

Abstract
ADAMTS13, a reprolysin-like metalloprotease, limits platelet-rich thrombus formation in the small arteries by cleaving von Willebrand factor (vWF) at the Tyr1605-Met1606 peptide bond. Deficiency of plasma ADAMTS13 activity, due to either an inherited or an acquired etiology, may lead to a potentially lethal syndrome, thrombotic thrombocytopenic purpura (TTP). Molecular cloning and characterization of the ADAMTS13 gene have provided further insight into the structure-function relationships, biosynthesis, and regulation of the ADAMTS13 protease, in addition to understanding the pathogenesis of TTP and perhaps other thrombotic disorders. ADAMTS13 consists of a short propeptide, a typical reprolysin-like metalloprotease domain, followed by a disinte-grin-like domain, first thrombospondin type 1 (TSP1) repeat, Cys-rich domain, and spacer domain. The carboxyl terminus of ADAMTS13 has seven more TSP1 repeats and two CUB domains. ADAMTS13 is synthesized mainly in hepatic stellate cells, but also in vascular endothelial cells. Recognition and cleavage of vWF require the proximal carboxyl terminal domains, but not the middle and distal carboxyl terminal domains. Cleavage of vWF appears to be modulated by shear force, binding to platelet or platelet glycoprotein-1bα, heparin, inflammatory cytokine (interleukin-6), and chloride ion. At the site of thrombus formation, the ADAMTS13 may be inactivated by thrombin, plasmin, and factor Xa. Having a sensitive and specific assay for ADAMTS13 activity is not only critical to understand the basic biology of ADAMTS13 protease, but also to facilitate a more timely and accurate clinical diagnosis of TTP, and to initiate potentially life-saving plasma exchange therapy. Although many assays have been developed and tested for clinical applications, the fluorescent resonance energy transfer-vWF73 assay appears to be the simplest and most promising assay to date.
Keywords: Thrombotic thrombocytopenic purpura (TTP), von Willebrand factor, microvascular thrombosis, ADAMTS13, metalloprotease, assays, clinical application
The 13th member of adisintegrin-like and metalloprotease with thrombospondin type 1 motifs (ADAMTS) family, ADAMTS13,1–7 limits platelet aggregation and microthrombi in small arteries by cleaving vWF at the Tyr1605-Met1606 bond.8,9 Deficiency of ADAMTS13 protease due to either an inherited5 or acquired etiology10 may result in formation of disseminated microvascular thromboses, exhibiting a potentially lethal syndrome, thrombotic thrombocytopenic purpura (TTP).10–13
TTP had classically been diagnosed using a pentad of criteria: thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, renal dysfunction, and neurological abnormalities.11 However, the diagnosis has been made recently in patients with only thrombocytopenia and MAHA.14–17 On a histological level, these symptoms are attributable to the presence of platelet- and von Willebrand factor (vWF) -rich micro-thrombi in the terminal arterioles and capillaries of distal organs, leading to organ failure, coma, and death.18 Until the late 1960s, TTP was virtually fatal, with a mortality rate of >90%,19 but with advances in plasma exchange, the survival rate from acute TTP approaches 80 to 90%.20,21
Several reviews have summarized the roles of vWF and ADAMTS13 in TTP.13,22–30 This article specifically focuses on certain aspects of ADAMTS13 biology: the molecular cloning, structure-function relationship, biosynthesis, regulation of proteolytic activity, ADAMTS13 mutations found in congenital TTP patients, and the diagnostic utility of assays for plasma ADAMTS13 proteolytic activity and inhibitors.
MOLECULAR CLONING OF ADAMTS13
In 1996, two groups attempted to isolate a protease from normal human plasma that cleaves vWF, although the attempts were not successful initially. However, much was learned about the biochemical properties of their partially purified products.8,9 The protease displays a greater proteolytic activity when vWF is subject to high shear stress or denatured by urea or guanidine in the presence of low ionic strength. Divalent cations such as Ba2+, Zn2+, Ca2+ or Co2+, are necessary to activate the protease.8,9 These early studies led to the development of several functional assays critical for determining the protease activity in human plasma or for tracking the activity in the fractions during subsequent chromatographic purification of the enzyme.1,2
In 2001, the vWF-cleaving metalloprotease was finally isolated and its partial amino-terminal sequence was determined.1,2 This led to the identification and classification of the vWF-cleaving metalloprotease as a new member of the ADAMTS family, designated ADAMTS13.1–4 By a completely different approach, the identical ADAMTS13 gene was also cloned.5
The ADAMTS13 gene is located on chromosome 9q34, spanning 37 kb in length and containing 29 exons.4,5 Several alternatively spliced variants were found during the cloning and sequencing.3–5 Analysis of the translated product showed that ADAMTS13 belongs to the metzincin superfamily of zinc metalloproteases, which, thus far, contains 19 family members that share the common structure of a hydrophobic signal sequence, a propeptide, a metalloprotease domain, first thrombo-spondin-1 (TSP1) repeat, a disintegrin-like domain, a cysteine-rich domain, and a spacer domain.3–5 The carboxyl terminus of ADAMTS13 contains seven more TSP1 repeats and two CUB domains, which are unique to ADAMTS13 and were previously identified in complement components c1r and c1s, urinary EGF, and bone morphogenetic protein.31
BIOSYNTHESIS OF ADAMTS13
Northern blotting detects a 4.7-kb mRNA in human liver,3–5 3.5- to 4.7-kb mRNAs in livers of different mouse strains,32,33 and a 2.4-kb mRNA in human placenta and skeletal muscle.4 In situ hybridization and immunohistochemical staining with a monoclonal antibody directed against the disintegrin domain of ADAMTS13 have localized the ADAMTS13 mRNA and protein, respectively, to hepatic stellate cells (the interstitial cells of liver parenchyma that are elongated and varied in size and shape).34,35 ADAMTS13-expressing cells are also positive for α-smooth muscle actin, suggesting only activated hepatic stellate cells or myofibroblasts produce ADAMTS13.34 Cell fractionation showed that full-length ADAMTS13 is synthesized and secreted from primary hepatic stellate cells from mouse35 and rat (M. Niiya and X. Zheng, unpublished data, 2005). Other cell types such as hepatocytes, sinusoidal endothelial cells, or Kupffer cells do not express detectable amounts of ADAMTS13 intracellularly or in the culture medium.35 Furthermore, cell lines derived from normal hepatocytes (THLE-3) or hepatocellular carcinoma (HuH-7) do not express ADAMTS13, but those derived from hepatic stellate cells of human (hTERT-HSC) and rat (CFSC-3H and -8B) do secrete active ADAMTS13 into the culture medium.35 These data further support that activated hepatic stellate cells are the major ADAMTS13-producing cells in the liver. Activated hepatic stellate cells have been implicated in the development of liver fibrosis and cirrhosis36–38 after various insults because of their ability to produce and to deposit extracellular matrix proteins such as fibril collagen36–38 in the injured area. Thus, upon activation, the enhanced proteolytic activity of ADAMTS13 in the hepatic stellate cells may be associated with either formation or resolution of liver fibrosis. More studies are needed to delineate the relationship between ADAMTS13 protease and liver fibrosis.
In addition to hepatic stellate cells, platelets or other tissues may also produce and secrete ADAMTS13. Reverse transcriptase polymerase chain reaction (RT-PCR) using a single-strand cDNA can detect the ADAMTS13 mRNA transcripts in almost all major organ tissues, including human heart, lungs, pancreas, brain, kidneys, adrenal glands, placenta, uterus, ovaries, and prostate.5–7 Such ubiquitous distribution of ADAMTS13 mRNA suggests that vascular endothelial cells may produce ADAMTS13. Our results (D. Shang, X.W. Zheng, and X. Zheng, unpublished data, 2005) showed that primary human umbilical cord vein endo-thelial cells, primary aortic endothelial cells, and microvascular endothelial cell lines (EVC304) all synthesize and secrete full-length ADAMTS13, that is, detected by RT-PCR, Western blotting with rabbit anti-ADAMTS13, and capable of proteolytic cleavage of vWF and vWF73 substrates. Considering the enormous surface area covered by endothelial cells, it is possible that the plasma pool of ADAMTS13 may be derived from the vascular endothelial cells, in addition to hepatic stellate cells.
In a pulse-chase experiment, ADAMTS13 is readily secreted and can be detected within 3 hours of chase in the conditioned medium of transiently trans-fected HeLa cells.39 The secreted recombinant ADAMTS13 migrates at 190 kd on a sodium dodecyl sulfate (SDS) polyacrylamide gel,39–41 similar to that of ADAMTS13 purified from plasma,1–3 although the calculated molecular weight is only 150 kd.3–5 The difference in molecular mass is attributed to glycosylation4,39,42 and perhaps other posttranslational modifications.4 Recombinant ADAMTS13 secreted into the conditioned medium is resistant to endoH digestion,39 but sensitive to N-glycanase F and sialidase,39 suggesting the presence of a complex-type structure of N-linked and O-linked oligosaccharides. ADAMTS13 is localized to the endoplasmic reticulum and Golgi apparatus, consistent with the distribution of secretory proteins.40 Despite structural homology to ADAMTS1, which interacts with the extracellular matrix via its spacer domain and thrombospondin type repeats,43,44 ADAMTS13 expressed from transfected COS7 and RFL-6 cells does not bind detectably to either cell membrane or extracellular matrix.40
The role of the propeptide in biosynthesis and secretion of ADAMTS13 has been investigated recently. The ADAMTS13 propeptide is unusually short (≈41 amino acid residues) compared with the typical ≈200 residues in other members of the ADAMTS or ADAM protease family.45–47 This domain terminates in a typical propeptide cleavage sequence, RQRR74. The furin consensus sequence is required for cleavage of ADAMTS13 propeptide,39 since changing this sequence to KQDR results in secretion of pro-ADAMTS13 with intact propeptide attached.39 For several related proteases, propeptide is required for folding and maintaining enzymatic latency during biosynthesis. However, deletion of the propeptide did not impair the secretion of active ADAMTS13 in transfected HeLa cells, suggesting that the ADAMTS13 propeptide is not required to maintain enzyme latency.39
CLEAVAGE OF vWF BY ADAMTS13
The mature form of the ADAMTS13 protease starts with the metalloproteases domain that has three His residues (HEXXHXXGXXHD) that coordinate the essential Zn2+ or Ca2+ binding.4 The conserved Met249 that forms a Metturn and the residues forming a predicted Ca2+ coordination site (Glu83, Asp173, Cys281, Asp284) are also present in the metalloprotease domain.4 The binding sites for Ca2+ and Zn2+ are consistent with data showing that the enzyme is inhibited by their chelation.40,41,48 At the concentration of 16 nM (three to four times higher than plasma concentration), metalloprotease domain alone does not cleave multimeric vWF or small peptidyl substrate GST-vWF73-H that contains 73 amino acid residues from the central A2 domain of vWF, flanked by a glutathione s-transferase (GST) at N-terminus and 6xHis epitope at C-terminus (Fig. 1).49 However, with a prolonged incubation (16 to 24 hours), the metalloprotease domain does cleave GST-vWF73-H substrate at a site other than the Tyr-Met bond, generating a N-terminal fragment ≈3 kd smaller than expected,49 suggesting the carboxyl-terminal domains are required for substrate recognition and specificity.
Figure 1.
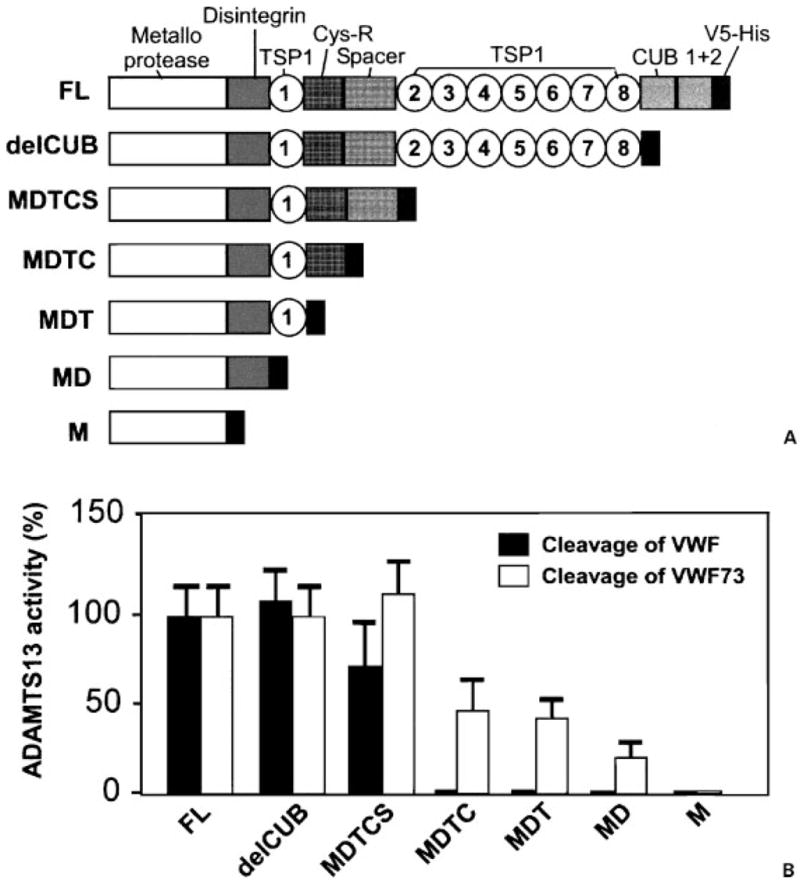
Cleavage of von Willebrand factor (vWF) and vWF73 by ADAMTS13 and variants. (A) Schematic representation of the construct of full-length (FL) ADAMTS13 and carboxyl-terminal truncated ADAMTS13 variants. (B) Cleavage of FL vWF and GST-vWF73-H by ADAMTS13 and C-terminal truncated variants. delCUB, aal-1191; MDTCS, aal-685; MDTC, aal-554; MDT, aal-439; MD, aal-385; M, metalloprotease. (Adapted from Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem 2003;278:30136–30141 and Ai J, Smith P, Wang S, et al. The proximal carboxyl terminal domains of ADAMTS13 determine substrate specificity and are all required for cleavage of von Willebrand factor. J Biol Chem 2005;280:29428–29434.)
Remarkably, addition of one or several proximal carboxyl terminal domains (including the disintegrin domain, first TSP1 repeat, the Cys-rich domain, and spacer domain) to the metalloprotease domain of ADAMTS13 restores the specific cleavage of Tyr-Met bond of GST-vWF73-H substrate.49 However, the proteolytic activity of the ADAMTS13 mutants toward both vWF and GST-vWF73-H (Fig. 1) or fluorescent resonance energy transfer (FRETS)-vWF73 is not fully restored until all the proximal carboxyl-terminal domains are added.49 These data suggest that all of the proximal carboxyl-terminal domains participate in substrate recognition and are required for cleavage of vWF. Further addition of the TSP1 2 to 8 domains and two CUB domains does not increase proteolytic cleavage of vWF,40,41,49 and GST-vWF73 or FRETS-vWF73.49 Several strains of mice (BALB/c, C3H/He, C57BL/6, and DBA/2) lack the carboxyl-terminal domains from the seventh TSP1 repeat to the CUB domain.33 These data support the notion that the middle and the distal carboxyl-terminal domains may be dispensable at least in vitro in humans and in vivo in mice.
ADAMTS13-vWF BINDING
Full-length ADAMTS13 can bind immobilized vWF with a stoichiometry of 1:2 (i.e., one ADAMTS13 molecule binds two vWF molecules), reaching an equilibrium in ≈2 hours, with a half-life of dissociation of ≈4 hours.39,49 The estimated Kd for ADAMTS13 binding to vWF is ≈14 nM, comparable to Km of 16 nM, determined independently.39 ADAMTS13 truncated after the spacer domain binds vWF comparably to full-length ADAMTS13, with a Kd of ≈24 nM (Table 1). Removal of the spacer domain significantly impairs its binding to vWF,39 suggesting that the spacer domain contributes substantially to ADAMTS13-vWF interaction. The metalloprotease domain at the highest concentration tested (≈220 nM) does not bind vWF detectably.39
Table 1.
Binding of ADAMT13 and Various Mutants to vWF and GST-vWF73-H
| Kd (nm)
|
||
|---|---|---|
| Constructs | vWF66 | vWF7349 |
| FL | 14 ±1.2 | 4.6 ± 1.3 |
| MDTCS | 23 ±0.9 | 7 ± 1.9 |
| DTCS | ND | 13 ± 2.1 |
| M | ND | ND |
| Dis | ND | 489 ±42 |
| TSP1 | ND | 136 ±4.0 |
| CysR | ND | 121 ±5.0 |
| Spa | ND | 108 ±14 |
Binding of FL ADAMTS13 and various ADAMTS13 mutants to the purified plasma vWF and GST-vWF73-H was determined by ELISA method and the values for Kd (s) were determined by fitting into a double-reciprocal version of the binding equation. MDTCS and DTCS indicate the ADAMTS13 mutant truncated after the spacer domain with and without the metalloprotease domain, respectively. M, D, TSP1, CysR, S were all tagged at the N-termini (except for M) by V5-His epitope derived from vector. M was tagged at the C-terminus by V5-His.
vWF, von Willebrand factor; FL, full-length; GST, glutathione s-transferase; MDTCS, XXX; ND, not determined; DTCS, XXX; M, metalloprotease; Dis, disintegrin domain; TSP1, thrombospondin type 1; CysR, Cys-rich domain; Spa, spacer domain; ELISA, enzyme-linked immunosorbent assay.
The binding interaction between ADAMTS13 and GST-vWF73-H has been determined recently. vWF73 (D1659-R1668) has been shown to be the minimal and essential peptide region of vWF being recognized and cleaved by ADAMTS13.50 Full-length ADAMTS13 and ADAMTS13 truncated after the spacer domain bound GST-vWF73-H with Kd (s) of ≈4.6 and ≈7.0 nM, respectively.49 The affinity of binding ADAMTS13 or the carboxyl-terminal truncated mutant to vWF73 is approximately three times higher than that to vWF,39,49 suggesting that the domains surrounding the central A2 subunit of vWF negatively regulate the interaction of ADAMTS13 with vWF.
The relative contribution of each of the proximal carboxyl-terminal domains in substrate binding has also been determined using recombinant domains of ADAMTS13 expressed from Escherichia coli and re-folded in vitro. The binding of ADAMTS13 domains to GST-vWF73-H was performed on an enzyme-linked immunosorbent assay (ELISA) plate and in solution. The Kd values for the disintegrin domain (D), the first TSP1 repeat (T), the Cys-rich domain (C), and the spacer domain (S) or DTCS fragment to bind GST-vWF73-H are ≈489, ≈136, ≈121, and ≈108 or 13 nM, respectively (Table 1).49 The metalloprotease domain does not bind detectably at the concentrations tested. These data suggest that each of the proximal carboxyl-terminal domains participates in substrate recognition and acts cooperatively to confer efficient binding, consistent with the proteolysis of vWF and vWF73 by various ADAMTS13 mutants described above.49
REGULATION OF ADAMTS13 ACTIVITY
Cleavage of vWF by ADAMTS13 may be modulated by flow shear stress,9,10,51 structural elements of vWF,48 heparin sulfate, platelets or platelet glycoprotein-1bα (GP1bα),48 sodium chloride,52 and inflammatory cytokines.53 vWF is not cleaved under physiological conditions due to structural inaccessibility of the binding or cleavage site on the vWF molecule by ADAMTS13. Modeling of the vWF-A2 domain suggests that the Tyr1605-Met1606 bond lies buried within the core β sheet of the native structure.54 Shear force and denaturing reagents, or binding of platelets that expose the ADAMTS13 binding and cleavage site on vWF, significantly enhance the cleavage of vWF by ADAMTS13.8–10 However, even in the presence of denaturing reagents, the ADAMTS13 binding site appears not fully exposed. For example, the rate of cleavage of the A2A3 mutant is ≈10-fold higher than that of the A1A2A3 mutant,48 suggesting that the A1 domain may block the access of ADAMTS13 to the cleavage site. This blockage can be removed by interaction with platelet GP1bα or unfractionated heparin.48
Other factors in the milieu may regulate ADAMTS13-vWF interaction, such as chloride ions52 and hemoglobin.55 The cleavage of vWF in the absence of NaCl at pH 8.0 occurs with apparent kcat/km ≈3.4 ×104 M −1s −1, but this value decreases ≈10-fold in the presence of 0.15 M NaCl.52 The inhibitory effect can be found with a relative potency of ClO–4 > Cl– >F–.52 Inhibition of ADAMTS13 activity has been observed by hemoglobin at concentrations exceeding 2 g/L, which occurred during severe hemolysis in a fatal case of congenital TTP.55 Unlike many other members of ADAMTS family that form SDS-stable complexes with α2-macroglobulin, a nonspecific protease inhibitor present in the serum of healthy individuals, ADAMTS13 does not bind α2-macroglobulin detectably.40 Inflammatory cytokines such interleukin-6, but not interleukin-8 and tumor necrosis factor-α, inhibit the cleavage of ultralarge vWF (ULvWF) under flow.53 The findings suggest a potential linkage between inflammation and thrombosis that may be of therapeutic importance.
Thrombin, factor Xa (FXa) and plasmin may regulate ADAMTS13 activity at the site of thrombus formation. ADAMTS13 is proteolytically cleaved following activation of coagulation in human plasma.56 Incubation of purified recombinant ADAMTS13 with various concentrations of human thrombin (0.9 to 90 nM) results in degradation of ADAMTS13 in a time- and concentration-dependent fashion.56 Such cleavage can be abrogated when thrombin binds soluble thrombomodulin, but not heparin, suggesting the involvement of thrombin exosite I, but not exosite II, in ADAMTS13 recognition. Plasmin and FXa also cleave ADAMTS13 and give rise to a similar but not identical fragmentation pattern, resulting in loss of ADAMTS13 activity.56 Plasmin cleaves ADAMTS13 faster than do thrombin and FXa,56 suggesting a role of ADAMTS13 and vWF in tissue repair.
MUTATIONS OF ADAMTS13
To date, more than 70 mutations on ADAMTS13 gene have been described in patients with congenital or familial TTP (Table 2).5,42,57–61 The majority of these mutations are missense mutations (≈50%), followed by splice site mutations, silent mutations, nonsense, and frameshifts.5,13,42 The mutations are distributed throughout the ADAMTS13 gene with no apparent clustering or hot spots. Twelve missense mutations involving cysteine residues have been identified in patients with familial TTP (Table 2), suggesting the importance of proper formation of disulfide bridges in ADAMTS13 function.
Table 2.
Location and Functional Consequence of ADAMTS13 Gene Mutation
| Exon/Intron | Nucleotide | Effect on Amino Acid | Domain | Activity in Human Plasma (unless “in vitro” is noted) | Secretion | Comment |
|---|---|---|---|---|---|---|
| 1 | 19C >T5,57 | R7W | SP | SNP57 | ||
| 2 | 130C >T57 | Q44X | proP | <3%80 | ||
| 3 | 237C >G100 | I79M | M | <5% | ||
| 3 | 286C >G5 | H96D | M | 2–7% | ||
| 3 | 304C >T5 | R102C | M | 2–7% | ||
| In 3 | 5′border In3 G >A63 | Splice | M | <3% | ||
| 4 | 354G >A5 | Silent | M | |||
| In 4 | 414 + 1G >A68 | Splice abolished | M | <3% | ||
| 5 | 420T >C568 | Silent | M | SNP5,42 | ||
| 546C >T | Silent | M | ||||
| 6 | 577C >T68 | R193W | M | <3% | 325% of wildtype68 | |
| 6 | 582C >T5 | Silent | M | |||
| 6 | 587C >T5 | T196I | M | 2–7% | ||
| 6 | 607T >C100 | S203P | M | <5% | ||
| In 6 | 686 + 1 >A68 or | Splice | M | <3% | ||
| 686 + 4T >G5 | ||||||
| 7 | 695T >A | L232Q | M | 2–6%70 | ||
| 7 | 703G >C58 | D235H | M | |||
| 7 | A250V63 | M | <3% | Decreased63 | ||
| 7 | 718–724Δ58 | Del G + C | M | |||
| 7 | 788C >G70 | S263C | M | <2–5% | ||
| 7 | 803G >C42 | R268P | M | <3%42; <5%100; in vitro: no activity42 | No42,68 | |
| 8 | 932G >A58 | C311Y | Dis | |||
| In 8 | 987 + 11C >T5 or+69C >T5 | Splice | Dis | |||
| 9 | 1058C >T58 | P353L | Dis | |||
| In 9 | 1092 +67G >A5 | Splice | Dis | |||
| 10 | 1169G >A70 | W390X | Tsp1–1 | |||
| 10 | 1170G >C101 | W390C | Tsp1–1 | <3% | ||
| 10 | 1193G >A5 | R398H | Tsp1–1 | 2–7% | ||
| In 10 | 1244 +2T >G68 | Splice abolished | Tsp1–1 | <3% | ||
| In 10 | 1245–32C >G5 | Splice | Tsp1–1 | |||
| 12 | 1342C >G5 | Q448E | Cys | <3%5,69 ; in vitro, full activity with↑ lower molecular weight vWF42 | yes42 | SNP5,42 |
| 12 | 1345C >T42 | Q449X | Cys | <3%69; in vitro: very low activity42 | yes42 | |
| 12 | 1370CT58 | P457L | Cys | |||
| 1423C >T5,42 | P475S | Cys | <3%5,42,69; in vitro: very low activity, with ↑ lower MW vWF subunits42 | yes42 | SNP5,42 | |
| 13 | 1520G >A | R507Q | Cys | <5%100 | ||
| 1523G >A | C508Y | Cys | <3%42; In vitro: no activity42 | No42,68 | ||
| 13 | 1582A >G | R528G | Cys | 2–7%5 | ||
| In 13 | 1584 +5G >A5, or +106C >G5, or + 236T >C5 | Splice | Cys | 2–7% | ||
| 15 | 1716G >A5 | Silent | Cys | SNP5,42 | ||
| 15 | 1783–1784 TT del71 | Frameshift | Sp | <0.1 U/mL | ||
| In 15 | 1787–26G >A5 | Splice | Sp | |||
| 16 | 1787C >T100 | A596V | Sp | <5% | ||
| 16 | 1852C >G5 | P618A | Sp | SNP57 | ||
| 16 | 1874G >A5 | R625H | Sp | SNP5 | ||
| 17 | 2017A >T | I673F | Sp | <3% | No68 | |
| 17 | 2074C >T5 | R692C | Tsp1–2 | 2–7% | ||
| 18 | 2195C >T5 | A732V | Tsp1–2 | SNP5,57 | ||
| 19 | 2272T >C100 | C758R | Tsp1–3 | <5% | ||
| 19 | 2279delG58 | Frameshift | Tsp1–3 | |||
| 19 | 2280 C >T68 | Silent | Tsp1–3 | SNP5,57 | ||
| 19 | 2376del26 (2376–2401Δ)5 | Frameshift | Tsp1–3 | 2–7% | ||
| 20 | 2549–2550delAT70 | Frameshift | Tsp1–4 | |||
| 21 | 2699C >T5 | A900V | Tsp1–5 | SNP5 | ||
| 21 | 2723G >A68 | C908Y | Tsp1–5 | <3% | No68 | |
| 21 | 2728C >T70 | R910X | Tsp1–5 | |||
| 22 | 2851T >G5 | C951G | Tsp1–5 | 2–7% | ||
| In 22 | 2861 +55C >T5 | Splice | Tsp1–6 | |||
| 23 | 2910C >T5 | Silent | Tsp1–6 | |||
| 24 | 3070T >G5 | C1024G | Tsp1–7 | 2–7% | ||
| 24 | 3097G >A5 | A1033T | Tsp1–7 | SNP5 | ||
| 24 | 3100A >T70 | R1034X | Tsp1–7 | <2% | ||
| 24 | 3108G >A5 | Silent | Tsp1–7 | |||
| 25 | Del 3252–3253CT | R1096X | Tsp1–8 | <5%100 | ||
| 25 | C3367T68 | R1123C | Tsp1–8 | <3% | No68 | |
| 26 | 3638G >A5 | C1213Y | CUB1 | 2–7%5 | ||
| 27 | 3735G >A101 | W1245X | CUB1 | <3% | ||
| 27 | 3769–3770insT5 | Frameshift | CUB1 | 2–7% | ||
| 28 | 4006C >T57 | R1336W | CUB2 | |||
| In 28 | 4077 +32T >C5 | Splice | CUB2 | |||
| 4143–4144insA60 | Frameshift | CUB2 | <10%; in vitro activity equals wildtype60 | 14% of wildtype60 | ||
| 29 | 4221 C >A60 | Silent | CUB2 | SNP5,42 |
The missense (change in amino acid residue), nonsense (introduction of premature stop), frameshift/deletion, alternatively splicing and silent (no amino acid change) mutations are indicated in column 3.
SNP, single nucleotide polymorphism; SP, signal peptide; ProP, propeptide; M, metalloprotease, Dis, disintegrin; Tsp, thrombospondin; Cys, cysteine-rich domain; Sp, spacer. The superscript numbers in columns 2 through 7 are reference citations.
Half of the patients with familial TTP experience their acute episodes during childhood and half during adulthood.62 In the latter patients, these episodes may be accompanied by pregnancy, infection, or other conditions that may result in the production of inciting factors.62 Although most of these mutations have been identified in patients with familial TTP, it is unclear if they also contribute to acquired TTP, which represents the majority of TTP cases.
Several missense mutations have been characterized by site-directed mutagenesis of the ADAMTS13 gene, followed by transfection into different expression systems, and assaying for their biosynthesis, secretion, and activity in the conditioned medium. Several recombinant mutants including R193W,59 A250V, R268P, and C508Y42 exhibit low or no secretion and little activity in the conditioned medium of transfected cells. The R193W59 and the A250V63 mutations exhibited decreased secretion as compared with the wild type ADAMTS13. The R193W59 is close to the active site of the metalloprotease domain, whereas the A250V is near the Met249 necessary for Zn2+ binding, suggesting that the small amount of protease secreted may also have compromised proteolytic activity. Similarly, the R268P substitution impairs secretion of the mutant protein from cells42,59 perhaps due to the disruption of the α-helix structure, which may account for its diminished activity in plasma. On the other hand, the recombinant P475S is secreted normally in transfected cells, but shows low proteolytic activity toward vWF.42 Interestingly, approximately 9.6% of Japanese may be heterozygous for this polymorphism, which may be responsible for reduced protease activity.42 However, despite of a high prevalence of P475S in the Japanese population, such a mutation with low proteolytic activity is rare in whites (0.5%)64 and in Chinese (1.7%).65 Similarly, the nonsense mutation Q449X is also secreted normally in transfected cells, but has very low activity,42 consistent with reports that the spacer domain is necessary for ADAMTS13 activity.40,41,49,66
Another recombinant missense, Q448E, which is secreted normally, has been reported as a single nucleotide polymorphism (SNP),5,42 although it retains full activity in transfected cells.42 Although numerous SNPs have been reported (Table 2), none have yet shown a clear relationship to TTP,28 although it is possible they may be associated with other conditions, such as coronary artery disease, in which ADAMTS13 activity is decreased.67
Several frameshift mutations have been identified, and these aberrantly carboxyl-terminal truncated mutants may demonstrate either impaired secretion and/or low proteolytic activity. The 4143 to 4144insA mutation that deletes the second CUB domain exhibits impaired secretion, but retains proteolytic activity if the same amount of protein is tested.60 Several ADAMTS13 gene mutations have been identified at splice sites, which disrupt the conventional GT-AG consensus splice sequences, and prevent production of normally spliced transcripts.5,68 Such alternatively spliced forms of ADAMTS13 are present in normal human plasma,2 although the biological functions of these variants are not known.
The in vitro expression studies indicate that ADAMTS13 mutations exhibit a heterogeneous phenotype, depending on the effects on biosynthesis, secretion, and protease bioactivity. These reflect the clinical heterogeneity seen among hereditary TTP patients, most of whom harbor compound heterozygous mutations. The parents of hereditary TTP patients (asymptomatic carriers) have about a 50% level of ADAMTS13 activity, although some asymptomatic carriers exhibit very low levels of ADAMTS13 activity (less than 10% of normal) who are compound heterozygotes such as R268P/P475S.69 Of note, most of the described homozygous mutations, such as R692C,5 Q449X,42 414 + 1G >A splice,68 L232Q,70 and 1783delTT,71 have some evidence of a consanguineous pedigree, and there are very few null alleles, suggesting that a complete lack of ADAMTS13 protease may be lethal.5,13,28,42,59
DIAGNOSTIC UTILITY OF ADAMTS13 ASSAYS
ADAMTS13 activity in normal plasma is reported to be 50 to 178%.14 Severe deficiency of plasma ADAMTS13 activity toward vWF has been considered to be the primary cause of congenital and acquired idiopathic TTP,5,10,11,72,73 although mildly to moderately reduced levels of ADAMTS13 have been observed in conditions such as inflammation, coronary artery disease,67 pregnancy, liver disease,74 disseminated intravascular coagulation,75 sepsis, and heparin-induced thrombocytopenia.76 The clinical utility of the ADAMTS13 proteolytic activity assays remains the subject of considerable debate in recent years,77–79 partly due to the variability of each assay. The assays can be categorized into direct versus indirect and static versus flow based, depending on the reaction conditions and how the cleavage of the substrate is determined.
Direct Assays
Direct assays focus on detection of cleavage products of a given substrate, whether it is a macromolecular vWF, a vWF A2 domain, or the small peptide GST-vWF-73 or FRETS-73. Agarose or polyacrylamide gel electrophoresis (PAGE), Western blotting, and FRETS techniques have been used to detect the cleavage products of the substrate. The FRETS-vWF73 detects proteolytic cleavage of the specific Tyr-Met peptide bond by ADAMTS13 in real time. By far, this assay may be the most sensitive and promising of all.
SDS-AGAROSE GEL ELECTROPHORESIS AND WESTERN BLOTTING
This assay was initially developed by Furlan et al8 to measure ADAMTS13 activity in plasma. After purified vWF is incubated with citrated plasma samples for approximately 24 hours, agarose gel electrophoresis and Western blotting with peroxidase-conjugated anti-vWF antibody are used to determine the proteolytic degradation of purified vWF multimers. The relative amount of ADAMTS13 activity is calculated from the reference of serially diluted normal human plasma.
This assay laid a strong foundation for understanding the pathogenesis of congenital or familial TTP72,80–82 and acquired TTP.80 It has also provided a tool to monitor the proteolytic activity of each fraction during the subsequent successful purification of ADAMTS13 protease from normal human plasma.2 However, this assay is very complex and requires technical expertise in handling agarose gel electrophoresis, transfer and Western blotting.
SDS-PAGE AND WESTERN BLOTTING
Tsai et al9,10,83 initially developed this assay based on detection of dimeric vWF fragments of 176 and 140 kd on denatured, but unreduced SDS-PAGE and Western blotting. This assay differs from the SDS-agarose method developed above by Furlan et al8,72,80 in these ways: (1) no barium chloride activation is required; (2) the incubation is relatively short (1 to 3 hours at 37°C); (3) it is easier to handle gels from SDS-PAGE than agarose-gel electrophoresis; and (4) there is direct visualization of the cleavage product, eliminating the possibility of nonspecific degradation of the multimers. This SDS-PAGE assay detected severe deficiency of ADAMTS13 activity (<5% of normal) in all 39 samples from 37 patients with idiopathic TTP.10 In addition, the inhibitory activity against ADAMTS13 protease was detected in 26 of 39 samples (67%).10 This assay is sensitive enough to differentiate patients with congenital TTP from asymptomatic carriers and healthy individuals, and allows for the precise mapping of the defective ADAMTS13 gene.5
Alternatively, the epitope-tagged vWF fragment (His-A2 domain or GST-vWF73) could be digested by plasma ADAMTS13 within 1 to 3 hours.49,50,84 The cleavage of the Tyr-Met bond is determined by Western blotting with an anti-His84 or anti-GST49,50 antibody. The assay using the vWF A2 domain detected 13 of the 16 samples (81%) with an activity of <6%.84 The GST-vWF73 substrate is not cleaved by plasma from patients with congenital or acquired TTP, but is cleaved by plasma from patients with hemolytic uremic syndrome, suggesting that GST-vWF73 is a specific substrate for ADAMTS13. However, truncated ADAMTS13 mutants after the Cys-rich domain may cleave GST-vWR73.49 Therefore, normal proteolytic cleavage does not rule out the possibility of the functional abnormality of ADAMTS13 protein due to mutations or acquired autoantibodies that are directed against the middle and distal carboxyl terminus. Again, the cleavage of a small recombinant domain or a peptidyl substrate does not require denaturing reagents and is free of contaminating ADAMTS13 in the substrate.
FRETS ASSAY
FRETS-vWF73 is a chemically modified vWF73 derived from the central A2 domain of vWF (D1596-R1668) containing A2pr (Nma) and A2Pr (Dnp; Fig. 2). It is readily cleaved by normal human plasma, but not by ADAMTS13-deficient plasma.85 It could differentiate congenital and acquired TTP from patients with hemolytic uremic syndrome (HUS) and heterozygous mutations of ADAMTS13 gene or healthy individuals.85 The proteolytic cleavage of the FRETS-vWF73 is completely blocked by ethylenediaminetetraacetic acid.85 Compared with the collagen-binding assay (described below) and the GST-vWF73 assay (described above), FRETS-vWF73 is superior. It is sensitive and specific, requiring only 2 to 5 μL of citrated plasma for each activity assay, and the cleavage of the substrate can be monitored in real time with a result in ≈1 hour.85 The FRETS-vWF73 can identify more TTP patients with severe ADAMTS13 deficiency and positive inhibitor, which can be verified independently by Technozyme-ELISA (Baxter, Orth, Austria) and immunoprecipitation and Western blotting methods (S. Shelat, P. Smith, J. Ai, and X. Zheng, unpublished data, 2005).
Figure 2.
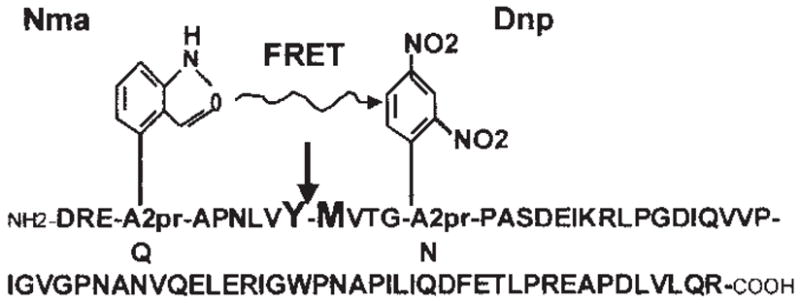
Schematic representation of fluorescent resonance energy transfer (FRETS)-von Willebrand factor (vWF) 73 substrate. The amino acid residues Q1599 and N1610 within the vWF peptide (D1595-R1668) are replaced by A2pr (Nma) and A2pr (Dnp), respectively. When the Nma group is excited at 340 nm, fluorescence resonance energy is transferred to the neighboring quencher, Dnp. If the Tyr-Met peptide bond (indicated by arrow) is cleaved by ADAMTS13, the energy transfer quenching the fluorescence does not occur, allowing the emission of fluorescence at 440 nm from Nma (Adapted from Kokame K, Nobe Y, Kokubo Y, et al. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol 2005;129:93–100.)
Indirect Assays
Indirect assays depend on measuring the residual substrate or disappearance of the substrates. This type of assay may be influenced by non-specific proteolysis of vWF by other enzymes in plasma, unless they are inactivated by incubation of plasma with Pefobloc73,80,81,86 or other serine protease inhibitors.17,87
COLLAGEN-BINDING ASSAY
Initially described by Gerritsen et al88 and modified by many others,17,74,87,89 this assay is based on preferential binding of large vWF multimers to type III human collagen. After incubation of purified vWF or ADAMTS13-depleted plasma with patient’s plasma or normal human plasma (as control) in the presence of 5 mM Tris-HCl (pH 8.0), 1.5 M urea, and 10 mM BaCl2 for 16 to 24 hours, the amount of residual vWF is determined by binding to human collagen type III. Bound vWF is quantified by peroxidase-conjugated anti-vWF immunoglobulin G (IgG) on an ELISA plate, followed by a densitometry measurement.17,88–90
This assay is relatively straightforward to perform and can accommodate many samples simultaneously. It is quite robust in the detection of severe deficiency of ADAMTS13 (16 of 20 patients) and high titer inhibitor (seven of 16 patients) in idiopathic TTP.17 It correlates with other indirect assays well.91 The presence of inhibitor detected by this assay correlates with more relapses,17,87 whereas patients with severe ADAMTS13 deficiency but no detectable inhibitor have a better response and outcome,17 suggesting that the assay may be used for making a diagnosis, monitoring therapy, and predicting outcome.
RISTOCETIN-INDUCED AGGREGATION
After digestion of the purified vWF in the buffer (similar to that in collagen-binding assay), the residual vWF is determined by a ristocetin cofactor activity in a platelet aggregometer. The method is reproducible as shown by low intra- and interassay coefficients of variation (2.8% and 7.5% for normal samples, respectively, and 8.7% and 12.9% for abnormal samples, respectively).92 Furthermore, it detects severe ADAMTS13 deficiency in patients with acute, classic TTP. The majority of patients with low titer inhibitor respond to plasma exchange treatment with increases in ADAMTS13 activity, whereas ADAMTS13 deficiency persists in some patients with high titer inhibitor despite clinical remission.92 This assay has not been widely used because only limited samples can be analyzed at one time on the aggregometer.
FLOW-BASED ASSAYS
Two types of assays use flow conditions for measuring ADAMTS13 activity. First, Shenkman et al93 used a cone and plate(let) analyzer to evaluate the ability of TTP plasma to increase deposition on a polystyrene surface from normal plasma under flow conditions. This assay is sensitive and specific for detection of ADAMTS13 deficiency and inhibitors.93 Increased deposition of normal platelets was observed following the addition of plasma in all 15 acute TTP patients, but only in three of 14 patients in remission.93 The IgG from plasma or serum of three of five acute TTP patients enhanced platelet deposition,93 suggesting the possibility of detecting autoantibodies against ADAMTS13 by this method. Second, Dong et al94 described that ULvWF multimers, upon release from cultured endothelial cells, form extremely long, platelet-decorated string-like structures on the surface of endothelial cells under fluid shear stress. When perfused with normal human plasma, the string-like structures disappear rapidly within a few minutes, indicating the cleavage of ULvWF by ADAMTS13. Under flow conditions, cleaving ULvWF occurs at 1000-fold faster kinetics than that observed under static conditions. However, plasma from patients with idiopathic TTP does not cleave these platelet-decorated strings.
ENZYME-LINKED IMMUNOSORBENT ASSAY
This assay uses epitope-tagged recombinant fragment (vWF A2 domain or vWF73) as a substrate.84,95 The substrate is immobilized to a microtiter plate with an antibody to one tag. After incubation of the immobilized substrate with plasma samples, an antibody to another tag detects the residual substrate. The proteolytic activity of ADAMTS13 is inversely proportional to the residual substrate concentration. This type of assay is relatively simple and sensitive. For example, the ADAMTS13 activity in patients with TTP detected by vWF A2 domain-ELISA is 2.4 ± 0.7%, compared with 40 ± 4.2% in normal human plasma.84 The vWF73-ELISA can differentiate TTP patients from carriers of ADAMTS13 gene mutation and normal healthy individuals.95
The ADAMTS13 Inhibitor Assay
ADAMTS13 autoantibodies may be classified into two groups: inhibitory antibodies or noninhibitory antibodies, depending on whether the autoantibodies block proteolytic cleavage of vWF (or other peptidyl substrate) in an in vitro assay. Antibodies that react proximally to ADAMTS13 may block proteolytic activity in vitro, whereas antibodies that target to the middle and distal carboxyl terminus of ADAMTS13 may not, based on the structure-function analysis of ADAMTS13.40,41 Thus far, all inhibitory antibodies react with the Cys-rich and spacer domains with some specificity toward the catalytic domain and other parts of the molecule,96 supporting the critical function of the Cys-rich and spacer domains in substrate recognition.40,41,49
All of the functional assays for ADAMTS13 activity are capable of detecting the inhibitory autoantibodies with a wide range of sensitivities. The FRETS-vWF73 appears, by far, the most sensitive test among three assays we evaluated (collagen-binding assay, GST-vWF73, and FRETS-vWF73) to identify the inhibitory autoantibodies (S. Shelat, P. Smith, J. Ai, and X. Zheng, unpublished observation, 2005). An ELISA assay along with Western blotting using the recombinant ADAMTS13 as an antigen has been developed recently, which can detect both inhibitory and noninhibitory autoantibodies.97 IgG antibodies are found in 97% of untreated patients with acute acquired TTP whose plasma levels of ADAMTS13 are below 10%.97 The corresponding prevalence of IgM antibodies is 11%.
The presence of ADAMTS13 autoantibodies are rather specific for making a diagnosis of acquired TTP.10,78 In addition, a high titer of inhibitory autoantibodies correlates with more relapses. Adjunct immune therapies such as rituximab, an anti-CD20 chimeric monoclonal antibody, or cyclophosphamide, may be considered once inhibitory autoantibodies are identified in patients with acute TTP who do not adequately respond to plasma exchange or are chronically relapsed.17,87,98,99 Therefore, a robust ADAMTS13 inhibitor assay is critical for understanding of the mechanism of TTP and for tailoring therapy.
In conclusion, molecular cloning and characterization of ADAMTS13 gene and protein structure have opened a new avenue for study of the biology and biochemistry of the ADAMTS13 protease. Development of a sensitive and specific assay for ADAMTS13 activity and inhibitor would not only help to understand the pathogenesis of TTP and other thrombotic disorders, but also to facilitate a more timely and accurate clinical diagnosis, which is crucial for initiating and tailoring therapy in patients with TTP.
Acknowledgments
This work is supported in part by grants from National Institute of Health (HL 079027 to XZ and HL 075246 to SS), American Heart Association Begin grant-in Aid, and National Blood Foundation.
Footnotes
Thrombotic Thrombocytopenic Purpura—2005; Editor in Chief, Eberhard F. Mammen, M.D.; Guest Editors, Hau C. Kwaan, M.D., Ph.D., Charles L. Bennett, M.D., Ph.D. Seminars in Thrombosis and Hemostasis, volume 31, number 6, 2005.
Objectives: On completion of the article, the reader should be able to (1) understand certain aspects of ADAMTS13 biology underlying thrombotic thrombocytopenic purpura, and (2) be familiar with current assays and clinical utility of ADAMTS13 activity and inhibitors.
Accreditation: Tufts University School of Medicine (TUSM) is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Credit: TUSM designates this educational activity for a maximum of 1 category 1 credit toward the AMA Physicians Recognition Award. Each physician should claim only those credits that he/she actually spent in the activity.
References
- 1.Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98:1662–1666. doi: 10.1182/blood.v98.6.1662. [DOI] [PubMed] [Google Scholar]
- 2.Gerritsen HE, Robles R, Lämmle B, Furlan M. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 2001;98:1654–1661. doi: 10.1182/blood.v98.6.1654. [DOI] [PubMed] [Google Scholar]
- 3.Soejima K, Mimura N, Hirashima M, et al. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? J Biochem (Tokyo) 2001;130:475–480. doi: 10.1093/oxfordjournals.jbchem.a003009. [DOI] [PubMed] [Google Scholar]
- 4.Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 5.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 6.Cal S, Obaya AJ, Llamazares M, et al. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene. 2002;283:49–62. doi: 10.1016/s0378-1119(01)00861-7. [DOI] [PubMed] [Google Scholar]
- 7.Plaimauer B, Zimmermann K, Volkel D, et al. Cloning, expression, and functional characterization of the von Willebrand factor-cleaving protease (ADAMTS13) Blood. 2002;100:3626–3632. doi: 10.1182/blood-2002-05-1397. [DOI] [PubMed] [Google Scholar]
- 8.Furlan M, Robles R, Lämmle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–4234. [PubMed] [Google Scholar]
- 9.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–4244. [PubMed] [Google Scholar]
- 10.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–1594. doi: 10.1056/NEJM199811263392203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 12.Furlan M. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. Adv Nephrol Necker Hosp. 2000;30:71–81. [PubMed] [Google Scholar]
- 13.Zheng X, Majerus EM, Sadler JE. ADAMTS13 and TTP. Curr Opin Hematol. 2002;9:389–394. doi: 10.1097/00062752-200209000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Veyradier A, Obert B, Houllier A, et al. Specific von Willebrand factor-cleaving protease in thrombotic micro-angiopathies: a study of 111 cases. Blood. 2001;98:1765–1772. doi: 10.1182/blood.v98.6.1765. [DOI] [PubMed] [Google Scholar]
- 15.Veyradier A, Obert B, Haddad E, et al. Severe deficiency of the specific von Willebrand factor-cleaving protease (ADAMTS 13) activity in a subgroup of children with atypical hemolytic uremic syndrome. J Pediatr. 2003;142:310–317. doi: 10.1067/mpd.2003.79. [DOI] [PubMed] [Google Scholar]
- 16.Vesely SK, George JN, Lämmle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102:60–68. doi: 10.1182/blood-2003-01-0193. [DOI] [PubMed] [Google Scholar]
- 17.Zheng XL, Richard KM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and non-idiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103:4043–4049. doi: 10.1182/blood-2003-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–839. doi: 10.5858/2003-127-834-TTPAHU. [DOI] [PubMed] [Google Scholar]
- 19.Shepard KV, Bukowski RM. The treatment of thrombotic thrombocytopenic purpura with exchange transfusions, plasma infusions, and plasma exchange. Semin Hematol. 1987;24:178–193. [PubMed] [Google Scholar]
- 20.Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–397. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- 21.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325:398–403. doi: 10.1056/NEJM199108083250605. [DOI] [PubMed] [Google Scholar]
- 22.Plaimauer B, Scheiflinger F. Expression and characterization of recombinant human ADAMTS-13. Semin Hematol. 2004;41:24–33. doi: 10.1053/j.seminhematol.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Kremer Hovinga JA, Studt JD, Lämmle B. The von Willebrand factor-cleaving protease (ADAMTS-13) and the diagnosis of thrombotic thrombocytopenic purpura (TTP) Pathophysiol Haemost Thromb. 2003;33:417–421. doi: 10.1159/000083839. [DOI] [PubMed] [Google Scholar]
- 24.Tsai HM. Molecular mechanisms in thrombotic thrombo-cytopenic purpura. Semin Thromb Hemost. 2004;30:549–557. doi: 10.1055/s-2004-835675. [DOI] [PubMed] [Google Scholar]
- 25.Moake JL. von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Hematol. 2004;41:4–14. doi: 10.1053/j.seminhematol.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology (Am Soc Hematol Educ Program) 2004;1:407–423. doi: 10.1182/asheducation-2004.1.407. [DOI] [PubMed] [Google Scholar]
- 27.George JN, Sadler JE, Lämmle B. Platelets: thrombotic thrombocytopenic purpura. Hematology (Am Soc Hematol Educ Program) 2002;1:315–334. doi: 10.1182/asheducation-2002.1.315. [DOI] [PubMed] [Google Scholar]
- 28.Kokame K, Miyata T. Genetic defects leading to hereditary thrombotic thrombocytopenic purpura. Semin Hematol. 2004;41:34–40. doi: 10.1053/j.seminhematol.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Veyradier A, Girma JP. Assays of ADAMTS-13 activity. Semin Hematol. 2004;41:41–47. doi: 10.1053/j.seminhematol.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Furlan M. Deficient activity of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura. Expert Rev Cardiovasc Ther. 2003;1:243–255. doi: 10.1586/14779072.1.2.243. [DOI] [PubMed] [Google Scholar]
- 31.Bork P, Beckmann G. The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol. 1993;231:539–545. doi: 10.1006/jmbi.1993.1305. [DOI] [PubMed] [Google Scholar]
- 32.Bruno K, Volkel D, Plaimauer B, et al. Cloning, expression and functional characterization of the full-length murine ADAMTS13. J Thromb Haemost. 2005;3:1064–1073. doi: 10.1111/j.1538-7836.2005.01246.x. [DOI] [PubMed] [Google Scholar]
- 33.Banno F, Kaminaka K, Soejima K, et al. Identification of strain-specific variants of mouse Adamts13 gene encoding von Willebrand factor-cleaving protease. J Biol Chem. 2004;279:30896–30903. doi: 10.1074/jbc.M314184200. [DOI] [PubMed] [Google Scholar]
- 34.Uemura M, Tatsumi K, Matsumoto M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–924. doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 35.Zhou W, Inada M, Lee TP, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005;85:780–788. doi: 10.1038/labinvest.3700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blomhoff R, Wake K. Perisinusoidal stellate cells of the liver: important roles in retinol metabolism and fibrosis. FASEB J. 1991;5:271–277. doi: 10.1096/fasebj.5.3.2001786. [DOI] [PubMed] [Google Scholar]
- 37.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 38.Okazaki I, Watanabe T, Hozawa S, et al. Molecular mechanism of the reversibility of hepatic fibrosis: with special reference to the role of matrix metalloproteinases. J Gastroenterol Hepatol. 2000;15(suppl):D26–D32. doi: 10.1046/j.1440-1746.2000.02185.x. [DOI] [PubMed] [Google Scholar]
- 39.Majerus EM, Zheng X, Tuley EA, Sadler JE. Cleavage of the ADAMTS13 propeptide is not required for protease activity. J Biol Chem. 2003;278:46643–46648. doi: 10.1074/jbc.M309872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278:30136–30141. doi: 10.1074/jbc.M305331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soejima K, Matsumoto M, Kokame K, et al. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood. 2003;102:3232–3237. doi: 10.1182/blood-2003-03-0908. [DOI] [PubMed] [Google Scholar]
- 42.Kokame K, Matsumoto M, Soejima K, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci USA. 2002;99:11902–11907. doi: 10.1073/pnas.172277399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. 1998;273:13912–13917. doi: 10.1074/jbc.273.22.13912. [DOI] [PubMed] [Google Scholar]
- 44.Kuno K, Terashima Y, Matsushima K. ADAMTS-1 is an active metalloproteinase associated with the extracellular matrix. J Biol Chem. 1999;274:18821–18826. doi: 10.1074/jbc.274.26.18821. [DOI] [PubMed] [Google Scholar]
- 45.Loechel F, Gilpin BJ, Engvall E, et al. Human ADAM 12 (meltrin alpha) is an active metalloprotease. J Biol Chem. 1998;273:16993–16997. doi: 10.1074/jbc.273.27.16993. [DOI] [PubMed] [Google Scholar]
- 46.Milla ME, Leesnitzer MA, Moss ML, et al. Specific sequence elements are required for the expression of functional tumor necrosis factor-alpha-converting enzyme (TACE) J Biol Chem. 1999;274:30563–30570. doi: 10.1074/jbc.274.43.30563. [DOI] [PubMed] [Google Scholar]
- 47.Cao J, Hymowitz M, Conner C, et al. The propeptide domain of membrane type 1-matrix metalloproteinase acts as an intramolecular chaperone when expressed in trans with the mature sequence in COS-1 cells. J Biol Chem. 2000;275:29648–29653. doi: 10.1074/jbc.M001920200. [DOI] [PubMed] [Google Scholar]
- 48.Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Ib alpha to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci USA. 2004;101:10578–10583. doi: 10.1073/pnas.0402041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ai J, Smith P, Wang S, et al. The proximal carboxyl terminal domains of ADAMTS13 determine substrate specificity and are all required for cleavage of von Willebrand factor. J Biol Chem. 2005;280:29428–29434. doi: 10.1074/jbc.M505513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kokame K, Matsumoto M, Fujimura Y, Miyata T. VWF73, a region from D1596 to R1668 of von Willebrand factor, provides a minimal substrate for ADAMTS-13. Blood. 2003;103:607–612. doi: 10.1182/blood-2003-08-2861. [DOI] [PubMed] [Google Scholar]
- 51.Tsai HM. Shear stress and von Willebrand factor in health and disease. Semin Thromb Hemost. 2003;29:479–488. doi: 10.1055/s-2003-44556. [DOI] [PubMed] [Google Scholar]
- 52.De Cristofaro R, Peyvandi F, Palla R, et al. Role of chloride ions in the modulation of the interaction between von Willebrand factor and ADAMTS-3. J Biol Chem. 2005;280:23295–23302. doi: 10.1074/jbc.M501143200. [DOI] [PubMed] [Google Scholar]
- 53.Bernardo A, Ball C, Nolasco L, et al. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104:100–106. doi: 10.1182/blood-2004-01-0107. [DOI] [PubMed] [Google Scholar]
- 54.Jenkins PV, Pasi KJ, Perkins SJ. Molecular modeling of ligand and mutation sites of the type A domains of human von Willebrand factor and their relevance to von Willebrand’s disease. Blood. 1998;91:2032–2044. [PubMed] [Google Scholar]
- 55.Studt JD, Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105:542–544. doi: 10.1182/blood-2004-06-2096. [DOI] [PubMed] [Google Scholar]
- 56.Crawley JT, Lam JK, Rance JB, et al. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. 2005;105:1085–1093. doi: 10.1182/blood-2004-03-1101. [DOI] [PubMed] [Google Scholar]
- 57.Antoine G, Zimmermann K, Plaimauer B, et al. ADAMTS13 gene defects in two brothers with constitutional thrombotic thrombocytopenic purpura and normalization of von Willebrand factor-cleaving protease activity by recombinant human ADAMTS13. Br J Haematol. 2003;120:821–824. doi: 10.1046/j.1365-2141.2003.04183.x. [DOI] [PubMed] [Google Scholar]
- 58.Assink K, Schiphorst R, Allford S, et al. Mutation analysis and clinical implications of von Willebrand factor-cleaving protease deficiency. Kidney Int. 2003;63:1995–1999. doi: 10.1046/j.1523-1755.63.6s.1.x. [DOI] [PubMed] [Google Scholar]
- 59.Matsumoto M, Kokame K, Soejima K, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. 2003;103:1305–1310. doi: 10.1182/blood-2003-06-1796. [DOI] [PubMed] [Google Scholar]
- 60.Pimanda JE, Maekawa A, Wind T, et al. Congenital thrombotic thrombocytopenic purpura in association with a mutation in the second CUB domain of ADAMTS13. Blood. 2004;103:627–629. doi: 10.1182/blood-2003-04-1346. [DOI] [PubMed] [Google Scholar]
- 61.Soejima K, Nakagaki T. Interplay between ADAMTS13 and von Willebrand factor in inherited and acquired thrombotic microangiopathies. Semin Hematol. 2005;42:56–62. doi: 10.1053/j.seminhematol.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 62.Furlan M, Lämmle B. Aetiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome: the role of von Willebrand factor-cleaving protease. Best Pract Res Clin Haematol. 2001;14:437–454. doi: 10.1053/beha.2001.0142. [DOI] [PubMed] [Google Scholar]
- 63.Uchida T, Wada H, Mizutani M, et al. Identification of novel mutations in ADAMTS13 in an adult patient with congenital thrombotic thrombocytopenic purpura. Blood. 2004;104:2081–2083. doi: 10.1182/blood-2004-02-0715. [DOI] [PubMed] [Google Scholar]
- 64.Bongers TN, De Maat MP, Dippel DW, et al. Absence of Pro475Ser polymorphism in ADAMTS-13 in Caucasians. J Thromb Haemost. 2005;3:805. doi: 10.1111/j.1538-7836.2005.01239.x. comment. [DOI] [PubMed] [Google Scholar]
- 65.Ruan C, Dai L, Su J, Wang Z. The frequency of P475S polymorphism in von Willebrand factor-cleaving protease in the Chinese population and its relevance to arterial thrombotic disorders. Thromb Haemost. 2004;91:1257–1258. [PubMed] [Google Scholar]
- 66.Majerus EM, Anderson PJ, Sadler JE. Binding of ADAMTS13 to von Willebrand factor. J Biol Chem. 2005;280:21773–21778. doi: 10.1074/jbc.M502529200. [DOI] [PubMed] [Google Scholar]
- 67.Yoo G, Blomback M, Schenck-Gustafsson K, He S. Decreased levels of von Willebrand factor-cleaving protease in coronary heart disease and thrombotic thrombocytopenic purpura: study of a simplified method for assaying the enzyme activity based on ristocetin-induced platelet aggregation. Br J Haematol. 2003;121:123–129. doi: 10.1046/j.1365-2141.2003.04257.x. [DOI] [PubMed] [Google Scholar]
- 68.Matsumoto M, Kokame K, Soejima K, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. 2004;103:1305–1310. doi: 10.1182/blood-2003-06-1796. [DOI] [PubMed] [Google Scholar]
- 69.Kinoshita S, Yoshioka A, Park YD, et al. Upshaw-Schulman syndrome revisited: a concept of congenital thrombotic thrombocytopenic purpura. Int J Hematol. 2001;74:101–108. doi: 10.1007/BF02982558. [DOI] [PubMed] [Google Scholar]
- 70.Schneppenheim R, Budde U, Oyen F, et al. von Willebrand factor cleaving protease and ADAMTS13 mutations in childhood TTP. Blood. 2003;101:1845–1850. doi: 10.1182/blood-2002-08-2399. [DOI] [PubMed] [Google Scholar]
- 71.Savasan S, Lee SK, Ginsburg D, Tsai HM. ADAMTS13 gene mutation in congenital thrombotic thrombocytopenic purpura with previously reported normal VWF cleaving protease activity. Blood. 2003;101:4449–4451. doi: 10.1182/blood-2002-12-3796. [DOI] [PubMed] [Google Scholar]
- 72.Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339:1578–1584. doi: 10.1056/NEJM199811263392202. [DOI] [PubMed] [Google Scholar]
- 73.Furlan M, Robles R, Solenthaler M, Lämmle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91:2839–2846. [PubMed] [Google Scholar]
- 74.Mannucci PM, Canciani MT, Forza I, et al. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001;98:2730–2735. doi: 10.1182/blood.v98.9.2730. [DOI] [PubMed] [Google Scholar]
- 75.Loof AH, van Vliet HH, Kappers-Klunne MC. Low activity of von Willebrand factor-cleaving protease is not restricted to patients suffering from thrombotic thrombocytopenic purpura. Br J Haematol. 2001;112:1087–1088. doi: 10.1046/j.1365-2141.2001.02622-5.x. [DOI] [PubMed] [Google Scholar]
- 76.Bianchi V, Robles R, Alberio L, et al. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710–713. doi: 10.1182/blood-2002-02-0344. [DOI] [PubMed] [Google Scholar]
- 77.Remuzzi G. Is ADAMTS-13 deficiency specific for thrombotic thrombocytopenic purpura? No. J Thromb Haemost. 2003;1:632–634. doi: 10.1046/j.1538-7836.2003.00170.x. [DOI] [PubMed] [Google Scholar]
- 78.Tsai HM. Is severe deficiency of ADAMTS-13 specific for thrombotic thrombocytopenic purpura? Yes. J Thromb Haemost. 2003;1:625–631. doi: 10.1046/j.1538-7836.2003.00169.x. [DOI] [PubMed] [Google Scholar]
- 79.Mannucci PM. Consistency of ADAMTS-13 activity assays: a moderately optimistic view. J Thromb Haemost. 2003;1:1880–1881. doi: 10.1046/j.1538-7836.2003.00356.x. [DOI] [PubMed] [Google Scholar]
- 80.Furlan M, Robles R, Solenthaler M, et al. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–3103. [PubMed] [Google Scholar]
- 81.Furlan M, Robles R, Morselli B, et al. Recovery and half-life of von Willebrand factor-cleaving protease after plasma therapy in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 1999;81:8–13. [PubMed] [Google Scholar]
- 82.Fujimura Y, Matsumoto M, Yagi H, et al. Von Willebrand factor-cleaving protease and Upshaw-Schulman syndrome. Int J Hematol. 2002;75:25–34. doi: 10.1007/BF02981975. [DOI] [PubMed] [Google Scholar]
- 83.Tsai HM, Sussman II, Ginsburg D, et al. Proteolytic cleavage of recombinant type 2A von Willebrand factor mutants R834W and R834Q: inhibition by doxycycline and by monoclonal antibody VP-1. Blood. 1997;89:1954–1962. [PubMed] [Google Scholar]
- 84.Whitelock JL, Nolasco L, Bernardo A, et al. ADAMTS-13 activity in plasma is rapidly measured by a new ELISA method that uses recombinant VWF-A2 domain as substrate. J Thromb Haemost. 2004;2:485–491. doi: 10.1111/j.1538-7836.2004.00601.x. [DOI] [PubMed] [Google Scholar]
- 85.Kokame K, Nobe Y, Kokubo Y, et al. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129:93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 86.Furlan M, Lammle B. Deficiency of von Willebrand factor-cleaving protease in familial and acquired thrombotic thrombocytopenic purpura. Baillieres Clin Haematol. 1998;11:509–514. doi: 10.1016/s0950-3536(98)80064-4. [DOI] [PubMed] [Google Scholar]
- 87.Zheng X, Pallera AM, Goodnough LT, et al. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med. 2003;138:105–108. doi: 10.7326/0003-4819-138-2-200301210-00011. [DOI] [PubMed] [Google Scholar]
- 88.Gerritsen HE, Turecek PL, Schwarz HP, et al. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP) Thromb Haemost. 1999;82:1386–1389. [PubMed] [Google Scholar]
- 89.Rick ME, Moll S, Taylor MA, et al. Clinical use of a rapid collagen binding assay for von Willebrand factor cleaving protease in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2002;88:598–604. [PubMed] [Google Scholar]
- 90.Owen J. Inappropriate use of positive predictive value in describing the rapid collagen binding assay for von Willebrand factor cleaving protease. Thromb Haemost. 2003;89:768–769. [PubMed] [Google Scholar]
- 91.Tripodi A, Chantarangkul V, Bohm M, et al. Measurement of von Willebrand factor cleaving protease (ADAMTS-13): results of an international collaborative study involving 11 methods testing the same set of coded plasmas. J Thromb Haemost. 2004;2:1601–1609. doi: 10.1111/j.1538-7836.2004.00879.x. [DOI] [PubMed] [Google Scholar]
- 92.Bohm M, Vigh T, Scharrer I. Evaluation and clinical application of a new method for measuring activity of von Willebrand factor-cleaving metalloprotease (ADAMTS13) Ann Hematol. 2002;81:430–435. doi: 10.1007/s00277-002-0502-3. [DOI] [PubMed] [Google Scholar]
- 93.Shenkman B, Inbal A, Tamarin I, et al. Diagnosis of thrombotic thrombocytopenic purpura based on modulation by patient plasma of normal platelet adhesion under flow condition. Br J Haematol. 2003;120:597–604. doi: 10.1046/j.1365-2141.2003.04119.x. [DOI] [PubMed] [Google Scholar]
- 94.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 95.Zhou W, Tsai HM. An enzyme immunoassay of ADAMTS13 distinguishes patients with thrombotic thrombocytopenic purpura from normal individuals and carriers of ADAMTS13 mutations. Thromb Haemost. 2004;91:806–811. doi: 10.1160/TH03-11-0675. [DOI] [PubMed] [Google Scholar]
- 96.Klaus C, Plaimauer B, Studt JD, et al. Epitope mapping of ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. Blood. 2004;103:4514–4519. doi: 10.1182/blood-2003-12-4165. [DOI] [PubMed] [Google Scholar]
- 97.Rieger M, Mannucce P, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood. 2005;106:1262–1267. doi: 10.1182/blood-2004-11-4490. [DOI] [PubMed] [Google Scholar]
- 98.Fakhouri F, Vernant JP, Veyradier A, et al. Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13 deficient-thrombotic thrombocytopenic purpura: a study of 11 cases. Blood. 2005;106:1932–1937. doi: 10.1182/blood-2005-03-0848. [DOI] [PubMed] [Google Scholar]
- 99.Fakhouri F, Teixeira L, Delarue R, et al. Responsiveness of thrombotic thrombocytopenic purpura to rituximab and cyclophosphamide. Ann Intern Med. 2004;140:314–315. doi: 10.7326/0003-4819-140-4-200402170-00028. [DOI] [PubMed] [Google Scholar]
- 100.Veyradier A, Lavergne JM, Ribba AS, et al. Ten candidate ADAMTS13 mutations in six French families with congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome) J Thromb Haemost. 2004;2:424–429. doi: 10.1111/j.1538-7933.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- 101.Licht C, Stapenhorst L, Simon T, et al. Two novel ADAMTS13 gene mutations in thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome (TTP/HUS) Kidney Int. 2004;66:955–958. doi: 10.1111/j.1523-1755.2004.00841.x. [DOI] [PubMed] [Google Scholar]