

Abstract
Huntington's disease (HD) is a fully penetrant autosomal-dominant inherited neurological disorder caused by expanded CAG repeats in the Huntingtin gene. Transcriptional dysfunction, excitotoxicity, and oxidative stress have all been proposed to play important roles in the pathogenesis of HD. This study was designed to explore the therapeutic potential of mithramycin, a clinically approved guanosine-cytosine-rich DNA binding antitumor antibiotic. Pharmacological treatment of a transgenic mouse model of HD (R6/2) with mithramycin extended survival by 29.1%, greater than any single agent reported to date. Increased survival was accompanied by improved motor performance and markedly delayed neuropathological sequelae. To identify the functional mechanism for the salubrious effects of mithramycin, we examined transcriptional dysfunction in R6/2 mice. Consistent with transcriptional repression playing a role in the pathogenesis of HD, we found increased methylation of lysine 9 in histone H3, a well established mechanism of gene silencing. Mithramycin treatment prevented the increase in H3 methylation observed in R6/2 mice, suggesting that the enhanced survival and neuroprotection might be attributable to the alleviation of repressed gene expression vital to neuronal function and survival. Because it is Food and Drug Administration-approved, mithramycin is a promising drug for the treatment of HD.
Keywords: mithramycin, Huntington's disease, HD, Huntingtin, Htt, histone methylation, neuroprotection, transcription
Introduction
Huntington's disease (HD) is an autosomal-dominant neurological disorder manifested by involuntary movements, cognitive dysfunction, and psychiatric disturbances. These symptoms are the result of expanded stretches of glutamines in the coding region of the huntingtin (htt) protein. Polyglutamine expansions lead to a number of cellular abnormalities, including dysregulation in transcriptional control (Cha, 2000). Indeed, the polyglutamine stretches in mutant htt interact specifically with a host of transcriptional activators and coactivators that contain distinct glutamine-rich activation domains, including CBP (CREB binding protein) (Steffan et al., 2001), TAFII130 (transcriptional activator factor II 130) (Dunah et al., 2002), and Sp1 (specificity protein 1) (Dunah et al., 2002; Li et al., 2002). These specific interactions have led to a model in which mutant htt, by harboring extra glutamines, becomes a hyperactive glutamine-containing corepressor (Freiman and Tjian, 2002). This model is corroborated by data from HD patients and HD rodent models that demonstrate transcriptional repression of a host of genes, including those encoding neurotransmitter receptors and pre-proenkephalin (Cha, 2000). Transcriptional repression may also be attributable to epigenetic modifications such as histone methylation and histone deacetylation (Hake et al., 2004). Indeed, levels of histone acetylation, a marker of gene activation, are decreased in models of HD (Steffan et al., 2001). Moreover, histone deacetylase inhibitors, which act to inhibit histone deacetylases and enhance transcription locally, can ameliorate the symptoms and pathology in models of HD (Ferrante et al., 2003). Despite these impressive advances, clinically approved agents that modulate transcription in a manner that would be favorable for HD patients have yet to be identified.
Mithramycin, an aureolic acid-type polyketide produced by the soil bacteria Streptomyces agrillaceus, is a clinically approved antibiotic used for the treatment of hypercalcemia and several types of cancers (Jones et al., 1995). Mithramycin binds to DNA sequences with guanosine-cytosine (GC) base specificity and has been shown to interfere with DNA binding proteins, such as Sp1 family transcription factors, which bind to GC-rich DNA sequences. In this regard, mithramycin has been shown to inhibit the expression of some proto-oncogenes, such as c-myc, and have an important role in the regulation of cell proliferation and differentiation (Jones et al., 1995). With regard to neuroprotection, previous studies from this laboratory have demonstrated that mithramycin can inhibit glutathione-depletion oxidative stress-induced death of cultured primary cortical neurons in culture (Chatterjee et al., 2001). These studies correlated the protective effects of mithramycin with its ability to bind to GC-rich DNA and displace Sp1 family transcription factors (Chatterjee et al., 2001).
Herein, we show that mithramycin prolongs survival, improves functional motor performance, and markedly improves brain neuropathological sequelae in the R6/2 transgenic mouse HD model. Furthermore, we show that methylation of lysine 9 in histone H3, a well established mechanism of gene silencing and transcriptional repression, is increased in R6/2 mice. Congruent with its protective effects, we show that mithramycin treatment prevents this histone H3 hypermethylation in the R6/2 line.
Materials and Methods
Animals. Male transgenic HD mice (R6/2 strain) were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained as a colony at the Bedford Veterans Medical Center. The male R6/2 mice were bred with females from their background strain (B6CBAFI/J), and offspring were genotyped using PCR. The mice were housed (five per cage) under standard conditions with access to water and food ad libitum. To ensure homogeneity of experimental cohorts, criteria for placement of mice into testing groups were standardized. Age-matched (within 4 d) mice from 31 litters of the same “F” generation were randomized. Any mice that had altered base-paired banding identified from PCR analysis were excluded from the study. All mice were weighed at 20 d of age and equally distributed according to weight within each cohort. Mice under 7 gm were not used in the experiments. Enrichment conditions were not applied to cages because of its effect on improving phenotype in R6/2 mice. All mice were handled under the same conditions by one investigator. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by both the Veterans Administration and Boston University Animal Care Committees.
Intraperitoneal dosing. Based on the study of Chatterjee et al. (2001), we completed a dose-response study, treating wild-type and R6/2 mice with 50, 100, 150, 300, and 600 μg/kg daily intraperitoneal injection of mithramycin A (Serva Feinbiochemica, Heidelberg, Germany) dissolved in PBS. Control groups were treated with PBS injection or untreated. Behavioral and survival data were obtained from ∼120 R6/2 and 120 littermate wild-type mice. During the temporal progress of the disease, the intraperitoneal injection was 100 μl per mouse until death.
Body weight. Body weights were recorded twice weekly at the same time of day in all groups.
Motor performance. Motor performance was assessed weekly from 21-63 d of age and twice weekly from 63 d of age in the mithramycin-treated (150 μg/kg) and untreated R6/2 mice. The mice were given two training sessions to acclimate them to the rotarod apparatus (Columbus Instruments, Columbus, OH). Mice were placed on a rotating rod at 16 rpm. The length of time on the rod was taken as the measure of competency. The maximum score was 60 sec, and each mouse performed three separate trials. The three results were averaged and recorded.
Survival. R6/2 mice were observed twice daily, mid-morning and late afternoon. Their motor performance and ability to feed was closely monitored and was the basis for determining when to kill the mice. The criteria for being killed was the point in time in which the HD mice were unable to right themselves after being placed on their back and initiate movement after being gently prodded for 30 sec. Two independent observers confirmed the criteria for being killed (R.J.F. and J.K.K.).
3-Nitropropionic acid administration. At 42 d, groups of 10 R6/2 mice were treated daily with 150 μg/kg mithramycin intraperitoneally or PBS for 2 weeks. At the beginning of the second week, 3-nitropropionic acid (3-NP) (Sigma, St. Louis, MO), freshly dissolved in PBS (pH adjusted to 7.4), was injected intraperitoneally nine times at 12 hr intervals (75 mg/kg) in R6/2 mice. Mice were deeply anesthetized 8-10 hr after the last 3-NP injection, transcardially perfused with buffered 4% paraformaldehyde, and then processed for histopathologic evaluation. These experiments were repeated twice, using cohorts of eight mice in the subsequent groups for a total of 52 mice. Glycerol cryoprotected brains were frozen sectioned at 50 μm and stained with cresyl violet for general cell staining. Quantitative analysis of lesion volumes was performed in serial sections as described below.
Histological evaluation. At 21 d, R6/2 transgenic mice and wild-type littermate control mice were treated with daily 150 μg · kg-1 52 d-1 intraperitoneal mithramycin or PBS. At 90 d of age, groups of 10 animals from each treatment paradigm were deeply anesthetized and transcardially perfused with 4% buffered paraformaldehyde. Approximately 40 mice were used for data collection in the neuropathological analysis and processed for histopathology, as described previously (Ferrante et al., 2003). Serially cut tissue sections were stained for Nissl substance and immunostained for htt, using a polyclonal rabbit antibody (EM48; dilution, 1:1000; Chemicon, Temecula, CA), and ubiquitin (rabbit polyclonal; dilution, 1:500; Dako, High Wycombe, UK), using a previously reported conjugated second antibody method in murine brain tissue samples (Ferrante et al., 2003). Specificity for the antisera used in this study was examined in each immunochemical experiment to assist with interpretation of the results. Preabsorption with excess target proteins, omission of the primary antibodies, and omission of secondary antibodies was performed to determine the amount of background generated from the detection assay.
Immunoblot analysis. For Western blotting, brain tissue was homogenized in 2.5× volume 50 mm Tris-HCl, 10% glycerol, 5 mm magnesium acetate, 0.2 mm EDTA, and 0.5 mm DTT, and protein concentration was determined using the DC protein assay kit (Bio-Rad, Hercules, CA). Ten micrograms of each sample were subjected to SDS-PAGE (10%) as described previously (Ferrante et al., 2003) using 1:5000 diluted anti-IC2 antibody (Chemicon) or anti-dimethyl-histone H3 (lysine 9) antibody (1:2000; Upstate Biotechnology, Lake Placid, NY). Protein loading was controlled by probing for actin (1:2500; Sigma) or α-tubulin (1:2500; Sigma) on the same membrane. Densitometric analysis was performed using Quantity One software (Bio-Rad).
Promoter activity analysis for Htt transgene expression. Htt-promoter analysis was performed using a reporter-construct containing -1032 to -15 of the 5′ regulatory region of the human htt gene as described previously (Coles et al., 1998). The SK-N-SH neuroblastoma cell line was used in the transient transfection assays and was treated with mithramycin at 100 or 200 nm for the indicated times. Reporter activity assays, at 48 hr after transfection, and data analysis were performed as described previously (Coles et al., 1998).
Stereology-quantitation. Serial-cut coronal tissue sections from the rostral segment of the neostriatum at the level of the anterior commissure (interaural 5.34 mm, bregma 1.54 mm to interaural 3.7 mm, bregma -0.10 mm) were used for htt aggregate analysis. Unbiased stereological counts of htt-positive aggregates (>1.0 μm) were obtained from the neostriatum in 10 mice each from mithramycin-treated and PBS-treated R6/2 mice at 90 d using Neurolucida Stereo Investigator software (MicroBrightField, Colchester, VT). The total areas of the neostriatum were defined in serial sections in which counting frames were randomly sampled. The optical dissector counting method was used in which htt-positive aggregates were counted in an unbiased selection of serial sections in a defined volume of the neostriatum. Striatal neuron areas were analyzed by microscopic video capture using a Windows-based image analysis system for area measurement (Optimas Bioscan, Edmonds, WA). The software automatically identifies and measures profiles. All computer-identified cell profiles were manually verified as neurons and exported to Microsoft (Seattle, WA) Excel. Cross-sectional areas were analyzed using Statview (Brain Power, Calabasas, CA). Lesion areas were quantitated from serial cut frozen sections from 3-NP-treated and mithramycin-3-NP-treated mice with the Optimas Bioscan microscopic video-capture system.
NMDA toxicity. Mixed cortical cell cultures containing astrocytes and neurons derived from postnatal and fetal mice, respectively, were prepared by a two-step plating method as described by Uliasz and Hewett (2000). Exposure to 100 or 200 mm NMDA (Sigma) for 5 min (fast-triggered excitotoxicity) or 15 mm NMDA for 20-24 hr (slow-triggered excitotoxicity) was performed either alone or in the presence of 3, 30, or 300 nm mithramycin. Delayed neuronal death was assessed 20-24 hr later by spectrophotometric measurement of lactate dehydrogenase activity and confirmed using trypan blue (Uliasz and Hewett, 2000). Mithramycin was present during and for 20-24 hr after NMDA exposure in the fast-triggered paradigm and was continuously present during the slow-triggered paradigm.
Low potassium-growth factor deprivation of granule neurons. Granule neurons were cultured from dissociated cerebella of 7- to 8-d-old rats as described previously (D'Mello et al., 1993). Treatments were performed 6-7 d after plating. Briefly, cells were rinsed once with serum-free BMEM [low-K+ (LK) medium] and then maintained in LK medium containing various doses of mithramycin. Viability was assayed 24 hr later using the MTT assay. Control cells received serum-free medium supplemented with 25 mm KCl [high-K+ (HK) medium].
Studies in isolated mitochondria. Four-month-old male, specific pathogen-free Fischer 344 × Brown Norway F1 rats were killed by decapitation in agreement with standard animal-usage guidelines as approved by the Institutional Animal Care and Use Committee of Weill Medical College of Cornell University. Liver mitochondria were isolated by differential centrifugation protocols essentially as described previously (Kristal and Brown, 1999), with the following modifications. After decapitation, livers were rapidly dissected out and placed in ice-cold 300 mm sucrose, 10 mm HEPES, and 1 mm EGTA, pH 7.4 with KOH and supplemented with 0.5% fatty acid-free bovine serum albumin (BSA) (A-6003; Sigma). Livers were homogenized with a motor-driven Teflon pestle. After homogenization, samples were centrifuged at 1000 × g for 10 min. Supernatants were removed and centrifuged at 8000 × g for 10 min. Pellets were washed twice before final resuspension. The first wash was in homogenization buffer; the final wash and resuspension buffer had no EGTA or BSA. Mitochondrial permeability transition (MPT) induction was monitored spectrophotometrically as described previously (Kristal and Brown, 1999). Changes in absorbance at 560 nm (A560) were followed for 1 hr using a SpectraMax Plate Reader (Molecular Dynamics, Sunnyvale, CA).
Statistics. The data are expressed as the mean ± SEM. Statistical comparisons of rotarod, weight data, and histology data were compared by ANOVA or repeated-measures ANOVA. Survival data were analyzed by the Kaplan-Meier survival curves.
Results
Mithramycin extends survival, enhances motor performance, and improves brain histopathology in R6/2 mice
Because the R6/2 transgenic mice, which express exon 1 of the human expanded CAG repeat-containing Huntingtin gene, display similar clinical and pathological features to those found in humans who have HD; they represent a reasonable model with which to identify candidate therapies for testing in humans. Mithramycin improved survival in a dose-dependent manner (Fig. 1A). The optimal mithramycin dose (150 μg · kg-1 · d-1, i.p.) extended survival in R6/2 mice by 29.1% (p < 0.001), greater than any other single neuroprotective agent reported to date (Fig. 1A). Doses at 300 and 600 μg · kg-1 · d-1 resulted in morbidity and death, respectively. Mithramycin (150 μg · kg-1 · d-1) also significantly improved rotarod performance, a complex locomotor test, at all times tested (4-17 weeks) during the lifespan of the R6/2 mice compared with vehicle-treated R6/2 mice (Fig. 1B). The combined improvement in rotarod performance was 42.6%. The weight curves of mithramycin-treated and vehicle-treated R6/2 mice closely paralleled one another, maintaining a plateau at 19-20 gm until death ensued within each treatment group. Significant differences in body weight occurred as an epiphenomenon of survival extension and not as a primary independent event related to mithramycin treatment.
Figure 1.

Clinical and neuropathological outcomes in mithramycin-treated R6/2 mice. Kaplan-Meier probability of survival analysis in R6/2 mice treated with 50, 100, and 150 μg · kg-1 52 d-1 mithramycin (Mith) (a GC-rich DNA binding agent) administered intraperitoneally (A). Mithramycin treatment at 300 and 600 μg · kg-1 52 d-1 resulted in morbidity and mortality, respectively. Rotarod performance in vehicle and mithramycin-treated R6/2 mice (B). Kaplan-Meier probability of survival analysis in R6/2 mice treated with a range of concentrations of the AT-rich binding drug distamycin (Dist; C) resulted in no improved extension in survival. Compared with wild-type littermate control mice (D), there is gross brain atrophy and ventricular hypertrophy in untreated R6/2 mice (E) at 90 d. Mithramycin significantly attenuated the neuropathological changes in R6/2 mice (F). Mithramycin reduced striatal neuron atrophy (H) compared with untreated R6/2 mice (I) at 90 d. Wild-type control mice (G) and mithramycin-treated R6/2 mice were not different. Scale bars: D, 2 mm; I, 100 μm.
A marked improvement in neuropathological sequelae was also observed in R6/2 mice treated with mithramycin. Mithramycin (150 μg · kg-1 · d-1) prevented brain atrophy, ventricular hypertrophy, and striatal neuronal atrophy seen in R6/2 mice (Fig. 1D-I). Indeed, gross brain atrophy with bilateral ventricular hypertrophy and flattening of the medial aspect of the striatum, present in the brains of R6/2 mice at 90 d of age, are essentially absent from R6/2 mice treated with mithramycin (Fig. 1E,F). There was a 21.3% reduction in brain weight in vehicle-treated R6/2 mice compared with littermate wild-type mice. In contrast, there was only a 2.8% brain weight loss in R6/2 mice treated with mithramycin (wild-type littermate mice, 456 ± 11 mg/kg; PBS-treated R6/2 mice, 359 ± 21 mg/kg, p < 0.001; 150 μg · kg-1 · d-1 mithramycin-treated R6/2 mice, 443 ± 16 mg/kg, p < 0.12). Similarly, there was no significant difference in neuronal size between mithramycin-treated R6/2 mice and littermate wild-type mice at 90 d (mithramycin-treated R6/2 mice, 127.3 ± 14.0 μm2; wild-type littermate control mice, 131.4 ± 12.7 μm2). In contrast to this, vehicle-treated R6/2 mice displayed a 41.9% decrease in neuronal size (55.1 ± 21.2 μm2, p < 0.001). These results clearly demonstrate that mithramycin is neuroprotective, enhances functional status, and improves survival in R6/2 mice.
Mithramycin has been shown to interact with the minor groove of DNA with a GC base specificity (Jones et al., 1995). To examine whether alternate DNA binding drugs had a similar neuroprotective effect to mithramycin, R6/2 mice were treated with distamycin. Distamycin, a drug that binds the minor groove with adenosine-thymidine (AT) base specificity, had no effect on survival or rotarod performance in the R6/2 mice (Fig. 1C), suggesting that not all DNA binding drugs are protective in the R6/2 model. In fact, this lack of effect suggests that GC base specificity may be a necessary component of the neuroprotective effects of mithramycin.
Mithramycin does not interfere with mutant htt transgene expression
The human htt promoter drives expression of the mutant htt transgene in the R6/2 model (Coles et al., 1998). This promoter has been found to have several essential GC-rich DNA binding sites that are legitimate binding sites for transcription factors such as Sp1 or BTEB (basic transcription element binding) (Coles et al., 1998). The established ability of mithramycin to displace transcriptional activators that bind to GC-rich binding sites (Jones et al., 1995) raised the possibility that mithramycin prevents neuronal dysfunction in the R6/2 model by decreasing expression of the toxic transgene. To evaluate this possibility, we examined the level of mutant htt exon 1 transgene expression in mithramycin-treated R6/2 mice compared with their vehicle-treated controls. The mean relative htt protein expression for treated versus untreated mice was 0.934 ± 0.171 (p = 0.28) (Fig. 2A), suggesting that mithramycin does not alter htt transgene expression in these mice. To examine the matter further, human SK-N-SH cells were treated with a supratherapeutic concentration of mithramycin (100 nmol/l), and mutant htt mRNA levels and htt promoter activity were examined. The results demonstrated that mithramycin did not influence either mutant htt mRNA levels or htt promoter activity in vitro (data not shown). To further demonstrate that protection was not a result of decreased htt expression, htt-positive or ubiquitin-positive striatal aggregates were examined in mithramycin-treated R6/2 mice at 90 d of age. Mithramycin treatment had no significant effect on either htt-positive or ubiquitin-positive striatal aggregates (Fig. 2B).
Figure 2.
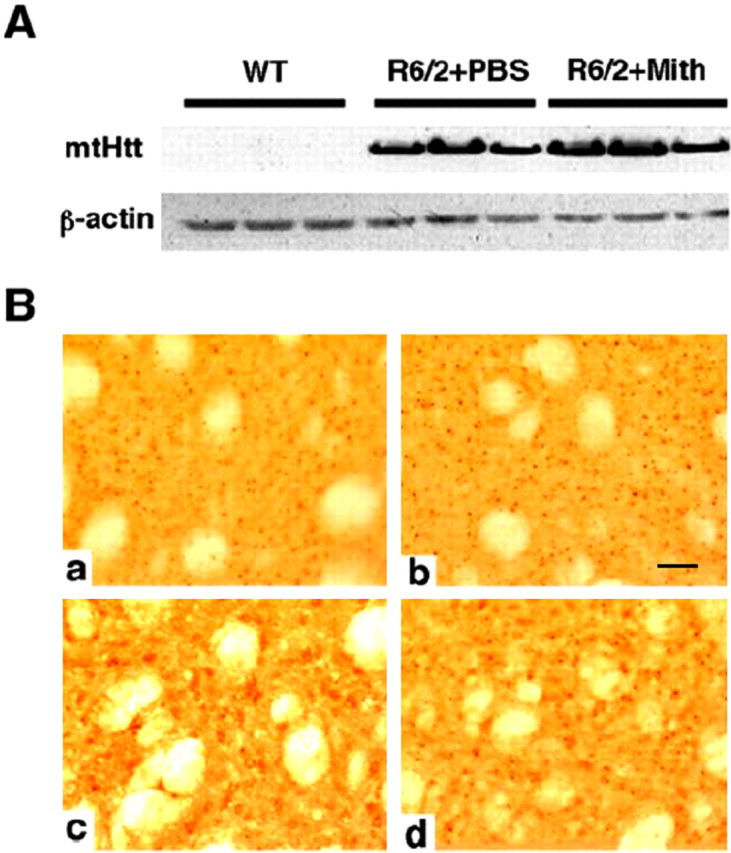
Effect of mithramycin on Htt transgene expression in R6/2 mice. A, Western blot analysis of mutant htt (mtHtt) expression in brains of wild-type (WT), control (PBS-treated) R6/2 mice, and mithramycin-treated (Mith) R6/2 mice. The mean relative htt protein expression of treated versus untreated from three separate Western blots was 0.934 ± 0.171 (p = 0.28). B, Huntingtin-immunostained tissue sections from the neostriatum in PBS-treated R6/2 mice (a) and mithramycin-treated R6/2 HD transgenic mice at 90 d (b). Huntingtin aggregates in mithramycin-treated R6/2 mice, 5.28 × 106± 1.34; PBS-treated R6/2 mice, 5.09 × 106± 1.52; F(2,20) = 1.67; p < 0.39. Ubiquitin-immunostained tissue sections from the neostriatum in PBS-treated R6/2 mice (c) and mithramycin-treated R6/2 HD transgenic mice at 90 d (d). Ubiquitin inclusions in mithramycin-treated R6/2 mice, 2.12 × 106± 0.78; PBS-treated R6/2 mice, 1.98 × 106± 0.63; F(2,20) = 1.45; p < 0.42. Scale bar (in b): a-d, 100 μm.
The treatment of mice with 3-NP, an irreversible inhibitor of succinate dehydrogenase that inhibits both the Krebs' cycle and complex II of the mitochondrial electron transport chain, produces selective striatal lesions that replicate some of the characteristic histopathological features of HD (Beal et al., 1993; Brouillet et al., 1995). Thus, 3-NP treatment presents an appropriate htt transgene-independent model by which to examine the protective effects of mithramycin. As expected, the administration of 3-nitropropionic acid in mice resulted in striatal toxicity and the development of striatal lesions (Fig. 3). In contrast, 3-NP-treated mice, which were treated with mithramycin, displayed lesion volumes that were markedly reduced or not present at all (mithramycin treated 3-NP R6/2 mice, 0.87 ± 0.41 mm3; PBS-treated 3-NP R6/2 mice, 10.6 ± 3.09 mm3; p < 0.001). There was a 91% reduction in lesion volume in the mithramycin-treated R6/2 mice (Fig. 3). Together, these findings demonstrate that mithramycin does not exert its salutary effects by reducing mutant Huntingtin transgene expression in mice. Rather, they argue that mithramycin acts downstream of mutant htt expression to stem neuronal injury and disease progression.
Figure 3.
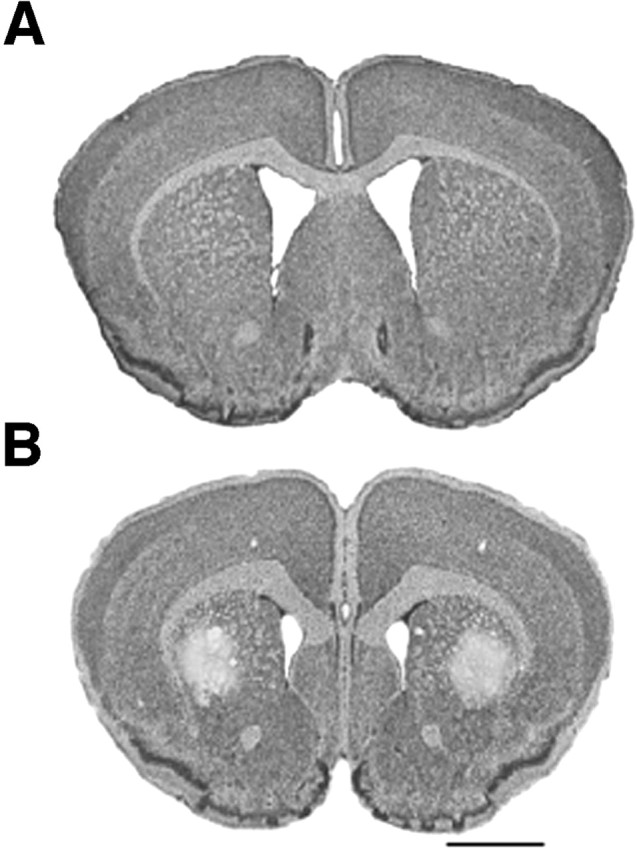
Mithramycin neuroprotection from 3-nitropropionic acid toxicity in R6/2 mice. Mithramycin treatment prevented striatal damage in R6/2 mice (top) compared with PBS-treated R6/2 mice (bottom). Histopathological evaluation of 3-NP-induced striatal lesions shows bilateral striatal lesions, areas of pallor, in PBS-treated R6/2 mice (bottom). Scale bar, 2 mm.
Mithramycin does not protect by altering glutamate receptor expression or activity
Because excitotoxicity is believed to be a contributor to neuronal dysfunction in Huntington's disease (Ferrante et al., 2002), it is possible that mithramycin enhances motor function and attenuates neuronal dysfunction by directly affecting the expression or activity of glutamate receptors. We therefore examined the effect of mithramycin on excitotoxic cell death in vitro. Cortical neurons, a cell type afflicted in HD, were treated with NMDA alone or in the presence of mithramycin. Mithramycin failed to protect cells from this NMDA-induced injury (Table 1). This lack of effect could not be attributed to loss of drug activity because oxidative neuronal cell death induced by mechanisms independent of ionotropic glutamate receptors was inhibited in a concentration-dependent manner (Chatterjee et al., 2001). The ability of mithramycin to prevent death during activity deprivation and growth factor deprivation in pure cerebellar granule neuron cultures (D'Mello et al., 1993) was examined. In this, another glutamate receptor-independent cell death model, mithramycin blocked neuronal death induced by both activity and growth factor deprivation (Fig. 4). Together, these results suggest that the salubrious effects of mithramycin are independent of glutamate receptors.
Table 1.
Effect of mithramycin on NMDA-induced neuronal death
|
Treatment |
Percentage cell death (mean ± SD) |
|
|
||
|---|---|---|---|---|---|
| MMA (nm) |
A |
B |
C |
D |
|
| 0 | 41.1 ± 5.6 | 78.3 ± 11.0 | 90.1 ± 7.5 | 98.5 ± 33.8 | |
| 3 | 52.6 ± 4.0 | 90.5 ± 16.2 | 96.9 ± 7.5 | 82.9 ± 14.0 | |
| 30 | 51.3 ± 1.9 | 89.3 ± 12.7 | 91.0 ± 6.1 | 90.4 ± 25.4 | |
| 300 |
51.6 ± 13.4 |
84.6 ± 12.1 |
92.7 ± 5.7 |
100.6 ± 19.7 |
|
Mouse cortical cell cultures were exposed to 75 mm NMDA (column A; n = 4-5 wells from 2 separate experiments), 100 μm NMDA (column B; n = 9-12 wells from 4 separate experiments), or 200 μm NMDA (column C; n = 5-7 wells from 3 separate experiments) for 5 min or 12-15 mm NMDA for 20-24 hr (column D; n = 12-15 wells from 5 separate experiments) alone or in the presence of increasing concentrations of mithramycin (3, 10, 30, 100, 300, and 1000 nm). Lactate dehydrogenase released into the bathing medium was assessed 20-24 hr later. Data are expressed as a percentage of total neuronal cell death (mean ± SD) that was determined in parallel sister cultures after a 24 hr exposure to 200 μm NMDA. Because there were no significance differences within any concentrations tested, as determined by ANOVA, results for 3, 30, and 300 nm only are shown.
Figure 4.
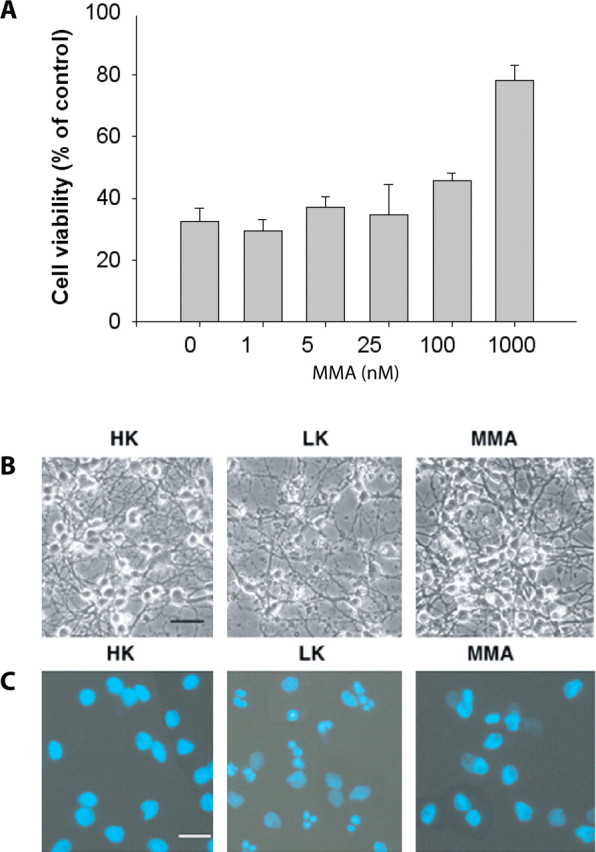
Mithramycin inhibits apoptosis induced by low extracellular potassium in cerebellar granule cells. A, Graph showing cell viability when cultured cerebellar granule neurons switched from HK medium to LK medium in the presence of increasing concentrations of mithramycin (MMA). Cell viability was measured by MTT reduction 24 hr after the switch to LK media and expressed as a percentage of control ± SEM. Phase contrast (B) and DAPI-stained fluorescence (C) microscopy of cultured cerebellar granule neurons. Cerebellar granule neurons undergo apoptosis when switched from HK medium to LK medium, as seen by the presence of classical apoptotic nuclear fragmentation. The addition of mithramycin (500 nm) to the LK medium significantly decreased the presence of these apoptotic nuclear fragments. Microscopy was performed 24 hr after the switch to LK or LK plus mithramycin medium. Scale bar, 10 μm.
Mithramycin does not modulate mitochondrial permeability transition induction threshold
Impaired mitochondrial function has also been implicated as an important contributor to HD pathogenesis. Mitochondria, isolated from lymphoblasts of HD patients and transgenic mice, have been shown to have an increased susceptibility to depolarize in response to proapoptotic stress (Sawa et al., 1999) and gradual calcium loads (Panov et al., 2002) compared with mitochondria from controls. Furthermore, mutant htt has been demonstrated to be associated with the outer mitochondrial membrane, and recombinant truncated mutant htt, but not wild type, can directly induce MPT pore opening in isolated mouse liver mitochondria (Choo et al., 2004).
To examine whether mithramycin exerts its protective effects by modulating MPT, rat liver mitochondria were incubated with phenylarsine oxide or Ca2+/tert-butyl hydroperoxide, two inducers of MPT. Treatment of isolated rat liver mitochondria with either of these MPT inducers resulted in pronounced mitochondrial swelling, a clear indicator of MPT (Fig. 5, trace B). Coincubation with the MPT inhibitor cyclosporine A (250 nm) reduced mitochondrial swelling (Fig. 5, trace A). In contrast to cyclosporine A, coincubation with increasing concentrations of mithramycin (20 to 10,240 nm) did not alter either phenylarsine oxide- or Ca2+/tert-butyl hydroperoxide-induced mitochondrial swelling (Fig. 5, traces C-L). These results clearly demonstrate that mithramycin does not protect by altering the threshold necessary for MPT induction.
Figure 5.
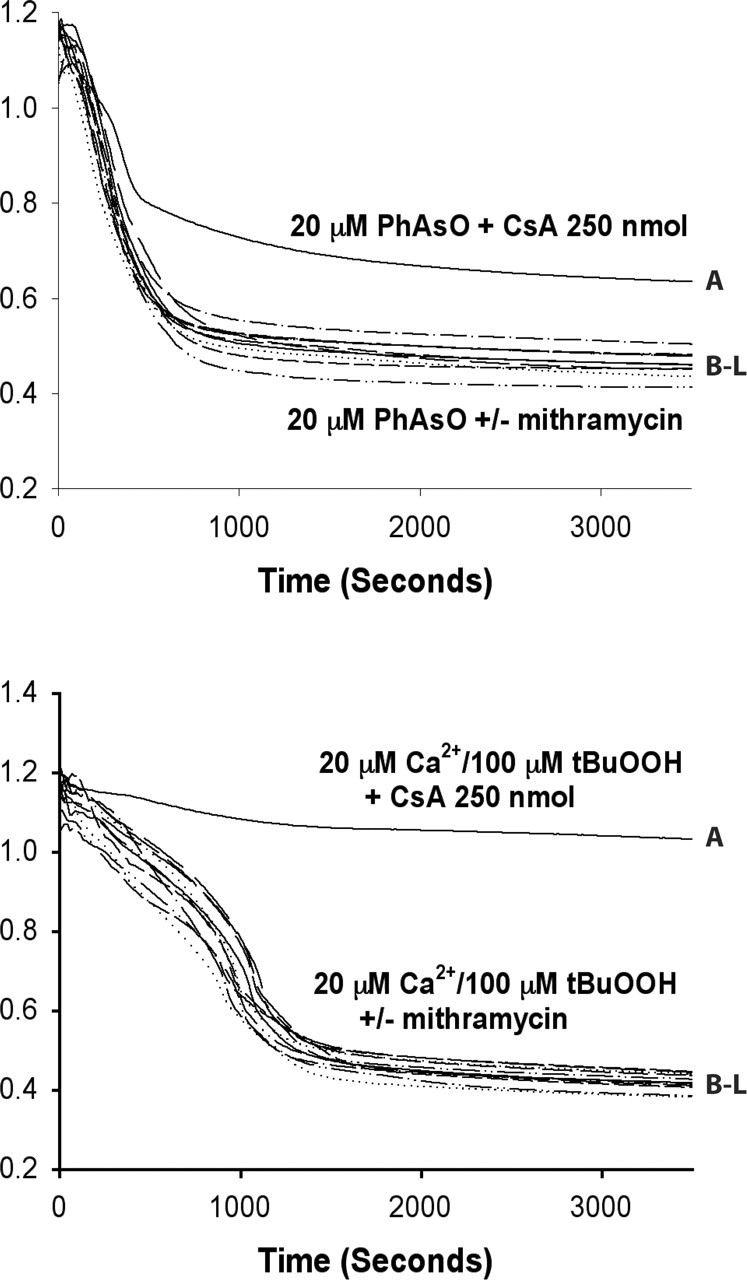
Mithramycin does not modulate rates of MPT. Representative traces from experiments on the effect of mithramycin on MPT induction. Isolated rat liver mitochondria (1 mg/ml) were incubated in 20 mm phenylarsine oxide (PhAsO; top) or 25 μm Ca2+ and 100 μm tert-butyl hydroperoxide (Ca2+/tBuOOH; bottom). Trace A in both panels had 250 nm cyclosporin A (CsA) added. Traces B-L had 0, 20, 40, 80, 160, 320, 640, 1280, 2560, 5120, or 10,240 nm mithramycin added, respectively.
Mithramycin prevents htt-specific histone H3 hypermethylation at lysine 9
Another determinant believed to be a contributor to neuronal debility and loss in Huntington's disease is transcriptional dysfunction (Cha, 2000). Part of this dysfunction comes from htt acting as a hyperactive glutamine-containing corepressor, interacting with transcriptional activators and coactivators, including Sp1, TAFII130, and CBP (Steffan et al., 2001; Dunah et al., 2002; Li et al., 2002). Indeed, several reports from different groups suggest that overexpression of Sp1 can counteract transcriptional repression induced by mutant htt and inhibit mutant htt- (Dunah et al., 2002; Freiman and Tjian, 2002; Li et al., 2002) or oxidative stress-induced neurotoxicity (Chatterjee et al., 2001). Although mithramycin is best known as an agent that displaces Sp1 from its GC-rich binding site, it is hard to reconcile Sp1 displacement with the neuroprotective ability of mithramycin. It is, therefore, likely that the mechanism of mithramycin action is independent of Sp1.
Because transcriptional dysfunction in HD has also been linked to decreased histone acetylation, we considered the possibility that mithramycin, a zinc chelator, acts to derepress transcription by inhibiting the zinc hydrolase activity of histone deacetylases. However, no evidence was found, in vitro, for mithramycin-induced increases in histone or transcription factor acetylation (data not shown). Another mechanism for gene silencing involves the methylation of histone H3 at lysine 9 (Hake et al., 2004). Examination of this histone H3 site in R6/2 mice revealed increased lysine 9 methylation relative to wild-type littermate controls (Fig. 6). When R6/2 mice were treated with mithramycin, histone 3 lysine 9 methylation was consistent with the levels seen in wild-type controls, suggesting that mithramycin treatment prevented the htt-induced H3 methylation increase (Fig. 6). Together, these findings are consistent with increased transcriptional dysfunction playing a neuropathogenic role in the R6/2 HD model and suggest that mithramycin improves this neuropathogenic outcome by partially restoring perturbed gene transcription.
Figure 6.

Mithramycin prevents the hypermethylation of histone H3 induced by htt in R6/2 mice. Immunoreactivity of dimethyl histone H3 (DiM-H3) in the striatum of wild-type mice (A) with increased immunoreactivity of DiM-H3 in R6/2 mice (B). Mithramycin treatment decreased dimethyl histone H3 in R6/2 mice to normal expression levels (C). Western blot analysis of dimethyl histone H3 protein levels (D). Mithramycin (Mith) significantly attenuated the level of dimethyl histone H3 in R6/2 mice (E). Densitometric values of dimethyl histone H3 were normalized to α-tubulin (α-tub) level. R6/2 mice were treated with 150 μg · kg-1 52 d-1 mithramycin administered intraperitoneally. p = 0.0168 for wild type (WT) versus R6/2. p = 0.0324 for nontreated R6/2 versus mithramycin-treated R6/2. Student's t test was used for statistical analysis.
Discussion
This study found that mithramycin treatment, given intraperitoneally at 150 μg · kg-1 52 d-1, was well tolerated and improved many of the Huntington's disease-associated symptoms in R6/2 transgenic mice. Specifically, mithramycin prolonged survival and improved functional motor performance. Furthermore, whereas the brains of untreated R6/2 mice displayed brain atrophy, ventricular hypertrophy, and striatal neuronal atrophy, histological sections from mithramycin-treated R6/2 mice and their wild-type littermates were virtually indistinguishable at 90 d of age. In agreement with these findings, mithramycin also markedly reduced lesion volumes in 3-NP-treated mice, another model in which HD-like striatal lesions are induced (Beal et al., 1993; Brouillet et al., 1995).
Previous studies in mice have shown that intraperitoneal administration of a single dose of 150 μg/kg is sufficient to inhibit expression of a c-myc promoter reporter construct in the brain by 90% (Jones et al., 1995). These findings suggest that mithramycin penetrates the blood-brain barrier to exert some of its beneficial effects. Furthermore, these protective effects appear to require GC DNA specificity, because an AT DNA binding antibiotic, distamycin, had no effect on either R6/2 transgenic mouse survival or rotorod performance. It is less clear, however, whether or not these protective effects are mediated through displacing the Sp1 family of transcriptional activators from their canonical GC DNA binding sites. It is plausible that Sp1 could transcriptionally activate a hitherto unknown gene or number of genes that are involved in promoting neuronal death. Alternatively, Sp1 could be acting directly or indirectly to repress transcription of a prosurvival gene(s). In either scenario, inhibiting Sp1 from binding DNA would result in reduced or delayed neuronal dysfunction and death. Investigations are currently underway to determine whether Sp1 displacement is critical for mediating the protective effects of mithramycin. However, based on reports demonstrating that Sp1 overexpression can reduce the cellular toxicity of htt (Dunah et al., 2002; Li et al., 2002) as well as protect neurons in models of oxidative stress (Ryu et al., 2003a), it seems unlikely that mithramycin protects by simply displacing Sp1. Given the ability of mithramycin to bind GC DNA, it is also possible that mithramycin treatment abates HD neuropathology in R6/2 mice by decreasing htt transgene expression. However, no evidence for downregulated htt transgene expression was found, despite multiple lines of experimental investigation. Indeed, the demonstration that mithramycin can reduce striatal lesions induced by the mitochondrial toxin 3-NP also suggests that the protective action of mithramycin is downstream of mutant htt.
Another possibility for the neuroprotective mechanism of mithramycin was that it alters glutamate receptor activity or expression. However, mithramycin had no effect on NMDA-induced excitotoxicity, despite protecting cerebellar granule neurons from activity and growth factor deprivation-induced death, and immature cortical neurons from oxidative stress-induced toxicity, two models of glutamate receptor-independent neuronal death.
Because mitochondrial dysfunction and impaired energy metabolism has been implicated in HD pathogenesis (Albin and Greenamyre, 1992; Beal, 1992), it was also necessary to rule out the possibility that mithramycin protects by modulating MPT. Indeed, minocycline, a second-generation tetracycline antibiotic that is protective in several neurological models including Huntington's disease, has been shown to be an inhibitor of MPT and consequent MPT-mediated cytochrome c release (Zhu et al., 2002; Wang et al., 2003; Teng et al., 2004). The incubation of rat liver mitochondria with mithramycin in the presence of two different MPT-inducing agents, phenylarsine oxide and Ca2+/tert-butyl hydroperoxide, clearly demonstrated that mithramycin has no direct effect on MPT threshold. These experiments suggest that mithramycin, unlike minocycline, acts in an MPT-independent manner to prevent degeneration induced by mutant htt. Of course, our experiments do not allow us to exclude the possibility that mithramycin alters the ability of mutant htt to induce MPT.
Because the etiology of HD pathogenesis has been linked to mechanisms of global transcriptional repression such as epigenetic histone modification, we focused on htt- and mithramycin-induced changes in histone acetylation and methylation. Consistent with repressed transcriptional activity in HD, we detected increased histone H3 lysine 9 methylation in R6/2 transgenic mice. This increased H3 methylation was prevented by mithramycin treatment. Posttranslational modifications of histone tails, including acetylation, phosphorylation, and methylation, play important roles in regulating transcription and chromatin structure. Methylation of histone H3 has been linked to distinct effects on transcription, depending on the enzyme and residue modified. That mithramycin prevented htt-induced hypermethylation is compatible with a model in which mithramycin could displace the GC-rich DNA binding of a histone methyltransferase (HMT) or a transcription factor that recruits an HMT and reduce associated gene silencing. Indeed, evidence that DNA binding transcription factors can recruit an HMT to specific gene promoters to repress transcription is established. For example, the histone H3-specific methyltransferase, G9a, responsible for H3 lysine 9 dimethylation and monomethylation within silent euchromatin (Rice et al., 2003), has been shown recently to interact with CCAAT displacement protein/cut homolog (CDP/cut) proteins (Nishio and Walsh, 2004), which have been characterized to function as transcriptional repressors in a large number of genes, including p21(waf1/cip1), gp91phox, c-myc, and histones (Skalnik et al., 1991; Dufort and Nepveu, 1994; el-Hodiri and Perry, 1995; Coqueret et al., 1998; Li et al., 1999). Consistent with its role as a transcriptional repressor, CDP/cut was demonstrated to recruit G9a to the human p21(waf1/cip1) promoter, in which H3 lysine 9 methylation and transcriptional repression occurs (Nishio and Walsh, 2004). Although no HMT-interacting GC-rich DNA binding proteins have been reported to date, the idea that mithramycin could displace such a factor is an appealing one. The ensuing enhanced gene expression at such sites may counteract the toxicity of the HD mutation either directly by opposing the hyperactive corepressor function of htt and/or by mediating a more generic protective response. This is analogous to what may occur with histone deacetylase inhibitors that protect against both polyglutamine-dependent and -independent insults (Ferrante et al., 2003; Ryu et al., 2003b).
The identification of the gene for HD has facilitated the development of rodent models to screen for novel therapeutics. These screens have elucidated a host of agents. Additional agents are needed, however, to complement or surpass the antioxidant and anti-glutamate strategies currently under investigation in humans with HD. Human clinical trials are expensive, and even the largest trials have only a limited power to detect a beneficial effect. The comparatively large effect of mithramycin, an Food and Drug Administration-approved drug, argues that it should be among the top priorities for testing in patients.
Footnotes
This work was supported by National Institutes of Health Grants NS40591 and NS46239 (R.R.R.) and NS35255, AT00613, NS045242, and NS045806 (R.J.F.), the Veteran's Administration (R.J.F.), the Huntington's Disease Society of America (H.R. and R.J.F.), the Hereditary Disease Foundation (H.R. and R.J.F.), the Jerry McDonald Fund in Huntington's Disease (H.R. and R.J.F.), and a Beth Israel Deaconess Medical Center Enterprise Award (R.R.R.).
Correspondence should be addressed to either of the following: Dr. Rajiv R. Ratan, Burke/Cornell Medical Research Institute, Weill Medical College of Cornell University, 785 Mamaroneck Avenue, White Plains, NY 10605, E-mail: rratan@burke.org; or Dr. Robert J. Ferrante, Geriatric Research and Education and Clinical Center, Veterans Administration Medical Center, Bedford, Department of Neurology, Pathology, and Psychiatry, Boston University School of Medicine, Boston, MA 02118, E-mail: rjferr@bu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410335-08$15.00/0
References
- Albin R, Greenamyre JT (1992) Alternative excitotoxic hypotheses. Neurology 42: 733-738. [DOI] [PubMed] [Google Scholar]
- Beal MF (1992) Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative disease? Ann Neurol 31: 119-130. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13: 4181-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF (1995) Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA 92: 7105-7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH (2000) Transcriptional dysregulation in Huntington's disease. Trends Neurosci 23: 387-392. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR (2001) Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol 49: 345-354. [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 13: 1407-1420. [DOI] [PubMed] [Google Scholar]
- Coles R, Caswell R, Rubinsztein DC (1998) Functional analysis of the Huntington's disease (HD) gene promoter. Hum Mol Genet 7: 791-800. [DOI] [PubMed] [Google Scholar]
- Coqueret O, Berube G, Nepveu A (1998) The mammalian Cut homeodomain protein functions as a cell-cycle-dependent transcriptional repressor which down modulates p21WAF1/CIP1/SDI1 in S phase. EMBO J 17: 4680-4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufort D, Nepveu A (1994) The human cut homeodomain protein represses transcription from the c-myc promoter. Mol Cell Biol 14: 4251-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296: 2238-2243. [DOI] [PubMed] [Google Scholar]
- el-Hodiri HM, Perry M (1995) Interaction of the CCAAT displacement protein with shared regulatory elements required for transcription of paired histone genes. Mol Cell Biol 15: 3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF (2002) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci 22: 1592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurode-generative phenotype in Huntington's disease mice. J Neurosci 23: 9418-9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman RN, Tjian R (2002) Neurodegeneration. A glutamine-rich trail leads to transcription factors. Science 296: 2149-2150. [DOI] [PubMed] [Google Scholar]
- Hake SB, Xiao A, Allis CD (2004) Linking the epigenetic “language” of covalent histone modifications to cancer. Br J Cancer 90: 761-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones Jr DE, Cui DM, Miller DM (1995) Expression of beta-galactosidase under the control of the human c-myc promoter in transgenic mice is inhibited by mithramycin. Oncogene 10: 2323-2330. [PubMed] [Google Scholar]
- Kristal BS, Brown AM (1999) Apoptogenic ganglioside GD3 directly induces the mitochondrial permeability transition. J Biol Chem 274: 23169-23175. [DOI] [PubMed] [Google Scholar]
- Li S, Moy L, Pittman N, Shue G, Aufiero B, Neufeld EJ, LeLeiko NS, Walsh MJ (1999) Transcriptional repression of the cystic fibrosis transmembrane conductance regulator gene, mediated by CCAAT displacement protein/cut homolog, is associated with histone deacetylation. J Biol Chem 274: 7803-7815. [DOI] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ (2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22: 1277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio H, Walsh MJ (2004) CCAAT displacement protein/cut homolog recruits G9a histone lysine methyltransferase to repress transcription. Proc Natl Acad Sci USA 101: 11257-11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci 5: 731-736. [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 12: 1591-1598. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR (2003a) Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci 23: 3597-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Olofsson BA, Mwidau A, Deodoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR (2003b) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci USA 100: 4281-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler Jr JF, Greenamyre JT, Snyder SH, Ross CA (1999) Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med 5: 1194-1198. [DOI] [PubMed] [Google Scholar]
- Skalnik DG, Strauss EC, Orkin SH (1991) CCAAT displacement protein as a repressor of the myelomonocytic-specific gp91-phox gene promoter. J Biol Chem 266: 16736-16744. [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila Nature 413: 739-743. [DOI] [PubMed] [Google Scholar]
- Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM (2004) Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci USA 101: 3071-3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliasz TF, Hewett SJ (2000) A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J Neurosci Methods 100: 157-163. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM (2003) Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci USA 100: 10483-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417: 74-78. [DOI] [PubMed] [Google Scholar]