

Abstract
Histone deacetylase (HDAC) inhibitors are currently in human clinical trials as antitumor drugs because of their ability to induce cell dysfunction and death in cancer cells. The toxic effects of HDAC inhibitors are also apparent in cortical neurons in vitro, despite the ability of these agents to induce significant protection in the cells they do not kill. Here we demonstrate that pulse exposure of cortical neurons (2 h) in an in vitro model of oxidative stress results in durable neuroprotection without toxicity. Protection was associated with transcriptional upregulation of the cell cycle inhibitor, p21waf1/cip1, both in this model and in an in vivo model of permanent ischemia. Transgenic overexpression of p21waf1/cip1 in neurons can mimic the protective effect of HDAC inhibitors against oxidative stress-induced toxicity, including death induced by glutathione depletion or peroxide addition. The protective effect of p21waf1/cip1 in the context of oxidative stress appears to be unrelated to its ability to act in the nucleus to inhibit cell cycle progression. However, although p21waf1/cip1 is sufficient for neuroprotection, it is not necessary for HDAC inhibitor neuroprotection, because these agents can completely protect neurons cultured from p21waf1/cip1-null mice. Together these findings demonstrate (1) that pulse inhibition of HDACs in cortical neurons can induce neuroprotection without apparent toxicity; (2) that p21waf1/cip1 is sufficient but not necessary to mimic the protective effects of HDAC inhibition; and (3) that oxidative stress in this model induces neuronal cell death via cell cycle-independent pathways that can be inhibited by a cytosolic, noncanonical action of p21waf1/cip1.
Keywords: oxidative stress, histone deacetylase inhibitors, HDAC, cell cycle, apoptosis, neuron(s), cyclin-dependent kinase, Cdk
Introduction
Low-molecular-weight inhibitors of class I and class II histone deacetylase (HDAC) enzymes are attracting growing attention as therapeutics for neurological diseases. The prototypical HDAC inhibitors, including hydroxamates such as trichostatin A (TSA), and short-chain fatty acids such as sodium butyrate, have been shown to ameliorate disease progression in numerous rodent models of neurodegeneration (Langley et al., 2005). In contrast to their broad neuroprotective effects in vivo, HDAC inhibitors have also received much attention as exciting additions to the arsenal of cancer therapeutics. Consistent with both of these properties, we found that HDAC inhibitors significantly prevent neuronal oxidative stress-induced death in vitro, but in doing so, induce small and reproducible toxicity. Such toxicity makes identification of biochemical pathways involved in neuroprotection challenging. Identifying strategies that disassociate the neuroprotective element of HDAC inhibition from the toxic element would forge opportunities for a better molecular understanding of their salubrious effects.
HDAC inhibitors induce gene expression by inhibiting the enzymes that deacetylate lysine and arginine-rich N-terminal domains of histones (Thiagalingam et al., 2003) so that acetylation by histone acetyltransferases is favored (Gregory et al., 2001; Roth et al., 2001). Enhanced histone acetylation of the cyclin-dependent kinase (Cdk) inhibitor p21cip/waf/sdi1 (hereafter p21) gene promoter and increased expression of p21 is believed to underlie some of the antiproliferative and antitumor effects of HDAC inhibitors (Richon et al., 2000). However, the role of p21 in postmitotic neuron biology during HDAC inhibition is unknown. Indeed, p21 could contribute to the neurotoxicity inherent to HDAC inhibition or contribute to neuroprotection. The latter assertion comes from two nonoverlapping models, both of which have established support. One model predicts that oxidative stress induces neuronal death by stimulating the aberrant reentry of postmitotic neurons into the cell cycle and that p21 inhibits this reentry. Corroborating this is evidence that postmitotic neurons die via a frustrated attempt to transition from the quiescent G1/0 to replicative S phase during neurodegeneration and injury (Herrup et al., 2004; Langley and Ratan, 2004). Furthermore, studies using molecular and pharmacological inhibitors of Cdk function have demonstrated causal relationships between cell cycle proteins and cell death in neurons (Park et al., 1996, 1997). The second model predicts that p21 contributes to HDAC inhibitor neuroprotection through its ability to interact with and inhibit prodeath factors, independent of the cell cycle. This model is supported by findings that p21 interactions inhibit apoptosis signal-regulating kinase-1 (ASK-1), an upstream activator of the prodeath stress-activated protein kinase (SAPK)/JNK pathway (Zhan et al., 2007), and caspase 3 activation (Suzuki et al., 1998). These interactions require a cytoplasmic localization of p21 and N-terminal binding domains that are independent of the Cdk or PCNA-binding domains (Child and Mann, 2006).
In this article, we define conditions under which HDAC inhibitors induce protection without apparent toxicity in vitro and in vivo. We use this strategy to invoke a role for cytosolic p21 in cell cycle-independent neuroprotection via the ability of this protein to interact with the prodeath signaling kinase, ASK-1. Finally, we provide evidence that glutathione depletion-induced oxidative stress does not cause aberrant cell cycle reentry of postmitotic neurons to induce death.
Materials and Methods
The HDAC inhibitors TSA, suberoyl bis-hydroxamic acid (SBHA), scriptaid, and nullscript were purchased from Biomol International (Plymouth Meeting, PA), whereas sodium butyrate was purchased from Sigma-Aldrich (St. Louis, MO). Homocysteate (HCA), camptothecin, and hydrogen peroxide were purchased from Sigma-Aldrich. Roscovitine and olomoucine were purchased from Biomol International. pEGFP, pEGFP-p21-full, and pEGFP-p21-ΔNLS constructs were kindly provided by Dr. Asada (International Medical Center of Japan, Tokyo, Japan). The hemagglutinin (HA)-tagged ASK-1 construct was kindly provided by Dr. Houghton (St. Jude Children's Research Hospital, Memphis, TN). B6:129S2-Cdkn1a (p21 knock-out) (Brugarolas et al., 1995) and B6:129SF2/J (wild-type control) mouse lines were obtained from the Jackson Laboratory (Bar Harbor, ME). Embryonic day 17 (E17) pregnant Sprague Dawley rats were obtained from Harlan Sprague Dawley (Frederick, MD). Adult male Sprague Dawley rats were obtained from Charles River Breeding Laboratories (Wilmington, MA).
Primary neurons and cell culture.
Cell cultures were obtained from the cerebral cortex of fetal Sprague Dawley rats (embryonic day 17) or B6:129S2-Cdkn1a (p21 knock-out) and B6:129SF2/J (wild-type control) mice (embryonic day 15) by a papain dissociation method as described previously (Murphy et al., 1990). Cultures were plated on poly-d-lysine (Sigma-Aldrich)-coated cell culture dishes and maintained in minimum essential medium (Invitrogen, Grand Island, NY) containing 5.5 g/L glucose, 2 mm l-glutamine, 100 μm cystine, and supplemented with 10% fetal bovine serum (FBS; Invitrogen). Cultures from the cortex of fetal rats or mice at this stage of development are ∼85% neuronal, the balance being predominantly glial. All experiments were initiated 24 h after plating unless stated otherwise. Under these conditions, the cells are not susceptible to glutamate-mediated excitotoxicity. Longer-term cultures for adenoviral infection were maintained by switching to minimum essential medium (Invitrogen) containing 5.5 g/L glucose, and supplemented with 2 mm GlutaMAX (Invitrogen) and 1× B27 (Invitrogen), at 3–4 d in vitro (DIV). The HT22 murine hippocampal cell line was a kind gift from D. Schubert (Salk Institute, La Jolla, CA). B35 neuroblastoma cell line was purchased from American Type Culture Collection (Manassas, VA). Both HT22 and B35 cell lines were maintained and cultured in DMEM (Invitrogen) with high glucose, l-glutamine, and pyridoxine hydrochloride, and supplemented with 10% FBS.
Viability assays.
For cytotoxicity studies, cells were rinsed with warm PBS and then placed in medium containing the glutamate analog HCA (5 mm, unless stated otherwise). HCA was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5. For HDAC inhibitor treatments except pulse treatments, HDAC inhibitors were added at the time of HCA treatment and present throughout experiment. Viability was assessed by calcein AM/ethidium homodimer-1 staining (live/dead assay) (Invitrogen) under fluorescence microscopy and the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method (Promega, Madison, WI).
Transfections and adenoviral infections.
HT22 hippocampal neuron cells were cotransfected with a puromycin cDNA construct (pPUR; Clontech, Mountain View, CA) and either pEGFP (Clontech) vector alone or pEGFP vector containing a p21 cDNA (pEGFP-p21-full) or pEGFP vector containing a p21-ΔNLS cDNA (pEGFP-p21-ΔNLS), using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's protocol. Stably transfected HT22 neurons were selected over several weeks by the addition of puromycin (4 μg/ml) to the culture medium. Puromycin-resistant clones were pooled to avoid confounds introduced by clonal selection, and p21 or GFP expression was verified by Western blot analysis and GFP immunofluorescence under an inverted fluorescence microscope (Axiovert 200M; Zeiss, Oberkochen, Germany). Primary mixed cortical neurons were infected with adenovirus (multiplicity of infection = 100) harboring both p21 and GFP cDNAs (Ad-p21+GFP), or GFP cDNA (Ad-GFP) alone. To generate adenoviruses, either p21 or GFP was subcloned into the multiple cloning site of pAd5-CMV-NpA vector (ViraQuest, North Liberty, IA) and verified by sequencing. Recombinant adenovirus generation and amplification was performed by ViraQuest using RAPAd technology. Additionally, recombination by ViraQuest included the addition of HSV promoter-GFP constructs so that, in addition to CMV promoter-p21 or CMV promoter GFP, adenoviruses also harbor HSV promoter-GFP. Primary mixed cortical neurons were infected at 5 DIV and cultured for an additional 4 d before exposure to hydrogen peroxide to allow for transgene expression.
Quantitative RT-PCR.
Total RNA was prepared from primary mixed cortical neurons using TriZOL (Invitrogen) and cDNA generating using a SuperScript III First-Strand Synthesis System for RT-PCR kit (Invitrogen), according to the manufacturer's protocol. Real-time PCRs were performed as a duplex reaction using p21 gene expression assay, which uses a FAM-labeled probe, and β-actin gene expression assay, which uses a VIC-labeled probe (Applied Biosystems, Foster City, CA), so that p21 amplification could be normalized to β-actin. Real-time PCRs were performed using a 7500 Real Time PCR System (Applied Biosystems) using standard PCR protocol and amplification conditions.
Immunoblot analysis.
Cell lysates were obtained by rinsing cortical neurons with cold PBS followed by lysis in NP-40 lysis buffer (Boston Bioproducts, Worcester, MA). Protein concentrations in lysates were quantified by Bradford assay (Bio-Rad, Hercules, CA). Nuclear and cytoplasmic protein extractions were obtained using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Samples were boiled in Laemmli buffer and electrophoresed under reducing conditions on 12% [or 7.5% for retinoblastoma protein (pRb) immunoblots] polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane (Bio-Rad) by electroblotting. Nonspecific binding was inhibited by incubation in Tris-buffered saline with Tween 20 (TBST: 50 mm Tris-HCl, pH 8.0, 0.9% NaCl, and 0.1% Tween 20) containing 5% nonfat milk for at least 1.5 h. Primary antibodies against p21 (BD Biosciences, San Jose, CA), p15 (Santa Cruz Biotechnology, Santa Cruz, CA), p16 (BD Biosciences), p27 (BD Biosciences), p57 (Millipore, Billerica, MA), pRb (BD Biosciences), GFP (Invitrogen), histone H4 (Millipore), acetyl histone H4 (Millipore), histone H3 (Millipore), phospho-JNK (Cell Signaling, Danvers, MA), total JNK (Cell Signaling), GAPDH (Millipore), NeuN (Millipore), α-tubulin (Sigma-Aldrich), and HA (Sigma-Aldrich) were diluted in TBST containing 5% milk overnight at 4°C followed by incubation with respective horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) for 2 h at room temperature. Immunoreactive proteins were detected according to the enhanced chemiluminescent protocol (GE Healthcare, Piscataway, NJ).
Animal preparation and monitoring for middle cerebral artery occlusion experiments.
Adult male Sprague Dawley rats (n = 6 per treatment) weighing 250–280 g were operated on to examine the effect of pre- and post-HDAC inhibitor treatment on infarct volumes after middle cerebral artery occlusion (MCAo). Twelve- to 14-week-old male mice, B6:129SF2/J (wild type; n = 9) and B6:129S2-Cdkn1a (p21 knock-out; n = 10) weighing 20–30 g were used to examine the endogenous role of p21 in the prevention of stroke damage. Animals were allowed ad libitum access to food and water before and after surgery. Rats were anesthetized by an intraperitoneal injection of 400 mg/kg chloral hydrate followed 45 min later by a maintenance intraperitoneal infusion at a rate of 120 mg/kg/h using a butterfly needle set. The animals were free breathing. Body temperatures were kept stable at 36.5 ± 0.5°C using a feedback-regulating heating pad and a rectal probe (Harvard Apparatus, Holliston, MA). In rats, the right femoral artery was cannulated for measurement of arterial blood gases, glucose, and mean arterial blood pressure. These physiological parameters were monitored before and after MCAo. In addition, laser Doppler flowmetry (Moor Instruments, Devon, UK) was used to monitor the regional cerebral blood flow through a burr hole 2 mm in diameter created in the right parietal bone (2 mm posterior and 6 mm lateral to bregma). Preparation and monitoring of mice were the same as for rats with the following changes. Mice were anesthetized by inhalation of a mixture of isoflurane (1.5–2%), oxygen (30%), and nitrogen (70%) via nosecone, and a laser Doppler flowmetry probe was attached directly to the parietal bone.
Surgery.
All rats and mice were subjected to right MCAo. Under the operating microscope, the right common carotid artery was exposed through a midline incision in the neck. A 4-0 (rats) or 6-0 (mice) nylon suture with its tip rounded by heating over a flame and subsequently coated with poly-l-lysine (Sigma-Aldrich) was introduced into the external carotid artery and then advanced into the internal carotid artery for a length of 18–19 mm (rats) or 9–10 mm (mice) from the bifurcation. This method placed the tip of the suture at the origin of the anterior cerebral artery, thereby occluding the MCA. The placement of the suture tip was monitored by laser Doppler flowmetry measurements of regional cerebral blood flow. MCAo caused a sharp drop in regional cerebral blood flow to <30% (20% for mice) of preischemic base line. The suture was left in place (rats) or removed after 20 min (mice), and the animals were allowed to awaken from the anesthesia after closure of the operation sites.
Drug administration.
Sodium butyrate was dissolved in PBS (vehicle) for a total volume of 100 μl and was administered intraperitoneally to animals (n = 6) at a dose of 1200 mg/kg of body weight. Sodium butyrate was administered 24 and 4 h before MCAo (pretreatment experiment) or 30 min after MCAo (posttreatment experiment). The control animals received an equivalent volume of the vehicle on an identical administration schedule.
Infarct measurement.
Twenty-four hours after MCAo, animals were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (50 mg/kg, i.p.) and decapitated. The brain was rapidly removed, sliced into seven 2 mm coronal sections using a rat matrix (RBM 4000C; ASI Instrument, Warren, MI), and stained according to the standard 2,3,5-triphenyltetrazolium chloride (TTC) method. Each slice was drawn using a computerized image analyzer (Scion, Frederick, MD). The calculated infarction areas were then compiled to obtain the infarct volumes per brain (in mm3). Infarct volumes were expressed as a percentage of the contralateral hemisphere volume to compensate for edema formation in the ipsilateral hemisphere.
In vivo experiments.
All experimental procedures were approved by the Harvard Medical Area Standing Committee on Animals and/or Weill Medical College of Cornell University Institutional Animal Care and Use Committee and meet the standards of the Federal and State reviewing organizations.
Statistics.
Statistical analyses were performed using the statistical analysis package Prism (GraphPad Software, San Diego, CA).
Results
HDAC inhibitors protect cortical neurons from oxidative stress-induced death
HDAC inhibitors are broadly neuroprotective in vivo, but the precise molecular mechanisms underlying their beneficial effects remain unclear. To elucidate further the mechanism of neuroprotection protection by HDAC inhibitors, we used the experimental leverage of an in vitro model of neuronal oxidative stress-induced death. In this model, cultured rat immature cortical neurons (E17) are exposed to high concentrations (1–5 mm) of the excitatory amino acid, glutamate, or its structural analogs such as HCA. In neurons at this stage of development, glutamate or HCA induces oxidative stress, not by acting on cell surface glutamate receptors, but rather via their ability to inhibit the uptake of the amino acid cystine into the cell via the plasma membrane transporting agency (system Xc−). Cystine uptake into neurons and/or glia is critical for cell survival as cystine is reduced intracellularly to cysteine, the rate-limiting amino acid in the synthesis of the versatile antioxidant tripeptide glutathione (gly-cys-glu). Cystine deprivation results in intracellular cysteine deprivation, reduced glutathione synthesis, and an imbalance between cellular oxidants and antioxidants. After cystine deprivation, neurons succumb to an antioxidant-sensitive form of death with features of apoptosis and necrosis (Murphy et al., 1989, 1990; Ratan et al., 1994). Thus, degeneration and death are not a result of ionotropic glutamate receptor activation and excitotoxicity, but rather accumulation of unopposed free radicals in the setting of reduced antioxidant defenses (Murphy et al., 1989, 1990; Ratan et al., 1994).
To verify that HDAC inhibition was sufficient to prevent oxidative stress-induced death, mixed immature cortical cultures were treated with HCA in the presence or absence of a range of structurally diverse, commercially available HDAC inhibitors. These HDAC inhibitors included the short-chain fatty acid sodium butyrate, as well as the hydroxamic acid-based inhibitors, TSA and SBHA. Treatment of mixed immature cortical cultures with HCA resulted in widespread cell death, as seen by calcein AM/ethidium homodimer (live/dead) staining and fluorescent microscopy (Fig. 1A) or quantified (90% death) by MTT assay (Fig. 1B) 24 h after HCA addition. In contrast to the widespread death seen in HCA-treated immature cortical cultures, the cotreatment with an HDAC inhibitor resulted in significant protection. Indeed, no difference was seen between immature cortical cultures treated separately with structurally diverse HDAC inhibitors and HCA, or treated with HDAC inhibitors alone (Fig. 1A,B).
Figure 1.

Structurally diverse HDAC inhibitors abrogate oxidative glutamate toxicity in embryonic cortical neuronal cultures and increase histone H4 acetylation. A, Representative micrographs showing live/dead staining of primary cortical neuronal cultures 24 h after treatment. a, nontreated control; b, 5 mm HCA; c, 0.66 μm TSA; d, HCA + TSA. Live cells are detected by uptake and trapping of calcein AM (green fluorescence). Dead cells fail to trap calcein but are freely permeable to the highly charged DNA-intercalating dye ethidium homodimer (red fluorescence). B, Graph showing primary cortical neuron viability, as determined using the MTT assay, after TSA (0.66 μm), SBHA (12.5 μm), scriptaid (6.13 μm), nullscript (6.13 μm), or sodium butyrate (1 mm) treatment in the presence (light gray) or absence (dark gray) of HCA (5 mm) for 24 h. Control is no HDAC inhibitor. C, Western blot analysis to detect relative levels of acetylated histone H4 and total histone H4 in acid-precipitated histone protein extracts from primary cortical neuronal cultures treated as in B. Antibodies against acetylated histone H4 or total histone H4 were used (see Materials and Methods). D, Graph showing cell viability in HT22 and B35, as determined using the MTT assay, after TSA (1.2 μm) treatment in the presence (light gray) or absence (dark gray) of HCA (5 mm) for 24 h. Control is no HDAC inhibitor. Graph bars in B and D depict mean ± SD. *Significant difference between non-HCA and HCA-treated groups, p < 0.0001, by two-way ANOVA. §Significant difference between non-HDAC inhibitor control and HDAC inhibitor alone-treated neurons, p < 0.05, by one-way ANOVA followed by Dunnett's multiple-comparisons test.
Crystal structure studies have revealed that the hydroxamic acid moiety of the HDAC inhibitor SAHA coordinates or binds the zinc ion at the catalytic core of the HDAC enzyme (Finnin et al., 1999). In order for this interaction to occur, it is necessary for the hydroxamic acid moiety to penetrate an 11-Å-deep tubular pocket; thus, the linker region of the HDAC inhibitor, at the end of which the hydroxamic acid is located, needs to be a critical length. The established ability of liberated or transported zinc to induce neuronal toxicity (Dineley et al., 2003) along with ability of hydroxamic acid-containing HDAC inhibitors to bind zinc raises the possibility that this class of HDAC inhibitors protects by simply binding zinc in the cytoplasm or mitochondria rather than by binding zinc in the catalytic core of HDAC enzymes. To exclude this possibility, we took advantage of two structural analogs, scriptaid and nullscript. These analogs are structurally similar molecules, differing only in the length of their aliphatic linker region; nullscript contains two less carbons than scriptaid in this region. Because of this difference, it is predicted that the hydroxamic acid moiety of both scriptaid and nullscript can bind zinc, but only scriptaid will do so within the HDAC enzyme, and that nullscript will therefore be less active as an HDAC inhibitor (Su et al., 2000). As expected, we found that scriptaid mimicked the protective effect of other HDAC inhibitors, whereas the HDAC inhibitor negative control, nullscript, failed to protect HCA-treated cultures from death (Fig. 1B). Indeed, we confirmed that all of our putative HDAC inhibitors, including scriptaid, result in increased histone H4 acetylation in cortical neuronal cultures, whereas nullscript does not (Fig. 1C). Together, these pharmacological studies support the notion that HDAC inhibition and not some other nonspecific effect, including zinc binding, of the agents examined is responsible for the protection we observe.
Because this neuronal model of oxidative stress-induced death is dependent on glutathione depletion by the competitive inhibition of cystine uptake at its plasma membrane transporter by glutamate or HCA (Murphy et al., 1989, 1990), it was necessary to determine whether HDAC inhibitors act to suppress HCA-induced death by preventing cystine deprivation and glutathione depletion. We therefore measured total glutathione levels [reduced glutathione (GSH) and oxidized glutathione (GSSG)] at several time points after HCA and HDAC inhibitor addition. The rate and level of glutathione loss were nearly identical in neurons treated with HCA, with or without HDAC inhibitors (data not shown). Together, these findings argue that HDAC inhibitors protect distal to glutathione depletion.
Prior studies have attributed the salutary effects of transcriptional activators such as nrf2 in preventing glutathione depletion-induced death to their ability to activate these transcriptional responses in the small subset of glial elements in the mixed cultures system we study (Shih et al., 2003). These findings raise the possibility that HDAC inhibitors may protect by enhancing transcription in glial cells rather than neurons. We therefore used two different homogeneous neuronal cell lines to investigate whether the protection mediated by HDAC inhibitors was dependent or independent of non-neuronal components. As seen for the mixed cortical cultures, the treatment of either B35 rat neuroblastoma cell line neurons (Schubert et al., 1974) or HT22 mouse hippocampal cell line neurons (Li et al., 1997) with HCA resulted in widespread cell death, whereas the cotreatment with an HDAC inhibitor resulted in significant protection (Fig. 1D). Again, no difference was seen between neurons treated with HDAC inhibitor and HCA, or treated with HDAC inhibitor alone (Fig. 1D). Together, these results suggest that glial elements are not necessary for the protection of neurons by HDAC inhibitors.
Toxicity of HDAC inhibitors limit biochemical analysis of pathways involved in survival
The biochemical analysis of the protective effects of HDAC inhibitors in vitro could be confounded because continuous exposures to HDAC inhibitors results in a small but reproducible level of toxicity in the cultures (Fig. 1). Recent studies suggest that HDAC activity is important for the repression of prodeath genes in some models of neuronal apoptosis (Liu and Greene, 2001). This model predicts that HDAC inhibitors cause toxicity by derepressing death gene expression and that the duration of derepression and thus toxicity would be dependent on the duration of HDAC inhibitor exposure. Consistent with this model, we observed that longer exposures of mixed immature cortical cultures to HDAC inhibitors alone (>2 DIV) caused a greater degree of cell loss (Fig. 2A, −24 to +24 h). We therefore investigated whether shorter exposures would result in no death but still maintain protection. Mixed cortical cells were treated with TSA for 8, 4, or 2 h before or at the time of HCA addition, and cell death was quantified after 24 h by MTT assay. A pulse treatment for 8, 4, or 2 h either before or at the addition of HCA was sufficient to protect the mixed immature cortical cultures from oxidative stress-induced death (Fig. 2A). Moreover, in contrast to a 24 h TSA treatment paradigm, no cell loss was observed with either a 4 or 2 h treatment of TSA (Fig. 2A). Consistent with this result, a 2 h pulse with a different HDAC inhibitor, scriptaid, followed by a 24 h HCA treatment was sufficient for protection, as seen by calcein AM/ethidium homodimer (live/dead) staining and fluorescent microscopy (Fig. 2B). Again no protection was observed for a 2 h pulse treatment with the HDAC inhibitor negative control, nullscript (Fig. 2B). These studies dissociate the neuroprotective effects of HDAC inhibitors from their prodeath effects.
Figure 2.
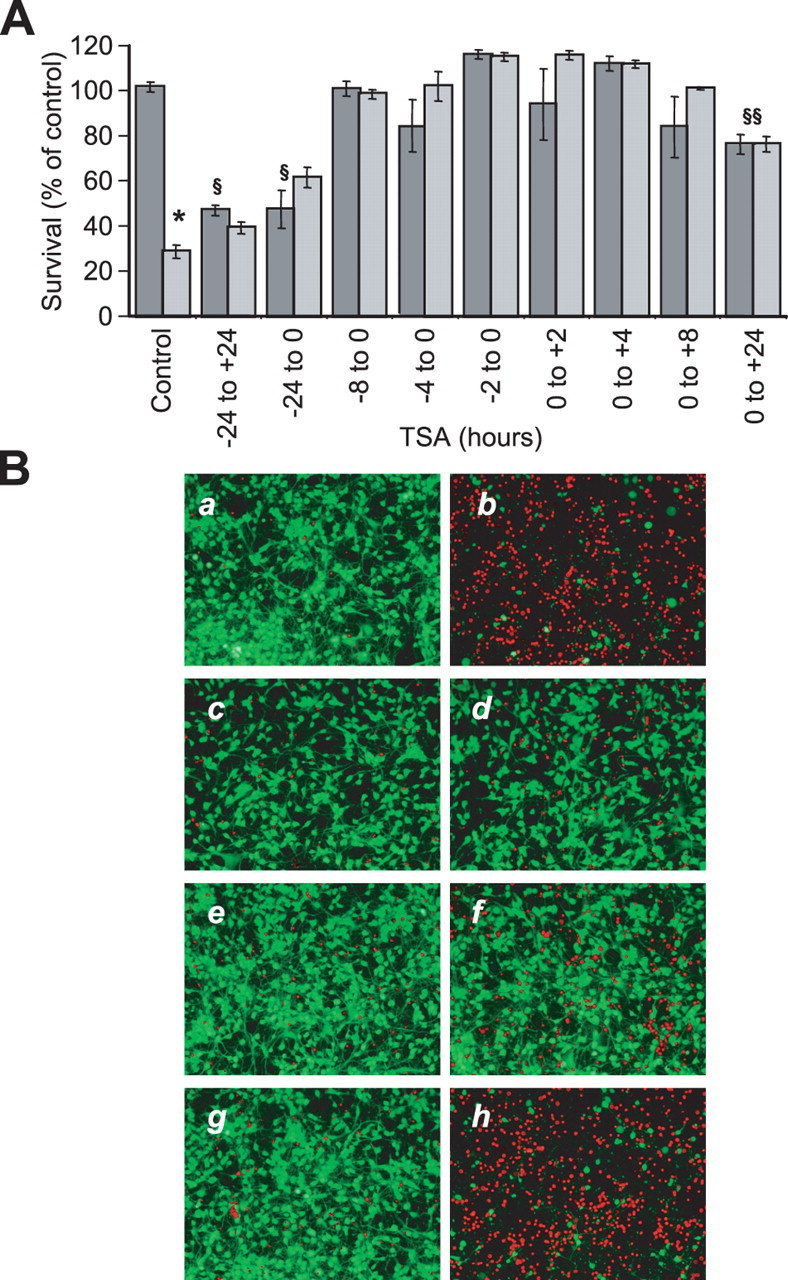
Pulse treatment of HDAC inhibitors abrogate oxidative glutamate toxicity in embryonic cortical neuronal cultures for up to 24 h. A, Graph showing cell viability, as determined using the MTT assay, after TSA (1.32 μm) treatment for different lengths of time before or after HCA treatment for 24 h. Values on the x-axis indicate hours of TSA exposure before (−) and after (+) HCA addition at time 0 (0). Light gray, Presence of HCA (5 mm); dark gray, absence of HCA. Note the high toxicity of TSA to cortical neuronal cultures with a 48 h exposure, whereas cells incubated with TSA for 2, 4, or 8 h show no significant death. Graph bars depict mean ± SD. *Significant difference between non-HCA and HCA-treated groups, p < 0.0001, by two-way ANOVA. Significant difference between non-HDAC inhibitor control and HDAC inhibitor alone-treated neurons is denoted by § (p < 0.01) and §§ (p < 0.05), analyzed by one-way ANOVA followed by Dunnett's multiple-comparisons test. B, Representative micrographs showing live/dead staining of primary cortical neuronal cultures after 2 h incubation with HDAC inhibitor followed by 24 h incubation with (b, d, f, h) or without (a, c, e, g) HCA (5 mm). HDAC inhibitors include the following: TSA (1.32 μm; c, d), scriptaid (12.3 μm; e, f) and nullscript (12.3 μm; g, h).
HDAC inhibition in cortical neurons induce p21 expression
Previous studies have demonstrated that HDAC inhibitors induce the accumulation of acetylated histones in both transformed and normal cells (Marks and Dokmanovic, 2005). A well characterized promoter in which the acetylation of histone proteins occurs in response to HDAC inhibition is the p21 promoter. Increased acetylation of this promoter is correlated with increased p21 expression. Indeed, numerous reports studying the cytostatic capacity of HDAC inhibitors in transformed and tumor cell lines have documented the robust induction of p21 (Xiao et al., 1999; Richon et al., 2000; Gui et al., 2004). Of note, p21 has also been shown to inhibit kinases that mediate apoptosis independent of the cell cycle (Shim et al., 1996; Suzuki et al., 1998, 1999; Asada et al., 1999; Xu and El-Deiry, 2000; Xue et al., 2003).
To examine whether p21 gene expression is increased in HDAC inhibitor-treated mixed cortical cells, and whether this correlates with protection from oxidative stress-induced death, cortical neuronal cultures were treated as before and p21 levels examined after 4, 8, or 12 h. The treatment with HCA alone at these time points resulted in no induction of p21 mRNA or protein relative to control cells as determined by quantitative RT-PCR and Western blot analysis, respectively. In contrast, treatment with the HDAC inhibitors, TSA, SBHA, and scriptaid, all induced a time-dependent increase in p21 mRNA and protein levels (Fig. 3A,B). As anticipated, no significant p21 induction was seen with the HDAC inhibitor negative control, nullscript (Fig. 3A,B).
Figure 3.
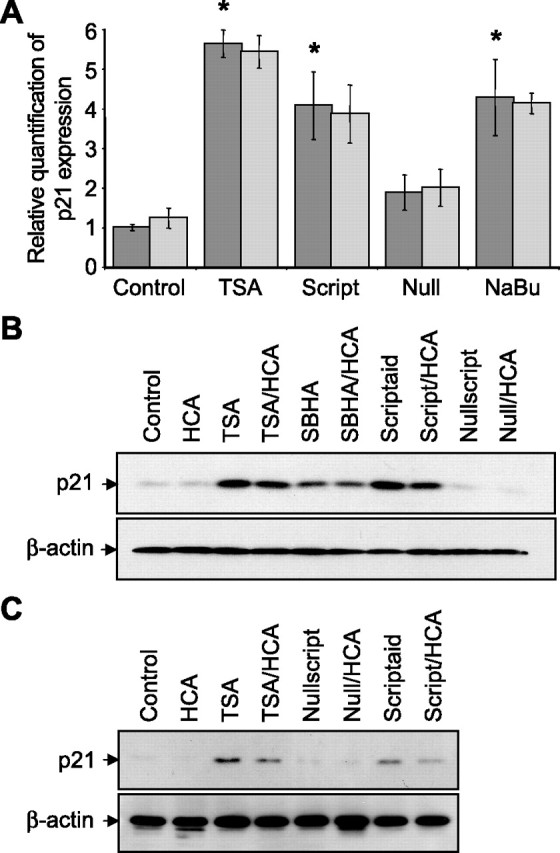
Structurally diverse HDAC inhibitors increase the expression of the cyclin-dependent kinase inhibitor, p21 in embryonic cortical neuronal cultures. A, Real-time PCR on cDNAs prepared from total RNA extracted from primary cortical neuronal cultures treated with TSA (0.66 μm), scriptaid (6.13 μm), nullscript (6.13 μm), and sodium butyrate (1 mm) treatments in the presence (light gray) or absence (dark gray) of HCA (5 mm) for 8 h. Control is no HDAC inhibitor. p21 amplification was normalized to β-actin in PCRs. Graph depicts mean fold increase in p21 expression relative to control ± SD. HCA has no significant effect on p21 expression by two-way ANOVA. *Significant difference from control, p < 0.01, by one-way ANOVA followed by Dunnett's multiple-comparisons test. B, Western blot analysis to detect relative levels of p21 in lysates from primary cortical neuronal cultures treated with TSA (0.66 μm), SBHA (12.5 μm), scriptaid (6.13 μm), and nullscript (6.13 μm). C, Western blot analysis to detect relative levels of p21 in lysates from primary cortical neuronal cultures after 2 h of incubation with HDAC inhibitor followed by 24 h of incubation with or without HCA (5 mm). HDAC inhibitors included TSA (1.32 μm), nullscript (12.3 μm), and scriptaid (12.3 μm). Relative levels of β-actin are shown to indicate loadings in Western blots. Antibodies against p21 or β-actin were used (see Materials and Methods).
Given the finding that a 2 h treatment of HDAC inhibitor is sufficient to protect immature cortical cultures from oxidative stress-induced death for 24 h without HDAC inhibitor-associated toxicity, we hypothesized that pulse inhibition might provide a strategy for identifying genes that contribute to neuroprotection but not toxicity. To take advantage of this strategy, we examined p21 gene expression 24 h after an HDAC inhibitor pulse treatment. Mixed cortical cells were treated with or without TSA for 2 h followed by incubation with or without HCA for 24 h. Western blot analysis showed that p21 protein is increased by a 2 h HDAC inhibitor pulse, and remains elevated for at least 24 h (Fig. 3C), providing good evidence that p21 upregulation correlates with HDAC inhibitor-mediated neuroprotection, but not toxicity.
HDAC inhibitors decrease cerebral ischemic infarct volumes in rats
MCAo in rodents has been developed and validated as a model of focal ischemia that is relevant to the clinical condition of stroke in humans (Bardutzky et al., 2005). MCAo infarction in rodents exhibits the complexity of evolving neuronal degeneration in human stroke, including rapid excitatory death in the ischemic core to more delayed apoptotic death in the penumbra (Love, 1999; Smith, 2004).
Using this model, we examined the ability of an HDAC inhibitor to protect against ischemic neuronal damage. Rats were treated with sodium butyrate (1200 mg/kg, i.p.) or PBS vehicle 24 h and again 4 h before permanently occluding the origin of the MCA with a filament. Histologically measured infarct volumes in brains of vehicle-treated rats (n = 6) killed 24 h after occlusion displayed infarction in the hemisphere ipsilateral to the occlusion (30.96 ± 5.20%) (Fig. 4). In contrast to this, infarct volumes in brains from sodium butyrate-treated rats (n = 6) killed 24 h after occlusion were significantly reduced (14.79 ± 3.29%; p < 0.025, by Student's t test) (Fig. 4). This >50% protection by sodium butyrate could not be attributed to independent affects of the drug on body temperature, plasma glucose levels, plasma pH, or blood flow, because these parameters were not different between treatment groups.
Figure 4.

The HDAC inhibitor sodium butyrate reduces infarct volumes induced by permanent focal ischemia in vivo and increases the expression of the cyclin-dependent kinase inhibitor, p21. Six rats were treated in parallel with sodium butyrate (NaBu) at a dose of 1200 mg/kg of body weight (intraperitoneal injection) 24 and again 4 h before MCAo. As controls, six rats received equal volume of vehicle (PBS) in a blinded study. Representative pictures show infarctions of PBS vehicle-treated (top) and NaBu-treated (bottom) brains after TTC staining.
This rodent stroke model was also used to examine the ability of an HDAC inhibitor to protect against ischemic damage if given after the permanent occlusion of the MCA. Rats were treated with sodium butyrate (1200 mg/kg, i.p.) or PBS vehicle 30 min after permanently occluding the MCA with a filament. Unlike the protection seen when HDAC inhibitors are given before occlusion, treating after permanently occluding the MCA resulted in no significant protection compared with vehicle treatment. Vehicle-treated rats killed 24 h after occlusion displayed infarct volumes of 33.08 ± 5.14%, whereas sodium butyrate-treated rats displayed infarct volumes of 34.77 ± 8.42%. Again, body temperature, plasma glucose levels, plasma pH, and blood flow were monitored throughout the MCAo procedure and were not significantly different between treatment groups. Additionally, to confirm that sodium butyrate treatment did not cause changes in body temperature that could significantly affect infarct volume size, body temperatures were monitored after surgery until the rats were killed. No significant change in body temperature was observed between sodium butyrate-treated and untreated rats.
To examine whether HDAC inhibitor protection against stroke is associated with an upregulation of p21 message, real-time PCR on cDNAs prepared from total RNA extracted from cortex of vehicle (n = 5) and sodium butyrate-treated animals (n = 5) was performed. As expected, the quantitative RT-PCR demonstrated an increase in p21 message levels, normalized to β-actin, in the cortex of sodium butyrate-treated animals compared with PBS-treated animals at the time point equivalent to when MCA occlusions were performed (4 h after last treatment). Fold inductions ± SD for PBS control-treatment and sodium treatment were 1.0 ± 0.8 and 3.6 ± 2.5, respectively (p < 0.03, by Student's t test).
Together, these findings suggest that HDAC enzymes could be considered as targets for protection against cerebral ischemia and that p21 upregulation accompanies HDAC inhibition and neuroprotection in vivo. The findings also support prior studies by several groups that HDAC inhibitors prevent neuronal damage in transient focal ischemia (Ren et al., 2004; Faraco et al., 2006).
p21 expression is sufficient for protection of neurons from oxidative stress-induced death
The robust induction of p21 in neurons and its correlation with protection, along with prior studies demonstrating the antideath effects of p21 in non-neural cells, made p21 an attractive candidate for mediating the neuroprotective effects of HDAC inhibition. To achieve high-efficiency expression of a p21 transgene in a cellular system sensitive to non-receptor-mediated glutamate toxicity, we turned our attention to using the mouse neuronal cell line, HT22, derived from immature hippocampal neurons (Li et al., 1997). HT22 cells were transfected to overexpress either GFP alone or a full-length p21-GFP fusion protein (Fig. 5A). The treatment of GFP-overexpressing HT22 cells with increasing concentrations of HCA resulted in increasing cell death, as quantified by MTT assay 24 h after HCA addition (Fig. 5B). In contrast to the cell death seen in HCA-treated GFP-overexpressing HT22 cells, significant protection from HCA-induced oxidative toxicity was seen in HT22 cells expressing the p21-GFP, even at high HCA concentrations (7.5 mm) (Fig. 5B). These results were confirmed by qualitative observations of HT22 cells using phase-contrast microscopy or live/dead staining. Together, these results suggest that p21 is sufficient to mimic the protective effect of HDAC inhibitors on glutathione depletion-induced death.
Figure 5.

p21 is sufficient for protection of neurons from oxidative stress-induced death. A, Western blot analysis to detect relative levels of GFP or p21-GFP fusion protein in lysates from HT22 murine hippocampal cells stably transfected with either pEGFP or pEGFP-p21 and treated with or without HCA (5 mm). Antibodies against p21 (top blot) or GFP (bottom blot) were used (see Materials and Methods). In addition to p21-GFP, the antibody for p21 detects endogenous p21, which is unchanged with HCA treatment. B, Graph showing viability of pEGFP- (dark gray) and pEGFP-p21- (light gray) transfected HT22 cells, as determined using the MTT assay, after treatment with increasing concentrations of HCA (2.5 mm to 7.5 mm) for 24 h. Graph bars depict mean ± SD. *Significant protection by p21 relative to GFP control, p < 0.001, by two-way ANOVA followed by Bonferroni posttests. C, Western blot analysis to detect relative levels p21 or GFP in lysates from cortical neuronal cultures infected with either Ad-GFP or Ad-p21+GFP adenoviruses and treated with or without hydrogen peroxide (10 μm). Antibodies against p21 or GFP were used (see Materials and Methods). D, Graph showing viability of Ad-GFP- (squares) and Ad-p21+GFP- (triangles) infected cortical neuron cultures, as determined using the MTT assay, after treatment with increasing concentrations of hydrogen peroxide (H2O2; 1–25 μm) for 24 h. Goodness of fit for GFP: R2 = 0.968; and for p21: R2 = 0.963. EC50GFP = 4 μm [95% confidence interval (CI), 3.3–4.7 μm]. EC50p21 = 12 μm (95% CI, 9.1–14.1 μm). EC50GFP and EC50p21 are significantly different (p < 0.0001).
To confirm that p21 overexpression was also protective against oxidative stress-induced death in primary neurons, mixed immature cortical cultures 5 DIV were infected with adenoviruses containing the expression cassettes for either GFP (Ad-GFP) or p21 and GFP (Ad-p21+GFP) (Fig. 5C). Mixed immature cortical cultures were infected at 5 DIV, because infections at earlier DIV resulted in poor transduction efficiency, as determined by GFP immunofluorescence (data not shown). After an additional 4 d in vitro, adenoviral-infected cortical cultures were exposed to increasing concentrations of hydrogen peroxide. Exogenous hydrogen peroxide was used instead of HCA as a source of oxidative stress in these studies because E17 neurons in culture over this time frame (9 DIV) can begin to express ionotropic glutamate receptors, making them susceptible to glutamate-induced excitotoxic death (Murphy and Baraban, 1990). Furthermore, hydrogen peroxide has been implicated in oxidative stress-induced cell death in a host of acute and chronic neurodegenerative conditions (Halliwell, 1992, 2006). The treatment of Ad-GFP-infected cortical cultures with increasing concentrations of hydrogen peroxide resulted in increasing cell death, as quantified by MTT assay 24 h after treatment (Fig. 5D). In contrast to the cell death seen in hydrogen peroxide-treated Ad-GFP-infected cells, significant protection from oxidative stress-induced toxicity was seen the Ad-p21+GFP-infected cells (Fig. 5D).
Altogether, these results suggest that p21 is sufficient to mimic the protective effect of HDAC inhibitors against oxidative death in neuronal cell lines and primary neurons. However, these experiments rely on the transgenic overexpression of p21, questioning whether p21 expressed at endogenous levels is sufficient to mediate a neuroprotective response. To ascertain whether p21 plays an endogenous role in neuroprotection, we asked whether deletion of p21 in vivo would exacerbate damage after transient focal ischemia. Wild-type and p21-null mice were subjected to a 20 min occlusion of the MCA with a filament, and infarct volumes were measured after the mice were killed at 72 h. Infarction in the hemisphere ipsilateral to the occlusion of wild-type mice was 46.2 ± 4.0 mm2 (n = 9). In contrast to this, p21-null mice displayed a significantly increased infarct volume of 60.2 ± 3.2 mm2 (n = 10; p < 0.05, by Student's t test). Thus, consistent with a role for endogenous p21 in mediating a protective response, we found that the p21-null mice have significantly larger infarcts than strain and age-matched wild-type mice.
p21 expression is not necessary for protection of neurons from oxidative stress-induced death
The observations that p21 is upregulated by HDAC inhibitors and that p21 overexpression is sufficient to protect cortical neurons against oxidative stress-induced death led us to explore whether p21 was an essential component of HDAC inhibitor-mediated neuroprotection.
Cultured immature cortical cultures prepared from p21-deficient or p21-wild-type embryonic day 15 mouse cortices were treated with HCA in the presence or absence of the hydroxamic acid-based inhibitors, trichostatin A and scriptaid, as well as the negative control HDAC inhibitor, nullscript. As seen previously with E17 wild-type rat mixed immature cortical cultures, the treatment of E15 p21-wild-type mouse immature cortical cultures with the HDAC inhibitor TSA resulted in a robust induction of p21. In contrast, neither a basal level nor TSA-induced level of p21 was detected in the p21-deficient mouse immature cortical cultures (Fig. 6B). As with wild-type rat mixed immature cortical cultures, the treatment with HCA resulted in widespread cell death in both p21-wild-type and p21-deficient mouse immature cortical cultures, as seen by calcein AM/ethidium homodimer (live/dead) staining and fluorescent microscopy (Fig. 6A) or quantified (90% death) by MTT assay (Fig. 6C) 24 h after HCA addition. In contrast to the widespread death seen in HCA-treated p21-deficient or p21-wild-type mouse immature cortical cultures, the cotreatment of p21-deficient or p21-wild-type mouse immature cortical cultures with an HDAC inhibitor resulted in significant protection (Fig. 6A,C). No difference was apparent between the protection seen in p21-wild-type mouse immature cortical cultures and p21-deficient immature cortical cultures. Also consistent with the wild-type rat mixed immature cortical cultures, the HDAC inhibitor negative control, nullscript, neither protected p21-deficient mouse cortical cultures nor p21-wild-type mouse cortical cultures from HCA-induced oxidative death (Fig. 6A,C). These findings suggest that p21 is not an essential component of the protective effect of HDAC inhibition on oxidative death.
Figure 6.

p21 expression is not necessary for protection of neurons from oxidative stress-induced death. A, Representative micrographs showing live/dead staining of p21-wild type (p21+/+; a–d) and p21-null (p21−/−; e–h) primary cortical neuronal cultures 24 h after treatment. a, e, Nontreated control; b, f, 5 mm HCA; c, g, 0.66 μm TSA; d, h, HCA + TSA. Live cells are detected by uptake and trapping of calcein AM (green fluorescence). Dead cells fail to trap calcein but are freely permeable to the highly charged DNA-intercalating dye ethidium homodimer (red fluorescence). B, Western blot analysis to detect relative levels of p21 in lysates from p21-WT and p21-null (KO) primary cortical neuronal cultures after TSA (0.66 μm) treatment in the presence or absence of HCA (5 mm) for 8 h. Control is no HDAC inhibitor. Relative levels of β-actin are shown to indicate loadings in Western blots. Antibodies against p21 or β-actin were used (see Materials and Methods). C, Graph showing viability of p21-wild type (dark gray) and p21-null (light gray) primary cortical neuronal cultures, as determined using the MTT assay, after TSA (0.66 μm), nullscript (6.13 μm), and scriptaid (6.13 μm) treatments in the presence or absence of HCA (5 mm) for 24 h. Control is no HDAC inhibitor. Graph bars depict mean ± SD. There is no statistical difference between p21-wild type and p21-null at each treatment by two-way ANOVA.
One of the reasons that p21 may not appear to be essential for HDAC inhibitor-mediated neuroprotection in p21-null mice is the potential compensation in function provided by other KIP family members, including p27 and p57 (Nakayama and Nakayama, 1998). Like p21, p27 and p57 can function in a nuclear context to inhibit cell cycle progression by constraining the activity of cyclin-dependent kinases (Nakayama and Nakayama, 1998). Immunoblot analysis of p27 and p57 showed that they are not upregulated in p21-wild-type mouse cortical neurons by HDAC inhibition, nor are they upregulated in p21-null mouse cortical neurons (Fig. 7). Whereas Cip/Kip inhibit a wide spectrum of Cdk/cyclin complexes, INK4 proteins (p15, p16, p18, and p19) are thought to be specific for cyclin-D-associated Cdks (Lees, 1995; Sherr and Roberts, 1999). Importantly, p15 and p16 have been demonstrated in some cell types to be upregulated by HDAC inhibition (Hitomi et al., 2003; Munro et al., 2004). Immunoblot analysis of the INK4 family proteins p15 and p16 showed that they are not upregulated by HDAC inhibitors in either wild-type (WT) or p21-null neurons (Fig. 7). Altogether, these findings make it unlikely that a failure to detect a reduction in viability in the absence of p21 is attributable to compensation by other cyclin-dependent kinase inhibitors.
Figure 7.
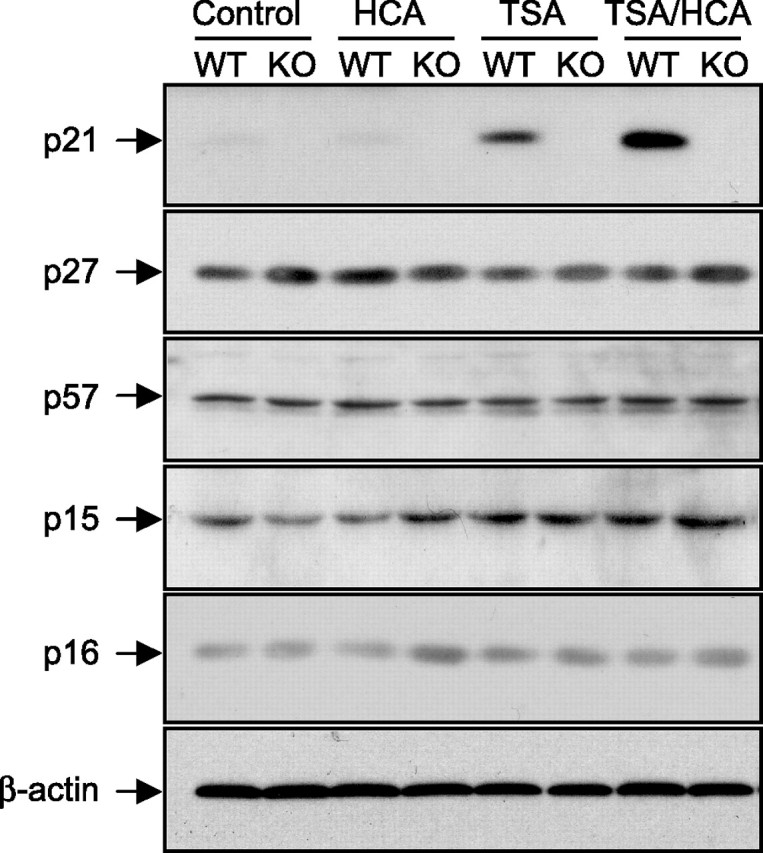
Other cyclin-dependent kinase inhibitors (CKIs) are not upregulated in p21-WT and p21-null (KO) primary cortical neurons by the HDAC inhibitor TSA. Shown is Western blot analysis to detect relative levels of the CKIs, p21, p27, p57, p15, and p16 in lysates from p21-WT and p21-null (KO) primary cortical neuronal cultures treated with TSA (0.66 μm) in the presence or absence of HCA (5 mm) for 8 h. Control is no HDAC inhibitor. Relative levels of β-actin are shown to indicate loadings in Western blots. Antibodies against p21, p27, p57, p15, p16, or β-actin were used (see Materials and Methods).
Oxidative glutamate toxicity in embryonic cortical neuronal cultures does not appear to induce aberrant cell cycle reentry
The lack of compensation by cyclin-dependent kinase inhibitors led us to go back and reexamine the notion that oxidative stress induces apoptosis by stimulating the aberrant reentry of postmitotic neurons into the cell cycle (Herrup et al., 2004; Langley and Ratan, 2004). To determine whether oxidative stress triggers reactivation of components of the cell cycle, we performed Western blot analysis to look for changes in the levels of cyclin D and Cdk4, which are active in early G1, as well as the levels of cyclin E and Cdk2, which are active in late G1 and early S (Lees, 1995; Sherr and Roberts, 1999). The treatment of mixed immature cortical cells with HCA alone resulted in no induction of cyclin D, cyclin E, Cdk2, or Cdk4 levels relative to control cells over the time course in which these cells commit to, and begin to die (Fig. 8A). Moreover, the treatment with the HDAC inhibitor, TSA, did not decrease the detectable levels of any of the cyclins or Cdks. In fact, the only change that was observed was a TSA-induced increase in cyclin E levels (Fig. 8A), consistent with that reported in human tumor cells by other groups (Sambucetti et al., 1999; Kim et al., 2006).
Figure 8.
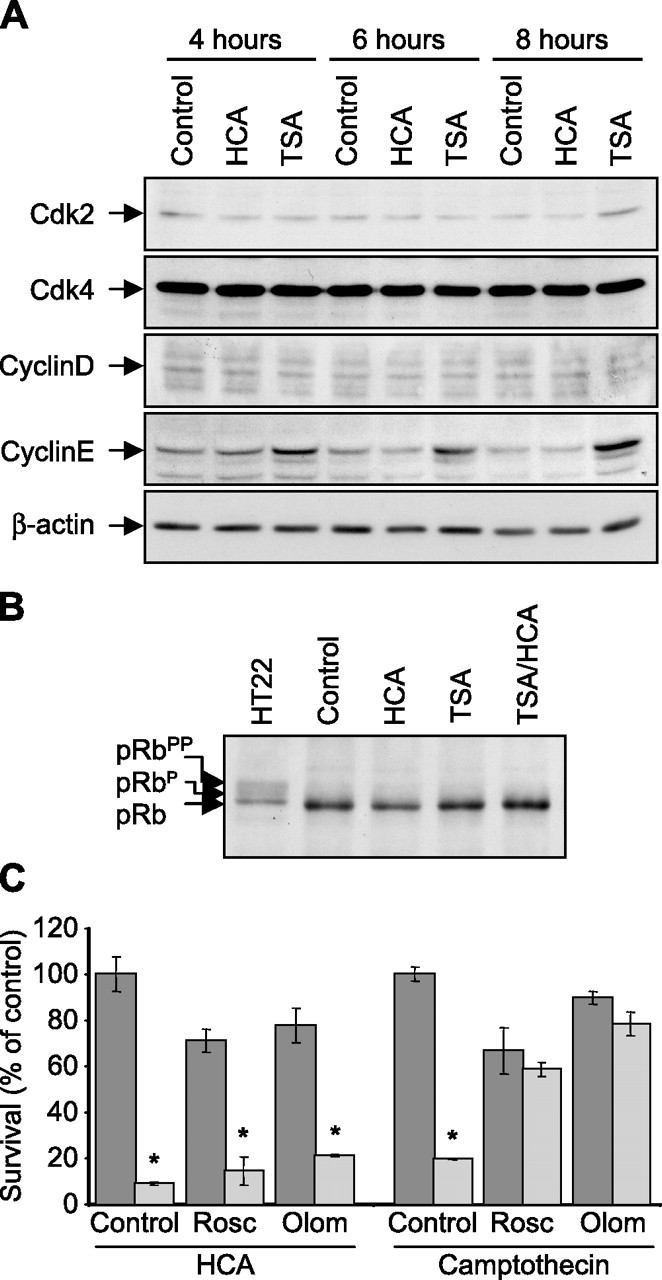
Oxidative glutamate toxicity in embryonic cortical neuronal cultures does not appear to induce aberrant cell cycle reentry. A, Western blot analysis to detect relative levels of the cyclin-dependent kinases, Cdk2 and Cdk4, and their respective cognate cyclins, cyclin D and cyclin E, in lysates from primary cortical neuronal cultures after HCA (5 mm) treatment in the presence or absence of TSA (0.66 μm) for 4, 6, or 8 h. Control is no HDAC inhibitor. Relative levels of β-actin are shown to indicate loadings in Western blots. Antibodies against Cdk2, Cdk4, cyclin D, cyclin E, and β-actin were used (see Materials and Methods). B, Western blot analysis to detect relative pRb phosphorylation in lysates from primary cortical neuronal cultures after HCA (5 mm) treatment in the presence or absence of TSA (0.66 μm) for 8 h. Control is no HDAC inhibitor. Lysates from the mitotically active HT22 murine hippocampal cell line were included to demonstrate phosphorylated species of pRb (shift in pRb migration). Antibodies against pRb were used (see Materials and Methods). C, Graph showing primary cortical neuron viability, as determined using the MTT assay, after roscovitine (Rosc; 10 μm) or olomoucine (Olom; 200 μm) treatments in the presence (light gray) or absence (dark gray) of HCA (5 mm) or camptothecin (10 μm) for 24 h. Graph bars depict mean ± SD. *Significant difference from controls, p < 0.0001, by two-way ANOVA followed by Bonferroni posttests.
pRb is a major substrate of cyclin-Cdk activity, and becomes phosphorylated during G1 and the G1–S transition of the cell cycle (Lees, 1995; Sherr and Roberts, 1999). Because pRb phosphorylation status has been used as a reliable marker of cell cycle reentry of postmitotic neurons in a host of neurodegenerative disease and injury states (Hayashi et al., 2000; Osuga et al., 2000; Ranganathan et al., 2001; Wang et al., 2002; Jordan-Sciutto et al., 2003; Nguyen et al., 2003), we examined by Western blot analysis whether oxidative stress induces pRb phosphorylation. Compared with the pRb and phosphorylated forms of pRb that can be detected in lysates from the proliferating hippocampal HT22 cell line, pRb in untreated control cell lysates was detected as a single hypophosphorylated band, consistent with E17 rat embryo-derived mixed immature cortical cells being predominantly postmitotic (Fig. 8B). Treatment of the mixed immature cortical cells with HCA alone resulted in no change in pRb phosphorylation status relative to control cells at a time when these cells have become committed to death (Fig. 8B). In fact, no change in pRb phosphorylation status was observed at even longer HCA exposure times of 12 and 16 h (data not shown). These observations are consistent with the possibility that glutathione depletion-induced oxidative stress does not promote neuronal death by stimulating the aberrant cell cycle reentry of postmitotic neurons.
A causative role for cell cycle proteins in activating death pathways in neurons is supported by the observations that pharmacological inhibitors of cell cycle proteins such as roscovitine, olomoucine, or flavopiridol, which inhibit Cdks via their ability to act as a competitive inhibitor for ATP binding (Vesely et al., 1994; De Azevedo et al., 1997; Meijer et al., 1997), are neuroprotective in vitro and in vivo (Park et al., 1996, 1997, 1998a,b; Osuga et al., 2000). To confirm our observations that oxidative stress does not induce neuronal death by stimulating the aberrant cell cycle reentry of postmitotic neurons, we examined whether the pharmacological Cdk inhibitors roscovitine and olomoucine were protective against HCA-induced oxidative death. Treatment of mixed immature cortical cultures with HCA resulted in cell death, as quantified 24 h after HCA addition by MTT assay (Fig. 8C). Oxidative stress-induced death was not abrogated by the addition of either of the pharmacological Cdk inhibitors, roscovitine or olomoucine. Camptothecin-induced neuronal toxicity is a well established model in which camptothecin, through its ability to inhibit topoisomerase I, results in the accumulation of DNA damage and death (Park et al., 1997). Whereas roscovitine and olomoucine did not protect against oxidative stress-induced death, both of these pharmacological Cdk inhibitors were able to protect mixed immature cortical cultures against camptothecin-induced death.
These results are consistent with findings by Park et al. (1998b), in which these inhibitors could protect against DNA-damaging agents but not against oxidative stress induced by superoxide dismutase depletion in sympathetic neurons. Furthermore, when taken together with the observations that G1 cyclin-Cdk expression and pRb phosphorylation status are not altered by oxidative stress, these findings make it unlikely that aberrant cell cycle reentry of postmitotic neurons is a predominant mechanism leading to death in this oxidative stress model.
p21 acts in the cytoplasm to protect against oxidative stress-induced death
The lack of evidence supporting a role for aberrant cell cycle reentry of postmitotic neurons in this model of oxidative stress-induced death suggests that the protective effects of p21 might be mediated via inhibition of a kinase or pathway involved in death signaling. Indeed, cytoplasmic protective roles for p21, which include inhibiting the proapoptotic proteins ASK-1 (Asada et al., 1999) and SAPK/JNK (Shim et al., 1996; Xue et al., 2003), as well as inhibiting caspase-8 and caspase-3 activation (Suzuki et al., 1998, 1999; Xu and El-Deiry, 2000) and maintaining antiapoptotic c-IAP1 (Steinman and Johnson, 2000) and Bcl-XL expression in various cell types outside the CNS have been described.
To explore this possibility, we examined whether HDAC inhibitor-induced p21 was present in the cytoplasm of immature cortical cells. Western blot analysis for p21 expression in nuclear and cytoplasmic fractionations of untreated immature cortical cells revealed that basal levels of p21 could be detected in the nuclear fraction but not the cytoplasmic fraction (Fig. 9A). As expected, this basal p21 level in the nuclear fraction was not increased by the treatment of these mixed immature cortical cultures with HCA for 8 h. In contrast, p21 protein levels in both the nucleus and cytoplasm were increased by treatment with the HDAC inhibitor TSA alone or in combination with HCA (Fig. 9A). These results demonstrate that HDAC inhibitor-induced p21 is indeed present in the cytoplasm of protected immature cortical cultures.
Figure 9.
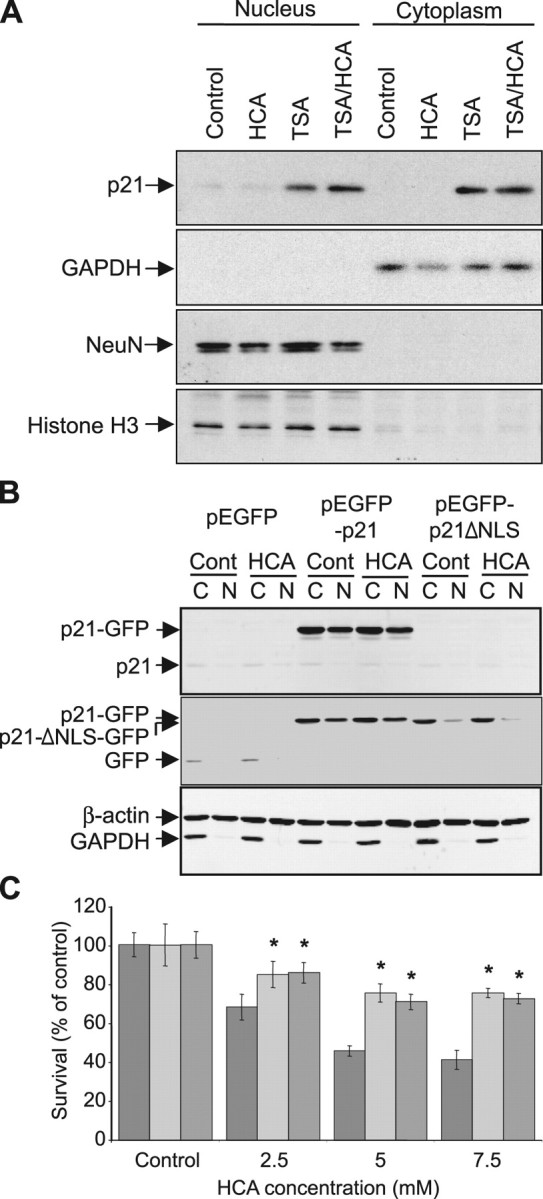
p21 acts in the cytoplasm to protect neurons from oxidative stress-induced death. A, Western blot analysis to detect relative levels of p21 in nuclear and cytoplasmic fractions from primary cortical neuronal cultures treated with TSA (0.66 μm) in the presence or absence of HCA (5 mm). Control is no TSA treatment. Relative levels of GAPDH or NeuN and Histone H3 are shown to indicate purity of cytoplasmic or nuclear fractions, respectively. Antibodies against p21, GAPDH, NeuN, and Histone H3 were used (see Materials and Methods). B, Western blot analysis to detect relative levels of GFP, p21-GFP fusion protein or p21ΔNLS-GFP fusion protein in cytoplasmic (C) and nuclear (N) fractions from HT22 murine hippocampal cells stably transfected with pEGFP, pEGFP-p21, or pEGFP-p21-ΔNLS and treated with or without HCA (5 mm). Antibodies against p21 (top) or GFP (middle) were used (see Materials and Methods). The antibody for p21 does not detect the p21ΔNLS-GFP fusion protein because the epitope this monoclonal antibody recognizes is located within the deleted nuclear localization signal (NLS). However, in addition to p21-GFP, this antibody detects endogenous p21, which is unchanged with HCA treatment. C, Graph showing viability of pEGFP- (dark gray), pEGFP-p21- (light gray), and pEGFP-p21-ΔNLS- (medium gray) transfected HT22 cells, as determined using the MTT assay, after treatment with increasing concentrations of HCA (2.5–7.5 mm) for 24 h. Graph bars depict mean ± SD. *Significant protection by p21 and p21-ΔNLS relative to GFP control, p < 0.001, by two-way ANOVA followed by Bonferroni posttests.
To test the hypothesis that p21 protects against oxidative stress via a cytoplasmic role, HT22 cells were transfected to overexpress either GFP, full-length p21-GFP, or a mutated p21-GFP construct in which p21 lacks a nuclear localization signal (p21-ΔNLS-GFP) (Asada et al., 1999) (Fig. 9B). Western blot analysis for p21 transgene expression in nuclear and cytoplasmic fractionations of HT22 neurons revealed that p21-GFP could be detected in both the nuclear and cytoplasmic fractions, whereas p21-ΔNLS-GFP was only detected in the cytoplasmic fraction (Fig. 9B). The treatment of GFP-overexpressing HT22 cells with increasing concentrations of HCA resulted in increasing cell death, as quantified by MTT assay 24 h after HCA addition (Fig. 9C). In contrast to the cell death seen in HCA-treated GFP expressing HT22 cells, significant protection from HCA-induced oxidative toxicity was seen with both full-length p21-GFP and p21-ΔNLS-GFP expression (Fig. 9C). These results were confirmed by qualitative observations of HT22 cells using phase-contrast microscopy or live/dead staining. Together, these results suggest that cytoplasmic p21 is sufficient to protect against glutathione depletion-induced death.
ASK-1 is a mammalian mitogen-activated protein kinase kinase kinase (MAPKKK), which is activated in response to various cytotoxic stresses including hydrogen peroxide, Fas ligand, TNF, serum withdrawal and some cancer chemotherapeutic agents, and activates the SAPK/JNK and p38 pathways (Ichijo et al., 1997; Tobiume et al., 1997; Chang et al., 1998; Gotoh and Cooper, 1998). Because cytoplasmic p21 has been reported to interact with and inhibit ASK-1 (Asada et al., 1999; Huang et al., 2003; Zhan et al., 2007), and HDAC inhibitor-induced p21 is present in the cytoplasm, we investigated p21 and ASK-1 interactions in neurons.
To first examine whether the stress-activated mitogen-activated protein kinase (MAPK) cascade is activated in response to oxidative stress and attenuated by HDAC inhibition, cortical neuronal cultures were treated for 16 and 18 h with HCA or TSA alone or with HCA and TSA. Relative to control levels, HCA treatment induced a robust increase in SAPK/JNK phosphorylation at 16 and 18 h, confirming prior reports that it is induced by oxidative stress (Fig. 10A). In contrast to this, no increased SAPK/JNK phosphorylation was observed at any time point when cortical neuronal cultures were cotreated with HCA and the HDAC inhibitor, TSA (Fig. 10A).
Figure 10.

HDAC inhibitors increase p21 interaction with ASK-1 and attenuate oxidative stress-induced SAPK/JNK signaling. A, Western blot analysis to detect phosphorylated JNK and total JNK after treatment with TSA (1.2 μm) in the presence or absence of HCA (5 mm) for 16 or 18 h. Antibodies against phospho-JNK and total JNK were used (see Materials and Methods). B, Western blot analysis to detect HA-ASK-1 in p21-immunoprecipitated lysates from B35 cells after treatment with TSA (1.2 μm) in the presence or absence of HCA (5 mm) for 16 h. B35 cells were transfected with HA-tagged ASK-1 expression construct 24 h before treatment. p21-immunoprecipitated lysates from HA-ASK-1 nontransfected cells served as negative control, and Western blot analysis of relative levels of β-actin is shown to confirm equal proteins in the lysates used for immunoprecipitation.
To examine whether an increased interaction between p21 and ASK-1 might account for this HDAC inhibitor-induced attenuation of SAPK/JNK activation, B35 neuroblastoma cell line neurons were transiently transfected to overexpress a hemagglutinin-tagged ASK-1 (HA-ASK-1), and p21·HA-ASK-1 coimmunoprecipitations (co-IPs) were performed on cellular lysates after treatment with HCA or TSA alone or with HCA and TSA. IP using a p21-specific antibody resulted in the co-IP of low levels of HA-ASK-1 in untreated control and HCA-treated cell lysates as determined by Western blot analysis (Fig. 10B). In contrast, p21 IP in B35 cells lysates after TSA or TSA and HCA treatment resulted in much greater co-IP of HA-ASK-1. As a control, p21 IP was performed on untransfected B35 lysates. As anticipated, no HA-ASK-1 co-IP was observed by Western blot analysis (Fig. 10B). Together, these results confirm that p21 interacts with ASK-1 in neurons and that the amount of interaction is increased by HDAC inhibitor-induced cytoplasmic p21. These observations, in addition to the finding that cytosolic p21 prevents oxidative death, are consistent with a model in which ectopic or HDAC inhibitor-mediated increases in cytosolic p21 result in inhibition of oxidative stress-induced stress kinase signaling via interaction and inhibition of ASK-1 (Fig. 11).
Figure 11.

Model for the role of p21 in HDAC inhibitor-mediated neuroprotection. Glutathione depletion-induced oxidative stress leads to neuronal death with both apoptotic and necrotic features. Dysregulation of the arachidonic acid-metabolizing enzyme 12-lipoxygenase results in the production of the eicosanoid 12-HETE and downstream activation of the MAPK-Erk signal cascade. Persistent phosphorylation and nuclear retention of Erk appears to be one of the late events before the commitment of neurons to death after glutathione depletion. In parallel, the MAPKKK ASK-1 is activated, which results in SAPK/JNK activation and neuronal death. Inhibiting either of these pathways promotes neuronal survival. Cytosolic p21 induced by transgenic overexpression or HDAC inhibition binds ASK-1 and inhibits its activation and the activation of downstream kinases that participate in neuronal death.
Discussion
HDAC inhibitors have generated a great deal of excitement as potential neurotherapeutics through their ability to prolong survival and enhance functional recovery in a host of neurological disease models in vivo. Progress in understanding the salutary effects of HDAC inhibitors at a molecular level has been hampered by toxicities associated with these agents in vitro, even in paradigms in which they impart significant protection (Fig. 1). The consideration of strategies that would optimize the salutary effects of HDAC inhibitors and diminish their toxicity to neurons in vitro were influenced by elegant observations suggesting that HDAC inhibition induces neuronal death via “derepression” of genes involved in cell death, including B-myb and Bim (Liu and Greene, 2001; Liu et al., 2004; Biswas et al., 2005). These studies imply that after the inhibitor is removed, HDAC activity at the prodeath gene promoter would resume and transcription would go back to a repressed state. Thus a short pulse of an HDAC inhibitor might be insufficient to activate a threshold level of prodeath gene expression. In contrast to this, the acetylation of certain transcription factors, such as Sp1, by HDAC inhibitors can mediate “activation” of genes involved in neuronal survival (Ryu et al., 2003). Unlike derepression, the recruitment of an activator and transcriptional activation could be sustained even after the inhibitor is removed. We were particularly drawn to this model because a pulse of HDAC inhibition more accurately mimics the pharmacodynamics of HDAC inhibitor delivery in vivo, where we found protection from permanent focal ischemia (Fig. 4). Consistent with both of these lines of investigation, we show that a 2 h pulse of an HDAC inhibitor is sufficient to impart durable protection without toxicity in a model of neuronal oxidative stress (Fig. 2A,B). If derepression of prodeath genes is responsible for the toxicity of these agents, then longer exposures of cortical neurons to HDAC inhibitors should increase the time of derepression and be increasingly toxic. Indeed, this was what we observed experimentally (Fig. 2). Future studies will build on this important advance to determine the precise identity of genes “derepressed” by prolonged HDAC inhibition in cortical neurons that mediate cell death.
The ability of pulse treatment to remove the toxicity associated with HDAC inhibition, without diminishing p21 induction, argues that p21 is not responsible for HDAC inhibitor toxicity in neurons. Indeed, neuroprotection in vivo from ischemia also correlated with increased p21 expression in the CNS. We found that the overexpression of p21 or a p21 mutant, in which the nuclear localization sequence has been deleted (p21-ΔNLS), was sufficient to mimic the effects of HDAC inhibition on neuronal protection. Because the cell cycle-inhibitory activity of p21 is intimately correlated with its nuclear localization (Jiang et al., 1994; Steinman et al., 1994; Halevy et al., 1995; Andres and Walsh, 1996), these findings dissociate the protective effects of p21 from its effects on the cell cycle. Consistent with this, we found no evidence of aberrant cell cycle regulation in response to glutathione depletion-induced oxidative death in our postmitotic neurons. We were also unable to observe protection with prototypical inhibitors of the cyclin-dependent kinases, roscovitine and olomoucine (Vesely et al., 1994; Meijer et al., 1997; Park et al., 1997). In contrast, and consistent with previous reports, we did find that these agents abrogate camptothecin-induced neuronal death (Park et al., 1996, 1997, 1998a), a model in which neuronal death by aberrant cell cycle reactivation has been clearly established. Together, these findings suggest that glutathione depletion-induced oxidative stress results in neuronal death via cell cycle-independent pathways and, in concert with previous studies, suggest that markers of cell cycle progression in neurological disease models might be mediated by stresses other than oxidant stress.
So how does cytoplasmic p21 prevent glutathione depletion-induced death? Glutathione depletion leads to a form of death with both apoptotic and necrotic features (Ratan et al., 1994). This death appears to involve dysregulation of the arachidonic acid-metabolizing enzyme, 12-lipoxygenase, because pharmacological or molecular deletion of 12-lipoxygenase prevents death (Li et al., 1997; Khanna et al., 2003, 2005). Indeed, 12-lipoxygenase appears to be an important mediator of neuronal injury after cerebral ischemia (Khanna et al., 2005). A major product of 12-lipoxygenase activity is the eicosanoid, 12-HETE (Szekeres et al., 2002). Downstream of 12-HETE is the activation of MAPK signal cascades. Persistent phosphorylation and nuclear retention of Erk appears to be one of the late events before the commitment of neurons to death after glutathione depletion (Fig. 11) (Stanciu et al., 2000; Stanciu and DeFranco, 2002). Another MAPK signaling cascade that is activated in response to oxidative stress is the stress-activated protein kinase SAPK/JNK and its upstream activator ASK-1 (Ichijo et al., 1997; Tobiume et al., 1997; Chang et al., 1998; Gotoh and Cooper, 1998). ASK-1 is tightly regulated in cells by its association with its physiological inhibitor thioredoxin. Under oxidative stress conditions, reactive oxygen species-dependent oxidation of thioredoxin results in ASK-1 dissociation (Liu et al., 2000) and activation by oligomerization-dependent autophosphorylation (Fig. 11) (Gotoh and Cooper, 1998). Here we report that HDAC inhibitor-mediated neuroprotection is associated with increased interaction of p21 with ASK-1 (Fig. 10B). As predicted from this interaction, HDAC inhibitors also inhibit activation of the SAPK/JNK pathway in response to oxidative stress (Fig. 10A). We also demonstrate that cytosolic p21 can prevent oxidative death (Fig. 9C). Together these findings are consistent with a model in which cytosolic p21 induced by transgenic overexpression or HDAC inhibition can inhibit ASK-1 and downstream kinases that participate in glutathione depletion-induced death (Fig. 11).
Despite being sufficient to mimic the protective effects of HDAC inhibitors on oxidative neuronal death, p21 does not appear to be essential for HDAC inhibitor-mediated neuroprotection. Indeed, like mouse cortical neuronal cultures from p21 wild-type mice, mouse cortical neuronal cultures from mice with homozygous deletion of p21 are completely protected from oxidative stress-induced death by HDAC inhibitors (Fig. 6). Moreover, this lack of difference between protection in the p21-wild-type and p21-null mice does not appear to be attributable to upregulation of other cyclin-dependent kinase inhibitors (e.g., p27, p57, p15, and p16) (Fig. 7). Thus, it is likely that the protective effects of HDAC inhibitors are p21 independent, or that HDAC inhibitor-mediated neuroprotection is the result of overlapping protective effects of a number of genes and signaling pathways. Supporting the latter, recent reports examining HDAC inhibition and neuroprotection against oxygen/glucose deprivation and focal ischemia have implicated gelsolin (Meisel et al., 2006), HSP70 (Ren et al., 2004; Faraco et al., 2006), and BclII (Faraco et al., 2006) as potential downstream targets that mediate neuroprotection. Studies are underway to define the precise HDAC whose inhibition allows upregulation of p21 expression by HDAC inhibitors. Molecular deletion of this HDAC isoform will allow us to determine whether putative overlapping protective capacities are controlled by a single HDAC isoform or represent the ability of HDAC inhibitors to broadly inhibit a number of isoforms from the family class I and II HDACs.
In this study, we show that the intraperitoneal administration of the short chain fatty acid HDAC inhibitor sodium butyrate can significantly decrease ischemic brain infarct volumes in a rat MCAo model of permanent focal ischemia (Fig. 4). With regard to the complexity and heterogeneity of ischemic stroke, the idea that HDAC inhibitors could potentially induce multiple neuroprotective proteins, in addition to p21, argues that HDAC inhibition might be a target for successful clinical therapy. Using intraperitoneal administration, we were not able to observe protection when sodium butyrate was delivered after stroke. Given the potential requirement for de novo gene expression in the salutary effects of HDAC inhibition, intraperitoneal administration may not provide the optimal pharmacodynamics to protect against acute stroke. Future studies using intravascular delivery with agents that rapidly cross in to the CNS may provide a stronger opportunity to realize neuroprotection in permanent ischemia. The use of HDAC inhibitors in clinical therapy will also depend on tolerability and toxicity. In this study, we show that HDAC inhibitor toxicity can be abrogated without lessening the neuroprotective effect, implying that the neuroprotective and toxic effects of HDAC inhibition are separable. Understanding the neuroprotective versus toxic mechanisms induced by HDAC inhibition, including which HDACs are involved in the different processes, will be important as this class of drugs are considered for and move toward the clinical treatment of stroke and neurodegenerative disease.
Footnotes
This work was supported by the National Institutes of Health Grant 5P01NS0452242-05 (R.R.R.), a Center of Excellence in Spinal Cord Injury grant funded by the New York State Department of Health (R.R.R.), and a Transition to Independence Award funded by the Goldsmith Foundation (B.L.). We thank Dr. Alvaro Estevez for helpful support with statistical analyses, Dr. Ambreena Siddiq for experimental support, Dr. Minoru Asada for providing the pEGFP, pEGFP-p21-full, and pEGFP-p21-ΔNLS constructs, and Dr. Houghton for providing the HA-tagged ASK-1 construct and expert advice.
References
- Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardutzky J, Meng X, Bouley J, Duong TQ, Ratan R, Fisher M. Effects of intravenous dimethyl sulfoxide on ischemia evolution in a rat permanent occlusion model. J Cereb Blood Flow Metab. 2005;25:968–977. doi: 10.1038/sj.jcbfm.9600095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH, et al. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Votyakova TV, Reynolds IJ. Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J Neurochem. 2003;85:563–570. doi: 10.1046/j.1471-4159.2003.01678.x. [DOI] [PubMed] [Google Scholar]
- Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J Biol Chem. 1998;273:17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- Gregory PD, Wagner K, Horz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265:195–202. doi: 10.1006/excr.2001.5187. [DOI] [PubMed] [Google Scholar]
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Sakai K, Sasaki C, Zhang WR, Abe K. Phosphorylation of retinoblastoma protein in rat brain after transient middle cerebral artery occlusion. Neuropathol Appl Neurobiol. 2000;26:390–397. doi: 10.1046/j.1365-2990.2000.00264.x. [DOI] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15(INK4b) in HDAC inhibitor-induced growth arrest. FEBS Lett. 2003;554:347–350. doi: 10.1016/s0014-5793(03)01186-4. [DOI] [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11:1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Jiang H, Lin J, Su ZZ, Collart FR, Huberman E, Fisher PB. Induction of differentiation in human promyelocytic HL-60 leukemia cells activates p21, WAF1/CIP1, expression in the absence of p53. Oncogene. 1994;9:3397–3406. [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, Achim CL. Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol. 2003;62:68–74. doi: 10.1093/jnen/62.1.68. [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL, Sen CK. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005;36:2258–2264. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kang JK, Kim YK, Seo DW, Ahn SH, Lee JC, Lee CH, You JS, Cho EJ, Lee HW, Han JW. Histone deacetylase inhibitor apicidin induces cyclin E expression through Sp1 sites. Biochem Biophys Res Commun. 2006;342:1168–1173. doi: 10.1016/j.bbrc.2006.02.081. [DOI] [PubMed] [Google Scholar]
- Langley B, Ratan RR. Oxidative stress-induced death in the nervous system: cell cycle dependent or independent? J Neurosci Res. 2004;77:621–629. doi: 10.1002/jnr.20210. [DOI] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Lees E. Cyclin dependent kinase regulation. Curr Opin Cell Biol. 1995;7:773–780. doi: 10.1016/0955-0674(95)80060-3. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001;32:425–438. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Liu DX, Biswas SC, Greene LA. B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J Neurosci. 2004;24:8720–8725. doi: 10.1523/JNEUROSCI.1821-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9:119–131. doi: 10.1111/j.1750-3639.1999.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Meisel A, Harms C, Yildirim F, Bosel J, Kronenberg G, Harms U, Fink KB, Endres M. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–1031. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res. 2004;295:525–538. doi: 10.1016/j.yexcr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Baraban JM. Glutamate toxicity in immature cortical neurons precedes development of glutamate receptor currents. Brain Res Dev Brain Res. 1990;57:146–150. doi: 10.1016/0165-3806(90)90195-5. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Nakayama K, Nakayama K. Cip/Kip cyclin-dependent kinase inhibitors: brakes of the cell cycle engine during development. Bioessays. 1998;20:1020–1029. doi: 10.1002/(SICI)1521-1878(199812)20:12<1020::AID-BIES8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Boudreau M, Kriz J, Couillard-Despres S, Kaplan DR, Julien JP. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J Neurosci. 2003;23:2131–2140. doi: 10.1523/JNEUROSCI.23-06-02131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda JE, Park DS. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA. 2000;97:10254–10259. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998a;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998b;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Scudiere S, Bowser R. Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer's disease and amyotrophic lateral sclerosis. J Alzheimers Dis. 2001;3:377–385. doi: 10.3233/jad-2001-3403. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci USA. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Lee H, Park J, Kim H, Choi EJ. A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature. 1996;381:804–806. doi: 10.1038/381804a0. [DOI] [PubMed] [Google Scholar]
- Smith WS. Pathophysiology of focal cerebral ischemia: a therapeutic perspective. J Vasc Interv Radiol. 2004;15:S3–12. doi: 10.1097/01.rvi.0000108687.75691.0c. [DOI] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Steinman RA, Johnson DE. p21WAF1 prevents down-modulation of the apoptotic inhibitor protein c-IAP1 and inhibits leukemic apoptosis. Mol Med. 2000;6:736–749. [PMC free article] [PubMed] [Google Scholar]
- Steinman RA, Hoffman B, Iro A, Guillouf C, Liebermann DA, el-Houseini ME. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene. 1994;9:3389–3396. [PubMed] [Google Scholar]
- Su GH, Sohn TA, Ryu B, Kern SE. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res. 2000;60:3137–3142. [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931–939. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Miura M, Akahane K. Caspase 3 inactivation to suppress Fas-mediated apoptosis: identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene. 1999;18:1239–1244. doi: 10.1038/sj.onc.1202409. [DOI] [PubMed] [Google Scholar]
- Szekeres CK, Trikha M, Honn KV. 12(S)-HETE, pleiotropic functions, multiple signaling pathways. Adv Exp Med Biol. 2002;507:509–515. doi: 10.1007/978-1-4615-0193-0_78. [DOI] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Inage T, Takeda K, Enomoto S, Miyazono K, Ichijo H. Molecular cloning and characterization of the mouse apoptosis signal-regulating kinase 1. Biochem Biophys Res Commun. 1997;239:905–910. doi: 10.1006/bbrc.1997.7580. [DOI] [PubMed] [Google Scholar]
- Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S, et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- Wang F, Corbett D, Osuga H, Osuga S, Ikeda JE, Slack RS, Hogan MJ, Hakim AM, Park DS. Inhibition of cyclin-dependent kinases improves CA1 neuronal survival and behavioral performance after global ischemia in the rat. J Cereb Blood Flow Metab. 2002;22:171–182. doi: 10.1097/00004647-200202000-00005. [DOI] [PubMed] [Google Scholar]
- Xiao H, Hasegawa T, Isobe K. Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J Cell Biochem. 1999;73:291–302. [PubMed] [Google Scholar]
- Xu SQ, El-Deiry WS. p21(WAF1/CIP1) inhibits initiator caspase cleavage by TRAIL death receptor DR4. Biochem Biophys Res Commun. 2000;269:179–190. doi: 10.1006/bbrc.2000.2247. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ramaswamy NT, Hong X, Pelling JC. Association of JNK1 with p21waf1 and p53: modulation of JNK1 activity. Mol Carcinog. 2003;36:38–44. doi: 10.1002/mc.10096. [DOI] [PubMed] [Google Scholar]
- Zhan J, Easton JB, Huang S, Mishra A, Xiao L, Lacy ER, Kriwacki RW, Houghton PJ. Negative regulation of ASK1 by p21Cip1 involves a small domain that includes serine 98 that is phosphorylated by ASK1 in vivo. Mol Cell Biol. 2007;27:3530–3541. doi: 10.1128/MCB.00086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]