

Abstract
Chitin, is a ubiquitous polysaccharide in fungi, insects and parasites. To test the hypothesis that chitin is an important immune modulator, we characterized the ability of chitin fragments to regulate murine macrophage cytokine production in vitro and induce acute inflammation in vivo. Here we show that chitin is a size-dependent stimulator of macrophage interleukin (IL)-17A production and IL-17A receptor (R) expression and demonstrate that these responses are Toll-like Receptor (TLR)-2 and MyD88-dependent. We further demonstrate that IL-17A pathway activation is an essential event in the stimulation of some but not all chitin-stimulated cytokines and that chitin utilizes a TLR-2, MyD88- and IL-17A-dependent mechanism(s) to induce acute inflammation. These studies demonstrate that chitin is a size-dependent pathogen-associated molecular pattern (PAMP) that activates TLR-2 and MyD88 in a novel IL-17A / IL-17AR-based innate immunity pathway.
Keywords: Monocytes/Macrophages, Cytokines, Inflammation, Lung, Rodent
INTRODUCTION
Chitin is a polymer of N-acetylglucosamine, which, after cellulose, is the second most abundant polysaccharide in nature. Although it does not have a mammalian counterpart, it is found in the walls of fungi, exoskeleton of crabs, shrimp and insects, the microfilarial sheath of parasitic nematodes, and the lining of the digestive tracts of many insects (1-9). In these locations, chitin is used by the organism to protect it from the harsh conditions in its environment and host anti-parasite / pathogen immune responses. In these settings, chitin accumulation is regulated by the balance of biosynthesis and degradation. The latter is mediated, in great extent, by chitinases which are endo-ß-1, 4-N-acetylglucosamidases (5). These enzymes are produced as part of immune responses to chitin containing pathogens where they induce chitin fragmentation (5, 10). Surprisingly, although chitin and chitin fragments are produced during pathogen invasion, very little is known about their ability to regulate local inflammatory cell function and the mechanisms of the effects that have been noted have not been adequately investigated.
Toll-like receptors (TLR) have recently been appreciated to function as sensors of microbial and parasitic invasion that initiate innate inflammatory and immune responses against these pathogens. They mediate these responses by recognizing conserved, often times repeating structures on these pathogens called pathogen-associated molecular patterns or PAMPs (11). The specificity of these TLRs for specific PAMPs including the recognition of peptidoglycans, lipopeptides and zymosan by TLR-2 and lipopolysaccharide by TLR-4 are now well described (11). Chitin has a repeating molecular pattern that is analogous, in many ways, to other TLR ligands, in particular the TLR-4 ligand hyaluronic acid (HA) (12, 13). However, the possibility that chitin and / or chitin fragments are ligands for TLRs has not been investigated.
Traditional concepts of adaptive immunity have focused on polarized type 1 immune responses which involve effectors like gamma interferon (IFN-γ) and type 2 immune responses which involve effectors such as IL-4, IL-13, IL-9 and IL-5 [for review, see (14)]. Recent studies have also revealed an additional adaptive effector pathway that involves IL-17A (IL-17) (15, 16). However, the production of IL-17 during TLR-stimulated innate immune responses has not been described.
We hypothesized that chitin is a pathogen associated molecular pattern (PAMP) that regulates IL-17 in vitro and in vivo. To test this hypothesis we characterized the ability of chitin to stimulate macrophage IL-17 production and induce acute inflammation. These studies demonstrate that chitin is a size-dependent PAMP that stimulates macrophage IL-17 production and induces acute tissue inflammation via a pathway(s) that involves TLR-2, IL-17A and MyD88.
MATERIAL AND METHODS
Preparation of Chitin Particles
Chitin fragments were generated as previously described (17). In brief, chitin powder (Sigma Chemical Co., St. Louis, MO) was suspended in sterile Phosphate Buffer Saline (PBS 1X: Gibco, Grand Island, NY) and sonicated at 25% output power three times for 5 min with a Branson sonicator (Sonifier 450, Branson Ultrasonics, Danbury, CT). The suspension was then filtered with 100 μm, 70 μm and 40 μm sterile cell strainers (BD Biosciences, Bedford, MA). Following centrifugation (2800 × g, 10 min), chitin pellets of different sizes (big chitin 70-100 μm, or chitin fragments: 40-70 μm) were suspended in the desired volume of sterile PBS and autoclaved. Particle sizes and size distribution were evaluated by flow cytometry by comparing the chitin to different sized latex bead controls (0.085, 11.156 and 42.0 μm in diameter; Polysciences, Warrington, PA). Endotoxin levels were below the limits of detection in a Limulus amebocyte lysate assay (Sigma). Prior to utilization, the chitin particles were concentrated by speed vacuuming.
Preparation and In Vitro Culture of Peritoneal Macrophages
To obtain peritoneal macrophages, 10-15 week old wild type and genetically manipulated mice received intraperitoneal (IP) 3%-thioglycollate medium (2 ml; Sigma Chemical). Five days later, peritoneal washings were undertaken with sterile PBS and the recovered cells were washed, counted, and plated in 6-well plates at 1.5 million cells per well. After an overnight incubation in complete medium [RPMI 1640 with L-glutamine, penicillin (50 U/ml) and streptomycin (50 mg/ml), supplemented with 10% heat-inactivated fetal calf serum (FCS) (all reagents from Gibco)] the cells were washed in complete medium with 0.3 % FCS. Fluorescent activated cell sorting (FACS) with antibodies against CD11b, F4/80, Gr1, CD3, and CD4 (BD Biosciences) were used to characterize the adherent cells. After washing, these cells were incubated with chitin (100 μg/ml unless otherwise noted) or its vehicle control. At the desired point in time, supernatants and cellular RNA were harvested. Experiments were realized in triplicate.
Preparation and In vitro Culture of Bone Marrow-Derived Macrophages
Femurs from 6 week-old wild-type mice were flushed with sterile PBS, using a 23-gauge needle. Bone-marrow cells were grown in complete medium [DMEM with 50 ng/ml M-CSF (Stemcell Technologies Inc., Vancouver, Canada), 10% heat-inactivated FCS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin] for 5-7 days until uniform monolayers of macrophages were established. Twelve hours prior to stimulation, cells were removed from the original plastic dishes using Versene (Invitrogen, Carlsbad, CA) and a total of 1 million cells per well were plated in 6-well plates in complete medium with 0.3% FCS. FACS with antibodies against CD11b, F4/80, Gr1, CD3, and CD4 was used to characterize these cells. Greater than 90-95% of these cells stained positively with F4/80 and CD11b and did not express Gr1, CD4 or CD3. Cell viability was assessed using trypan blue dye exclusion and by staining with annexin V and propidium iodide. After washing, these cells were incubated with chitin (100 μg/ml unless otherwise noted) or its vehicle control. At the desired point in time, supernatants and cellular RNA were harvested. Experiments were realized in triplicate.
Cytokine Quantification
The levels of supernatant or bronchoalveolar lavage (BAL) IL-17, IL-23 (p19/p40), TNF, RANTES/CCL5, MIP-2/CXCL2, IL-12/23p40 and IL-12p70 were measured using commercial ELISAs (R&D Systems, Minneapolis, MN for IL-17, TNF, RANTES/CCL5, IL-12/23p40, IL-12p70 and MIP-2/CXCL2 and eBioscience, San Diego, CA for IL-23) according to the instructions provided by the manufacturer. These assays detected cytokine quantities as low as 10.9, 11.7, 7.8, 15.6, 23.4, 7.8 and 7.8 pg/ml for IL-17, IL-12/23p40, IL-12p70, IL-23, TNF, RANTES/CCL5 and MIP-2/CXCL2 respectively.
IL-17 Replacement Experiments
In selected experiments we determined if IL-17 replacement therapy could restore the ability of chitin to stimulate IL-23 production by macrophages from IL-17 null mice. In these experiments macrophages were obtained from wild type and IL-17 null mice, incubated with recombinant IL-17 (100 ng/ml, R&D Systems) or vehicle for 15 min. They were then treated with chitin or vehicle for 8 hours and supernatant IL-23 was evaluated by ELISA as described above.
Assessment of mRNA
Total RNA was extracted from the control and test macrophages with Trizol® (Invitrogen) and real time RT-PCR was undertaken as described previously by our laboratory (18). These experiments were realized in triplicate. The primers that were employed are described below:
| IL-17AR–isoform A sense primer: | 5′-GGGCAACCTTACATCAAGGA-3′ |
| IL-17AR-isoform A anti-sense primer: | 5′-ACTAGAAGGCCCAGCTCCTC-3′, |
| β actin sense primer: | 5′-TGAGAGGGAAATCGTGCGTGAC-3′ |
| β actin anti-sense primer: | 5′-AAGAAGGAAGGCTGGAAAAGAG-3′, |
The ratios of the levels of mRNA encoding IL-17AR versus β-actin were calculated for each sample and expressed as relative copy number fold increase.
Flow Cytometry
Thioglycollate elicited peritoneal macrophages, bone marrow-derived macrophages or BAL fluid macrophages were suspended in FACS buffer (PBS, 2% BSA, 2% FCS) and incubated for 30 minutes at room temperature with purified rat anti-mouse CD16/CD32 mAb (1 μg/105 cells; mouse Fc-Block; BD Biosciences) to prevent non-specific Fc receptor binding. Macrophages were identified by characteristic surface marker expression (F4/80+, CD11b+, CD3∊−, CD3−, CD4−, Gr1−)(BD Biosciences). Macrophage IL-17AR surface receptor expression was measured using rabbit anti-mouse IL-17AR (40 minutes, 4°C, Santa Cruz Biotechnology, Santa Cruz, CA) followed by staining with a goat anti-rabbit-FITC secondary antibody (30 minutes, 4°C, Santa Cruz). In selected experiments, the specificity of this antibody was evaluated. This was done by comparing the staining that was seen in parallel experiments, that contained and did not contain the primary antibody. In others the primary antibody was incubated with a 5 fold excess of the peptide that the antibody was raised against (kindly supplied by the Customer Service Department of Santa Cruz Inc.).
For intracellular cytokine staining, harvested macrophages were treated with Brefeldin A (2 μM, Sigma-Aldrich), fixed with 0.5 ml of ice-cold 2% paraformaldehyde, permeabilized using 0.5% saponin (Sigma) and stained with anti–IL-17A FITC (eBioscience) or the appropriate FITC isotype control (eBioscience) to assess non-specific staining. Saturating concentrations of the respective antibodies were used as determined by titration experiments prior to the study. Non-specific antibody binding was assessed by rabbit anti-mouse isotype staining utilizing the secondary antibody only. After staining, macrophages were washed, suspended in 0.5 ml of iced 2% paraformaldehyde in PBS and analyzed by flow cytometry (FACSCalibur, Becton-Dickinson, Heidelberg, Germany). Ten thousand macrophages / sample were analyzed. Isotype controls were subtracted from the respective specific antibody expression and the results are reported as mean fluorescence intensity (MFI). Calculations were performed with Cell Quest analysis software (Becton-Dickinson). Experiments were performed in triplicate.
Mice
TLR-2, TLR-4, and MyD88 null mice on a C57BL/6 background were generated and characterized as described (19). The IL-17 null mice were generated, characterized by and obtained from Dr. Yoichiro Iwakura (University of Tokyo) (20). Rag2/γC double null mice on a C57BL/6 background were purchased from Taconic Inc. (Hudson, NY). Control C57BL/6 mice were from Jackson Laboratories (Bar Harbor, Maine). All mice were housed and cared for in the animal facilities at Yale University and all experiments were approved by the Institutional Animal Care and Use Committee at Yale University School of Medicine.
Chitin Induction of Acute Inflammation
WT and genetically manipulated mice were anesthetized with intra peritoneal (IP) ketamine hydrochloride (Hospira Inc., Lake Forest, IL). A single dose of varying concentrations of chitin fragments (25 μg unless otherwise indicated) or vehicle control were then applied to the nares (30 μl) and aspirated into their lungs. The mice were sacrificed at intervals after the intranasal challenge. Experiments were realized with 10 mice per group.
Evaluation of Lung Inflammation
Mice were sacrificed and BAL and tissue samples were obtained as described previously (21). Each BAL sample was centrifuged, and the supernatants were stored at −70°C until used. The total number and differential of the recovered inflammatory cells were evaluated after Diff-quick staining (Dade Behring, Dudingen, Switzerland) of BAL pellets in a cytospin preparation. For histologic evaluations, the entire lung was inflated to 25 cm with Streck solution (Streck Laboratories, La Vista, Nebraska, USA).
Histologic Evaluations
Hematoxylin & Eosin (H&E), Myeloperoxidase (Rabbit polyclonal anti-mouse MPO; ab15484, Abcam Inc., Cambridge, MA) and Major Basic Protein (Rabbit polyclonal anti-mouse MBP1, kindly provided by Dr. J. Lee, Mayo Clinic- Scottsdale, AZ) stains were performed in the Research Histology Laboratory of the Department of Pathology at the Yale University School of Medicine. Images of lung section were captured at ×40 final magnification on an Olympus (Tokyo, Japan) BH-2 microscope using a Sony DXC-760 MD camera attached to a computer.
Expression of Results and Statistical Analysis
Data are expressed as means ± SEM unless otherwise indicated. Data were assessed for significance using the Student's t test or ANOVA as appropriate.
RESULTS
Chitin Regulation of Macrophage IL-17A and IL-17A Receptor
To begin to determine if chitin regulated macrophage function, macrophages were incubated with big chitin (BC) (70-100 μm) and chitin fragments (40-70 μm) or their vehicle controls. The production of IL-17 and the expression of the IL-17A receptor (R) were then compared. These studies demonstrated an important size-dependent effect of chitin. BC had no effect on macrophage IL-17 production and did not alter the expression of the IL-17AR (data not shown). In contrast, chitin fragments were potent stimulators of macrophage IL-17 production (Figure 1). The stimulation of soluble IL-17 was seen after as little as 2 hours and peaked after 8 hours of macrophage-chitin incubation (Figure 1A). It was dose-dependent, being seen with doses of chitin as low as 20 μg/ml (Figure 1B). Chitin-induced increases in intracellular IL-17 protein levels were also readily apparent (Figure 1C). This effect was not specific for peritoneal cells because chitin also stimulated bone-marrow derived macrophage IL-17 production (Figure 1D). Macrophages from Rag2/γC double null also contained enhanced quantities of intracellular IL-17 after chitin stimulation further reinforcing the macrophage as the source of this cytokine (Figure 1E). Interestingly, the induction of IL-17 was also associated with increased levels of mRNA encoding the IL-17AR (Figure 2A) and enhanced IL-17AR expression (Figure 2B). These studies demonstrate that chitin fragments are potent stimulators of macrophage IL-17 production that simultaneously enhance the expression of the IL-17AR.
Figure 1. Chitin regulation of macrophage IL-17.

3% thioglycollate-elicited peritoneal macrophages (A-C, E) and bone-marrow derived macrophages (D) from wild-type (A-D) or Rag2/γC null mice (E) were incubated with chitin (40-70μm; 100μg/ml unless otherwise indicated) or vehicle controls for 8h unless otherwise noted. The effects of these treatments on the levels of supernatant IL-17 protein (A, B, Chitin: solid squares, controls: solid circles) were assessed by ELISA. In panel C macrophage intracellular IL-17 protein was assessed using FACS analysis. In this evaluation, the upper panel compares the expression of the intracellular IL-17 in cells incubated in medium alone (dark) or medium plus chitin (line) (mean fluorescent intensity; MFI). The staining with the isotype control is shown in light gray. The lower panel illustrates the MFI values (mean ± SEM) quantitating the intracellular IL-17 protein expression of cells incubated in medium alone (hatched bars) and medium plus chitin (solid bars). In panels D and E, cell debris was excluded by light-scatter cell gating (D, E; first column) and this gate was used for all other analysis. Macrophages were identified by surface marker staining using F4/80 and CD11b co-staining which revealed a macrophage purity greater than 90% for all tested conditions (D, E; second column). To specifically assess intracellular IL-17A, macrophages were fixed and permeabilized, Fc blocking and isotype controls were used to exclude unspecific staining (D, E; third column). In control (D, E; fourth column) and chitin (D, E; fifth column)-treated macrophages intracellular IL-17A staining is depicted with F4/80 co-staining. * p < 0.05, ** p < 0.01, *** p < 0.001 for chitin-treated versus control-treated cells.
Figure 2. Chitin regulation of macrophage IL-17AR.

Peritoneal macrophages were incubated with chitin (40-70μm; 100μg/ml) or vehicle control for 8h or other times when indicated. The effects of these treatments on IL-17R mRNA (A) and IL-17R protein expression (B) were evaluated. IL-17R mRNA was quantified using real-time RT-PCR and is expressed as a % of the control (Ct) values. Macrophage intracellular IL-17R protein was assessed using FACS analysis. The upper panel compares the expression of the IL-17R in cells incubated in medium alone (dark) or medium plus chitin (line) (mean fluorescent intensity; MFI). The staining with the isotype control is shown in light gray. The lower panel illustrates the MFI values (mean ± SEM) quantitating the IL-17R protein expression on cells incubated in medium alone (hatched bars) and medium plus chitin (solid bars). * p < 0.05, ** p < 0.01, *** p < 0.001.
Mechanism of Chitin Stimulation of Macrophage IL-17A
Studies were next undertaken to determine if chitin stimulated macrophage IL-17 production via a pathway that involves TLR. This was done by comparing the levels of intracellular IL-17 and IL-17 production by chitin-stimulated macrophages from WT mice and mice with null mutations of MyD88, TLR-2 or TLR-4. These experiments demonstrated that null mutations of MyD88 completely abrogated the ability of chitin fragments to stimulate macrophage IL-17 accumulation and production (Figure 3A and B). This effect appeared to be at least partially TLR-2-dependent because the ability of chitin fragments to stimulate IL-17 was significantly diminished in cells from TLR-2 null animals (Figure 3A and B). Similar alterations were not seen with cells from TLR-4 null animals (Figure 2A and B) highlighting the TLR specificity of this finding. MyD88 and TLR-2 played similarly important roles and TLR-4 did not play a significant role in the ability of chitin to stimulate IL-17AR (Figure 3C). When viewed in combination these studies demonstrate that chitin fragments stimulate macrophage IL-17 production and enhance the expression of the IL-17AR via a MyD88-dependent pathway that involves TLR-2, but does not involve TLR-4.
Figure 3. Mechanisms of chitin regulation of macrophage IL-17 and IL-17R.

Peritoneal macrophages were obtained from wild-type (WT), MyD88, or TLR sufficient (+/+; +) or deficient (−/−; −) mice and were incubated with chitin (40-70 μm; 100 μg/ml) or vehicle control for 8h. The effects of these interventions on intracellular IL-17 protein (A), soluble IL-17 protein (B) and IL-17R protein production (C) were evaluated. Macrophage intracellular IL-17 and IL-17R protein were assessed using FACS analysis. For intracellular IL-17 staining, cell debris was excluded by light-scatter cell gating (A; first column) and this gate was used for all subsequent evaluations. Macrophages were identified by surface marker staining using F4/80 and CD11b co-staining which revealed a macrophage purity higher than 90% for all tested conditions (A; second column). To specifically stain intracellular IL-17A, macrophages were fixed and permeabilized. Fc blocking and isotype controls were used to exclude unspecific staining (A; third column). In control (A; fourth column) and chitin (A; fifth column)-treated macrophages intracellular IL-17A staining is depicted with F4/80 co-staining. In (B), supernatant IL-17 was assessed by ELISA. In C, macrophage IL-17AR expression was assessed using FACS analysis and is expressed as the MFI of the evaluation. *** p < 0.001.
Role of IL-17A in Chitin-Induced Cytokine Elaboration
To further define the spectrum of and the pathways involved in chitin fragment stimulation of macrophage cytokine production, studies were undertaken to determine if chitin-stimulated macrophage produce other inflammatory cytokines and the roles of IL-17 in these responses were evaluated. These studies demonstrate that chitin fragments are potent stimulators of macrophage IL-12/23p40 and IL-23 (p19/p40) but did not stimulate IL-12p70 (Figure 4, A and B and data not shown). Chitin also stimulated macrophage TNF, RANTES/CCL5 and MIP-2/CXCL2 production (Figure 4, C-E). Chitin fragment stimulation of macrophage IL-12/23p40 and IL-23 was mediated by a MyD88-dependent, TLR-2-dependent mechanism(s) (Figure 4, A and B). Interestingly, this induction was IL-17-dependent because the ability of chitin fragments to stimulate IL-23 was also totally abolished in experiments with cells from IL-17 null mice and could be restored when rIL-17 was added to cultures containing chitin and macrophages from IL-17 null mice (Figure 4, A, B and F). Chitin fragments also stimulated TNF production via a MyD88-dependent, TLR-2-dependent, IL-17-dependent and TLR-4-independent pathway(s) (Figure 4C). In contrast, the ability of chitin fragments to stimulate macrophage RANTES/CCL5 and MIP-2/CXCL2 was not decreased in experiments using cells from IL-17 null animals (Figure 4, D and E). When viewed in combination, these studies demonstrate that chitin fragments stimulate macrophage cytokine elaboration via IL-17-dependent and -independent mechanisms and highlight the importance of IL-17 in the production of IL-23 and TNF by chitin-stimulated cells.
Figure 4. Roles of TLR(s), MyD88 and IL-17 in chitin-induced cytokine and chemokine production.

Peritoneal macrophages were obtained from TLR, MyD88 and IL-17A sufficient (+/+; +) and deficient (−/−; −) mice. These cells were incubated with chitin (solid bars, 100 μg/ml) or vehicle control (hatched bars) for 8h. The effects of these treatments on IL-12/IL-23p40 (A), IL-23 (B), TNF (C), RANTES/CCL5 (D) and MIP-2/CXCL2 (E) protein production were assessed by ELISA. In panel F, cells were obtained from wild type and IL-17 null mice and incubated with rmIL-17 (100 ng/ml) or vehicle. They were then treated with chitin or vehicle for 8 hours and the levels of supernatant IL-23 were assessed by ELISA. ** p < 0.01, *** p < 0.001.
Role of TLR-2 and IL-17A in Chitin-Induced Inflammation
To determine if the TLR-2-IL-17 pathway described above is operative in vivo, we characterized the effects of acutely administered chitin in WT mice and mice with null mutations of MyD88, TLRs and IL-17. In these experiments a single dose of chitin (based on dose-response and kinetic studies, data not shown) in WT mice caused an acute inflammatory response characterized by an increase in BAL and tissue total cell and macrophage recovery (Figure 5A-C). An increase in neutrophil accumulation was also seen in tissue and BAL evaluations and MPO stains (Figure 5 A-C and data not shown). These BAL and tissue inflammatory responses were significantly decreased in mice with null mutations of MyD88 and TLR-2, but were not altered by null mutations of TLR-4 (Figure 5A and B). They were also significantly decreased in mice with null mutations of IL-17 (Figure 5B and C). In keeping with these findings, chitin fragments stimulated IL-17 in macrophages from WT mice and Rag2/γC null mice (Figure 6 A and B and data not shown) and induced IL-17AR accumulation (Figure 6C). They also stimulated the accumulation of IL-12/23p40 and IL-23, but not IL-12p70, via a MyD88 and TLR-2-dependent mechanism(s) (Figure 6D and E). Thus, in accord with our in vitro studies, chitin utilizes a MyD88-dependent, TLR-2-dependent, IL-17-dependent and TLR-4-independent pathway(s) to induce acute inflammation and stimulate the accumulation of IL-12/23p40 and IL-23 in the lung.
Figure 5. Roles of TLR(s), MyD88 and IL-17 in chitin-induced acute inflammation.
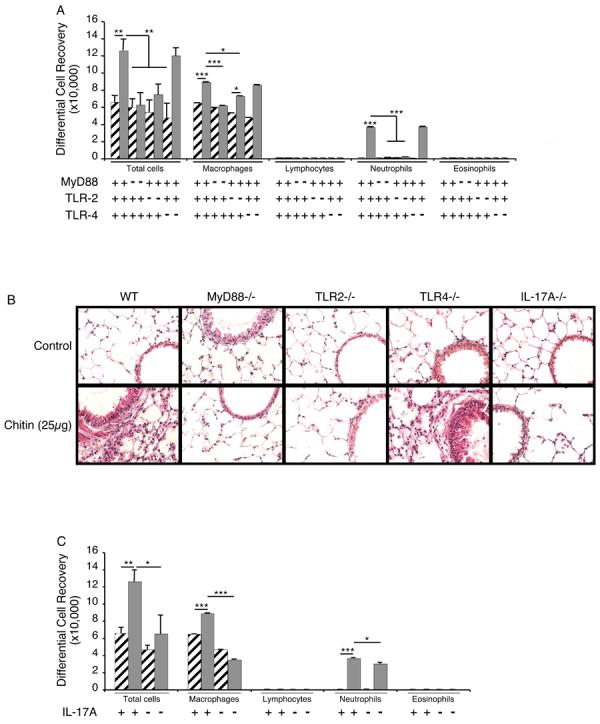
TLR, MyD88 and IL-17 sufficient (+/+; +) and deficient (−/−; −) mice were treated with intranasal chitin (solid bars, 25 μg) or vehicle controls (hatched bars). Six hours later the animals were sacrificed, BAL was undertaken and lung sections were obtained. The effects of these treatments on differential BAL cell recovery (A, C) and lung histology (H & E staining) (B) were evaluated. * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 6. Roles of TLR(s), MyD88 and IL-17 in chitin-induced cytokine production in vivo.

TLR, MyD88 and IL-17 sufficient (+/+; +) and deficient (−/−; −) as well as Rag2/γC null mice (B) mice were treated with intranasal chitin (solid bars or dot plots) or vehicle controls (hatched bars or dot plots). Six hours later the animals were sacrificed and BAL was undertaken. The effects of these treatments on intracellular IL-17 protein (A and B), IL-17AR protein (C), BAL IL-12/IL-23p40 protein accumulation (D) and BAL IL-23 protein accumulation (E) were evaluated. IL-12/IL-23p40 and IL-23 were assessed by ELISA. IL-17A and IL-17AR expression were assessed using FACS analysis. These results are shown as dot plots or expressed in MFI values (mean ± SEM) comparing the IL-17A or IL-17AR protein on cells incubated with medium alone (hatched bars) and medium plus chitin (solid bars). * p < 0.05, ** p < 0.01, *** p < 0.001.
DISCUSSION
To determine if chitin regulates innate immune and inflammatory responses in the lung, we evaluated the effects of chitin fragments on macrophage function in vitro and acute tissue inflammatory responses in vivo. These studies demonstrate that appropriately sized chitin fragments stimulate macrophage production of IL-17, macrophage expression of the IL-17AR, macrophage production of IL-12/23p40, IL-23 and TNF and acute inflammation. They also demonstrate that these responses are mediated via MyD88- and TLR2-dependent mechanisms and highlight the TLR-2 specificity of these responses by demonstrating that chitin fragments do not interact in a similar fashion with TLR-4. When viewed in combination these studies demonstrate that chitin is a size-dependent PAMP that activates TLR-2 and MyD88. They also highlight a novel IL-17 / IL-17AR-based innate immunity pathway that regulates macrophage activation and induces acute inflammation.
CD4+ T cells can be divided into different subsets with distinct cytokine profiles and functional characteristics (22, 23). Th1 cells, which are characterized by their ability to produce gamma-interferon (IFN-γ) and Th2 cells, which produce IL-4, IL-5, IL-9 and IL-13, have been implicated in the pathogenesis of responses against intracellular pathogens and parasites, respectively (22). Recent studies have defined an IL-23-dependent T cell population that produces IL-17 but not IFN-γ or IL-4. These studies have also provided mounting evidence that the IL-17 that is produced by these cells plays a key role in inflammation, autoimmunity and tissue injury (22, 24). Interestingly, although IL-17 is produced predominantly by this adaptive arm of the immune system, it is now known to be an activator of innate immunity (22, 25, 26). This has been interpreted to be a mechanism whereby the adaptive immune response communicates with the innate immune system to promote inflammation (22). Our laboratory and others have observed that chitin treatment in vivo induces a neutrophil-rich inflammatory response (27). Given the crucial roles of both IL-17 and its receptor in pulmonary neutrophilic inflammation (28, 29), we studied the roles of IL-17 and IL-17R in chitin-induced acute airway inflammation. Our studies add to our knowledge of IL-17 by demonstrating, for the first time, that it is produced by TLR-2-activated macrophages. In so doing, they define a novel TLR-2- and IL-17-based innate immune pathway that contributes to a variety of in vitro and in vivo immune and inflammatory responses.
The cytokine IL-23 is a heterodimeric molecule that shares the p40 subunit of IL-12 (30). As noted above, IL-23 is known to be intimately associated with IL-17. The majority of studies have focused on the role of IL-23 in the production of IL-17. These studies have demonstrated that IL-23 is an essential survival factor for Th17 cells (31, 32) and that mice lacking IL-23 have significantly decreased numbers of Th17 cells (33). Our studies demonstrate that chitin-stimulated macrophages produce IL-23 and that this inductive event is markedly diminished in cells with null mutations of IL-17. Thus, in addition to the ability of IL-23 to contribute to the production of IL-17, IL-17 can contribute to macrophage IL-23 elaboration. Interestingly, this effect was at least partially IL-23-specific because IL-12p70 was not regulated in a similar manner. When viewed in combination, one can see how these interactions can lead to an amplification loop that can contribute to the magnitude, severity and / or duration of the inflammation and injury that are induced by these cytokines.
At sites of infection with chitin-containing agents anti-infectious immune responses and local chitinases are believed to induce chitin fragmentation. To determine if these chitin fragments have immune-regulatory effects, a number of investigators have characterized the effects of chitin in the lung and other tissues. These studies have, however, not provided uniform results with chitin being reported to induce (27) and inhibit (9) tissue inflammatory responses. To address this issue we elected to evaluate the effects of size-selected chitin fragments. These investigations demonstrate that appropriately sized chitin (40-70 μm) induces acute pulmonary inflammation. The ability of chitin to induce an acute inflammatory response is in accord with recent studies by Reese et al. that utilized acetylated chitosan-coated beads (27). However, unlike Reese et al., our studies demonstrate that chitin fragments induce a macrophage- and neutrophil-rich inflammatory response with only a modest degree of eosinophil infiltration. We believe these differences are the result of technical differences because we used a single dose of unbound, size-selected chitin and an early time point while Reese et al. used 2 doses of chitin beads and a later time point of assessment. In accord with this contention we have also noted a transition to an eosinophil-rich tissue response when chitin was administered repeatedly over a 2-week interval (Lee and Elias, unpublished observation). When viewed in combination these studies suggest that chitin induces an inflammatory response that is initially neutrophilic and becomes eosinophilic over time. Our studies also add to our understanding of the mechanisms of these alterations by demonstrating that the early response is mediated, at least in part, by an innate immune response pathway that involves TLR-2 and MyD88. They also demonstrate that this tissue response is IL-17-dependent. This is in keeping with the well-established ability of IL-17 to induce neutrophilic tissue responses (28). Interestingly, macrophage infiltration was also significantly decreased in the absence of IL-17, suggesting that IL-17 plays a role in macrophage tissue homeostasis as well.
In summary, our studies demonstrate that appropriately sized chitin fragments are TLR-2 ligands that utilize a MyD88-dependent pathway to induce IL-17 elaboration and enhance the expression of the IL-17AR. These studies also demonstrate that this novel innate immune pathway plays an essential role in the regulation of macrophage cytokine production and the induction of acute inflammation. Additional investigations of the mechanisms that chitin uses to regulate IL-17, IL-17AR and innate immunity, and the utility of therapies that alter these pathways in the treatment of pathogen / parasite and other inflammatory diseases, is warranted.
ACKNOWLEDGEMENTS
The authors thank Ruslan Medzhitov, Ph.D., for the TLR-2, TLR-4 and MyD88 null mice as well as Yoichiro Iwakura, D. Sc., for the IL-17 null mice that were used and Ms. Kathleen Bertier for her excellent secretarial assistance.
Work supported by NIH grant # HL081639
REFERENCES
- 1.Araujo AC, Souto-Padron T, de Souza W. Cytochemical localization of carbohydrate residues in microfilariae of Wuchereria bancrofti and Brugia malayi. J Histochem Cytochem. 1993;41:571–578. doi: 10.1177/41.4.8450196. [DOI] [PubMed] [Google Scholar]
- 2.Boot RG, Blommaart EF, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, Place A, Aerts JM. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;276:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 3.Boot RG, Renkema GH, Verhoek M, Strijland A, Bliek J, de Meulemeester TM, Mannens MM, Aerts JM. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273:25680–25685. doi: 10.1074/jbc.273.40.25680. [DOI] [PubMed] [Google Scholar]
- 4.Debono M, Gordee RS. Antibiotics that inhibit fungal cell wall development. Annu Rev Microbiol. 1994;48:471–497. doi: 10.1146/annurev.mi.48.100194.002351. [DOI] [PubMed] [Google Scholar]
- 5.Elias JA, Homer RJ, Hamid Q, Lee CG. Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol. 2005;116:497–500. doi: 10.1016/j.jaci.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 6.Fuhrman JA, Piessens WF. Chitin synthesis and sheath morphogenesis in Brugia malayi microfilariae. Mol Biochem Parasitol. 1985;17:93–104. doi: 10.1016/0166-6851(85)90130-6. [DOI] [PubMed] [Google Scholar]
- 7.Neville AC, Parry DA, Woodhead-Galloway J. The chitin crystallite in arthropod cuticle. J Cell Sci. 1976;21:73–82. doi: 10.1242/jcs.21.1.73. [DOI] [PubMed] [Google Scholar]
- 8.Shahabuddin M, Kaslow DC. Plasmodium: parasite chitinase and its role in malaria transmission. Exp Parasitol. 1994;79:85–88. doi: 10.1006/expr.1994.1066. [DOI] [PubMed] [Google Scholar]
- 9.Shibata Y, Foster LA, Bradfield JF, Myrvik QN. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol. 2000;164:1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 11.Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112:521–530. doi: 10.1111/j.1365-2567.2004.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 13.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 14.Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol. 2001;54:577–589. doi: 10.1136/jcp.54.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–356. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Wynn TA. T(H)-17: a giant step from T(H)1 and T(H)2. Nat Immunol. 2005;6:1069–1070. doi: 10.1038/ni1105-1069. [DOI] [PubMed] [Google Scholar]
- 17.Shibata Y, Foster LA, Metzger WJ, Myrvik QN. Alveolar macrophage priming by intravenous administration of chitin particles, polymers of N-acetyl-D-glucosamine, in mice. Infect Immun. 1997;65:1734–1741. doi: 10.1128/iai.65.5.1734-1741.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000;10:1139–1142. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- 20.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 21.Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, Waxman AB, Elias JA. IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest. 2000;106:783–791. doi: 10.1172/JCI9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi Y, Liu G, Yang R. Th17 cell induction and immune regulatory effects. J Cell Physiol. 2007;211:273–278. doi: 10.1002/jcp.20973. [DOI] [PubMed] [Google Scholar]
- 23.Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- 24.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 25.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 26.Schnyder B, Schnyder-Candrian S, Pansky A, Schmitz ML, Heim M, Ryffel B, Moser R. IL-17 reduces TNF-induced Rantes and VCAM-1 expression. Cytokine. 2005;31:191–202. doi: 10.1016/j.cyto.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 29.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 31.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 32.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]