

Abstract
FKBP52 and FKBP51 are tetratricopeptide repeat (TPR) proteins found in steroid receptor complexes and FKBP51 is an androgen receptor (AR) target gene. Although in vitro studies suggest that FKBP52 and FKBP51 regulate hormone-binding and/or subcellular trafficking of receptors, the roles of FKBP52 and FKBP51 in vivo have not been extensively investigated. Here, we evaluate their physiological roles in FKBP52-deficient and FKBP51-deficient mice. FKBP52-deficient males developed defects in select reproductive organs (e.g., penile hypospadias, prostate dysgenesis, but normal testis), pointing to a role for FKBP52 in AR-mediated signaling and function. Surprisingly, ablation of FKBP52 did not affect AR hormone-binding or nuclear translocation in vivo and in vitro. Molecular studies in MEF cells uncovered that FKBP52 is critical to AR transcriptional activity. Interestingly, FKBP51 expression was down-regulated in FKBP52-deficient males but only in affected tissues, providing further evidence of tissue-specific loss of AR activity and suggesting that FKBP51 is an AR target gene essential to penile and prostate development. However, FKBP51-deficient mice were normal, showing no defects in AR-mediated reproductive function. Our work demonstrates that FKBP52 but not FKBP51 is essential to AR-mediated signaling, and provides evidence for an unprecedented FKBP52 function – direct control of steroid receptor transcriptional activity.
Androgen receptor (AR) is a hormone-induced transcription factor that controls male sexual development and other important physiologies. Similar to other members of the nuclear receptor family (1,2), AR has three major functional domains: an N-terminal transactivation domain, a DNA-binding domain, and a C-terminal ligand-binding domain (3), (4), (5). Mutations found in each of these domains lead to a series of AR functional defects associated with androgen insensitivity syndrome (AIS) or partial androgen insensitivity syndrome (PAIS) in humans (6,7). The majority of AIS and PAIS patients have developmental defects in the male reproductive system. Loss-of-function AR mutations in mice recapitulate many of the reproductive defects found in AIS patients. For example, the AR-deficient (ARKO) mouse (8) and the tfm (testicular feminization mutant) mouse (9) both develop severe defects of testicular development and an overall lack of male sexual differentiation, including hypospadias and penile agenesis. The tfm male mouse demonstrates many female secondary structures, including vagina and teats (10).
Molecular regulation of AR function can be achieved at several levels, such as spatial-temporal expression of the receptor, modulation of ligand binding, cytoplasm to nucleus translocation, and DNA binding and transcriptional activities (11,12). Prior to hormone binding, steroid receptors form large protein complexes containing the molecular chaperone heat shock protein 90 (Hsp90), as well as various co-chaperone tetratricopeptide repeat (TPR) proteins (13-15). These co-chaperones include FK506-binding proteins 52 (FKBP52) and 51 (FKBP51), cyclophilin 40 (Cyp40) and protein phosphatase 5 (PP5). FKBP52 and FKBP51 are ubiquitously expressed proteins with peptidyl prolyl cis/trans isomerase (PPIase) activity that is inhibited by the binding of FK506 immunosuppressant ligand (16-18). Each protein enters into steroid receptor complexes through a direct and competitive binding at the C-terminus of Hsp90 via their essential TPR domains (19-21). Although FKBP52 and FKBP51 share a similar domain structure, as well as 60% sequence identity and 75% similarity, they do differ in that FKBP51 is missing a C-terminal calmodulin-binding domain.
To date, most studies on TPR control of SR action have been done using conventional molecular and cellular approaches, and using the glucocorticoid (GR) and progesterone (PR) receptors as models. It has been shown that FKBP52 is localized to both cytoplasm and nucleus, but that the cyptoplasmic fraction co-localizes with microtubules in a complex containing dynein (22,23). For these reasons, it was proposed that FKBP52 serves as an adaptor between the GR/Hsp90 complex and the microtubule transport machinery (24). Indeed, various groups have shown that FKBP52 contributes to both the dynein interaction and hormone-induced translocation of GR (25-27). Meanwhile, a second line of inquiry has provided evidence for reciprocal control of GR and PR hormone-binding function by FKBP52 and FKBP51, with FKBP52 causing potentiation and FKBP51 attenuation of this activity (15,28,29). These observations suggest a model in which differential incorporation of TPR proteins into SR complexes forms the basis for selective control of hormone binding and subcellular trafficking.
Although evidence for interaction of FKBP52 with AR is fairly common (21,30,31), reports of FKBP51 in AR complexes are limited (32). However, it has recently become clear that FKBP51 is a highly-sensitive AR-regulated gene, at least in the prostate cancer cell line LnCAP, where androgen-induced expression of FKBP51 can be blocked by the Hsp90 inhibitor galdanamycin (32-34). These observations would suggest that FKBP52 and FKBP51 play essential and perhaps distinct roles in AR function, yet little is known on this matter. Interestingly, a recent report by Febbo et al (32) suggests that over-expression of FKBP51 has a stimulatory effect on AR-mediated transcriptional activity, rather than inhibition as known for GR and PR. Thus, it is possible that FKBP52 and FKBP51 exert distinct and diverse effects on various members of the steroid receptor family.
With the above considerations in mind, we set out to generate mice genetically deficient in FKBP52 and FKBP51. Although it was fully expected that alterations to GR-regulated physiology would be a prominent feature of FKBP52 loss, we were surprised to find no such effect. Instead, the primary phenotype of FKBP52 ablation was infertility in both male and female mice. Female FKBP52-deficient mice were sterile due to a selective loss of activity by the PR-A isoform in the uterus, leading to a complete failure of implantation (35). Here, we report that FKBP52 is critical to male fertility by controlling AR-mediated signaling and physiology. Loss of FKBP52 resulted in aberrant penile development causing hypospadias, as well as prostate dysgenesis, while leaving other AR-regulated organs unaffected. Molecular studies showed that FKBP52 loss had a dramatic effect on AR-mediated gene expression that, surprisingly, was not due to altered AR hormone-binding and/or nuclear translocation functions. Analysis of FKBP51 as an AR target gene showed it to be down-regulated only in FKBP52-deficient tissues exhibiting altered AR activity. By generating and analyzing FKBP51-deficient mice, we were able to assess its direct contribution to AR activity in vivo and its role as a target gene in AR-mediated development of the penis and prostate. Interestingly, no alterations to male (or female) physiology were noted in the FKBP51-deficient mice, showing that FKBP51 is not essential to AR signaling in vivo.
Materials and Methods
Generation of FKBP52-deficient mice and FKBP51-deficient mice
The generation of FKBP52-deficient mice was described in a previous report (35). To generate FKBP51-deficient mice, a promoter trapped ES cell line RRC236 containing an insertional mutation in the mouse FKBP51 gene was identified and obtained from BayGenomics (http://baygenomics.ucsf.edu) (36). The gene-trap vector (pGT1Lxf) contains a splice-acceptor sequence upstream of the reporter gene βgeo. Using genomic PCR, Southern blot, and sequencing analyses, we confirmed a single genomic insertion at intron 4 of the mouse FKBP51 gene. The chimeric male mice were generated from RRC236 ES cell line and were further bred to C57BL/6J females to generate F1 offspring. Genotypes were determined by Southern blot analysis. Northern blot, qRT-PCR and Western blot analyses confirmed that there was no FKBP51 leaky expression in FKBP51-deficient mice. The SC11518 antibody (Santa Cruz Biotechnology) was used to detect FKBP51 by Western blotting. FKBP52/FKBP51 compound mutants were generated by interbreeding heterozygous of FKBP52 and FKBP51 mutant mice. We also used PCR analysis of the sry gene to determine genders of embryos. All animal experiments were carried out using a protocol approved by the Indiana University School of Medicine Institutional Animal Care and Research Advisory Committee.
Morphology, histology and immunohistochemistry
We examined the morphology and histology of multiple tissues and organs from male mice and age-matched littermate control mice. Embryos were harvested from timed-mating females by cesarean section. Isolated embryos and tissue samples were fixed in 10% neutral-buffered formalin, paraffin embedded, and sectioned (6μm), and stained with haematoxylin and eosin. Three-dimensional reconstruction was carried out as previously described (37). Immunohistological analysis was carried out as previously described (38). Antibody against AR, SC7305 is from Santa Cruz Biotechnology.
Gel Electrophoresis and Western Blotting
Samples were resolved on denaturing SDS gels (39). Transfer of samples to Immobilon-P® membranes and immuno-blotting were performed as previously described (26). The SC7305 (Santa Cruz Biotechnology) antibody against AR was used to probe for receptor, while various antibodies were used to probe for FKBP52 (SC1803; Santa Cruz Biotechnology), FKBP51 (SC11518; Santa Cruz Biotechnology), Cyp40 (PA3-022; Affinity BioReagents), and PP5 [gift from Dr. Michael Chinkers (40). The blots were then incubated with appropriate peroxidase-conjugated counter antibodies, followed by detection of bands by enhanced chemiluminescence.
Serum hormone measurement
Afternoon blood samples were drawn (between 1:00 to 2:00 pm) from the right ventricle of adult mice (2-3 months of age). Serum levels of testosterone and DHT were analyzed by using RIA kits (DSL-4000 for testosterone, DSL-9600 for DHT, Diagnostic Systems Laboratories, INC., Webster, TX). Steroid measurements were performed at the Endocrinology Core Laboratory of Indiana University School of Medicine.
Reporter gene assays in mouse embryonic fibroblast cells
Mouse embryonic fibroblasts (MEF) were isolated from wild-type and FKBP52-deficient E13.5 embryos. MEF cells were cultured in DMEM with 15% FBS until confluence. To generate immortalized MEF cells, primary cells were transfected with SV40. Pooled transformed cells were maintained in DMEM supplemented with 10% FBS. Both wild-type and FKBP52-deficient MEF cells maintain normal fibroblast morphology with high proliferative activity. MEF cells were either transiently or permanently transfected with expression vectors for human AR (kind gift of Lirim Shemshedini). The MMTV-CAT reporter and PSA-luciferase reporter (kind gift of Marianne Sadar) constructs were used to assay for AR activity (41,42). CAT enzyme activity was measured as described by Nordeen et al (43) using [3H]acetyl-CoA as substrate, while luciferase activity was measured using a commercial kit (Promega). All values were normalized for transfection efficiency by co-transfection with a CMV-driven -galactosidase reporter.
AR hormone binding assay
Wild-type and FKBP52-deficient MEF cells were transiently transfected with human AR expression plasmid (pSG5). Forty eight hours following transfection, cell pellets were washed twice with PBS and resuspended in ice cold homogenization buffer (10 mM HEPES, 1.5 mM EDTA and 10 mM sodium molybdate, pH 7.4) with protease inhibitors, followed by Dounce homogenization and centrifugation for 10 min at 16,000 × g. Supernatants (cytosol fraction) were used for binding assay without freezing. In a typical binding assay, 150 μl of cytosol (∼2.0 mg/ml) were incubated with 10.0 nM 3[H]mibolerone, (70.0 Ci/mmol) for 20 h on ice. Mibolerone is a synthetic high-affinity AR ligand. Nonspecific binding was determined in the presence of 1000-fold excess unlabeled mibolerone (10 μM). Protein-bound radioactivity was isolated using 1% dextran-coated charcoal in 10 mM HEPES buffer (pH 7.4). Specific binding was normalized for AR expression as determined by Western blot and expressed as DPM per milligram of cytosol protein.
Quantitative RT-PCR
Total RNAs were isolated from mouse tissues or cells using TRIzol (invitrogen). First strand cDNAs were synthesized by the iScript cDNA synthesis kit (Bio-Rad) using 1 μg RNA as template according to manufacturer's instruction. Real time PCR was performed using iCycler iQ (Bio-Rad) with iQ SYBR Green supermix (Bio-Rad). The relative expression was normalized to GAPDH. The sequences of specific primers are listed as following: human AR: 5′-TGGAAG CCATTGAGCCAGGTGTAG-3′, and 5′-CGTCCACGTGTAAGTTGCGGA AG-3′; FKBP52: 5′-GCCTCTCGAAGGAGTGGACATCAG-3′, and 5′-CGGTCCAGACTGG AGTCAAACTT TG-3′; FKBP51: 5′-TGCAGATCTCCATGTGCCAGAGG-3′ and 5′-GCTCC TTCTACAGCC TT CTTGCTCC-3′; GAPDH 5′-TCCTGGTATGACAATGAATACGGC-3′ and 5′-TCTTGCT CAGTGTCCTTGCTGG-3′.
Results
Reduced fertility in FKBP52-deficient males
FKBP52-deficient mice were generated as previously described (35). FKBP52-deficient mice were viable. However, mutant males showed a rate of growth equivalent to that of wild type females (Fig. 1A). By breeding to wild type female mice, we found that FKBP52-deficient males had greatly reduced fertility. Only 5% of FKBP52-deficient males were able to plug females. Analysis of plugged females showed significantly smaller litters from FKBP52-deficient males (mean = 2.1 ± 0.9; N=7) compared to wild type males (mean = 6.8 ± 1.6, N=16, P < 0.001). FKBP52-deficient females were sterile due to implantation failure. Detailed analysis of FKBP52-deficient females and the role of FKBP52 in regulating progesterone receptor function at the uterus was reported elsewhere (35).
Figure 1.
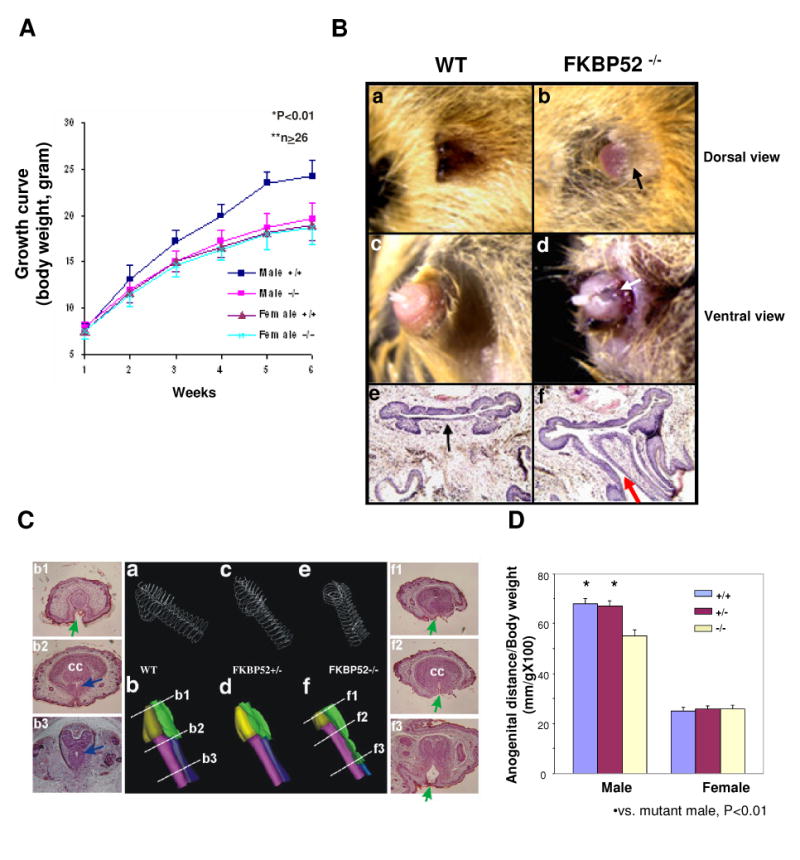
Morphological and histological analysis of FKBP52-deficient males. (A) Growth curves of WT and FKBP52-deficient mice; (B) Comparison of adult wild-type and FKBP52-deficient male external genitalia at dorsal (a and b) and ventral surfaces (c and d). Arrows (in b and d) indicate the under-developed foreskin and ectopic urethral opening at the ventral aspect compared to normal morphology in controls (a and c). Histological sections of wild-type (e) and FKBP52-deficient (f) male genitalia. A red arrow indicates ectopic urethral opening at the ventral aspect compared to normal morphology in controls (black arrow in e). (C) Three-dimensional reconstruction of the mouse E18.5 penises. Wire frame images of outer penile skin and urethra of the three FKBP52 genotypes are shown in a, c and e. In b, d and f, the skins had been artificially removed. The glans penis is colored in gold, while the urethral opening is green, corpus cavernosum (cc) is purple, and the closed urethra is blue. Note that the urethral opening persisted throughout the FKBP52-deficient penile shaft, while normal controls only have a temporary urethral opening at distal end. Side panels show representative sections of wild-type and mutant penises. Green arrows indicate open urethra, blue arrows indicate closed urethra. (D) Comparison of anogenital distances in wild-type, heterozygous and FKBP52-deficient mice. The anogenital distance, normalized by the animal body weight, in FKBP52-deficient males was significantly shorter compared to littermate wild-type and heterozygous males.
Selective reproductive defects in adult FKBP52-deficient males
The most striking morphological defect observed in adult FKBP52-deficient males was the presence of hypospadias with 100% penetrance in the external penile genitalia (Fig. 1B). Similar to hypospadias in humans, the foreskin was under-developed and the penile glans and anterior portion of tubercle were exposed in the majority of FKBP52-deficient mutant males, compared to the normal male genital tubercle that was surrounded and covered by foreskin (compare panels a, b). All FKBP52-deficient males showed an ectopic opening on the ventral side of the penis (panels c, d), which was confirmed by histological staining (panels e, f). Three-dimensional reconstruction of serial histological sections of embryonic genital tubercles confirmed the hypospadias phenotype to be a developmental defect resulting from failure to form urethral seams (Fig. 1C). These defects mimic the typical clinical features of hypospadias patients. In addition, the length and weight of FKBP52-deficient penises were significantly less when compared to littermate controls (Table 1). The anogenital distance was shortened in FKBP52-deficient males (3 to 10 weeks of age) (Fig. 1D), a feminization phenotype similar to that seen in androgen receptor knockout (ARKO) and testicular feminization (tfm) mice, albeit to a lesser degree (8,10).
Table 1. Comparison of reproductive organs and functions in male mice.
| Wild-type | Heterozygous | Homozygous | |
|---|---|---|---|
| Body Weight (g) | 29.1±3.1 (n=32) * | 28.8±2.0 (n=38)* | 25.2±2.4 (n=32) |
| Testis/Body (mg/g) | 7.00±1.19 (n=32) | 7.10±1.35 (n=38) | 7.71±1.72(n=32) |
| Epididymis/Body (mg/g) | 2.52±0.44 (n=32) | 2.48±0.38 (n=33) | 2.47±0.39(n=28) |
| Seminal vesicular/Body (mg/g) | 7.40±1.31 (n=32) * | 7.63±1.28 (n=38)* | 5.28±1.58(n=32) |
| Penile/Body (mg/g) | 0.77±0.13 (n=32) * | 0.83±0.13 (n=36)* | 0.56±0.16(n=31) |
| Penile length (mm) | 6.1±0.16 (n=5) * | 6.1±0.19 (n=8)* | 4.4±0.33(n=8) |
| Sperm count (×106/epididymis) | 12.6±2.9 (n=10) | 12.1±1.3 (n=10) | 9.6±2.9(n=10) |
| Sperm motility | 69.0±6.8 (n=10) * | 66.2±2.5 (n=10)* | 47.7±8.8(n=10) |
| Serum Testosterone (ng/ml) | 1.89±0.85 (n=17)* | ND | 3.62±0.46 (n=19) |
| Serum DHT (pg/ml) | 175.4±56.3 (n=17)* | ND | 264.3±74.8 (n=19) |
vs homozygous (−/−), p<0.01
Interestingly, primary sex organs appeared to be unaffected by FKBP52 loss. FKBP52-deficient males developed normal size of testes and epididymis (Table 1), and had normal scrotal sacs and inguinal canals (data not shown). Histological analysis confirmed seminiferous tubules, spermatogenesis, Sertoli cells and Leydig cells all to be normal in the FKBP52 mutant testis (data not shown). Sperm motility rates were slightly lower in mutant males compared to wild type and heterozygous littermates (Table 1). In contrast, the majority of reproductive tissues affected in FKBP52-deficient males were the secondary sex organs. Seminal vesicles (SV) were present but significantly smaller in FKBP52-deficient mice (Table 1 and Fig. 2A). Prostate glands were initially formed through embryonic developmental process, but lacked further growth at puberty and eventually became dysgenic in FKBP52-deficient adult males (Fig. 2B). This phenomenon is another example of compromised AR-mediated function due to FKBP52-deficiency, as AR activity is required for the formation of mature prostate glands (44).
Figure 2.
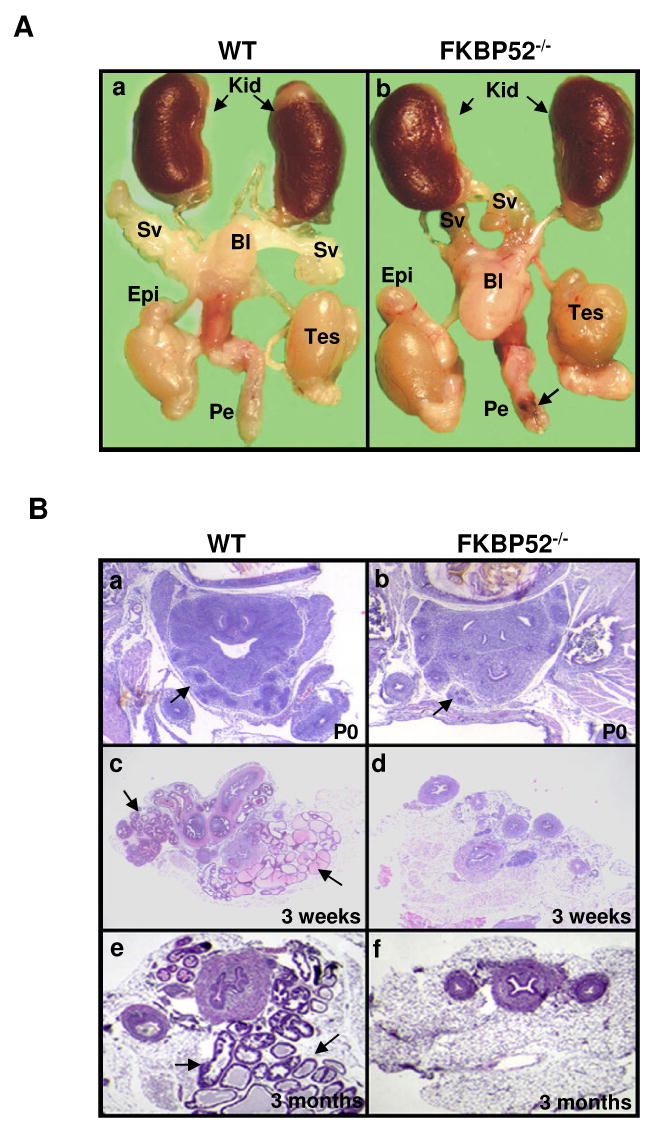
(A) Morphological comparison of male internal reproductive organs in wild-type (a) and FKBP52-deficient adult mice (b). FKBP52-deficient males have overall normal testis (Tes) formation and normal epididymis (Epi), but significantly smaller seminal vesicles (SV). Kid: kidney; Bl: bladder; Pe: penis; Black arrow indicates urethral opening. (B) Histological analysis of prostate gland development in FKBP52-deficient and age-matched littermate control mice at birth (P0) to 3 month old. Prostate glands are initially formed during embryonic developmental in both wild-type (a) FKBP52-deficient mutant (b), but lack further growth after the birth (d) and eventually become dysgenic in FKBP52-deficient adult males (f) compared to littermate wild-type mice (c and e). Prostate glands are indicated by black arrows.
Since secondary sex characteristics are determined by steroidal sex hormones, we measured serum testosterone and DHT levels in FKBP52-deficient males. Levels of each hormone were slightly but significantly elevated in FKBP52-deficient adult males (Table 1), demonstrating that lack of androgen production is not the cause of the abnormal phenotypes seen in FKBP52-deficient males.
Normal androgen receptor expression, hormone binding and nuclear translocation in FKBP52-deficient mice
Because FKBP52-deficient males showed growth curves similar to females and had selective defects in reproductive organs, we reasoned that altered AR signaling might be the underlying cause of the defects. To test whether FKBP52 ablation led to reduced levels of AR, we performed Northern blot and Western blot analyses. In both penis and testis, FKBP52 loss had no effect on AR mRNA and protein levels (data not shown). Thus, FKBP52 is not essential to AR expression or AR stability. To assess possible impairment of AR nuclear translocation, immunohistochemical staining using antibody against AR was performed on genital tubercles isolated from embryos at day E18.5 (Fig. 3A). In agreement with our Western blot data, overall staining of AR was equal in both wild type and homozygous embryonic tubercles. In wild type tubercle cells, AR was primarily located in the nucleus (Fig. 3A-b), presumably due to activation by circulating testosterone. Because reports for GR have shown a role for FKBP52 in nuclear translocation (26,45), we were surprised to see nuclear localization of AR in FKBP52-deficient tubercle cells (Fig. 3A-d). To confirm this behavior, we analyzed AR nuclear translocation in FKBP52-deficient and wild type mouse embryonic fibroblast (MEF) cells stably transfected with AR. Fig. 3B shows the complete absence of FKBP52 in FKBP52-deficient MEF cells and that loss of FKBP52 has no compensatory effect on expression of FKBP51, Cyp40 and PP5 in these cells. Prior to treatment with androgen agonist R1881, AR was primarily located in the cytoplasm of both wild type and FKBP52-deficient MEF cells (Fig. 3C-a and -b). After R1881 treatment, AR in both cells lines moved to the nucleus efficiently (Fig. 3C-c and -d). It therefore appears that AR nuclear translocation is not dependent on the presence of FKBP52. Consistent with this finding, AR hormone-binding activity in FKBP52-deficient MEF cells was also normal (Fig. 3D). Thus, the principal role for FKBP52 in AR-mediated signaling must be a novel effect downstream of the hormone binding and nuclear translocation events.
Figure 3.
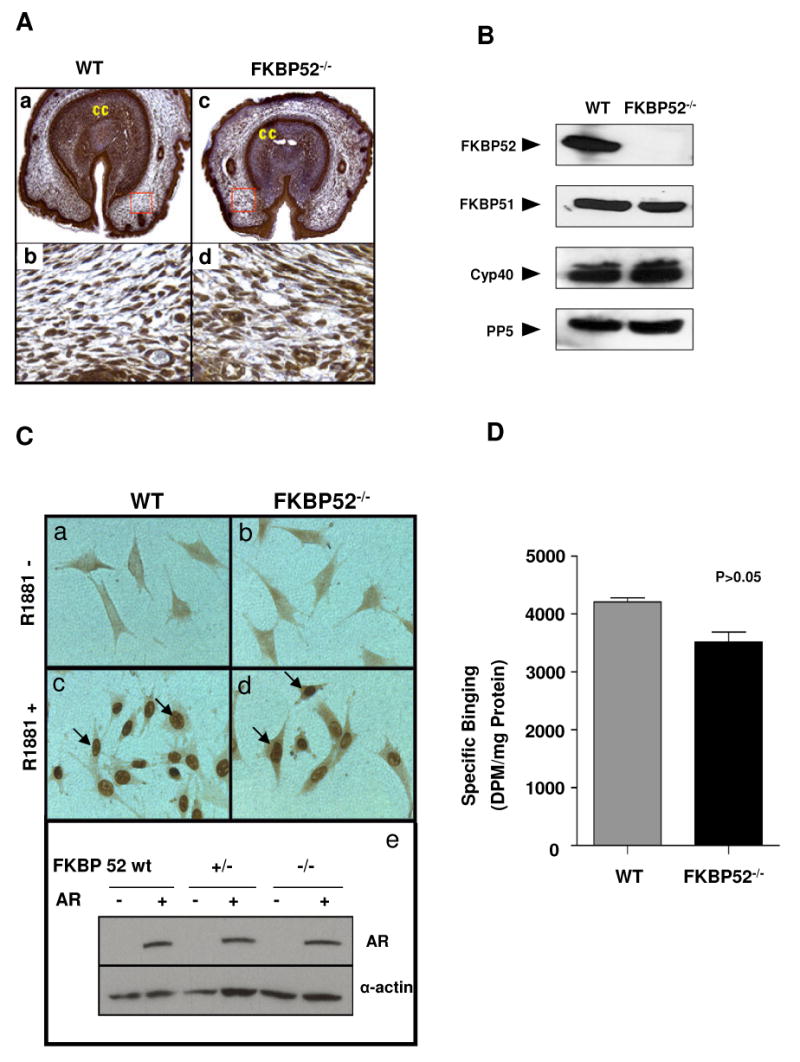
Analysis of FKBP52 deficiency on AR nuclear translocation and hormone binding. (A) Immunochemical staining shows that AR is highly expressed in all cell types of wild-type (a) and FKBP52-deficient genital tubercles (c), and that AR nuclear localization is not altered in FKBP52-deficient mutants (d) compared to wild-type controls (b). (B) Ablation of FKBP52 has no compensatory effect on expression of FKBP51, Cyp40 and PP5 in FKBP52-deficient MEF cells. (C) Using AR-stably transfected MEF cells to determine the AR nuclear translocation activity in FKBP52-deficient cells. Without hormone (R1881) treatment (a and b), ARs are mainly localized in cytoplasm. Upon R1881 treatment, ARs in both wild-type (c) and FKBP52-deficient (d) MEF cells translocate to nuclei in a similar fashion. The overall AR expression levels in AR-transfected cells are comparable among these cell lines, as evaluated by Western blot analysis (e). Genotypes of the cells are as indicated. (D) Measurement of AR hormone-binding capacity in cytosols from AR transfected wild-type and FKBP52-deficient MEF cells using [3H]mibolerone. The results shown are the means +/- SEM of two independent experiments, each performed in triplicate. No significant effect of FKBP52 ablation is seen on AR hormone-binding function.
Compromised AR transcriptional activity in FKBP52 (−/−) MEF cells
To determine if FKBP52 controlled AR transcriptional activity, we measured AR activity at two heterologous reporter genes. When the AR transcriptional activity was measured using the MMTV-CAT reporter, a dramatic loss of ligand-induced AR activity was seen in the FKBP52-deficient cells (Fig. 4A). To confirm this result, we used another AR-specific promoter, PSA-luciferase. Interestingly, in some cell systems this promoter is known to have androgen-independent AR activity (42,46). This was the case in the MEF cells, as AR activity did not increase with addition of hormone (Fig. 4B). More importantly, however, this activity was also dependent on FKBP52 since the PSA promoter activity was completely inhibited in FKBP52-deficient MEF cells. To confirm that loss of AR activity was due to FKBP52 rather than a non-specific property in the FKBP52-deficient MEF cells, we reintroduced FKBP52 into the FKBP52-deficient MEF cells. Fig. 4C shows complete re-activation of PSA promoter activity following FKBP52 re-expression. As a whole, these data show FKBP52 to be critical to AR transcriptional activity.
Figure 4.

Analysis of AR transcriptional activity in FKBP52-deficient MEF cells. (A) Transcriptional enhancement activity by hormone at MMTV-CAT is inhibited in FKBP52-deficient cells. (B) Transcriptional enhancement activity at the PSA-luciferase reporter is also inhibited. (C) Analysis of AR transcription activity in FKBP52-deficient MEF cells with re-introduction of human FKBP52. Values represent the means +/− SEM of four independent experiments.
Tissue selective loss of AR activity in FKBP52-deficient males
The results of Fig. 4 suggest that FKBP52 is essential to AR activity and provide the basis for the aberrant development seen in the prostate and penile tissues of FKBP52-deficient mice. However, FKBP52 cannot be exerting a global effect on AR activity since FKBP52-deficient males show normal testicular and epididymis development, two organs that are severely affected in AR knockout (ARKO) mouse models (8,47). Thus, a major unanswered question is the mechanism by which AR of the testes, for example, gets around the loss of FKBP52. One possibility is that other members of the SRA-TPR family compensate for FKBP52, at least on a tissue-specific basis. To test, we compared patterns of TPR protein expression in testicular and penile tissues in both wild type and FKBP52-deficient animals. Analysis by qRT-PCR showed no major effect of FKBP52 loss on Cyp40 or PP5 expression in both testes and penis (data not shown), suggesting that compensatory up-regulation is not a likely mechanism for normal AR activity in the testes of FKBP52-deficient animals. Interestingly, a dramatic down-regulation of FKBP51 expression by qRT-PCR was observed in the penis of FKBP52-deficient animals, but not in the testis (Fig. 5A). This tissue-selectivity was confirmed by Western-blotting for FKBP51 protein (Fig. 5B). It was not surprising that FKBP51 expression was down-regulated in the FKBP52-deficient penis, since it has been shown that FKBP51 expression is controlled by AR (32,48). Thus, these results appear to confirm that AR activity is indeed reduced in the penis but not in the testes.
Figure 5.

Analyses of FKBP51 expression in the testis and penis. (A) Quantitative RT-PCR analysis of FKBP51 in testis and penis of wild-type and FKBP52-deficient males (values represent the means +/− SEM of four independent experiments). In FKBP52-deficient penile tissues, FKBP51 is dramatically down-regulated. (B) The tissue-selectivity of FKBP51 expression was confirmed by Western-blotting.
Loss FKBP51 expression in the aberrant penile tissue of FKBP52-deficient males raises the possibility that FKBP51 is either directly involved in normal penile development, or indirectly involved in further AR actions necessary for development. Support for the latter comes from Magee et al (48) who showed recently that FKBP51 is a positive regulator of AR transcriptional activity in the prostate cancer cell line LnCAP. Thus, AR control of FKBP51 expression may form a positive-feedback loop designed to maximize AR activity, perhaps in a developmental or tissue-specific fashion. Such a mechanism may, therefore, be responsible for the tissue-selective abnormalities seen in FKBP52-deficient males.
Generation and characterization of FKBP51-deficient and compound FKBP52/FKBP51-deficient mice
To determine the contribution of FKBP51 to AR signaling and male reproduction, we generated FKBP51-deficient mice by using the BayGenomic Gene Trapping Resource (36). We confirmed a single insertion site in intron 4 in the mouse FKBP51 gene (Fig. 6A). The trapped allele yielded an FKBP51-βgeo fusion protein that lacked peptidyl-prolyl cis-trans isomerase like (PPlase-like) domain and all 3 TPR domains that are known to be critical to FKBP51 function (49). Therefore, this gene-trapped allele was an FKBP51 null allele. Northern, Western and PCR analyses further confirmed the absence of leaky FKBP51 expression in the mutant mouse strain (Fig. 6BC). FKBP51 heterozygous mutant mice were fertile and were intercrossed to generate FKBP51-deficient mice. Surprisingly, FKBP51-deficient mice (male and female) appeared to have normal growth and fertility. We carefully analyzed the male reproductive system. FKBP51-deficient males had normal formation of external genital tubercle, prostate, and the other male reproductive tissues (Fig. 7). FKBP52 expression also remained normal in all the FKBP51-deficient tissues (data not shown). Taken together, these findings show that FKBP51 is not essential to AR-regulated physiology. We can also conclude that development of hypospadias and prostate dysgenesis in the FKBP52-deficient males is not attributable to the loss of AR-mediated FKBP51 expression.
Figure 6.

Generation of FKBP51-deficient mice. (A) Genomic structure of the mouse FKBP51 gene, gene trap vector, and FKBP51 mutant allele. (B) Southern blot, (C) Western blot, and (D) qRT-PCR analyses confirm the FKBP51 mutant allele to be null. For Southern blot, the genomic DNA was digested by Bgl II (New England Biolabs). The probe indicated in (A) reveals a 6.9kb fragment from wild type allele and a 5.5kb fragment from FKBP51 mutant allele. Primers for qRT-PCR analysis are indicated by a pair of triangles in (A).
Figure 7.
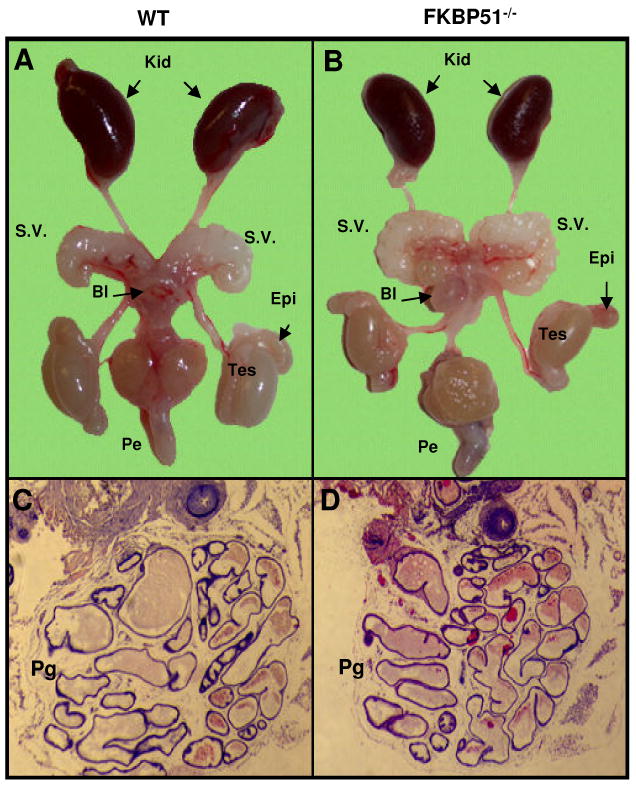
Morphological comparison of male internal reproductive organs in wild-type (A) and FKBP51-deficient adult mice (B). FKBP51-deficient males have overall normal testis (Tes) formation, normal epididymis (Epi) normal seminal vesicles (SV). Kid: kidney; Bl: bladder; Pe: penis; (C and D) Histological analysis of prostate glands in adult FKBP51-deficient and age-matched littermate control mice. FKBP51 mutant adult males have normal prostate glands (Pg).
The above data suggest that FKBP52 and FKBP51 are not functionally redundant with respect to androgen control of male reproduction. However, given the high sequence homology between these two proteins (50), it remained possible that they shared a common function with respect to other SRs or, indeed, unknown client proteins. To address this question, we generated a series of compound FKBP52/FKBP51 mutant mice. FKBP52/FKBP51 double heterozygous mice were normal and were intercrossed to generate FKBP52+/−/FKBP51−/−, FKBP52−/−/FKBP51+/− and FKBP52−/−/FKBP51−/− mice. FKBP52+/−/FKBP51−/− males were fertile and normal, while FKBP52−/−/FKBP51+/− males developed hypospadias similar to FKBP52-deficient males (data not shown). However, we were not able to obtain FKBP52−/−/FKBP51−/− mice at birth (data not shown), suggesting that compound FKBP52/FKBP51-deficient animals die in utero. Timed-pregnancy studies further indicated that FKBP52/FKBP51-deficient mice died at an early embryonic stage (before E7.5) that predates the start of the sex differentiation (detailed analyses of FKBP52/FKBP51-deficient mice will be reported elsewhere). These findings suggest that, although FKBP52 appears to be the major co-chaperone in regulating AR transcriptional activity, FKBP52 and FKBP51 are functionally redundant with respect to an early cellular function that is critical to embryonic development.
In summary, our current study demonstrates that FKBP52, but not FKBP51, is essential to AR mediated signaling and physiology. FKBP52 is not required for AR to bind androgen or for its hormone-induced nuclear translocation. Instead, FKBP52 appears to play a novel, yet critical role in AR transcriptional activity.
Discussion
In recent years, the complexity of molecular regulation of steroid receptor function has become apparent, with most investigators focusing on the mechanism by which co-regulatory proteins control tissue- and ligand-specific transcription activity by SRs (51-53). However, the discovery that inactive SRs exist as heterogeneous complexes based on TPR protein content (54), suggests that the early stages of SR signaling may also contribute to diversity of action. To address this question, we have generated both FKBP52-deficient and FKBP51-deficient mice. In a prior report (35), we showed that the FKBP52-deficient female is sterile due to a selective attenuation of some PR-regulated physiologies, in particular, the uterine receptivity to implantation. In the current work, we show a similar selectivity of FKBP52 action in the male. Male FKBP52-deficient mice showed dysgenic prostate and seminal vesicle development and penile hypospadias, yet had apparently normal development and function of other AR-regulated tissues, such as the testes. Interestingly, loss of FKBP52 had no obvious effect on GR-mediated physiology. Meanwhile, loss of FKBP51 appears to be neutral with respect to AR, PR and GR actions in vivo. Viewed as a whole, we believe that these observations usher in a new concept in which TPR proteins serve as agents for tissue- and receptor-specific control of steroidal actions.
Clearly, a major unanswered question that derives from our work is why loss FKBP52 or FKBP51 appears to leave the in vivo actions of GR unaffected. Because the GR knock-out mouse is a peri-natal lethal (55), we expected loss of FKBP52 or FKBP51 to have a similar phenotype if either of these proteins exerted an essential and global effect on GR actions. A possible explanation for this lack is the stress nature of cortisol secretion in which the main function of activated GR is to attenuate over-activity by “first responder” stress pathways, such as inflammation (56,57). Thus, a defect of GR signaling in the FKBP52-deficient animals may only be seen following a prior stress event, such as inflammatory challenge. Another explanation may lie with the concept of functional redundancy within the family of TPRs that regulate SR action. For example, it is possible that FKBP51 compensates for loss of FKBP52, with FKBP52 reciprocating for loss of FKBP51. With this in mind, we expected to see ablation of GR function in the double knock-out FKBP52/FKBP51 animals. Unexpectedly, the compound mutant turned out to be an embryonic lethal, suggesting that FKBP52 and FKBP51 are functionally redundant for an unknown factor/s, perhaps an orphan receptor, essential to early development. The FKBP52/FKBP51 compound mutant will now be an important reagent for identifying this unique developmental function and signal pathway. Of course, it is also possible that either PP5 or Cyp40 compensates for FKBP52 and FKBP51 in GR signaling. Because PP5 and Cyp40 are both known to interact with the motor protein dynein (25), either protein may provide at least the mechanism by which GR and AR (see below) translocate to the nucleus. We have previously shown PP5 to be found in the GR heterocomplexes of L cells and that this interaction increases when FKBP52 and FKBP51 are removed from GR/HSP90 using FK506 (58). Thus, one of our future goals is to analyze the role of PP5 in SR signaling by generating appropriate PP5 mutant mice.
In addition to lack of abnormal GR-related phenotypes, other unexpected results were found. For example, although FKBP52 is clearly essential to AR activity in the MEF cells, no overt defect of testicular development was observed in FKBP52-deficient males. As studies in the ARKO mouse have shown AR activity to be essential for testis development (8,47), our results must mean that AR activity in the testis is not dependent on FKBP52, even though this tissue expressed more FKBP52 than penis or prostate (data not shown). Thus, we speculated that another TPR, perhaps FKBP51, might fill this role in the testis. In the course of analyzing TPR expression levels in the tissues of wild type and FKBP52-deficient mice, we found a dramatic down-regulation of FKBP51 in FKBP52-deficient penile tissue, but not in the FKBP52-deficient testis. Because FKBP51 is a known target gene of AR action (29,32,33), this result was further evidence of compromised AR activity in the penis but not the testis. Moreover, since there is at least one report that FKBP51 over-expression in prostate cells enhances AR activity (32), the interesting possibility was raised that a genetic interplay may exist between FKBP52 and FKBP51, perhaps for the purpose of regulating the tissue-selective functions of AR. However, by generating and analyzing FKBP51-deficient mice, we have ruled out this possibility. It is now clear that FKBP51 is not a target gene essential to AR control of prostate or penis development, nor is it as important as FKBP52 to the intrinsic regulation of AR signaling in vivo. In short, down-regulation of FKBP51 is likely just a simple reflection of compromised AR function.
An important question that arises from our study is the mechanism by which FKBP52 regulates AR transcriptional activity. Our results in the MEF cells show that AR transactivity is severely compromised in the absence of FKBP52, yet AR in these cells exhibited normal hormone binding and nuclear translocation. Thus, the principal role of FKBP52 must be to control either the DNA recognition or transactivation functions of AR, perhaps by controlling recruitment of co-activators. Interestingly, we obtained similar results for the PR of FKBP52-deficient females: loss of transactivity but normal hormone-binding function (35). Although Cyp40 or PP5 may account for the normal hormone-binding and translocation activities of these receptors, it is clear that FKBP52 exerts an unexpected function on both AR and PR that, for the moment, leaves only room for speculation. The most likely mechanism is a lasting, down-stream effect of FKBP52 PPIase activity on the AF-1 or AF-2 domains of AR that is necessary for co-activator recruitment. Such a mechanism would be unprecedented for a TPR protein and will be the focus of future studies.
Our results have some similarities to those of D.F. Smith and colleagues who independently made FKBP52-deficinet mice and reported infertility due to dysgenic development of the penis and prostate (21). Our work extends upon their observations and provides more systematic and thorough analyses at the histological and molecular levels. Most notably we demonstrate a critical role for FKBP52 in regulating AR transcriptional activity and by assessing the contribution of FKBP51 to the FKBP52-deficient defect. More importantly, we herein provide the first report of FKBP51-deficient animals, allowing us to conclude that FKBP51 does not play an obvious or significant role in AR signaling in vivo, in spite of cellular and molecular evidence to the contrary. Finally, our studies have uncovered a novel and potentially unique function for FKBP52 in the regulation steroid receptor signaling. This discovery has important clinical implications, such as the identification of a new pathogenetic pathway that may explain androgen insensitivity syndrome (AIS) in humans.
Acknowledgments
We wish to thank Dr. Shaolian Jing and Mr. William Carter of Indiana University Mouse Core for their superb assistance. We also thank Drs. Lirim Shemshedini and Marianne Sadar for hAR expression vector and PSA-Luc reporter, respectively, Dr. Theo Rein for human FKBP52 cDNA and Dr. Michael Chinkers for PP5 antibody. This study was supported in part by National Institute of Health grants DK73402 (W.S., E.S), DK70127 (E.S., W.S.), DK43867 (E.S.), and the Riley Children's Foundation (W.S.).
References
- 1.Tsai MJ, O'Malley BW. Annual review of biochemistry. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 2.Beato M, Klug J. Hum Reprod Update. 2000;6(3):225–236. doi: 10.1093/humupd/6.3.225. [DOI] [PubMed] [Google Scholar]
- 3.Danielian PS, White R, Lees JA, Parker MG. The EMBO journal. 1992;11(3):1025–1033. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM. Mol Cell. 2004;16(3):425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 6.Batch JA, Williams DM, Davies HR, Brown BD, Evans BA, Hughes IA, Patterson MN. Human molecular genetics. 1992;1(7):497–503. doi: 10.1093/hmg/1.7.497. [DOI] [PubMed] [Google Scholar]
- 7.Nakao R, Haji M, Yanase T, Ogo A, Takayanagi R, Katsube T, Fukumaki Y, Nawata H. The Journal of clinical endocrinology and metabolism. 1992;74(5):1152–1157. doi: 10.1210/jcem.74.5.1569163. [DOI] [PubMed] [Google Scholar]
- 8.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charest NJ, Zhou ZX, Lubahn DB, Olsen KL, Wilson EM, French FS. Molecular endocrinology. 4. Vol. 5. (Baltimore, Md: 1991. pp. 573–581. [DOI] [PubMed] [Google Scholar]
- 10.Lyon MF, Hawkes SG. Nature. 1970;227(5264):1217–1219. doi: 10.1038/2271217a0. [DOI] [PubMed] [Google Scholar]
- 11.Tilley WD, Marcelli M, Wilson JD, McPhaul MJ. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(1):327–331. doi: 10.1073/pnas.86.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song CS, Her S, Slomczynska M, Choi SJ, Jung MH, Roy AK, Chatterjee B. Biochem J. 1993;294(Pt 3):779–784. doi: 10.1042/bj2940779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. Cell stress & chaperones. 1996;1(4):237–250. doi: 10.1379/1466-1268(1996)001<0237:apomci>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratt WB, Toft DO. Endocrine reviews. 1997;18(3):306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 15.Davies TH, Sanchez ER. The international journal of biochemistry & cell biology. 2005;37(1):42–47. doi: 10.1016/j.biocel.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Yem AW, Tomasselli AG, Heinrikson RL, Zurcher-Neely H, Ruff VA, Johnson RA, Deibel MR., Jr The Journal of biological chemistry. 1992;267(5):2868–2871. [PubMed] [Google Scholar]
- 17.Peattie DA, Harding MW, Fleming MA, DeCenzo MT, Lippke JA, Livingston DJ, Benasutti M. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10974–10978. doi: 10.1073/pnas.89.22.10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baughman G, Wiederrecht GJ, Campbell NF, Martin MM, Bourgeois S. Molecular and cellular biology. 1995;15(8):4395–4402. doi: 10.1128/mcb.15.8.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens-Grillo JK, Hoffmann K, Hutchison KA, Yem AW, Deibel MR, Jr, Handschumacher RE, Pratt WB. The Journal of biological chemistry. 1995;270(35):20479–20484. doi: 10.1074/jbc.270.35.20479. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Sullivan WP, Toft DO, Smith DF. Cell stress & chaperones. 1998;3(2):118–129. doi: 10.1379/1466-1268(1998)003<0118:diopat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung-Flynn J, Roberts PJ, Riggs DL, Smith DF. The Journal of biological chemistry. 2003;278(19):17388–17394. doi: 10.1074/jbc.M300955200. [DOI] [PubMed] [Google Scholar]
- 22.Ruff VA, Yem AW, Munns PL, Adams LD, Reardon IM, Deibel MR, Jr, Leach KL. The Journal of biological chemistry. 1992;267(30):21285–21288. [PubMed] [Google Scholar]
- 23.Czar MJ, Owens-Grillo JK, Yem AW, Leach KL, Deibel MR, Jr, Welsh MJ, Pratt WB. Molecular endocrinology. 12. Vol. 8. (Baltimore, Md: 1994. pp. 1731–1741. [DOI] [PubMed] [Google Scholar]
- 24.Pratt WB, Czar MJ, Stancato LF, Owens JK. The Journal of steroid biochemistry and molecular biology. 1993;46(3):269–279. doi: 10.1016/0960-0760(93)90216-j. [DOI] [PubMed] [Google Scholar]
- 25.Galigniana MD, Harrell JM, Murphy PJ, Chinkers M, Radanyi C, Renoir JM, Zhang M, Pratt WB. Biochemistry. 2002;41(46):13602–13610. doi: 10.1021/bi020399z. [DOI] [PubMed] [Google Scholar]
- 26.Davies TH, Ning YM, Sanchez ER. The Journal of biological chemistry. 2002;277(7):4597–4600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- 27.Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T. The Journal of biological chemistry. 2005;280(6):4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds PD, Ruan Y, Smith DF, Scammell JG. The Journal of clinical endocrinology and metabolism. 1999;84(2):663–669. doi: 10.1210/jcem.84.2.5429. [DOI] [PubMed] [Google Scholar]
- 29.Hubler TR, Denny WB, Valentine DL, Cheung-Flynn J, Smith DF, Scammell JG. Endocrinology. 2003;144(6):2380–2387. doi: 10.1210/en.2003-0092. [DOI] [PubMed] [Google Scholar]
- 30.Tai PK, Maeda Y, Nakao K, Wakim NG, Duhring JL, Faber LE. Biochemistry. 1986;25(18):5269–5275. doi: 10.1021/bi00366a043. [DOI] [PubMed] [Google Scholar]
- 31.Veldscholte J, Berrevoets CA, Zegers ND, van der Kwast TH, Grootegoed JA, Mulder E. Biochemistry. 1992;31(32):7422–7430. doi: 10.1021/bi00147a029. [DOI] [PubMed] [Google Scholar]
- 32.Febbo PG, Lowenberg M, Thorner AR, Brown M, Loda M, Golub TR. The Journal of urology. 2005;173(5):1772–1777. doi: 10.1097/01.ju.0000155845.44729.ba. [DOI] [PubMed] [Google Scholar]
- 33.Zhu W, Zhang JS, Young CY. Carcinogenesis. 2001;22(9):1399–1403. doi: 10.1093/carcin/22.9.1399. [DOI] [PubMed] [Google Scholar]
- 34.Vanaja DK, Mitchell SH, Toft DO, Young CY. Cell stress & chaperones. 2002;7(1):55–64. doi: 10.1379/1466-1268(2002)007<0055:eogoar>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, Wolf IM, Chen H, Periyasamy S, Chen Z, Yong W, Shi S, Zhao W, Xu J, Srivastava A, Sanchez ER, Shou W. Molecular endocrinology. (Baltimore, Md: 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanford WL, Cohn JB, Cordes SP. Nature reviews. 2001;2(10):756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 37.Yucel S, Cavalcanti AG, Desouza A, Wang Z, Baskin LS. BJU international. 2003;92(9):1016–1021. doi: 10.1111/j.1464-410x.2003.04511.x. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W. Development. 9. Vol. 131. (Cambridge, England): 2004. pp. 2219–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laemmli UK. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Chen MS, Silverstein AM, Pratt WB, Chinkers M. The Journal of biological chemistry. 1996;271(50):32315–32320. doi: 10.1074/jbc.271.50.32315. [DOI] [PubMed] [Google Scholar]
- 41.Wise SC, Burmeister LA, Zhou XF, Bubulya A, Oberfield JL, Birrer MJ, Shemshedini L. Oncogene. 1998;16(15):2001–2009. doi: 10.1038/sj.onc.1201697. [DOI] [PubMed] [Google Scholar]
- 42.Sadar MD. The Journal of biological chemistry. 1999;274(12):7777–7783. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 43.Nordeen SK, Green PP, 3rd, Fowlkes DM. DNA (Mary Ann Liebert, Inc. 1987;6(2):173–178. doi: 10.1089/dna.1987.6.173. [DOI] [PubMed] [Google Scholar]
- 44.Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Endocrine reviews. 1995;16(3):271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 45.Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB. The Journal of biological chemistry. 2001;276(18):14884–14889. doi: 10.1074/jbc.M010809200. [DOI] [PubMed] [Google Scholar]
- 46.Huang ZQ, Li J, Wong J. Molecular endocrinology. 5. Vol. 16. (Baltimore, Md: 2002. pp. 924–937. [DOI] [PubMed] [Google Scholar]
- 47.Tan KA, De Gendt K, Atanassova N, Walker M, Sharpe RM, Saunders PT, Denolet E, Verhoeven G. Endocrinology. 2005;146(6):2674–2683. doi: 10.1210/en.2004-1630. [DOI] [PubMed] [Google Scholar]
- 48.Magee JA, Chang LW, Stormo GD, Milbrandt J. Endocrinology. 2006;147(1):590–598. doi: 10.1210/en.2005-1001. [DOI] [PubMed] [Google Scholar]
- 49.Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(3):868–873. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nair SC, Rimerman RA, Toran EJ, Chen S, Prapapanich V, Butts RN, Smith DF. Molecular and cellular biology. 1997;17(2):594–603. doi: 10.1128/mcb.17.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu X, Lazar MA. Trends Endocrinol Metab. 2000;11(1):6–10. doi: 10.1016/s1043-2760(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 52.Smith CL, O'Malley BW. Endocrine reviews. 2004;25(1):45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 53.Lonard DM, O'Malley BW. Cell. 2006;125(3):411–414. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 54.Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Essays in biochemistry. 2004;40:41–58. doi: 10.1042/bse0400041. [DOI] [PubMed] [Google Scholar]
- 55.Cole TJ, Blendy JA, Monaghan AP, Schmid W, Aguzzi A, Schutz G. Steroids. 1995;60(1):93–96. doi: 10.1016/0039-128x(94)00009-2. [DOI] [PubMed] [Google Scholar]
- 56.Sapolsky RM, Romero LM, Munck AU. Endocrine reviews. 2000;21(1):55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 57.Yeager MP, Guyre PM, Munck AU. Acta anaesthesiologica Scandinavica. 2004;48(7):799–813. doi: 10.1111/j.1399-6576.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- 58.Davies TH, Ning YM, Sanchez ER. Biochemistry. 2005;44(6):2030–2038. doi: 10.1021/bi048503v. [DOI] [PubMed] [Google Scholar]