

Abstract
The early events that occur rapidly after injury trigger signal cascades that are essential for proper wound closure of corneal epithelial cells. We hypothesize that injury releases ATP, which stimulates purinergic receptors and elicits the phosphorylation of epidermal growth factor receptor (EGFR) tyrosine residues and subsequent cell migration by a MMP and HB-EGF dependent pathway. We demonstrated that the inhibition of purinergic receptors with the antagonist, Reactive Blue 2, abrogated the phosphorylation of EGFR and ERK. Preincubation of cells with the EGFR kinase inhibitor, AG1478, and subsequent stimulation by injury or ATP resulted in a decrease in phosphorylation of EGFR and migration. Furthermore, downregulation of EGFR by siRNA, inhibited the EGF induced intracellular Ca2+ wave. However, the response to injury and ATP was retained indicating the presence of 2 signaling pathways. Inhibition with either CRM197 or TIMP-3 decreased injury and nucleotide induced phosphorylation of both EGFR and ERK. Incubation in the presence of a functional blocking antibody to HB-EGF also resulted in a decrease in the phosphorylation of EGFR. In addition, cell migration was inhibited by CRM197 and rescued when cells were incubated with HB-EGF. We showed that injury induced phosphorylation of specific tyrosine residues and found that a similar pattern of phosphorylation was induced by trinucleotides. These studies indicate that injury induced purinergic receptor activation leads to phosphorylation of EGFR, ERK and migration.
Keywords: wound healing, epithelium, P2YR, ERK, EGFR, cell migration
1. Introduction
Corneal epithelial wound repair requires remodeling of cytoskeletal and extracellular matrix proteins as well as changes in signal cascades, gene regulation and migration. Nucleotides may be released from cells via physical injury, pores in channels, or exocytotic release upon ligand binding causing a rapid elevation in intracellular Ca2+ (Burnstock 1997; Abbracchio and Burnstock, 1998; Neary et al.,1999; Schwiebert and Zsembery, 2003; Yang et al., 2004; Klepeis et al., 2001; Klepeis et al., 2004; Weinger et al., 2005; Pintor et al., 2004). The extracellular nucleotides stimulate cells by activation of P2 purinergic receptors present on the cell surface. The receptors are divided into two families, P2X and P2Y receptors. The P2X receptors play an important role in excitable cell types and are ligand gated channels that permit Ca2+ entry from extracellular stores after nucleotide binding (Burnstock 1997; Abbracchio and Burnstock, 1998). The P2Y receptors are G-protein coupled receptors (GPCRs) that stimulate the release of intracellular Ca2+ by phospholipase C β-mediated hydrolysis and activation of the IP3 pathway. We have shown that epithelial cells express P2Y receptors (1,2,4,6 and 11) and that Reactive Blue 2 (RB-2), an antagonist of P2YR, impairs wound closure (Klepeis et al., 2004). In live cell imaging experiments, injury induces the propagation of a Ca2+ wave, which occurs in the absence of extracellular Ca2+ but is inhibited with thapsigargin or BAPTA (Klepeis et al., 2001; Weinger et al., 2005). Furthermore, when the wound medium or ATP is treated with apyrase and then applied to cells, both cell migration and the Ca2+ wave are inhibited, while untreated medium elicits a typical Ca2+ wave. ATP and UTP have been shown to play a critical role in the propagation of injury induced Ca2+ waves while ADP, UDP and BzATP have not (Weinger et al., 2005). The data indicate that the trinucleotide P2Y receptors play a role in the Ca2+ response that occurs immediately after injury.
Epidermal growth factor receptors (EGFRs) are present in epithelium and play a critical role in cell migration and wound repair (Klepeis et al., 2001; Schultz et al., 1994; Song et al., 2001; Song et al., 2003; Wilson et al., 1999; Zieske et al., 2000). Injury to the epithelium can occur via physical trauma through accidents, surgery, or viral and bacterial infections. We hypothesize that stimulation of the P2Y receptors and activation of IP3 induces the phosphorylation of EGFR. While both nucleotides and epidermal growth factor (EGF) induce the propagation of a Ca2+ wave, the characteristics of the wave and the time course are distinct. In addition, inhibition of the EGF induced wave with AG1478 does not inhibit the injury-induced wave (Klepeis et al., 2001). When cells were either prestimulated with nucleotides or injured and then stimulated with EGF, the Ca2+ response was significantly lowered (Weinger et al., 2005).
Nucleotides, injury to cells and the conditioned medium collected from wounded cultures induce activation of ERK. In addition, GPCR-induced stimulation of cell proliferation and cell motility has been shown to require phosphorylation of EGFR (Gschwind et al., 2001). While EGF is not detected after injury (Yang et al., 2004), release of heparin binding epidermal-like growth factor (HB-EGF) has been detected in the extracellular matrix during development or injury in a number of cell types (Xu et al., 2004; Prenzel et al., 1999; Prenzel et al., 2000; Wetzker and Bohmer, 2003; Gschwind et al., 2001; Fischer et al., 2004). Furthermore, stimulation of GPCRs can cause activation of metalloproteases leading to cleavage of pro-HB-EGF on the cell surface (Prenzel et al., 2000; Wetzker and Bohmer, 2003). In addition, P2Y receptors have been shown to recruit Src for induction of downstream signaling events such as activation of EGFR (Liu et al., 2004).
In this study we tested the hypothesis that inhibition of purinergic receptors decreases the injury induced phosphorylation of specific residues of EGFR. Neither phosphorylation of ERK nor propagation of Ca2+ waves was detected when cells transfected with siRNA to EGFR were stimulated with EGF. However, the cells did respond to injury or ATP indicating the presence of two distinct but complimentary pathways. Treatment of cells with inhibitors of HB-EGF prior to injury resulted in a decrease in phosphorylation of both EGFR and ERK indicating that transactivation is critical. In addition live cell imaging experiments showed that HB-EGF was able to rescue the inhibition caused by CRM197. We hypothesize that the signaling is proposed to occur via a MMP and HB-EGF dependent pathway. In this study we present the novel finding that rapid release of ATP with injury induces purinergic receptors that mediate the activation of the EGFR signaling pathway.
2. Materials and methods
2.1 Reagents
Antibodies directed against total ERK1/2 and active MAPK were purchased from Promega (Madison, WI). Antibodies directed against different phosphorylated tyrosine sites on EGFR (845, 1068, 1086, 1148 and 1173) were obtained and used in the presence and absence of control peptides from BioSource (Camarillo, CA). Antibodies and control peptides were evaluated in preliminary experiments with different concentrations of EGF to assess specificity and to determine optimization. HB-EGF and the functional blocking antibody were purchased from R and D (Minn). Fluo-3/AM, and pluronic acid were purchased from Molecular Probes (Eugene, OR). The pan antibody to EGFR, PY20, secondary antibodies (HRP-conjugated goat anti-rabbit and anti-mouse IgG for immunoblot analysis), and protein A were from Santa Cruz (Santa Cruz, CA) while the BCA Protein Assay kit was purchased from Pierce (Rockford, IL). Adenosine triphosphate (ATP), uridine tri-phosphate (UTP), adenosine diphosphate (ADP), uridine diphosphate (UDP), Reactive Blue-2 and other routine chemicals were obtained either from Sigma (St. Louis, MO) or from American Bioanalytical (Natick, MA). EGFR siRNA was purchased from Ambion (Austin, TX), while the mismatched control sequences were purchased from Dharmacon (Lafayette, CO). CRM197 and TIMP-3 were purchased from Calbiochem (San Diego, CA). The ATP determination kit was purchased from Molecular Probes-Invitrogen Detection Technologies (Eugene OR).
2.2 Cell Culture
A human corneal epithelial cell line established by Araki-Sasaki (Araki-Sasaki et al., 1995) was developed using a recombinant SV40 adenovirus vector. Previous experiments have been performed comparing primary epithelial cells and the corneal epithelial cell line (Weinger et al., 2005). Cells were seeded at a density of 104 cells/cm2 cultured in keratinocyte-SFM medium (K-SFM) supplemented with 30 μg/ml bovine pituitary extract, 0.1 ng/ml EGF, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco Invitrogen, Carlsbad,CA) (Klepeis et al., 2001). Twenty-four hours prior to the experiment, EGF and bovine pituitary extract were removed. Porcine aortic endothelial (PAE) cells lacking the EGFR or transfected with it were established previously and used as model cells for evaluating the role of the EGFR (Riese et al., 1995; Meyer et al., 2002). Phosphorylation of ERK and EGFR were evaluated after cells were either wounded or stimulated with nucleotides.
2.3 siRNA Transfection
Transfection of 21-nucleotide siRNA targeting EGFR was performed. Briefly, EGFR siRNA (siRNA ID # 644 and the sequence with 2 mismatches (GGUAGAAAAGAUGAAACUATG) was transfected using siPORT amine (Ambion, Austin TX) after the optimum conditions for siPORT were established. Live cell imaging and western blot analysis were performed on the same cohort of cells to demonstrate the efficacy of transfection. Cells were plated on 2 well chambered coverslips, siPORT amine and siRNA (5 nM) were added and cultured for 16 hours, at which time medium was added and cells were cultured for an additional 24 hours. Cells were evaluated for the ability to propagate a Ca2+ wave in response to EGF followed by ATP. Cell lysates were immediately harvested and evaluated for total EGFR, pERK and total ERK.
2.4 Calcium Imaging
Epithelial cells (HCE-Ts) were grown to confluency and Ca2+ imaging was performed as previously described (Klepeis et al., 2001). Briefly, cells were incubated in an HEPES-buffered saline solution containing 137 mM NaCl, 5 mM KCl, 4 mM MgCl2, 3 mM CaCl2·2H2O, 25 mM glucose and 10 mM HEPES (Cornell-Bell et al., 1990) and loaded with the Ca2+ indicator dye fluo-3/AM (5 μM). The cells were imaged using a Zeiss LSM 510 Axiovert confocal laser scanning microscope equipped with an Argon and 2 HeNe lasers. All perturbations were made while continuously scanning the cells every 789 milliseconds. Cells were perfused with HEPES-buffered saline prior to stimulation or injury to establish a base line fluorescence reading. Cells were stimulated with agonist prepared in HEPES-buffered saline and washed with HEPES-buffered saline. Ca2+ dynamics were evaluated as described (Klepeis et al., 2001; Cornell-Bell et al., 1990).
2.5 Scrape Wound Assay for biochemical analysis
Cells were grown to confluency. For each biochemical experiment, cells were washed once with phosphate buffered saline (PBS pH7.2) and scrape wounds were made at a density of 2.5 mm/wound in P100 culture dishes (Haq and Trinkaus-Randall, 1998). To determine the injury response, lysates were collected at various times after injury and analyzed using immunoprecipitation and/or western blot analysis.
2.6 Migration Assays
The transwell migration assay was performed as described (Weinger et al 2005, Klepeis et al 2004). Briefly, cells were pre-incubated in the presence or absence of AG1478 (10 nM), trypsinized, and resuspended to a concentration of 125,000 cells/100 μl in binding buffer in the presence or absence of AG1478. Binding buffer, with or without stimuli was placed in the wells of a 24 well plate. Costar Transwell inserts were placed into the wells, and 125,000 cells were added to the inserts. Experiments were conducted for 8 hours at 37°C.
The directed migration assay (scratch wounds) was performed on the LSM 510. Confluent cells in 8 well chamber slides were treated with control medium, inhibitors and/or agonists and then placed on the heated microscope stage. Cells were maintained at 37°C and 5% CO2 using the Zeiss Environmental Chamber. Two wounds were made per well and contiguous regions along the wound were imaged every 20 minutes. The autofocus function was set to focus at each time point prior to imaging. After 20 hours the images were compiled using the Zeiss LSM 510 Multi-Time Software, and the LSM 510 software was used to measure changes in the wound area over time. Percent change in area was calculated. Repeats were performed within runs and between runs to assess consistency. Statistical significance was determined using the one way ANOVA followed by Tukey's post hoc test.
2.7 Measurement of ATP Released with Injury
The release of ATP after injury was assayed using a luciferin-luciferase bioluminescence assay according to the protocol of Molecular Probes. The assay is based on the requirement of ATP for luciferase to produce light. Cells were cultured to confluency in P-35's and the medium was replaced with 250 μl HEPES. Cultures were wounded and 10 μl of the medium was removed immediately after injury and measured using a luminometer (Luminoskan Ascent 2.5, Thermo Lab Systems Waltham, MA). The ATP released into the medium of injured cells was compared to control cells (medium change only). Experiments were performed five times with an internal replicate of three each time.
2.8 Immunoprecipitation
Cells were cultured to confluency, washed with PBS, placed on ice and lysed in 10 mM Tris-HCl (pH 7.4) containing 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM ethylenediamine-tetraacetic acid (EDTA), 2 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM sodium orthovanadate (Na3VO4), 1μg/ml aprotinin, 1μg/ml leupeptin and 1μg/ml pepstatin. The lysates were centrifuged at 10,000g for 15 minutes at 4°C. The supernatant was precleared with protein A beads and mouse IgG, and the primary antibody was added to the supernatant at the concentration of 5μg/ml, followed by overnight incubation at 4°C. Washed protein A slurry was added to the supernatant and rocked at 4°C for 4 hours. The mixture was centrifuged at 3,000g for 30 seconds at 4°C, and the pellet was washed with lysis buffer and prepared for SDS-PAGE. The resulting phosphorylation was normalized to total EGFR.
2.9 SDS PAGE and Western Blot Analysis
Lysates were collected and sheared as described previously (Yang et al., 2004). The protein concentration of the supernatant was determined using the BCA assay. Equivalent amounts of protein from each lysate (40 μg) were subjected to SDS-PAGE and transferred to polyscreen PVDF membrane (PerkinElmer, Boston, MA). Blots were blocked in a Tris buffer (10 mM Tris, 100 mM NaCl, 0.1% Tween-20) containing 0.2% I-block (Applied Biosystems, Foster City, CA) and membranes were incubated with appropriate primary antibodies, washed and incubated with appropriate secondary antibodies and rinsed with TBST. Immunodot blot assays were performed on the lysates collected from the 2 well slides and probed for pERK and ERK. Visualization was performed by enhanced chemiluminescence (PerkinElmer, Boston MA) and quantified with the Kodak Imaging system. Responses were normalized to control.
2.10 Immunohistochemical Analysis
Cells were grown to confluency and either stimulated with EGF or scrape wounded. A parallel group of cells were incubated with TIMP-3 for one hour prior to stimulation. After 5 min, cells were rinsed with PBS, fixed for 20 min with 3.7% formalin (pH 7.2). Cells were prepared for immunohistochemical staining as described previously (Klepeis et al., 2004). Cells were blocked with 5% BSA/PBS and then incubated overnight at 4°C with the antibody of interest in 3% BSA/PBS. Cells were rinsed with PBS, blocked and incubated with Alexa 488 anti-mouse IgG (Invitrogen, Eugene OR). Negative controls were incubated without the primary antibody. Cells were imaged and analyzed on a Zeiss LSM 510 Axiovert confocal laser scanning microscope as described previously (Klepeis et al., 2004; Song et al., 2003).
3. Results
3.1 Injury Induced Response is Mediated by Purinergic and EGF Receptors
Our goal was to determine if the injury induced phosphorylation of EGFR and ERK and resulting wound repair is mediated by cross-talk between purinergic and growth factor receptors. We demonstrated that ATP is the active component released with injury, which induces biological responses (i.e. migration, mobilization of Ca2+ or phosphorylation of ERK). We have shown that migration was induced when medium collected from injured cultures was added to control cells and we demonstrated that the response was inhibited with apyrase (Klepeis et al 2001; 2004; Yang et al 2004; Weinger et al 2005). To determine the concentration of ATP released from injury, cells were cultured to confluency, the medium was removed, HEPES was added and scratch wounds were made. Control cells received a medium change without injury. The concentration of ATP was determined using a bioluminescence assay. We found that the mean ATP released with injury was 0.63 μM compared to control (0.08 μM) (Figure 1A). There was a consistent 8 fold increase over control. While the concentration of ATP released into the medium is less than that used in the experimental assays, it is capable of inducing a biological response and is detected on our dose response curve (Weinger et al 2005). The concentration used elicits a strong, consistent non-saturating response, which elicits optimal phosphorylation of the EGFR.
Fig. 1. Role of ATP in injury induced response.
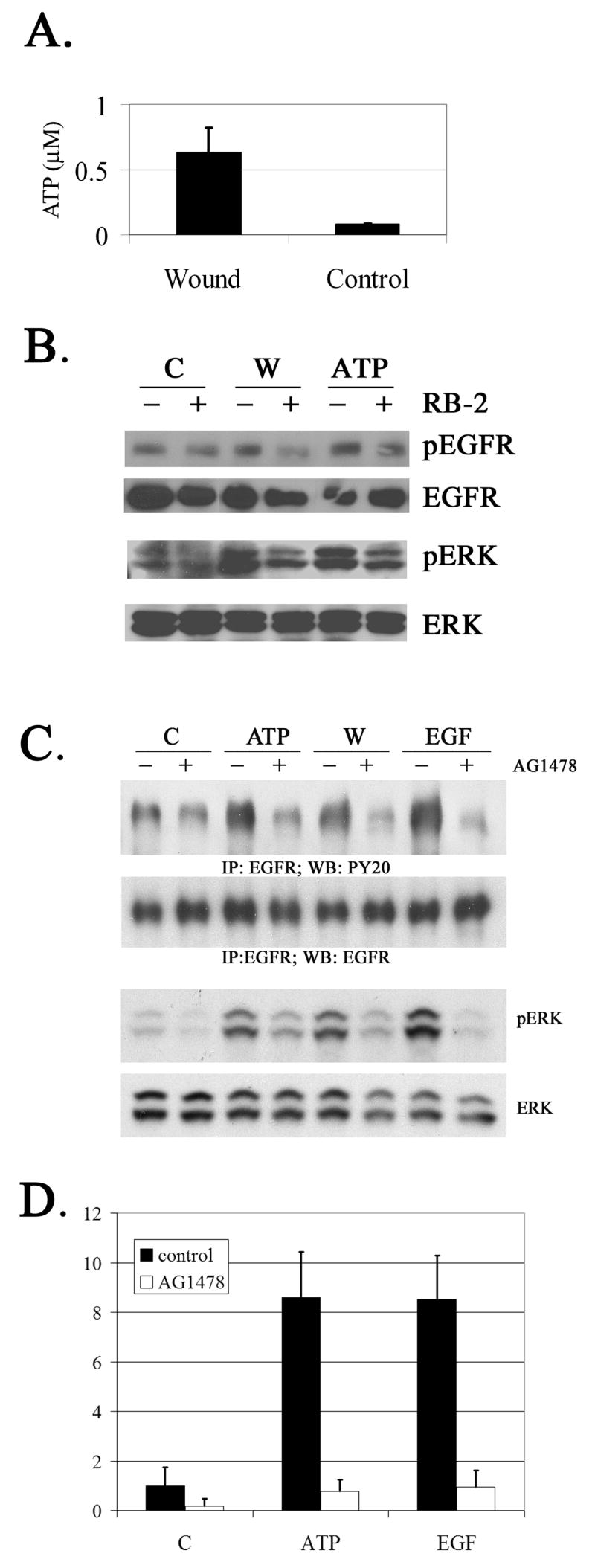
A. ATP bioluminescence assay of injured cells compared to control cells. N=6 independent cultures. B. Cells were incubated in the presence or absence of Reactive Blue 2 (RB-2) (100 μM) and injured (W) or stimulated with ATP and incubated for 5 min. Untreated cells (medium change +/− RB-2) were used as control. Lysates of equivalent protein concentration were resolved in SDS-PAGE and immunoblotted with antibodies directed against phosphotyrosine 1173 (pEGFR), pan-EGFR, total and phosphoERK. N=3 independent experiments. C. Cells were preincubated with AG1478, and subjected to injury, ATP, or EGF for 5 min. Untreated cells (medium change) were used as control. Lysates of equivalent protein concentration were immunoprecipitated with EGFR and immunoblotted with PY20 or with EGFR. In parallel experiments equivalent amounts of protein were resolved on SDS-PAGE and immunoblotted with antibodies directed against total and phosphoERK. N=5 independent experiments. D. Cells were incubated in the presence or absence of AG1478 (10 nM) and stimulated with ATP, EGF or binding buffer (−). Transwell migrations were performed for 8 hr at 37°C. The migrated cells were stained with propidium iodide, counted in six randomly chosen fields, averaged and normalized to unstimulated control. N=3 independent experiments.
To determine if inhibiting the purinergic receptors results in a decrease in the phosphorylation of EGFR and ERK after injury, cells were incubated with Reactive Blue 2 (RB-2), an antagonist of purinergic receptors. Cells were incubated in the presence or absence of RB-2 (100 μM) and the cell cultures were subjected to a medium change. The cells were injured or stimulated with ATP (25 μM) and incubated for 5 min. Lysates were collected, subjected to SDS-PAGE and immunoblotted with antibodies to phosphoEGFR, total EGFR, phosphoERK and total ERK. A reduction in pEGFR and pERK was detected in the cell cultures that were incubated with RB-2 and injured or stimulated with ATP (Figure 1B). No change in phosphorylation was detected in controls (medium change) in the presence or absence of RB-2. These indicate that injury induced release of ATP activates purinergic receptors, which initiates down stream signaling cascades resulting in the phosphorylation of EGFR and ERK.
To determine if injury induced phosphorylation of ERK is mediated by cross talk with EGF, cells were injured in the presence or absence of the EGFR kinase inhibitor, AG1478, (10 nM). Cells were injured and lysates were collected after 5 min and compared to lysates from cells treated with ATP (50 μM) or EGF (0.5 nM) for 5 min. Lysates were probed with antibodies directed against ERK and pERK, or immunoprecipitated with an antibody directed against EGFR and immunoblotted with antibodies directed against EGFR or PY20. The phosphorylation of EGFR induced by injury or ATP resembled that induced by EGF (Figure 1C). When cells pretreated with AG1478 were stimulated with ATP or injured and probed for phosphorylated EGFR there was a decrease in phosphorylation to control levels (Figure 1C). Cells incubated under the same treatment regimen displayed a consistent decrease in pERK in the presence of AG1478. As predicted, when cells were pretreated with AG1478 and stimulated with EGF, inhibition was almost complete. Controls (uninjured and/or untreated medium change) displayed no change in phosphorylation (Figure 1C). The role of the EGFR in ATP induced migration was verified in chemotactic Transwell migration assays (Figure 1D). In these experiments cells were pre-incubated in the presence or absence of AG1478, and stimulated with ATP or EGF. Controls were incubated in binding buffer. Cell migration in the presence of the binding buffer and inhibitor was negligible. Cells stimulated with either ATP or EGF in binding buffer migrated 8 fold more than cells incubated in the presence of binding buffer containing AG1478. Together these data indicate active cross talk between the P2Y and EGFR signaling pathways.
To evaluate the role of the EGFR in injury and wound repair, cells were transfected with EGFR siRNA or a control sequence with 2 mismatched nucleotides. Western blot analysis of transfected and control cells demonstrated a 60% decrease in EGFR expression (Figure 2A). Transfected cultures were stimulated by injury, ATP or EGF and live cell confocal imaging of the cells was followed by immunoblot analysis. Prior experiments demonstrated that injury, ATP and EGF induced an increase in intracellular Ca2+ (Klepeis et al., 2001; Klepeis et al., 2004; Weinger et al., 2005). When transfected cells were stimulated with EGF there was only a negligible increase in intracellular Ca2+, while the controls (siPORT and sequence with 2 mismatches) responded to EGF in a significantly greater manner (t-test p≤0.001) (Figure 2B). In contrast, there was no significant difference in response to injury and ATP between transfected and control cells (Figure 2B) indicating that the cells retained the ability to activate purinergic receptors. To assay if the EGFR was required for phosphorylation of ERK, the same cells that were analyzed by live cell confocal imaging were lysed and probed for pERK. The phosphorylation of ERK in the transfected cells was significantly decreased compared to control cells (t-test p≤0.05) (Figure 2C). These results indicate that the injury induced mobilization of Ca2+ is upstream of EGFR. Confirmatory experiments were performed with porcine aortic endothelial cells (PAE), a cell line that lacks EGFR family members. PAE cells lacking the EGFR exhibited negligible ERK phosphorylation in response to injury and EGF (Figure 2D). However, ATP induced a detectable response, which was similar to that detected when cells pre-incubated with AG1478 were stimulated with ATP (Figure 1C). When cells stably transfected with the EGFR were stimulated by injury, ATP or EGF, phosphorylation of ERK was present. These results indicate that there was an interaction between the two signaling pathways.
Fig. 2. siRNA to EGFR does not reduce Ca2+ mobilization but reduces pERK.

A. Lysates from cells transfected with siRNA to EGFR were probed for EGFR. Equivalent amounts of protein were resolved in SDS-PAGE and blotted with a pan-EGFR antibody. B. Cells transfected with the siRNA and control sequences were incubated in fluo-3/AM and imaged using live cell confocal microscopy. Cells were stimulated with EGF, ATP or subjected to injury (W). The data is presented as relative fluorescence. ** t-test (p<0.001). C. The cells imaged were lysed and equivalent protein was immunoblotted onto membranes and probed with antibodies to pERK and ERK. * t-test (p<0.05). D. PAE cells (lacking or stably transfected with EGFR) were stimulated with injury (W), ATP or EGF for 5 min and lysates were probed as described above. (N.D -not detected). Phosphorylation was normalized to total ERK and a representative blot was graphed as fold change over control. N= 3 independent experiments for A, B, C and D.
3.2 Mechanism of Phosphorylation of EGF Receptor by Injury and Nucleotides
While ATP was detected in the wound medium, we did not detect EGF in the wound medium (ELISA assay; data not shown). This finding was supported by previous observations in our system where EGF induced a response that had different temporal characteristics than the injury response (Klepeis et al 2001). We predict that the activation of the EGFR by purinergic receptors occurs after HB-EGF is released from the cell membrane. In our model system, HB-EGF was detected in the medium in the picomolar range (three fold over unwounded control) by ELISA after cells were injured (data not shown). To determine if picomolar concentrations of HB-EGF can induce a wound healing response, transwell migrations were performed. While maximal migration of cells occurred at 1 nM, migration at 10 and 100 pM was greater than 3.5 fold over control (binding buffer) (Figure 3A). Therefore, while the concentration of HB-EGF released into the medium is less than that used in some of the experimental assays presented here, it is capable of inducing a biological response. As previously shown (Klepeis et al 2004), high concentrations did not induce migration. To further test the role of HB-EGF, injured and control cells were incubated in the presence or absence of the functional blocking antibody to HB-EGF. While injury induced the phosphorylation of the EGFR, the presence of the functional blocking antibody caused a reduction in the phosphorylation of EGFR (Figure 3B).When HB-EGF was added to cells EGFR was phosphorylated. Control experiments were conducted where HB-EGF and antibody were preincubated prior to the addition to cells and phosphorylation was reduced (Figure 3B).
Fig. 3. Injury induced phosphorylation of EGFR is mediated by HB-EGF.

A. Cells were incubated in the presence or absence of the functional blocking antibody to HB-EGF (10 μg/ml) and subjected to control medium, injury (W), or HB-EGF (10 nM). Equivalent amounts of protein were immunoprecipitated with pan-EGFR antibody, resolved on SDS-PAGE and immunoblotted with PY20. Phosphorylation was normalized to EGFR and the graph is presented as relative phosphorylation. B. Cell migration to HB-EGF is concentration dependent. Cells were stimulated with HB-EGF, EGF or binding buffer (−). Transwell migrations were performed for 8 hr at 37°C. The migrated cells were stained with propidium iodide, counted in six randomly chosen fields, averaged and normalized to unstimulated control. N=3 independent experiments.
To test if injury utilizes a cross-activation pathway (P2YR-EGFR), CRM197, a nontoxic mutant of diphtheria toxin that binds and inactivates pro-HB-EGF, was used to block its release (Fischer et al 2004; Pierce et al 2001; Prenzel et al 1999). Epithelial cells were pre-incubated with CRM197 (40 μg/ml), stimulated with injury or nucleotide, immunoprecipitated with a polyclonal antibody directed to EGFR and immunoblotted with the antibody, PY20. When cells were pre-incubated with CRM197 and injured or stimulated with ATP, the phosphorylation of EGFR was similar to control untreated cells (Figure 4A). There was a minor change in the EGF treated cells, and in the control cells (pre-incubated with CRM197 but treated with only a medium change). To determine if the release of pro-HB-EGF occurred by metalloprotease-mediated cleavage, experiments were conducted where cells were pre-incubated with TIMP-3 (50 ng/ml). When cells preincubated with TIMP-3 were injured there was over a 64% decrease in phosphorylation (Figure 4B). A similar decrease was detected when cells were stimulated with ATP.
Fig. 4. Injury and ATP induced phosphorylation of EGFR is mediated by cleavage of the HB-EGF.

A. Cells were incubated in the presence or absence of CRM197 (40 μg/ml) and then subjected to control medium, injury (W), ATP or EGF. Equivalent amounts of protein were immunoprecipitated with pan-EGFR antibody, resolved in SDS-PAGE and immunoblotted with either PY20 or EGFR. B. Cells were incubated in the presence or absence of TIMP-3 (50 ng/ml) and then subjected to control medium, injury (W), ATP or EGF. Equivalent amounts of protein were immunoprecipitated with pan-EGFR, resolved on SDS-PAGE and blotted with either PY20 or EGFR. Densitometric analysis was performed and graphs are presented as normalized to EGFR for each condition in A. and B. C. Cells were incubated in the presence or absence of CRM197 (40 μg/ml) and subjected to control medium, injury (W), ATP or EGF. Lysates were harvested after 5 min. Equivalent amounts of protein were resolved in SDS-PAGE and immunoblotted with anti-ERK. Membranes were probed with antibodies directed against phospho and total ERK. D. Cells were incubated in the presence or absence of TIMP-3 (50 ng/ml) and subjected to control medium, injury (W), ATP or EGF as described above. Equivalent amounts of protein were resolved in SDS-PAGE and blotted with antibodies directed against pERK and total ERK. Graphs are normalized to total ERK for C. and D. N=3 independent experiments for A, B, C, and D.
Parallel experiments were performed as described above and lysates were subjected to SDS-PAGE and immunoblotted with antibodies to pERK and total ERK. When pretreated cells were injured, there was a 92% reduction in phosphorylation of ERK (Figure 4C). A smaller decrease (66%) was observed when cells were stimulated with ATP. Unstimulated cells did not induce phosphorylation of ERK (Figure 4C). Cells that were pre-incubated with TIMP-3 and injured did not display any detectable phosphorylation (Figure 4D). When cells were stimulated with ATP there was a similar reduction. In control experiments, when cells were preincubated with TIMP-3 and stimulated with EGF, phosphorylation of ERK was present. Together these data demonstrate that injury induced phosphorylation of EGFR and ERK requires the release of HB-EGF by metalloproteases.
3.3 Wound Closure is Mediated through HB-EGF
The role of HB-EGF in migration assays was determined by using live cell imaging to monitor the closure of scratch wounds. Cells were cultured to confluency in 8-well cover slips in the presence or absence of CRM 197 or CRM 197 and HB-EGF and wounds were made in each well. Adjacent images were taken along the wound margin so that a large contiguous region could be analyzed. Images were collected every 20 minutes at each site for 20 hrs and a representative time course for each condition is shown (Figure 5A). In the initial period, cells in all of the experimental groups migrated at a similar rate. Cells that were incubated in the presence of CRM 197 showed decreased migration after two to three hours. However, this decrease was rescued when cells were incubated in medium containing CRM197 and HB-EGF (Figure 5A). At 20 hours, cells incubated in CRM197 only closed 19%, while controls closed 45%. However, cells cultured in the presence of CRM197 and HB-EGF closed 37% (Figure 5B). A similar trend was detected in the presence of TIMP-3 (data not shown). These indicate that wound closure requires the release of HB-EGF.
Fig. 5. CRM197 inhibits wound repair.
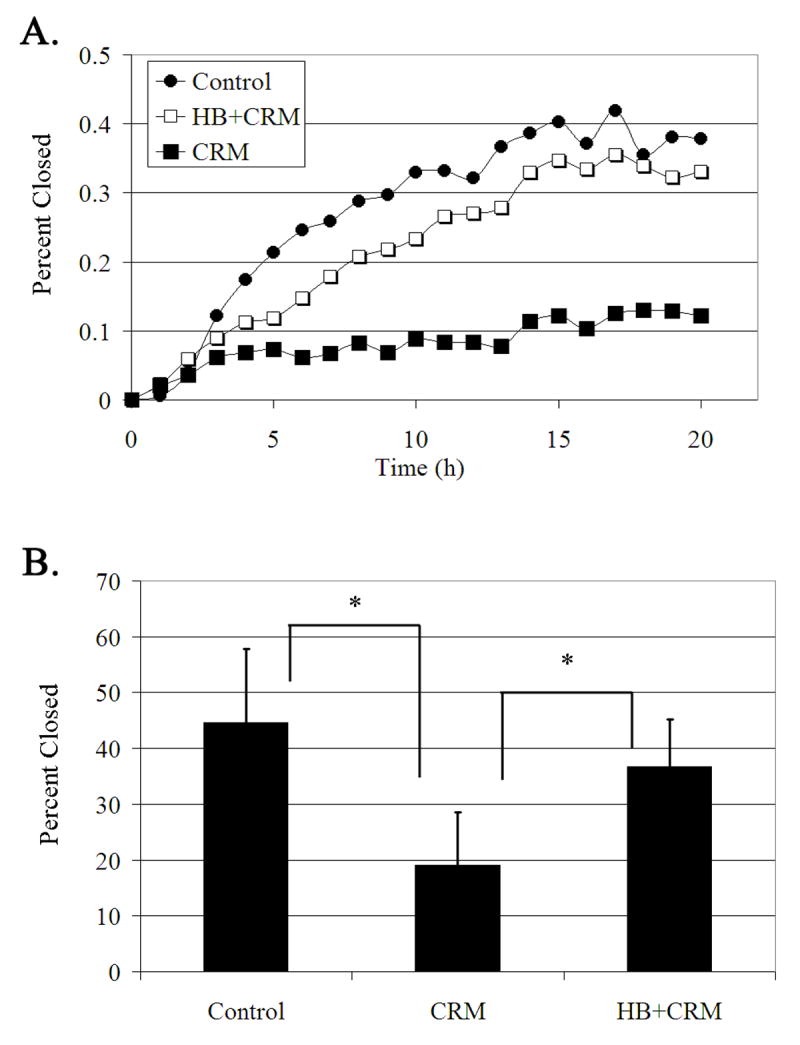
Confluent cells in 8 well chamber slides were incubated overnight in medium lacking growth factors. Cells were pre-incubated in the presence or absence of CRM197 (40 μg/ml) or CRM197 (40 μg/ml) and HB-EGF (10 nM), placed on a heated microscope stage, wounded and incubated in an environmental chamber at 37°C and 5% CO2 for 20 hours. The wounds were demarcated and contiguous regions were tiled and imaged every 20 min. A. Migration time course of a representative run. B. Percent closure at 20 hours. *p<0.01 Significance was determined by a one-way ANOVA followed by Tukey's post hoc test. N=3 independent experiments.
3.4 Injury and ATP induce tyrosine phosphorylation of EGF receptors
Our goal was to determine which tyrosine residues on EGFR are phosphorylated by injury or ATP, compared to EGF. The cell lysates were probed with antibodies generated against phosphotyrosine residues (845,1068, 1086, 1148 and 1173). Phosphorylation of 1068, 1086 and 1173 was detected in response to injury and ATP (Figure 6A). In these experiments, a low concentration of EGF (0.5 nM) was used to achieve levels of phosphorylation comparable to that induced by ATP. Preliminary experiments demonstrated that concentrations greater than 0.5nM showed detectable cross phosphorylation of control cells lacking specific tyrosine residues. Immunohistochemical studies where cells were pre-incubated in the presence or absence of TIMP-3 and then injured and probed with the antibodies against phosphotyrosine residues were performed. Expression was greatest for 1068, 1086 and 1173 (Figure 6B), which supported the western blot analysis (Figure 6A). Cells pretreated with TIMP-3 showed a general decrease in fluorescence compared to controls (untreated and lack of primary antibody). Interestingly, 1173 did not decrease.
Fig. 6. Injury and nucleotides elicit phosphorylation of EGFR.

A. Cells were cultured and subjected to control (medium change), injury (W), ATP, or EGF. Equivalent amounts of protein were resolved on SDS-PAGE and immunoblotted with polyclonal antibodies directed against specific phosphotyrosine sites on EGFR (845, 1068, 1086, 1148 and 1173). Blots were probed also for EGFR. Densitometric analysis was performed and graphs are presented as fold change over control. B. Immunohistochemical analysis. Cells were cultured, a linear wound was made and cells were washed and fixed at five min. In fifty percent of the cultures, cells were preincubated for one hour with serum free medium containing TIMP-3 while the other half were incubated in serum free medium. Cells were permeabilized, blocked and probed with polyclonal antibodies directed against the phosphotyrosine sites and detected with Alexa 488-conjugated IgG. Fluorescent and phase images were taken and merged. Negative control lacks primary antibody. Confocal micrographs represent single optical sections of 3μm. Micrographs are representative of two independent experiments (scale bar equals 100 μm). C. Differential phosphorylation of EGFR in response to ATP, UTP, ADP, UDP compared to control. Equivalent amounts of protein were resolved in SDS-PAGE and blotted with antibodies directed against phosphotyrosine specific sites on EGFR (845, 1068, 1086, 1148 and 1173). Blots were probed also for total EGFR. Densitometric analysis was performed and graphs are presented as fold change over control. N- 3 independent experiments.
To determine if the response was limited to ATP or was common for di- and trinucleotides, cells were stimulated with either trinucleotides ATP, UTP or dinucleotides ADP and UDP. Phosphorylation of EGFR upon stimulation by UTP was comparable to stimulation with ATP at sites 1068, 1086 and 1173 (Figure 6C). ADP induced phosphorylation of 1068 and 1173 but not 1086. Interestingly, UDP, which did not induce migration (Weinger et al., 2005), did not induce phosphorylation of any of the sites evaluated (Figure 6C). These indicate that the injury induced response caused a specific phophorylation profile that correlated with stimulation by ATP or UTP.
4. Discussion
The epithelial wound model provides an excellent system to understand the role of the EGFR in injury induced migration. We hypothesized that the injury of corneal epithelial cells releases ATP and activates purinergic receptors, which induce downstream signaling events including the mobilization of Ca2+, release of HB-EGF from the membrane, phosphorylation of EGFR and ERK and cell migration. The release of ATP from injury was demonstrated and the concentration is supported by investigators who studied its released by mechanical stimulation (Okada et al 2006, Lazarowski et al 1997). Conversely, RB-2 inhibited injury induced phosphorylation of EGFR and ERK indicating that the release of ATP and activation of purinergic receptors was a critical event in signaling after injury. We have shown that injury or treatment with nucleotides induced the phosphorylation of ERK and the propagation of an intercellular Ca2+ wave that spread rapidly to adjacent cells (Yang et al., 2004; Klepeis et al., 2001; Klepeis et al., 2004; Weinger et al., 2005). Furthermore, desensitization was induced by stimulation with nucleotides prior to injury. The events were inhibited when wound media pre-incubated with apyrase was added to cells (Yang et al., 2004; Klepeis et al., 2004). The response to the release of nucleotides and activation of nucleotide receptors has been shown to elicit activation of PLC-β, production of IP3 and DAG, and proliferation in other cell systems (Neary et al., 1999). Cell migration was abrogated when the media was either preincubated with apyrase or incubated in the presence of RB-2 (Klepeis et al., 2004; Weinger et al 2005). Together these data indicate that the initial release of ATP via injury stimulates the onset of a complex signaling pathway.
We hypothesize that the EGFR acts as an integrator of the signals that are induced by injury and activation of the purinergic receptors, and those where growth factors directly activate the EGFR. These studies extend the previous results demonstrating that the response to epithelial injury involved the upregulation of EGFR (Zieske et al 2000, Schultz et al 1992 and 1994, Song et al 2003). Our hypothesis is supported by studies where the addition of exogenous ATP to cells transfected with EGFR siRNA resulted in a decrease in pERK indicating that its phosphorylation required the activation of the EGFR. Furthermore, injury and the addition of exogenous ATP induced the mobilization of Ca2+, while the addition of EGF to the transfected cells did not elicit a response. These data indicate that the mobilization of Ca2+ is upstream of the EGFR while the phosphorylation of ERK is downstream (Figure 7). In proof-of-principle experiments, PAE cells lacking endogenous EGFR failed to exhibit an injury induced phosphorylation of ERK. However, ATP did induce an abrogated pERK. The reduction in the nucleotide induced phosphorylation of ERK in cells lacking the EGFR agrees with the partial decrease detected when cells were incubated in the presence of CRM197 or TIMP-3, which inactivate HB-EGF. The lack of complete inhibition in the presence of ATP could be due to the fact that it is in excess compared to the concentration released by injury. Alternatively, other studies have shown that phosphorylation of ERK may occur via P2YRs and other GPCRs and be independent of EGFR (Andreev, et al., 2001; Montiel, et al., 2006) (Figure 7).
Fig.7. Schematic of cellular response to injury and trinucleotides.

A number of investigators have shown that stimulation of GPCRs leads to release of HB-EGF and activation of the EGFR (Block et al., 2004; Pierce et al., 2001; Prenzel et al., 1999; Schafer et al 2004b; Shah et al 2004). Activation of the GPCRs can stimulate the ADAM family of matrix metalloproteases that release the EGFR ligands (Fischer et al., 2004; Yan et al., 2002; Schafer et al., 2004b). The use of metalloprotease inhibitors has been shown to block the release of EGFR ligands from cells (Tokumaru et al., 2000). HB-EGF release has been detected in a number of systems including acute renal injury and wounded keratinocytes (Sakai et al., 1997; Tokumaru et al 2000). The use of functional blocking antibodies inhibited the responses induced by this release (Shah et al 2004, Block et al 2004, Xu et al 2004 and 2006). In our system HB-EGF blocking antibody inhibited wound induced EGFR phosphorylation (Figure 3). In addition, CRM197 was an effective inhibitor of cell migration in scratch wound assays (Figure 5). The observation that the wound repair can be rescued with the addition of HB-EGF indicates that migration depends on the availability and/or release of HB-EGF into the medium. These data support our hypothesis that the purinergic receptors are one member of the family of GPCRs that are hypothesized to participate in the cross talk with EGFR via release of HB-EGF (Schafer et al 2004a).
Previously, cross talk between the purinergic and EGF receptors was detected in live cell imaging studies when cells pre-stimulated with ATP or UTP showed a decreased response to EGF (Weinger et al., 2005). This response was specific and did not occur when cells were pre-stimulated with BzATP or di-nucleotides. These indicate that the tri-nucleotide receptors (P2Y2 and P2Y4) played a critical role in the initial wound response. Adenosine and its specific receptors were not evaluated as it did not elicit a Ca2+ wave, induce phosphorylation of ERK or play a role in cell migration in corneal epithelial cells (Klepeis et al., 2004). In cardiomyocytes, the nucleotide UTP has been shown to transactivate EGFR (Morris et al., 2004). Still other work has shown that activation of P2Y2 receptors in PC12 cells induced EGFR in a PKC dependent manner (Soltoff 1998). In yet another cell line, the mechanism of activation has been evaluated using 1321N1 astrocytoma cells, where mutant P2Y2 receptors lacking the PXXP motif stimulate changes in intracellular Ca2+ but fail to stimulate phosphorylation of Src and the EGFR (Liu et al., 2004).
To examine the injury and nucleotide induced phosphorylation of EGFR, the phosphorylation of a number of tyrosine residues was evaluated. Injury, ATP and UTP induced the phosphorylation of 1068, 1086 and 1173. Interestingly, ADP phosphorylated a subset of residues, which may correlate with the lower capacity that it has to induce migration (Weinger et al., 2005). UDP was not found to play a role in initiating this signal cascade, which correlates with its inability to stimulate migration. However, UDP can induce Ca2+ release and the phosphorylation of paxillin and ERK (Klepeis et al 2004). Together our data indicate that injury induces a pattern of EGFR phosphorylation distinct from that elicited by direct activation of the EGFR by EGF suggesting distinct pathways. These results are supported by studies that have shown that differential phosphorylation of specific tyrosine residues on EGFR is observed in a number of systems. The αv integrin subunit induced the phosphorylation of 845, 1068, 1086, and 1173 (Moro et al., 2002). In addition, ionizing radiation induced phosphorylation of 992 and 1173 while betacellulin induced 1068 (Sturla et al., 2005; Saito et al., 2004). While these are major sites of phosphorylation in response to EGF, it is interesting that differential phosphorylation occurs in response to specific biologic activation. Future studies will include examining the role of specific residues in deletion experiments.
In summary, we demonstrated that interaction between the P2YR and EGFR families is a component of the injury and wound repair response, and both receptors play a critical role. The results indicate that injury induced purinergic receptor activation may lead to the cleavage of HB-EGF and phosphorylation of EGFR and ERK. However, there may be components that are independent of the EGFR signaling pathway where injury can stimulate ERK and migration. Future studies are aimed at investigating EGFR independent signaling pathways, as well as modulating specific tyrosine residues on the EGFR and downregulating specific P2YR members as potential targets for effective clinical modalities.
Acknowledgments
We thank Drs. Nader Rahimi and Matthew Nugent for discussions and review of the manuscript. This work was supported by a grant from the National Institute of Health (EY06000, V.T-R) and departmental grants from the Massachusetts Lions Eye Research Fund, and the New England Corneal Transplant Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbracchio MP, Burnstock G. Purinergic signalling: pathophysiological roles. Jpn J Pharmacol. 1998;78:113–45. doi: 10.1254/jjp.78.113. [DOI] [PubMed] [Google Scholar]
- Andreev J, Galisteo ML, Kranenburg O, Logan SK, Chiu ES, Okigaki M, Cary LA, Moolenaar WH, Schlessinger J. Src and Pyk2 Mediate G-protein-coupled Receptor Activation of Epidermal Growth Factor Receptor (EGFR) but are not Required for Coupling to the Mitogen-activated Protein (MAP) Kinase Signaling Cascade. J Biol Chem. 2001;276:20130–20135. doi: 10.1074/jbc.M102307200. [DOI] [PubMed] [Google Scholar]
- Araki-Sasaki K, Ohashi Y, Sasabe T, Hayashi K, Watanabe H, Tano Y, Handa H. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest Ophthalmol Vis Sci. 1995;36:614–621. [PubMed] [Google Scholar]
- Block ER, Matela A, SundarRaj N, Iszkula ER, Klarlund JK. Wounding induces motility in sheets of corneal epithelial cells through loss of spatial constraints: role of heparin-binding epidermal growth factor-like growth factor signaling. J Biol Chem. 2004;279:24307–24312. doi: 10.1074/jbc.M401058200. [DOI] [PubMed] [Google Scholar]
- Burnstock G. The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology. 1997;36:1127–39. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Fischer OM, Hart S, Gschwind A, Prenzel N, Ullrich A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol Cell Biol. 2004;24:5172–5183. doi: 10.1128/MCB.24.12.5172-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- Haq F, Trinkaus-Randall V. Injury of stromal fibroblasts induces phosphorylation of focal adhesion proteins. Curr Eye Res. 1998;17:512–523. doi: 10.1076/ceyr.17.5.512.5188. [DOI] [PubMed] [Google Scholar]
- Klepeis VE, Cornell-Bell A, Trinkaus-Randall V. Growth factors but not gap junctions play a role in injury-induced Ca2+ waves in epithelial cells. J Cell Sci. 2001;114:4185–95. doi: 10.1242/jcs.114.23.4185. [DOI] [PubMed] [Google Scholar]
- Klepeis VE, Weinger I, Kaczmarek E, Trinkaus-Randall V. P2Y receptors play a critical role in epithelial cell communication and migration. J Cell Biochem. 2004;93:1115–33. doi: 10.1002/jcb.20258. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Homolya L, Boucher RC, Harden K. Direct demonstration of mechanically induced release of cellular UTP and its implicatin for uridine nucleotide receptor activation. J Biol Chem. 1997;272:24348–24354. doi: 10.1074/jbc.272.39.24348. [DOI] [PubMed] [Google Scholar]
- Liu J, Liao Z, Camden J, Griffin KD, Garrad RC, Santiago-Perez LI, Gonzalez FA, Seye CI, Weisman GA, Erb L. Src homology 3 binding sites in the P2Y2 nucleotide receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2, and growth factor receptors. J Biol Chem. 2004;279:8218–8218. doi: 10.1074/jbc.M312230200. [DOI] [PubMed] [Google Scholar]
- Meyer RD, Dayanir V, Majnoun F, Rahimi N. The presence of a single tyrosine residue at the carboxyl domain of vascular endothelial growth factor receptor-2/FLK-1 regulates its autophosphorylation and activation of signaling molecules. J Biol Chem. 2002;277:27081–7. doi: 10.1074/jbc.M110544200. [DOI] [PubMed] [Google Scholar]
- Montiel M, de la Blanca EP, Jimenez E. P2Y Receptors Activate MAPK/ERK Through a Pathway Involving PI3K/PDK1/PKC-ς- in Human Vein Endothelial Cells. Cell Physiol and Biochem. 2006;18:123–134. doi: 10.1159/000095180. [DOI] [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Erba EB, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- Morris JB, Pham TM, Kenney B, Sheppard KE, Woodcock EA. UTP transactivates epidermal growth factor receptors and promotes cardiomyocyte hypertrophy despite inhibiting transcription of the hypertrophic marker gene, atrial natriuretic peptide. J Biol Chem. 2004;279:8740–8746. doi: 10.1074/jbc.M310012200. [DOI] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Bu Y, Yu E, Akong K, Peters CM. Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway. J Neurosci. 1999;9:4211–20. doi: 10.1523/JNEUROSCI.19-11-04211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. Physiological regulation of ATP release at the apical surface of human airway epithelia. 2006;281:22992–2223002. doi: 10.1074/jbc.M603019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ. Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J Biol Chem. 2001;276:23155–23160. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- Pintor J, Bautista A, Carracedo G, Peral A. UTP and diadenosine tetraphosphate accelerate wound healing in the rabbit cornea. Ophthalmic Physiol Opt. 2004;24:186–93. doi: 10.1111/j.1475-1313.2004.00182.x. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Leserer M, Ullrich A. Tyrosine kinase signalling in breast cancer. Epidermal growth factor receptor: convergence point for signal integration and diversification. Breast Cancer Res. 2000;2:184–190. doi: 10.1186/bcr52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese DJ, II, van Raaij TM, Plowman GD, Andrews GC, Stern DF. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol. 1995;15:5770–6. doi: 10.1128/mcb.15.10.5770. Erratum in: Mol Cell Biol 1996. 16, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Okada S, Ohshima K, Yamada E, Sato M, Uehara Y, Shimizu H, Pessin J, Mori M. Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by betacellulin and EGF. Endocrinol. 2004;145:4232–4243. doi: 10.1210/en.2004-0401. [DOI] [PubMed] [Google Scholar]
- Sakai M, Zhang M, Homma T, Garrick B, Abraham JA, McKanna JA, Harris RC. Production of heparin binding epidermal growth factor-like growth factor in the early phase of regeneration after acute renal injury. Isolation and localization of bioactive molecules. J Clin Invest. 1997;99:2128–2138. doi: 10.1172/JCI119386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer B, Gschwind A, Ullrich A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene. 2004a;23:991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- Schafer B, Marg B, Gschwind A, Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004b;279:47929–47938. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- Schultz G, Chegini N, Grant M, Khaw P, MacKay S. Effects of growth factors on corneal wound healing. Acta Ophthalmol. 1992;(Suppl):60–66. doi: 10.1111/j.1755-3768.1992.tb02170.x. [DOI] [PubMed] [Google Scholar]
- Schultz G, Khaw PT, Oxford K, MaCauley S, Van Setten G, Chegini N. Growth factors and ocular wound healing. Eye. 1994;8:184–7. doi: 10.1038/eye.1994.43. [DOI] [PubMed] [Google Scholar]
- Schwiebert EM, Zsembery A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim Biophys Acta. 2003;1615:7–32. doi: 10.1016/s0005-2736(03)00210-4. [DOI] [PubMed] [Google Scholar]
- Shah BH, Farshori MP, Catt KJ. Neuropeptide-induced transactivation of a neuronal epidermal growth factor receptor is mediated by metalloprotease-dependent formation of heparin-binding epidermal growth factor. J Biol Chem. 2004;279:414–420. doi: 10.1074/jbc.M309083200. [DOI] [PubMed] [Google Scholar]
- Soltoff SP. Related adhesion focal tyrosine kinase and the epidermal growth factor receptor mediate the stimulation of mitogen-activated protein kinase by the G-protein-coupled P2Y2 Receptor. J Biol Chem. 1998;273:23110–23117. doi: 10.1074/jbc.273.36.23110. [DOI] [PubMed] [Google Scholar]
- Song QH, Gong H, Trinkaus-Randall V. Role of epidermal growth factor and epidermal growth factor receptor on hemidesmosome complex formation and integrin subunit beta4. Cell Tissue Res. 2003;312:203–220. doi: 10.1007/s00441-002-0693-x. [DOI] [PubMed] [Google Scholar]
- Song QH, Singh RP, Trinkaus-Randall V. Injury and EGF mediate the expression of alpha6beta4 integrin subunits in corneal epithelium. J Cell Biochem. 2001;80:397–414. doi: 10.1002/1097-4644(20010301)80:3<397::aid-jcb140>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Sturla L, Amorino G, Alexander M, Mikkelsen R, Valerie K, Schmidt-Ullrich R. Requirement of Tyr-992 and Tyr-1173 in phosphorylation of the epidermal growth factor receptor by ionizing radiation and modulation by SHP2. J Biol Chem. 2005;280:14597–14604. doi: 10.1074/jbc.M413287200. [DOI] [PubMed] [Google Scholar]
- Tokumaru S, Higashiyama S, Endo T, Nakagawa T, Miyagawa J, Yamamori K, Hanakawa Y, Ohmoto H, Yoshino K, Shirakata Y, Matsuzawa Y, Hashimoto K, Taniguchi N. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J Biol Chem. 2000;151:209–220. doi: 10.1083/jcb.151.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinger I, Klepeis VE, Trinkaus-Randall V. Tri-nucleotide receptors play a critical role in epithelia wound repair. Purinergic Signalling. 2005;1:281–292. doi: 10.1007/s11302-005-8132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4:651–657. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- Wilson SE, Chen L, Mohan RR, Lian Q, Liu J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp Eye Res. 1999;68:377–399. doi: 10.1006/exer.1998.0603. [DOI] [PubMed] [Google Scholar]
- Xu KP, Ding Y, Ling J, Dong Z, Yu FSX. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:813–820. doi: 10.1167/iovs.03-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KP, Yin J, Yu FS. SRC-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:2832–2839. doi: 10.1167/iovs.05-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Shirakabe K, Werb Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J Cell Biol. 2002;158:221–226. doi: 10.1083/jcb.200112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Cranson D, Trinkaus-Randall V. Cellular injury induces activation of MAPK via P2Y receptors. J Cell Biochem. 2004;91:938–50. doi: 10.1002/jcb.10774. [DOI] [PubMed] [Google Scholar]
- Zieske JD, Takahashi H, Hutcheon AE, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1346–55. [PubMed] [Google Scholar]