Xie, X., Sheth, S. A., Chang, B., Goldberger, O. A., Spinazzola, A., Zeviani, M., Carr, S. A., and Mootha, V. K. ( 2006) Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 38, 576–582 [DOI] [PubMed] [Google Scholar]
- 18.Picotti, P., Aebersold, R., and Domon, B. ( 2007) The implications of proteolytic background for shotgun proteomics. Mol. Cell. Proteomics 6, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 19.Yi, E. C., Marelli, M., Lee, H., Purvine, S. O., Aebersold, R., Aitchison, J. D., and Goodlett, D. R. ( 2002) Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis 23, 3205–3216 [DOI] [PubMed] [Google Scholar]
- 20.Bodenmiller, B., Mueller, L. N., Mueller, M., Domon, B., and Aebersold, R. ( 2007) Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat. Methods 4, 231–237 [DOI] [PubMed] [Google Scholar]
- 21.Pedrioli, P. G., Eng, J. K., Hubley, R., Vogelzang, M., Deutsch, E. W., Raught, B., Pratt, B., Nilsson, E., Angeletti, R. H., Apweiler, R., Cheung, K., Costello, C. E., Hermjakob, H., Huang, S., Julian, R. K., Kapp, E., McComb, M. E., Oliver, S. G., Omenn, G., Paton, N. W., Simpson, R., Smith, R., Taylor, C. F., Zhu, W., and Aebersold, R. ( 2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 22.Mueller, L. N., Rinner, O., Schmidt, A., Letarte, S., Bodenmiller, B., Brusniak, M. Y., Vitek, O., Aebersold, R., and Muller, M. ( 2007) SuperHirn—a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 23.Elias, J. E., and Gygi, S. P. ( 2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 24.Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. ( 2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 25.Bodenmiller, B., Mueller, L. N., Pedrioli, P. G., Pflieger, D., Junger, M. A., Eng, J. K., Aebersold, R., and Tao, W. A. ( 2007) An integrated chemical, mass spectrometric and computational strategy for (quantitative) phosphoproteomics: application to Drosophila melanogaster Kc167 cells. Mol. Biosyst. 3, 275–286 [DOI] [PubMed] [Google Scholar]
- 26.Williams, D. K., Jr., and Muddiman, D. C. ( 2007) Parts-per-billion mass measurement accuracy achieved through the combination of multiple linear regression and automatic gain control in a Fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 79, 5058–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masselon, C., Tolmachev, A. V., Anderson, G. A., Harkewicz, R., and Smith, R. D. ( 2002) Mass measurement errors caused by “local” frequency perturbations in FTICR mass spectrometry. J. Am. Soc. Mass Spectrom. 13, 99–106 [DOI] [PubMed] [Google Scholar]
- 28.Haas, W., Faherty, B. K., Gerber, S. A., Elias, J. E., Beausoleil, S. A., Bakalarski, C. E., Li, X., Villen, J., and Gygi, S. P. ( 2006) Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell. Proteomics 5, 1326–1337 [DOI] [PubMed] [Google Scholar]
- 29.Lam, H., Deutsch, E. W., Eddes, J. S., Eng, J. K., King, N., Stein, S. E., and Aebersold, R. ( 2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 30.Larsen, M. R., Thingholm, T. E., Jensen, O. N., Roepstorff, P., and Jorgensen, T. J. ( 2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 [DOI] [PubMed] [Google Scholar]
- 31.Schroeder, M. J., Shabanowitz, J., Schwartz, J. C., Hunt, D. F., and Coon, J. J. ( 2004) A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 76, 3590–3598 [DOI] [PubMed] [Google Scholar]
- 32.Li, X., Gerber, S. A., Rudner, A. D., Beausoleil, S. A., Haas, W., Villen, J., Elias, J. E., and Gygi, S. P. ( 2007) Large-scale phosphorylation analysis of α-factor-arrested Saccharomyces cerevisiae. J. Proteome Res. 6, 1190–1197 [DOI] [PubMed] [Google Scholar]
- 33.Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. ( 2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 34.Reya, T., and Clevers, H. ( 2005) Wnt signalling in stem cells and cancer. Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 35.Segditsas, S., and Tomlinson, I. ( 2006) Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 25, 7531–7537 [DOI] [PubMed] [Google Scholar]
- 36.Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., Diemer, K., Muruganujan, A., and Narechania, A. ( 2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 38.Old, W. M., Meyer-Arendt, K., Aveline-Wolf, L., Pierce, K. G., Mendoza, A., Sevinsky, J. R., Resing, K. A., and Ahn, N. G. ( 2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 39.Goodlett, D. R., Bruce, J. E., Anderson, G. A., Rist, B., Pasa-Tolic, L., Fiehn, O., Smith, R. D., and Aebersold, R. ( 2000) Protein identification with a single accurate mass of a cysteine-containing peptide and constrained database searching. Anal. Chem. 72, 1112–1118 [DOI] [PubMed] [Google Scholar]
- 40.Rinner, O., Seebacher, J., Walzthoeni, T., Mueller, L., Beck, M., Schmidt, A., Mueller, M., and Aebersold, R. ( 2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods 5, 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]