

Abstract
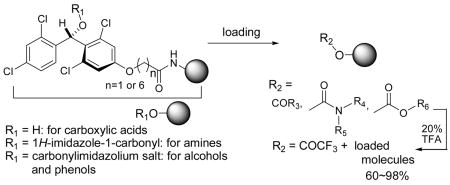
An acid and base stable hydroxytetrachlorodiphenylmethyl (HTPM) linker is developed for polymer-supported organic synthesis. The linkers reported here are utilized for loading carboxylic acids, amines, alcohols, and phenols, and are stable to Brønsted and Lewis acids, Brønsted bases and a wide variety of nucleophiles. However, the HTPM linkers can conveniently be cleaved by the solvolytic displacement reactions with 20% TFA.
Since introduction of the concept of solid-phase synthesis in late 1950s,1 a variety of linkers for immobilizing organic molecules (the first building blocks or natural product derivatives) and organic reactions amenable to polymer-supported chemistries have been developed, greatly advancing research in biochemistry, molecular biology, pharmacology, and drug discovery.2 Although recent advances in analysis and purification methods dramatically enhanced the usefulness of solid-phase synthesis,3 development of new linker which allows delivering target molecules in high yield and purity is still required. The linkers should be stable against a planned set of reaction conditions, but be cleaved under mild conditions that do not degrade the products. To date, many useful linkers for solid-phase synthesis have been developed.4 However, the choice of spacer and linker requires careful consideration when applying diverse organic reactions on the solid phase.4f
In connection with the ongoing studies on the development of novel MraY inhibitors5, we have delivered a set of small optimized libraries based on uridine-β-hydroxyamino acid (Scheme 1).6 In order to efficiently generate such libraries in solution or on polymer-support, we sought a protecting group or a linker for the carboxylic acid which can be cleaved simultaneously with the acetonide by a volatile and mild acid such as TFA. In addition, a protecting group (or a linker) for the carboxylic acid should have susceptibility to relatively strong Brønsted and Lewis acids, and a wide variety of nucleophiles. Although a large number of acid cleavable protecting groups (i.e. trityl, TBDPS, methoxymethyl, tetrahydropyranyl, 2-(trimethylsilyl)ethyl, t-butyl, p-methoxybenzyl, ortho esters, and their related protecting groups) for carboxylic acids have been utilized in organic syntheses, these protecting groups did not meet our needs; trityl, silyl, and MPM protecting groups are too labile to acids being utilized in the library production and MOM, SEM, t-butyl, and ortho esters require harsh acidic conditions for the regeneration of the carboxylic acids.7 Because of the reasons mentioned above we have developed a new protecting group for carboxylic acid functional groups which can be selectively cleaved by TFA but is stable to a variety of acids.
Scheme 1.

General structures of MraY inhibitors i.
Diphenylmethyl ester is an acid labile functional group and has been utilized as a temporary protecting group for carboxylic acids. In our experiments diphenylmethanol exhibited similar nucleophilicity to allylic alcohol and is not esterified efficiently with carboxylic acids via conventional carboxylic acid activation methods (i.e. DCC, BOPCl, and mixed anhydride). In order to stabilize diphenylmethyl esters by tuning electronic properties of dibenzene moieties, several chlorosubstituted-diphenylmethyl esters were synthesized and tested for stability against representative acids such as TsOH·H2O (20% in CH2Cl2-THF), HF (10% in CH3CN), BF3·OEt 2 (10% in CH2Cl2), and La(OTf)3 (10% in aq THF). Interestingly, as summarized in Scheme 2 all (4-methoxyphenyl) (chlorophenyl)methanols 4a–d, conveniently synthesized by Friedel-Crafts reactions followed by NaBH4 reductions, could be efficiently esterified by using EDCI, DCC or acid chloride methods. The esters 4a–c regenerated the corresponding acids by the treatment of 20% TsOH within 1 h and were also not stable under 10% HF, 15% TFA, 10% BF3·OEt 2, and 10% La(OTf)3.
Scheme 2. Syntheses of chlorosubstituted diphenylmethyl esters and their stability against the representative acids.

a 20% in CH2Cl2-THF (1/1).;b 10% in CH3CN.; c 15% in CH2Cl2.;d10% in aq THF.;H indicates the protecting group is readily cleaved.; M indicates that the protecting group is cleaved very slowly.; L indicates that the protecting group is stable.; e~5% of regeneration of the carboxylic acids was observed after 1 h.
However, the tetrachloro-substituted 4-methoxydiphenylmethyl esters 4d showed an unusual acid stability; no regeneration of the acids from the esters 4d was observed under 20% TsOH for over 20 h. The esters 4d also exhibited excellent stablility to 15% TFA, 10% HF, and a variety of Lewis acids such as AlCl3, B(C6F5)3, BCl3, TMSOTf, and La(OTf)3. Moreover, the esters 4d 1) were photolytically stable; no change by the irradiation at 200~350 nm in DMF for 72 h, 2) showed stability under basic conditions; no saponifications were observed under 40% NH4OH in aq THF, 10% LiOH in aq THF-MeOH, 10% KOH in MeOH-THF, and 10% DBU in aq THF at rt for over 12 h, and 3) showed excellent stability to nucleophiles; the esters 4d were not susceptible to the nucleophilic attacks of primary and secondary amines (in aq THF at 80 °C), NH2NH2 (in aq THF at rt), alkylthiols (in THF at 80 °C), and NaN3 (90 °C in DMF) for over 12 h.8 However, the esters 4d slowly reacted with 10% BF3·OEt 2 to furnish the carboxylic acids (~5% after 1 h) and 1,3-dichloro-2-((2,4-dichlorophenyl)fluoromethyl)-5-methoxybenzene. The esters 4d could conveniently be cleaved by using 20% TFA in CH2Cl2 to afford the corresponding acids and the trifluoroacetate (R1, R2, and R3 = Cl, R4 = CF3 in 4d).9 Thus, we succeeded in stabilizing diphenylmethyl ester, enabling a wide range of organic reactions for the generation of small optimized libraries of MraY inhibitors in solution (Scheme 1). Taking advantage of excellent chemical stability of esters of (2,6-dicholoro-4-methoxyphenyl)(2,4-dichlorophenyl)methanol, we have developed a new linker to immobilize carboxylic acids, amines, and alcohols which can, however, be cleaved by 20% TFA.
As illustrated in Scheme 3 the 3,5-dichloro-4-((2,4-dichlorophenyl)(hydroxy)methyl)phenol group could be efficiently linked with (aminomethyl)polystyrene 7a and aminomethyl-Lantern™ 7b10 through C2 and C7 spacers without using sophisticated procedures. Available alcohol-linkers on the polymer surface after derivatization of the polymers 7a (~1.2 mmol/g) and 7b (~15μmol/Lantern) were determined to be 1.0~1.2 mmol/g for 8a-C2 and 8a-C7, and 12~15μmol/Lantern for 8b-C2 and 8b-C7 by coupling of the linkers with Fmoc-β-Ala-OH and subsequent release of Fmoc chromophore and elemental analyses of the Cl atoms for 8a and 8b.
Scheme 3.

Synthesis of the hydroxy-tetrachlorodiphenylmethyl (HTPM) linkers.
We first investigated reactivity of the hydroxytetrachlorodiphenylmethyl (HTPM) linkers 8a and 8b to carboxylic acids. We have not observed noticeable differences in the rates of esterification with N-blocked amino acids between 8a-C2 and 8a-C7 or between 8b-C2 and 8b-C7; a variety of Cbz or Boc-protected amino acids could be loaded onto 8a-C2, 8a-C7, 8b-C2, and 8b-C7 within 6 h in >95% yields via a 4~5 fold excess of N-protected amino acids and EDCI or DICI.11 It is worthwhile mentioning that Cbz-D-alanyl esters of 8a-C2 and 8b-C2 showed excellent stability to BF3·OEt2; on the other hand, ~10% of the same esters of 8a-C7 and 8b-C7 were cleaved after 2 h. Thus, we utilized 8a-C2 and 8b-C2 for further studies. Robustness of the linkers 8a-C2 and 8b-C2 to an acid (20% TsOH) was demonstrated by synthesizing the pentapeptide moiety of Park’s nucleotide ( UDP-N-acetylmuramyl-L-alanyl-D-isoglutamyl-L-lysinyl-D-alanyl-D-alanine)12 using the Boc-strategy. One gram of the Boc-D-Ala-HTPM resin (~1.0 mmol/g) was used for the following sequential ligation reactions with Boc-D-Ala-OH, Boc-L-Lys(COCF3)-OH, Boc-γ-D-Glu(OMe)-OH, Cbz-L-Ala-OH. The generated pentapeptide on the HTPM resin was cleaved with 20% TFA in CH2Cl2 for 1 h followed by methylation with CH2N2 to afford Cbz-L-Ala-γ-D- Glu(OMe)-L-Lys(COCF3)-D-Ala-D-Ala-OMe in over 90% overall yield after SiO2-plug filtration (CHCl3:MeOH=3:1).13 As observed in the esters 4d, 8a-C2 could be regenerated by ammonolysis of the recovered resins and the regenerated HTPM linker was reused two times for the synthesis of Cbz-L-Ala-γ-D-Glu(OMe)-L- Lys(COCF3)-D-Ala-D-Ala-OMe; the overall yields using the regenerated resins were 90% (1st reuse) and 89% (2nd reuse), respectively.14
Solid phase peptide synthesis has proven to be a practical method of producing synthetic oligo- to longer peptides and the Boc-strategy is a popular method currently used for oligo peptide synthesis.15 However, the Boc-solid phase peptide synthesis suffers from low overall yield and low purity due to 1) partial cleavage of the anchor peptides in the cycle of the deprotection of Boc groups, and 2) inefficient cleavage of the products when applied to photolabile linkers4g. We could demonstrate the synthesis of the oligopeptide using the HTPM linker 8a-C2 with excellent yield and purity. In addition, the appropriately protected uridine-β-hydroxyamino acid 9 could also be loaded completely via 1.3 eq of the HTMP resin 8a-C2. The ester linker was stable to the basic conditions such as Triton B or NH4OH in aq THF-MeOH. The acetonide deprotection and concomitant cleavage of the linker under 20% TFA provided the fully deprotected uridine-β-hydroxyamino acid 10 in over 90% (Scheme 4).
Scheme 4. a A demonstration of loading onto and cleavage from the HTPM-resin using the carboxylic acid 9.

a 1.3 equiv of the HTPM resin against 9 was used.
The HTPM linker 8a-C2 could be converted to the carbonylimidazole linker 8a-C2-CI by the reaction with CDI, which could further be converted to the carbonylimidazolium salts 8a-C2-CI-Me by the treatment with MeI in CH3CN or MeOTf in CH2Cl2. The resin 8a-C2-CI16 exhibited excellent reactivity against primary and secondary amines but much less reactivity towards alcohol nucleophiles; the primary alcohols did not react with the carbonylimidazole resin even in the presence of tertiary amines at elevated temperatures. Thus, the resin 8a-C2-CI can be utilized in loading amino groups selectively in the presence of alcohol groups in the same molecule. On the other hand, the corresponding carbonylimidazolium salts 8a-C2-CI-Me could be utilized in the immobilization of alcohols and phenols in the presence of DMAP in CH2Cl2 at rt; we did not observe difference in the loading efficiency of 8a-C2-CI-Me due to the difference of the counter ion (I vs OTf). As summarized in Scheme 5 benzylamine (11), N-methylbenzylamine (12), 2-aminopropan-1-ol (13), (2-methoxyphenyl)hydrazine (14) were successfully loaded onto and cleaved from the 8a-C2-CI linker; overall yields for two steps were over 98%.11 3-Phenylpropan-1-ol (15), 4-phenylbutan-2-ol (16), the partially protected uridine 17, (4-chlorophenyl)(4-hydroxyphenyl)methanone (18), and estrone (19) were also loaded onto and cleaved from 8a-C2-CI-Me in 60~70% yield. Interestingly, 3-formylrifamycin (20) could be completely loaded using an excess of 8a-C2-CI-Me.17 The yields for loading the alcohols and phenols were lower than those achieved for amines using the 8a-C2-CI linker. It may be explained by the following reasons: 1) the imidazolium salt formation may be incomplete due to the lower swelling nature of 8a-C2-CI-Me; which would significantly slow down the imidazolium salt formation on the polymer surface in the course of the reaction, and 2) adventitious water in the reaction is also activated by DMAP to hydrolyze the carbonylimidazolium salts. Nonetheless, we could achieve a useful level of loading of alcohols and phenols onto the 8a-C2-CI-Me. As expected the carbamate and the carbonate linkers exhibited excellent stability to acids, bases, and a variety of nucleophiles as demonstrated for the esters 4d.
Scheme 5. The linkers 8a-C2-Cl and 8a-C2-Cl-Me for the immobilizations of amines, alcohols, and phenols.

a 5 equiv of the amine was used in CH2Cl2. b 5equiv of the alcohol and DMAP were used in CH2Cl2. c 2 equiv of the resin was used.
In conclusion, we have developed acid and base stable hydroxytetrachlorodiphenylmethyl (HTPM) linkers, which have significant advantages over currently utilized linkers for loading carboxylic acids in that: 1) the ester linker is stable to a wide variety of nucleophiles, 2) the loaded molecules can be cleaved by solvolytic cleavage with 20% TFA within 1 h, and 3) the linker can be regenerated by treatment with aq NH3 in THF. Utility of the HTPM linker was also demonstrated for loading amines, alcohols, and phenols using a carbonylating reagent. These new linkers should be a valuable asset for polymer-supported organic synthesis. Applicability of these linkers to the generation of diverse structures of libraries defining the scope and limitations of the linkers will be reported elsewhere.
Supplementary Material
Experimental procedures and copies of NMRs. This is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This paper is dedicated to Professor Yoshito Kishi (Harvard University) on the occasion of his 70th birthday. We thank the National Institutes of Health (NIAID grants AI049151, AI018357, and AI06357) and Colorado State University for generous financial support. We also thank Dr. Narayanasamy (Colorado State University) for providing the intermediates for the synthesis of MraY inhibitors.
References
- 1.(a) Merrifield RB. Science. 1965;150:178. doi: 10.1126/science.150.3693.178. [DOI] [PubMed] [Google Scholar]; (b) Merrifield RB. J Am Chem Soc. 1963;85:2149. [Google Scholar]; (c) Merrifield RB. Science. 1958;128:238. doi: 10.1126/science.128.3318.238. [DOI] [PubMed] [Google Scholar]
- 2.(a) Park KH, Kurth MJ. Drugs of the Future. 2000;25:1265. [Google Scholar]; (b) Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (c) Ellingboe JW. Curr Opin Drug Dis Dev. 1999;2:350. [PubMed] [Google Scholar]
- 3.(a) Letot E, Koch G, Falchetto R, Bovermann G, Oberer L, Roth HJ. J Comb Chem. 2005;7:364. doi: 10.1021/cc049850r. [DOI] [PubMed] [Google Scholar]; (b) Stewart JT. Drug Dev Ind Pharm. 2004;30:793. doi: 10.1081/ddc-120039509. [DOI] [PubMed] [Google Scholar]
- 4.(a) McAllister LA, McCormick RA, Procter DJ. Tetrahedron. 2005;61:11527. [Google Scholar]; (b) DiBlasi CM, Macks DE, Tan DS. Org Lett. 2005;7:1777. doi: 10.1021/ol050370y. [DOI] [PubMed] [Google Scholar]; (c) Wu A, Ede NJ. Org Lett. 2003;5:2935. doi: 10.1021/ol035153g. [DOI] [PubMed] [Google Scholar]; (d) Tallarico JA, Depew KM, Pelish HE, Westwood NJ, Lindsley CW, Shair MD, Schreiber SL, Foley MA. J Comb Chem. 2001;3:312. doi: 10.1021/cc000107i. [DOI] [PubMed] [Google Scholar]; (e) Guiller F, Orain D, Bradley M. Chem Rev. 2000;100:2091. doi: 10.1021/cr000014n. [DOI] [PubMed] [Google Scholar]; (f) James IW. Tetrahedron. 1999;55:4855. [Google Scholar]; (g) Wang SS. J Org Chem. 1976;41:3258. doi: 10.1021/jo00882a010. [DOI] [PubMed] [Google Scholar]
- 5.Dini C. Curr Top Med Chem. 2005;5:1221. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]
- 6.Kurosu M, Narayanasamy P, Crick DC. Heterocycles. 2007;72 in press. [Google Scholar]
- 7.(a) Pearson AJ, Roush WR. Handbook of Reagents for Organic Synthesis: Activating and Agents and Protecting Groups. Wiley, Chichester; UK: 1999. pp. 1–513. [Google Scholar]; (b) Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. John Wiley and Sons, Inc; New York: 1999. pp. 369–453. [Google Scholar]
- 8.The MM2 calculation of 4d (R4 = Me) indicated that the dihedral angle formed by two planar chlorosubstituted-benzenes is 76.4°. The -CO-O- linkage and the ether methine proton would preferentially be in a common plane. Thus, the chloro atoms at o-positions of two benzenes hinder the nucleophilc attack on the carbonyl group of the esters.
- 9.For solvolytic displacement reactions with TFA, see. Milne JB. J Chem No-Aqueous Solvents. 1978;5B:1.Nordlander JE, Gruetzmacher RR, Miller F. Tetrahedron Lett. 1973;12:927.
- 10.Lantern™ (trademark of Mimotopes Pty, Ltd) is cylindrical in appearance and polystyrene grafted surface.
- 11.The yield was determined by 1H-NMR analysis using an internal standard (1-bromo-4-methoxybenzene).
- 12.(a) Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Tetrahedron Lett. 2007;48:799. [Google Scholar]; (b) Kurosu M, Crick DC. Tetrahedron Lett. 2006;47:5325. and references therein. [Google Scholar]
- 13.In our hands, the synthesis of the same pentapetide with the HMBA linker employing the Fmoc-strategy afforded the desired product in ~15%.
- 14.The same experiments were performed using 8b-C2 and we obtained compatible results that were observed with 8a-C2.
- 15.Shin DS, Kim DH, Chung WJ, Lee YS. J Bio Mol Biol. 2005;38:517. doi: 10.5483/bmbrep.2005.38.5.517. [DOI] [PubMed] [Google Scholar]
- 16.The resin 8a-C2-Cl could be stored for over 20 days without decrease of the loading efficiency.
- 17.Phenolic alcohols reacted with 8a-C2-Cl-Me much faster than alkanols; greater than 90% selectivity was observed in the competition experiments using 15 and 19.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and copies of NMRs. This is available free of charge via the Internet at http://pubs.acs.org.