

Abstract
We previously showed that antral gastric tumors develop in gastrin-deficient (Gas−/−) mice. Therefore Gas−/− mice were studied sequentially over 12 months to identify molecular mechanisms underlying gastric transformation. Fundic atrophy developed by 9 months in Gas−/− mice. Antral mucosal hyperplasia developed coincident with the focal loss of TFF1 and Muc5AC. Microarray analysis of 12 month Gas−/− tumors revealed an increase in follistatin, an activin/BMP antagonist. We found that elevated follistatin expression occurred in the proliferative neck zone of hyperplastic antrums, in antral tumors of Gas−/− mice, and also in human gastric cancers. Follistatin induced cyclin D1 and the trefoil factors TFF1 and TFF2 in a gastric cancer cell line. We concluded that antral hyperplasia in Gas−/− mice involves amplification of mucous cell lineages due to follistatin, suggesting its role in the development of antral gastric tumors.
Keywords: Gastrin, intestinal metaplasia, TFF1, atrophic gastritis, mucous neck cell, gastric cancer, Muc6, activins, follistatin
Introduction
Gastric cancer is the fourth most common cancer and the second most common cause of cancer-related deaths in the world [1, 2]. The molecular mechanisms underlying the sequential phenotypic changes in the gastric mucosa from chronic inflammation (gastritis) to atrophy, metaplasia then eventually cancer are not well defined [3]. An increasing number of signaling pathways and regulatory molecules have been suggested to play a role in gastric transformation, e.g., TFF1 [4] Smad4, [5] Runx3 [6], STAT3 [7] [8] and Klf4 [9]. Several of these pathways implicate epithelial-mesenchymal crosstalk with TGFβ family members, e.g., activins, Smad4, Runx3. Gordon and coworkers showed that high levels of activin A in the α-inhibin null mouse induces expansion of the mucous neck and surface pit lineages, suggesting that activin-dependent signaling affects the proliferative activity and differentiation of gastric stem cells [10]. Follistatin is an antagonist of the activin and BMP ligands. Moreover, an increase in follistatins correlates with an increase in proliferation and counteracts activin-induced apoptosis [11]. Prior studies have suggested that increased follistatin levels partially block the wasting syndrome observed under activin overexpression [12]. However, little is known regarding the role of follistatin in gastric neoplasia.
Reports from tumor registries and more recent histological mapping studies show that approximately 50% of intestinal-type gastric cancers are located in the antrum, 38% are in the corpus and 12% are located at the incisura [13]. Therefore, corpus atrophy appears to correlate with tumors developing in the distal rather than the acid-secreting portion of the stomach. This observation raises the possibility that the linkage is not arbitrary but rather may reveal mechanisms that influence cancer pathogenesis. We previously demonstrated that Gas−/− develop antral tumors by 12 months of age and exhibit reduced TFF1 expression and elevated phosphorylated STAT3 expression [14]. Therefore, we used a microarray analysis of Gas−/− tumors to identify molecules involved in tumor pathogenesis, and based on the role of the activin pathway in gastric epithelial lineages differentiation demonstrated in transgenic mice [10], we found that follistatin was significantly uprregulated, supporting the importance of this pathway in tumorigenesis.
Materials and Methods
Human samples
Human normal and tumor protein extracts and tissue sections without patient identifiers were obtained from Bio Chain Institute, Inc. (Hayward, CA).
Animals
Gastrin deficient (Gas−/−) and strain-matched wild-type (WT) control mice were maintained on a mixed 129/Sv-C57/BL6 background [15]. The animals were housed in microisolator, solid-bottom polycarbonate cages in non-barrier mouse rooms. The study was performed with the approval of the University of Michigan Animal Care and Use Committee, which maintains an American Association for the Assessment and Accreditation of Laboratory Animal Care facility.
Immunohistochemical Analysis
A longitudinal section of the mouse stomach (spanning both fundus and antrum) was fixed in 4% paraformaldehyde in PBS, and paraffin-embedded. Four µm sections were subsequently prepared. Antigen retrieval was performed for all antibodies by microwaving in 10 mM citrate buffer (pH 6.0) for 10 min. Sections were incubated with the primary antibodies at 4°C overnight; 1:500 dilution of biotin-conjugated secondary antibodies was then added for 30 min at 25°C and visualized with avidin-biotin complexes (Vectastain Elite ABC) and diaminobenzidine (DAB) (Vector Laboratories). For immunofluorescence, Alexa labeled secondary antibodies (Invitrogen) at 1:500 dilution were incubated for 30 min at 25°C, before staining with 4',6-diamidino-2-phenylindole (DAPI).
The primary antibodies used were: rabbit anti-TFF2 (1:1000 dilution, a gift from Dr. Andy Giraud, University of Melbourne, Australia), polyclonal goat anti-TFF1 (pS2) (1:100 dilution, Santa Cruz); Muc5AC (1:50 dilution, Santa Cruz), mouse monoclonal anti-Muc6 (1:200 dilution, Novocastra), goat polyclonal anti-Ki67 (1:500 dilution, Pharmingen), anti-β subunit of the H+, K+-ATPase (1:1000 dilution, BCL), plant lectin Griffonia Simplicifolia II (GSII), [16] at a 1:1000 dilution (Vector Laboratories), and rabbit polyclonal anti-follistatin (1:50 dilution, Santa Cruz).
Immunoblots
Stomachs removed from WT and Gas−/− mice were opened along the greater curve then cut along the incisura to separate the antrum from the corpus. Protein was extracted from antral and fundic tissues, using T-per lysis buffer (Pierce) supplemented with protease inhibitors (Roche). After centrifuging the lysate for 10 min at 10,000 × g, 50 µg of protein extract were resolved using 4–20% gradient SDS-PAGE and then transferred onto nitrocellulose membranes. Immunoblot analysis was performed at 4°C overnight, using anti-Muc5AC and mouse monoclonal anti-N-myc (Novus). The signals were visualized with an ECL detection system (Amersham). The membrane was stripped and re-blotted against GAPDH to normalize the input.
NCI-N87, AGS, and MKN-45 cells were grown to confluence and serum starved for 24 h. The cells were lysed using RIPA buffer (Sigma) and proteins were resolved on SDS-PAGE gels and analyzed for follistatin (Santa Cruz), as described above. Additionally, whole cell extracts of follistatin treated NCI-N87 cells were blotted for cyclin D1 (rabbit polyclonal, Santa Cruz), TFF1 and TFF2 (both goat polyclonal antibodies, Santa Cruz).
Quantitative RT-PCR
The PCR primer sequences used to amplify Muc6 and GAPDH cDNA were as previously described [17]. The fluorogenic probe (100 nM, Research Genetics Inc., Huntsville, Al, USA) was used for all quantitative PCR analysis. The reactions were carried out in a total volume of 25 µl in triplicate wells in a BioRad iCycler (Hercules, CA). The fold change for each G−/− mouse was calculated as suggested by Livak and Schmittgen [18]: 2ΔΔCT=fold change, and ΔΔCT=(CT, Target, -CT,GAPDH)G−/− (CT, Target,-CT,GAPDH)WT, where CT=threshold cycle and (CT,Target,-CT,GAPDH) WT=mean value for seven WT mice.
Primary cell preparation and Flow cytometry
Epithelial cells were isolated from the gastric mucosa from individual mice. Briefly, the corpus was separated from the antrum and dissected into 2-mm size pieces. The pieces were incubated in 20 ml of Hank’s balanced salt solution containing 5% bovine serum albumin, 1 mM dithiothreitol, and 1mM EDTA for 1 h with vigorous shaking at 37°C to release the epithelial cell population. This first cell suspension was filtered (50 µm Filcon filter; Dako), collected, and washed twice with RPMI1640 medium with 5% fetal calf serum. The stripped mucosa was then subjected to enzymatic digestion in 20 ml of RPMI medium with 1 mg/ml dispase II (Roche) for two 60 min incubations at 37°C with vigorous shaking. The total number of cells isolated from each WT corpus was 1.5~2 × 106, and 0.9 ~ 1.2 × 106 from each 12 mo old Gas−/− stomach. Mucous neck cells were detected with the FITC-conjugated lectin GSII, after permeabilization. Labeled cells were analyzed using FACScan (Beckman Dickson). Data were analyzed using CellQuest Software (Becton Dickinson). The number of GSII positive mucous neck cells per corpus was calculated as follows: (Total cell number/corpus) × (%GSII positive cells).
Microarray Analysis
Total RNA was extracted from three antrums from WT mice and Gas−/− mice, all 12 mo for generation of cDNA and probe generation by the University of Michigan Microarray Core. The quantity and quality of the RNA was verified on an Agilent 2100 BioAnalyzer. The mouse Affymetrix Gene Chip was probed after fluorescent labeling.
Follistatin treatment
NCI-N87 (ATCC) and AGS (ATCC) cells were grown to confluence in RPMI-1640 media supplemented with 10% FBS and 1% streptomycin. Cell cultures were serum starved for 24 h and then treated with 0.5 µg/ml human recombinant-follistatin (R&D Systems) in serum free media for 24 h. Whole cell extracts were obtained, resolved on SDS-PAGE gels, transferred onto nitrocellulose membranes then immunoblotted as described above.
Statistical Analysis
The mean ± the standard error of the mean (SEM) was calculated and compared to controls using the unpaired t-test from the commercially available software Prism 3 (GraphPad Software, San Diego, CA, USA). A P-value of <0.05 was considered significant.
Results
Gastric atrophy and mucous cell hyperplasia develops in 9 month old Gas−/− mice
We have previously shown that parietal cell atrophy and distal gastric tumors occur in 12 month old Gas−/− mice [14]. Yet, early molecular changes correlating with the parietal cell loss, i.e., corpus atrophy, are not clearly described. Therefore, we examined histologic markers defining the glandular stomach prior to 12 months in Gas−/− mice. Immunodetection of the H+K+-ATPase beta subunit demonstrated a significant reduction in the number of parietal cells even as early as 9 months (Fig. 1A–C). There was an increase in the surface mucous compartment documented by immunostaining for Muc5AC (Fig. 1D–F). Enumeration of Ki-67-positive cells in the Gas−/− corpus compared to WT mice revealed a 2.5-fold increase (Fig. 1G–I). The decrease in parietal cells demonstrated by reduced H+,K+-ATPase coincided with an increase in mucous neck cells demonstrated by increased GSII and Muc6 expression (Fig. 1J, I). Indeed, quantification of the total number of neck cells in the corpuses of 12 month Gas−/− mice by flow cytometry confirmed that there was an ~18% increase in the Gas−/− mice compared to WT mice (data not shown). Collectively, these data demonstrated hyperplasia of both the surface mucous and neck cell compartments at the expense of parietal cells.
Figure 1. Corpus atrophy in 9 month Gas−/− mice.
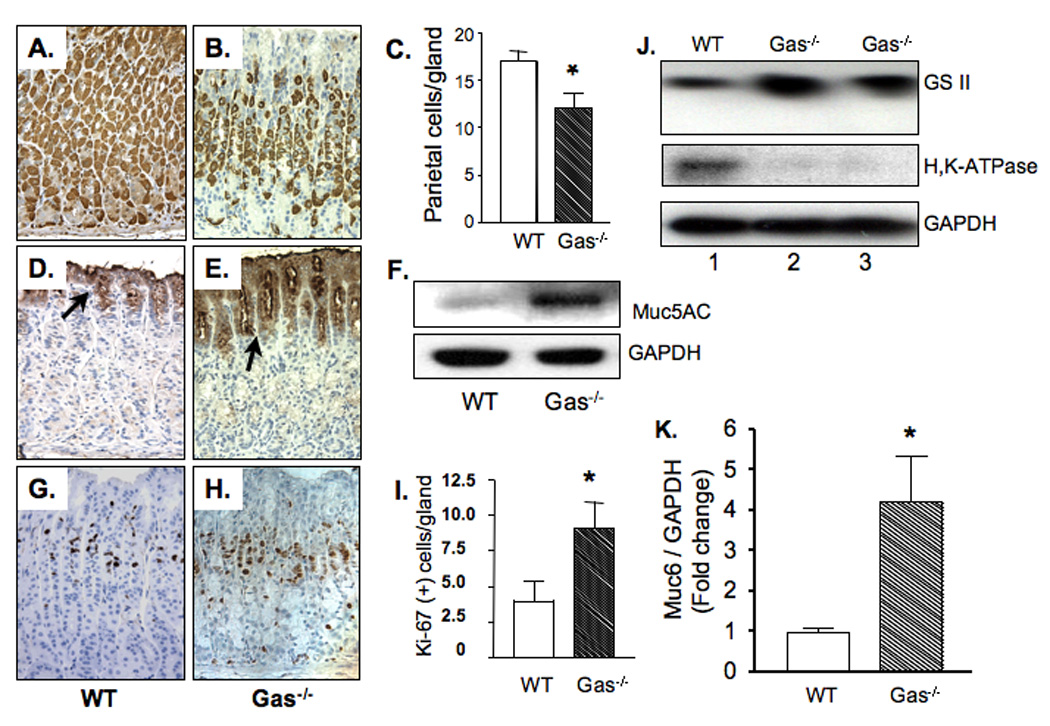
(A) Representative images of immunohistochemical staining of parietal cells with H+,K+-ATPase in 9 month WT (A) and Gas−/− mice (B) corpus. Positive cells were quantified by morphometry and expressed as the number of parietal cells/gland in Gas−/− mice (C). The surface pit cell population (arrows) were labeled using Muc5AC in 9 month WT (D) and Gas−/− mice (E). Immunoblotting was used to quantify the amount of Muc5AC protein in the WT and Gas−/− corpuses (F). Proliferating cells were stained for Ki67 in the WT (G) and Gas−/− (H) corpus mucosa and quantified by morphometry (I). N=9 for 9 month WT mice. N=12 for 9 month Gas−/− mice. *P<0.05 vs WT mice. Magnification, 400X. An increase in GSII mucin and a decrease in H+K+-ATPase protein are shown in a 9 month WT mouse and two 9 month Gas−/− mice (J). Muc6 mRNA was measured by qRT-PCR. N=9 for the WT group, N=12 for Gas−/− mice 9 month. *P<0.05 vs. WT mice (K).
Antral hyperplasia in Gas−/− mice
At the same time that parietal cell atrophy and expansion of the mucous cell compartments was observed, the antrum became hyperplastic. In the antrum, the proliferating cells expressing Ki-67 correlated with mucosal elongation (Suppl. Fig. 1A and B). By 12 months, the Ki-67 staining cells overlapped with the GSII-positive pyloric gland cells that were also TFF2 positive (data not shown) demonstrating that the antral mucous neck cell lineage was also expanding (Suppl. Fig. 1C and D).
The antral surface pit cell lineage was examined using TFF1 immunostaining. TFF1 was detected above the hyperplastic mucous neck cell compartment in WT and 9 month Gas−/− mice (Suppl. Fig. 1E and F). We also observed a distinct cell boundary separating normal TFF1-positive areas from the neighboring hyperplastic cell population that exhibited reduced TFF1, Muc5AC and PAS staining (Suppl. Fig. 1G–I) in Gas−/− antrums by 12 months. These results suggested an increase in the neck cell lineage, and reduced surface mucous cell differentiation at the transition from hyperplastic to dysplastic antrums.
Follistatin expression increased in gastric tumors
To identify the molecular pathways that were modulated in the gastric antrum during transformation, we performed a microarray analysis on three tumors from 12 month old Gas−/− mice and compared the results to age-matched controls. Table 1 shows relevant genes that were up- or down-regulated >2 fold in the antral tumors. Gastrin and H+K+-ATPase mRNA levels were >20-fold depressed in the microarray, confirming that the samples used for the analysis were gastrin-deficient and had little or no corpus tissue, which would have been H+K+-ATPase positive. A number of pathways were represented in the up-regulated set of genes including tissue metalloproteases, IL-1β and related molecules, EGF receptor ligands, e.g., amphiregullin, epiregulin; TGFβ family members, Inhibin bA and follistatin; and the Notch ligand jagged. Interestingly, several of the genes regulated in this Gas−/− mouse model, e.g., metalloproteases and amphiregulin, are also modulated in the H. felis-infected INS-GAS model [19]. To our knowledge, this is the first report of TGFβ family members inhibin bA and follistatin overexpression in gastric tumors.
Table 1.
Antral Changes in Gastrin-deficient mice
| Up-regulated | Down-regulated | |
|---|---|---|
| MMP 13 (59x) | Jagged1 (2.5x) | Angiogenin1 (−43x) |
| MMP 10 (17x) | Runx2 (2.5x) | H,K-ATPase (−29x) |
| IL 1R-like (13x) | Inhibin bA (2.5x) | Gastrin (−20x) |
| Mast cell protease (9x) | TNFrsf (2x) | Sfrp1 (−6x) |
| Ceacam1 (9x) | Sox 18 (2x) | GABAr (−5x) |
| Epiregulin (7x) | CyclinD2 (2x) | Necdin (−5x) |
| Vanin (7x) | EphRb2 (2x) | Msi2h (−4x) |
| IL 1β (7x) | HB-EGF (1.8x) | FGFbp 1 (−4x) |
| CXCl1 (7x) | Klf5 (1.6x) | TGFbR3 (−3x) |
| Follistatin (7x) | Nephronectin (4x) | Frzd 7 (−3x) |
| IGFbp3 (6x) | Claudin 4 & 23 (3x) | Barx1 (−2.5x) |
| Cathepsin E (6x) | Egr1 (3x) | Frzd 2 (−2.5x) |
| Clusterin (3x) | Sox 2 (3x) | Supervillin (−2x) |
| IL1rn (4x) | Amphiregulin (3x) | |
The expression of follistatin was dramatically increased in the antral mucosa of 9 month Gas−/− compared to age-matched WT mice (Suppl. Fig. 2A–C) and correlated with an expansion of the mucous neck cells (GSII staining in Suppl. Fig. 2B). Follistatin-expressing cells overlapped with the proliferation marker Ki-67 in both the hyperplastic antrum (Suppl. Fig. 2E), and Gas−/− tumor (Suppl. Fig. 2F).
Follistatin is expressed in normal human antrum, and its expression is significantly increased in tubular adenocarcinoma (Fig. 2A,B), and signet ring carcinoma (Fig. 2C), suggesting a participation of this protein in the development of human gastric cancer. To determine whether elevated levels of follistatin induced proliferation, three human gastric cell lines were screened for follistatin expression. Interestingly, only the mucopeptic cell line NCI-N87 expressed follistatin. Treatment of this cell line with follistatin for 24h induced the proliferation marker cyclin D1 and the mucous lineage specific proteins TFF1 and 2 suggesting that follistatin promotes mucous cell proliferation and differentiation. The less differentiated AGS cells were not responsive to follistatin treatment (data not shown), suggesting that effect of follistatin is cell type dependent.
Figure 2. Follistatin expression elevated in human gastric cancer.
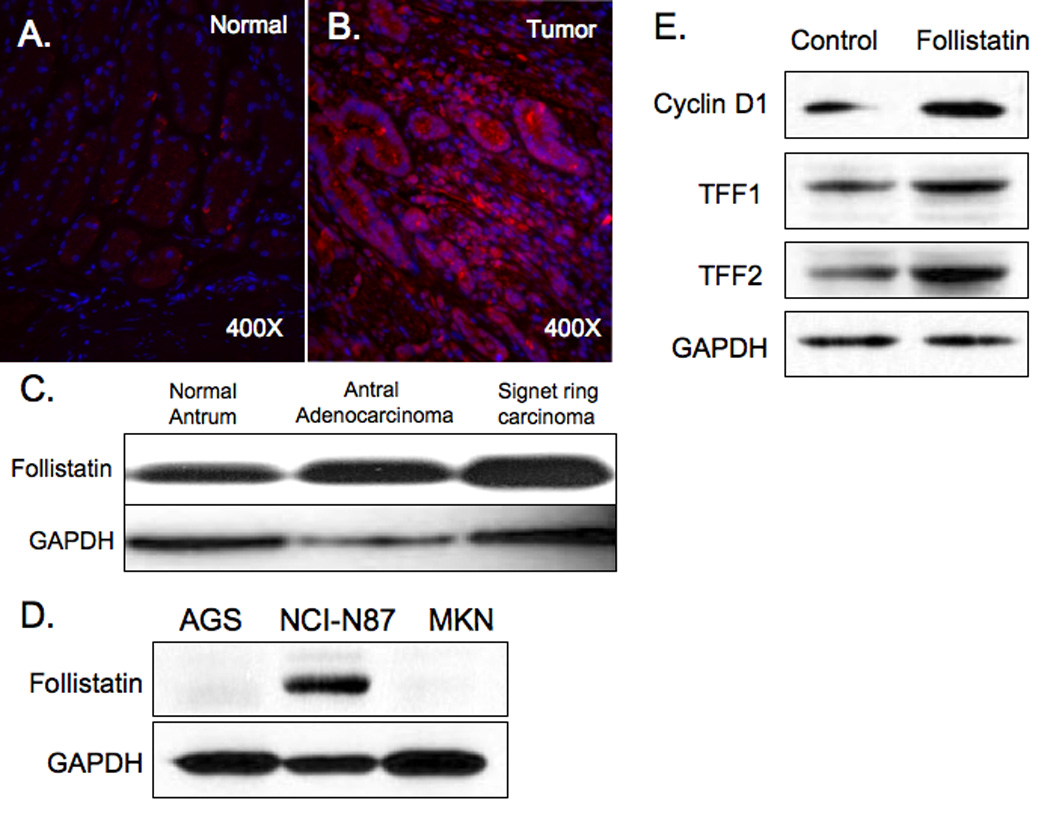
Follistatin (red) expression in normal human antrum (A) and a tubular carcinoma (B) (magnification 400X) and by immunoblot in normal human antrum, antral adenocarcinoma and signet ring cell carcinoma (C). AGS, NCI-N87 and MKN45 gastric cancer cell line extracts immunoblotted for follistatin protein (D). NCI-N87 cells were treated with follistatin (0.5 µg/ml) for 24h then immunoblotted for cyclin D1, TFF1 and TFF2 (E). Control = untreated cells.
Discussion
The spatial and cellular aspects of tumor development observed in Gas−/− mice [14] is very similar to that of the TFF1 knockout mouse [4] and the gp130757F/F mouse [8]. In this study, further analysis demonstrated that well-defined regional loss of surface mucous cells (TFF1 expression) occurred in the hyperplastic antrum. Consistent with the TFF1-deficient mice model, there were apparently lower levels of TFF1 than TFF2 in hiperplastic Gas−/− antrums (data not shown). This result suggested that more of the hyperplastic cells were phenotypically of the mucous neck cell lineage.
Follistatin is a single-chain glycoprotein that was first isolated from ovarian follicular fluids, and inhibits follicle stimulating hormone (FSH) secretion in the pituitary [20], but it also binds activins preventing receptor activation. Activins have been shown to exert regulatory effects on growth, proliferation and differentiation in different cell types [11, 21, 22]. In addition to activins, follistatin also binds bone morphogenetic proteins (BMP), including BMP-2,-4,-7 and 11 [23], in a cell specific way.
Activin receptor activation is necessary for foregut development, and it has been reported that activin A signaling promotes differentiation of pancreatic into intestinal tissue [24]. In 12 month Gas−/− mice, we observed overexpression of follistatin, but not activin. Previously, Becker et al. reported low levels of activin A expression in normal gastric samples, but a significant increase in activin A within the tissue surrounding experimentally-induced ulcers in rats [25]. Follistatin expression is induced by several signal transduction pathways including, EGF, PKA, PKC and steroid hormones [20] and strongly induced by activin A through Smad proteins [22]. Thus, the elevated levels of follistatin in the Gas−/− mouse antrums and human tumors, in absence of activin A, might represent induction by EGF or PKA, PKC. Interestingly, activin receptor II gene expression in Gas−/− mice was down-regulated (1.53-fold), suggesting muted activin signaling in the hyperplastic and dysplastic Gas−/− mice.
Follistatin expression was observed in areas of highly proliferative mucous cells in Gas−/− mice antrums. NCI-N87 is a mucous producing-cell line of well-differentiated gastric tumor origin, while AGS and MKN cells are non-mucous, poorly differentiated gastric adenocarcinoma cell lines. Only NCI-N87 cells express and response to follistatin. To our knowledge, this is the first report of follistatin activity in absence of activins. The pathways involved in the differential cell response are currently under study.
In summary, the hyperplasia and dysplasia in the antrums of Gas−/− mice is comprised mainly of mucous cells and includes both neck and surface cell lineages. The presence of elevated follistatin levels in mouse antral tumors and human gastric cancer suggests that neutralizing the TGF-β signaling pathway plays a role in mucous cell proliferation in the stomach.
Supplementary Material
Antral hyperplasia coincides with expanded mucous neck and focal loss of surface pit lineages in Gas−/− mice. Immunohistochemical staining for Ki67 (brown) and GSII-lectin (red) in the antral mucosa of WT (A) and Gas−/− mice (B), showing the expansion of antral neck mucous cells at 9 months. At 12 months, the antral hyperplasia is evident (C and D). The distribution of TFF1-positive surface cells in the antrum of WT (E), 9 month Gas−/− mice (F), and 12 month Gas−/− mice (G) is altered in the transition zone between the hyperplastic and dysplastic antrum, evidenced by Muc5AC (H) and periodic acid Schiff (PAS) (I) staining on serial sections. Dotted lines indicate the boundaries between the positive-staining versus reduced staining antral areas for the two cell markers TFF1 and Muc5AC. Original magnifications: 400X in A-C, E, F, H, and I; 1000X in D; and 200X in G.
Follistatin expressed in antral hyperplasia and tumors of Gas−/− mice. Follistatin immunostaining in WT (A, D) and Gas−/− mouse antrums costained with the mucous neck cell marker GSII (B,C) or with Ki-67 (E,F) in 9 month hyperplastic (E) or 12 month antral tumors (F). Magnification: 100X in B, 400X in A, C-F.
Acknowledgements
This work was supported in part by Public Health Service Grant R01-DK61410 to J.L.M. and the Michigan Gastrointestinal Peptide Research Center P30-DK34933.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pisani P, Parkin DM, Bray F, Ferlay J. Estimates of the worldwide mortality from 25 cancers in 1990. Int J Cancer. 1999;83:18–29. doi: 10.1002/(sici)1097-0215(19990924)83:1<18::aid-ijc5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 3.Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975;2:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre O, Chenard MP, Masson R, Linares J, Dierich A, LeMeur M, Wendling C, Tomasetto C, Chambon P, Rio MC. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science. 1996;274:259–262. doi: 10.1126/science.274.5285.259. [DOI] [PubMed] [Google Scholar]
- 5.Xu X, Brodie SG, Yang X, Im YH, Parks WT, Chen L, Zhou YX, Weinstein M, Kim SJ, Deng CX. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene. 2000;19:1868–1874. doi: 10.1038/sj.onc.1203504. [DOI] [PubMed] [Google Scholar]
- 6.Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima T, Bae SC, Ito Y. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 7.Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F, Heath JK, Ernst M. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 8.Judd LM, Alderman BM, Howlett M, Shulkes A, Dow C, Moverley J, Grail D, Jenkins BJ, Ernst M, Giraud AS. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 9.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Li Q, Karam SM, Coerver KA, Matzuk MM, Gordon JI. Stimulation of activin receptor II signaling pathways inhibits differentiation of multiple gastric epithelial lineages. Mol Endocrinol. 1998;12:181–192. doi: 10.1210/mend.12.2.0060. [DOI] [PubMed] [Google Scholar]
- 11.Stove C, Vanrobaeys F, Devreese B, Van Beeumen J, Mareel M, Bracke M. Melanoma cells secrete follistatin, an antagonist of activin-mediated growth inhibition. Oncogene. 2004;23:5330–5339. doi: 10.1038/sj.onc.1207699. [DOI] [PubMed] [Google Scholar]
- 12.Cipriano SC, Chen L, Kumar TR, Matzuk MM. Follistatin is a modulator of gonadal tumor progression and the activin-induced wasting syndrome in inhibin-deficient mice. Endocrinology. 2000;141:2319–2327. doi: 10.1210/endo.141.7.7535. [DOI] [PubMed] [Google Scholar]
- 13.El-Zimaity HM, Ota H, Graham DY, Akamatsu T, Katsuyama T. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer. 2002;94:1428–1436. doi: 10.1002/cncr.10375. [DOI] [PubMed] [Google Scholar]
- 14.Zavros Y, Eaton KA, Kang W, Rathinavelu S, Katukuri V, Kao JY, Samuelson LC, Merchant JL. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene. 2005;24:2354–2366. doi: 10.1038/sj.onc.1208407. [DOI] [PubMed] [Google Scholar]
- 15.Friis-Hansen L, Sundler F, Li Y, Gillespie PJ, Saunders TL, Greenson JK, Owyang C, Rehfeld JF, Samuelson LC. Impaired gastric acid secretion in gastrin-deficient mice. Am J Physiol. 1998;274:G561–G568. doi: 10.1152/ajpgi.1998.274.3.G561. [DOI] [PubMed] [Google Scholar]
- 16.Lyer PN, Wilkinson KD, Goldstein LJ. An -N-acetyl-D-glycosamine binding lectin from Bandeiraea simplicifolia seeds. Arch Biochem Biophys. 1976;177:330–333. doi: 10.1016/0003-9861(76)90444-6. [DOI] [PubMed] [Google Scholar]
- 17.Kang W, Rathinavelu S, Samuelson LC, Merchant JL. Interferon gamma induction of gastric mucous neck cell hypertrophy. Lab Invest. 2005 doi: 10.1038/labinvest.3700260. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Takaishi S, Wang TC. Gene expression profiling in a mouse model of Helicobacter-induced gastric cancer. Cancer Sci. 2007;98:284–293. doi: 10.1111/j.1349-7006.2007.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michel U, Farnworth P. Follistatin steady state messenger ribonucleic acid levels in confluent cultures of a rat renal mesangial cell line are regulated by multiple factors. Endocrinology. 1992;130:3684–3693. doi: 10.1210/endo.130.6.1375907. [DOI] [PubMed] [Google Scholar]
- 21.van Eyll JM, Pierreux CE, Lemaigre FP, Rousseau GG. Shh-dependent differentiation of intestinal tissue from embryonic pancreas by activin A. J Cell Sci. 2004;117:2077–2086. doi: 10.1242/jcs.01067. [DOI] [PubMed] [Google Scholar]
- 22.Bartholin L, Maguer-Satta V, Hayette S, Martel S, Gadoux M, Corbo L, Magaud JP, Rimokh R. Transcription activation of FLRG and follistatin by activin A, through Smad proteins, participates in a negative feedback loop to modulate activin A function. Oncogene. 2002;21:2227–2235. doi: 10.1038/sj.onc.1205294. [DOI] [PubMed] [Google Scholar]
- 23.Iemura S, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, Sugino H, Ueno N. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc Natl Acad Sci U S A. 1998;95:9337–9342. doi: 10.1073/pnas.95.16.9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SK, Hebrok M, Li E, Oh SP, Schrewe H, Harmon EB, Lee JS, Melton DA. Activin receptor patterning of foregut organogenesis. Genes Dev. 2000;14:1866–1871. [PMC free article] [PubMed] [Google Scholar]
- 25.Becker JC, Hertel M, Markmann A, Shahin M, Werner S, Domschke W, Pohle T. Dynamics and localization of activin A expression in rat gastric ulcers. Scand J Gastroenterol. 2003;38:260–267. doi: 10.1080/00365520310000636a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antral hyperplasia coincides with expanded mucous neck and focal loss of surface pit lineages in Gas−/− mice. Immunohistochemical staining for Ki67 (brown) and GSII-lectin (red) in the antral mucosa of WT (A) and Gas−/− mice (B), showing the expansion of antral neck mucous cells at 9 months. At 12 months, the antral hyperplasia is evident (C and D). The distribution of TFF1-positive surface cells in the antrum of WT (E), 9 month Gas−/− mice (F), and 12 month Gas−/− mice (G) is altered in the transition zone between the hyperplastic and dysplastic antrum, evidenced by Muc5AC (H) and periodic acid Schiff (PAS) (I) staining on serial sections. Dotted lines indicate the boundaries between the positive-staining versus reduced staining antral areas for the two cell markers TFF1 and Muc5AC. Original magnifications: 400X in A-C, E, F, H, and I; 1000X in D; and 200X in G.
Follistatin expressed in antral hyperplasia and tumors of Gas−/− mice. Follistatin immunostaining in WT (A, D) and Gas−/− mouse antrums costained with the mucous neck cell marker GSII (B,C) or with Ki-67 (E,F) in 9 month hyperplastic (E) or 12 month antral tumors (F). Magnification: 100X in B, 400X in A, C-F.