

Abstract
Eukaryotic cells have evolved cell cycle checkpoints to maintain genomic stability and integrity. Protein phosphatase 5 (Ppp5), a tetratricopeptide repeat domain protein, has been implicated in multiple cellular functions, including cellular proliferation, migration, differentiation and survival, and cell cycle checkpoint regulation via the ATM/ATR signal pathway. However, the physiological functions of Ppp5 have not been reported. To confirm the role of Ppp5 in cell cycle checkpoint regulation, we generated Ppp5-deficient mice and isolated mouse embryonic fibroblast (MEF) cells from Ppp5-deficient and littermate control embryos. Although Ppp5-deficient mice can survive through embryonic development and postnatal life and MEF cells from the Ppp5-deficient mice maintain normal replication checkpoint induced by hydroxyurea, Ppp5-deficient MEF cells display a significant defect in G2/M DNA damage checkpoint in response to ionizing radiation (IR). To determine whether this defect in IR induced-G2/M checkpoint is due to altered ATM-mediated signaling, we measured ATM kinase activity and ATM-mediated downstream events. Our data demonstrated that IR-induced ATM kinase activity is attenuated in Ppp5-deficient MEFs. Phosphorylation levels of two known ATM substrates, Rad17 and Chk2, were significantly reduced in Ppp5-deficient MEFs in response to IR. Furthermore, DNA damage-induced Rad17 nuclear foci were dramatically reduced in Ppp5-deficient MEFs. These results demonstrate a direct regulatory linkage between Ppp5 and activation of the ATM-mediated G2/M DNA damage checkpoint pathway in vivo. Thus, the Ppp5 null mouse should now provide a useful animal model for further study of Ppp5 in checkpoint regulation.
In eukaryotic cells, DNA repair and cell cycle checkpoints are highly integrated processes that are critical for maintaining genomic stability and integrity (1). In the event of DNA damage, two closely related checkpoint kinases ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3 related) are thought to be master controllers of cell cycle checkpoint signaling that coordinate multiple cellular process, such as cell cycle arrest, G2/M checkpoint, intra-S-phase checkpoint, DNA repair, and cellular apoptosis (2). Defects in cell cycle checkpoints will result in genetic mutations, chromosomal abnormalities and aneuploidy, all of which contribute to tumorigenesis (3). Although ATM and ATR appeared to have an overlapped biological function, ATM was thought to be more relevant in sensing DNA double-stranded breaks induced by ionizing radiation (4), and ATR was responsible for the checkpoint activation in response to DNA damage induced by a variety of broader stressors, such as ultraviolet (UV) light, HU and other types of genotoxic insults (2). Interestingly, AT cells or ATM-deficient cells often display IR-resistant DNA synthesis in S phase (2), suggesting that ATM may also function in DNA replication checkpoint in S-phase.
Protein phosphatase 5 (Ppp5, also known as PP5) is serine/threonine phosphatase involved in several important cellular functions. Ppp5 is unique among phosphatases in that it contains a series of 34-amino acid tetratricopeptide repeat (TPR) motifs that serve as a protein-protein interaction domain (5). Ppp5 is ubiquitously expressed, and is able to form heterocomplexes with glucocorticoid receptors (GR) via heat shock protein 90 (HSP90) (6). In addition to steroid signaling, Ppp5 has been implicated in cellular growth, proliferation, differentiation, migration, and survival via several intracellular signaling complexes, including CDC16 and CDC27 (7), apoptosis signal-regulating kinase 1 (ASK1) (8), Gα12/Gα13 (9), Rac GTPase (10), and Raf-MEK-ERK mediated pathways (11). Recently, several lines of evidence have suggested a critical role for Ppp5 in both the ATM/ATR and DNA-PK pathways controlling cell cycle checkpoints and DNA double-strand break repair (12-14). Down-regulation of Ppp5 via siRNA or over-expression of a catalytically inactive Ppp5 mutant in cells inhibits ATM activation and ATM autophosphorylation on Serine 1981, leading to a checkpoint defect in DNA-damaged cells (14). Another report suggests that Ppp5 is also required for ATR or ATM activation in response to HU-induced replication block (12). Taken as a whole, these studies strongly hint at the important contribution of Ppp5 to various basic cellular functions.
To confirm the above activities of Ppp5 in a physiological context, we undertook a loss-of-function study by generating Ppp5-deficient mice. Surprisingly, Ppp5-deficiency did not give rise to dramatic growth defects during embryonic development and postnatal life. To investigate the potential function of Ppp5 in ATM-mediated cell cycle checkpoint signaling induced by double-strand breaks, we examined the checkpoint response to ionizing radiation (IR) in mouse embryonic fibroblasts (MEFs) isolated from wild-type and Ppp5-deficient embryos. We demonstrate that Ppp5 is essential for the activation of G2/M checkpoint induced by IR exposure, but not the S phase replication checkpoint that can be triggered by HU treatment. Our results support a regulatory linkage between Ppp5 and ATM kinase-mediated G2/M checkpoint activation in response to radiation-induced DNA damage. Together, these data strongly suggest that Ppp5 is a crucial regulator in the DNA damage checkpoint pathway.
Material and Methods
Generation of Ppp5-deficient mice
A promoter trapped ES cell line XG029 containing an insertional mutation in the mouse ppp5 gene was obtained from BayGenomics. The gene-trap vector (pGT1Lxf) contains a splice-acceptor sequence upstream of the reporter gene β-geo. Using PCR mapping approach, Southern blot, and sequencing analyses, we confirmed and identified the location of a single genomic insertion in the first intron of the mouse ppp5 gene. The genotyping primer sequences and probes for Southern blot analysis were designed based on this insertion site at +3893 from the transcription initiation site (Fig. 1A). Chimeric male mice were then generated and were further bred to C57BL/6J females to generate F1 offspring. Primers for genotyping are: Ppp5 geno F: TACAGAGCAGGGAAGTGGGGTCAG; Ppp5 geno R: AGGTTGGAGGACCATGTGCCAG; Ppp5-geo R: TTCAGTCTTCCTTGGTGGCCTGTC.
Figure 1.
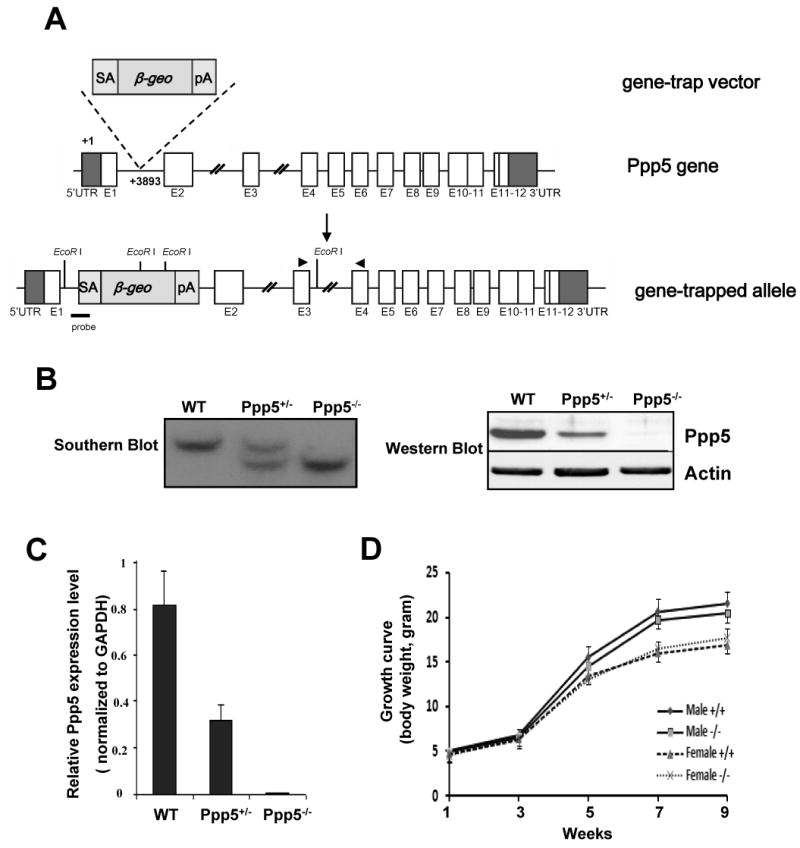
Generation of Ppp5-deficient mice. (A) Genomic structure of the mouse Ppp5 gene, gene trap vector, and Ppp5 mutant allele. (B) Southern blot, Western blot, and (C) qRT-PCR analyses confirm the Ppp5 mutant allele to be null. For Southern blot, the genomic DNA was digested by EcoR I (New England Biolabs). (D) Growth curves of wild-type and Ppp5-deficient mice. The probe indicated in (A) reveals a 11.4kb fragment from wild-type allele and a 7.1kb fragment from Ppp5 mutant allele. Real time PCR primers are indicated in (A).
Quantitative RT-PCR
Total RNA was extracted from various tissues using TRizol (Invitrogen). First strand cDNA was synthesized by the iScript cDNA synthesis kit (Bio-Rad) using 1 ug of total RNA as a template according to the protocol provided by the manufacturer. Real time PCR was performed using iCycle iQ (Bio-Rad). The relative expression was normalized to the reference gene ribosomal protein L7 (RPL7). Sequences of specific primers are as follows: q-Ppp5 F: CACAGACGCTCTGTCGTGGACTCTC; q-Ppp5 R: GCACTTCCGGTGCAGTTTCTTCTG; RPL7 F: AGTTGAAGGTGAAGCGCCTGAGG; RPL 7 R: TGCCATCCTAGCCATCCGAATC.
Cell culture, IR treatments and FACS analysis
The Ppp5 wild-type (wt), Ppp5 heterozygous (+/-) mutant and Ppp5 homozygous (-/-) mutant mouse embryonic fibroblasts were isolated from E13.5 embryo and cultured in DMEM with 10% FBS. Cells were irradiated with 3Gy ionizing radiation in the AGFA X-RAD320 Irradiation System. 24h after IR (3Gy), IR-treated or untreated MEF cells were fixed with 70% ethanol and incubated for 30 min with RNaseA (100 ug/ml) and propidium iodide (50 ug/ml) at 37 C. Cell-cycle distributions were analyzed by flow cytometry. Mitotic indexes in wild-type and Ppp5-deficient MEF cells before and after IR treatment were determined as previously described (15).
Western blot analysis and in vitro ATM kinase assay
Irradiated MEF cells were harvested at 1 h post-IR or as indicated. Western blot analysis of phospho-Rad17 (pRad17), total Rad17, phospho-Chk2 (pChk2), total Chk2 and Ppp5 was performed as previously described (14). Phospho-specific antibody for Rad17 S645 (α-pS645-Rad17) was obtained from GeneTex (TX), and the phospho-specific antibody for Rad17 S635 (α-pS635-Rad17) has been previously characterized (12,15). The phospho-specific antibody for Chk2 and Chk1 and the antibody for total Chk2 and Chk1 were from Cell Signaling Inc. Antibody for total Rad17 was purchased from Santa Cruz Co, and antibody for Ppp5 was a generous gift from Dr. Michael Chinker. The ATM complex was immunoprecipitated from untreated or irradiated wild-type and Ppp5-decifient MEF cells, and the in vitro ATM kinase assays were performed as previously described (15). ATM kinase activities were assessed using 1μg GST-hRad17 fusion protein containing the C-terminal 185 amino acids of hRad17 as substrate.
Immunofluorescent Staining
MEF cells isolated from wild-type or Ppp5-deficient mice were cultured on coverslips in DMEM with 10% FBS followed by irradiation. Cells were fixed at 3 h post-IR with 4% paraformaldehyde and permeablized with 0.1% Triton-100 in PBS. After blocking with 3% bovine serum albumin, cells were incubated with the α-pS645-Rad17 antibody (diluted according to the manufacturer's instructions) at 4°C overnight. Following three washes with PBS, cells were incubated with rhodamine-conjugated anti-rabbit IgG secondary antibody for 1 h. After washing with PBS, cell nuclei were stained with DAPI (Sigma). Pictures were taken under a fluorescent microscope (Axiovert 200, Zeiss).
Results and Discussion
Ppp5 heterozygous mutant mice were viable and fertile and were inter-crossed to obtain Ppp5 homozygous mutants. Genotyping analysis of 153 F2 offspring at 2 weeks of age demonstrated normal Mendelian 1:2:1 distribution of all three genotypes (+/+: 42; +/-: 74; -/-: 37), indicating that Ppp5-deficient mice are viable in utero through the course of embryonic development. Western blotting and real time quantitative RT-PCR analysis of Ppp5 expression in wild-type, Ppp5 heterozygous and Ppp5 homozygous mutant mice demonstrated a 50% reduction in Ppp5 heterozygous and a complete absence of Ppp5 protein and mRNA in Ppp5 homozygous mutant (Fig. 1B, C). There was no leaky Ppp5 expression found in this Ppp5 mutant mouse strain. Therefore, this promoter-trapped allele is a Ppp5 null allele. Both Ppp5-deficient male and female mice were fertile and showed a rate of growth equivalent to that of wild-type (Fig. 1D). Analysis of potentially compromised GR activity in Ppp5-deficient mice is on the way. Here, as our first report on the generation of Ppp5-deficient mice, we focus on our initial analysis of the role of Ppp5 in ATM mediated cell cycle checkpoint pathway.
ATM is important in cell-cycle checkpoint activation triggered by ionizing radiation and radiomimetic agents (15). Recently, several reports using siRNA have suggested a role for Ppp5 in ATM kinase activity and its subsequent checkpoint activation (12,14). To investigate whether Ppp5 is essential to cell cycle checkpoints, we isolated mouse embryonic fibroblasts from wild-type and Ppp5-deficient E13.5 embryos. The MEF cells were exposed to ionizing radiation (3 Gy) and HU, respectively. In the case of IR, cell cycle profiles were analyzed 24h post-irradiation using flow cytometry (FACS). Our analysis revealed that wild-type MEF cells displayed a typical and distinct G2/M cell cycle arrest after IR exposure (43% arrest at G2/M phase) (Fig. 2). In contrast, Ppp5-deficient MEF cells significantly failed to accumulate in G2/M phase after IR exposure (23% arrest at G2/M). These data strongly indicate a defect in the IR-induced G2/M checkpoint in Ppp5-deficient cells. This conclusion was further confirmed by determining the mitotic indexes in wild-type and Ppp5 null MEF cells before and after IR treatment. As shown in Fig. 2C, at 24 hr after IR, Ppp5 null MEF cells display a significant higher mitotic index than the wild-type cells, indicating that Ppp5 null cells have a defect in G2 arrest in response to IR, which further demonstrates that Ppp5 null cells are defective in the IR-induced G2 checkpoint. In the case of HU treatment, however, Ppp5-deficient MEF cell did not appear significantly different when compared with wild type MEF cells (data not shown).
Figure 2.
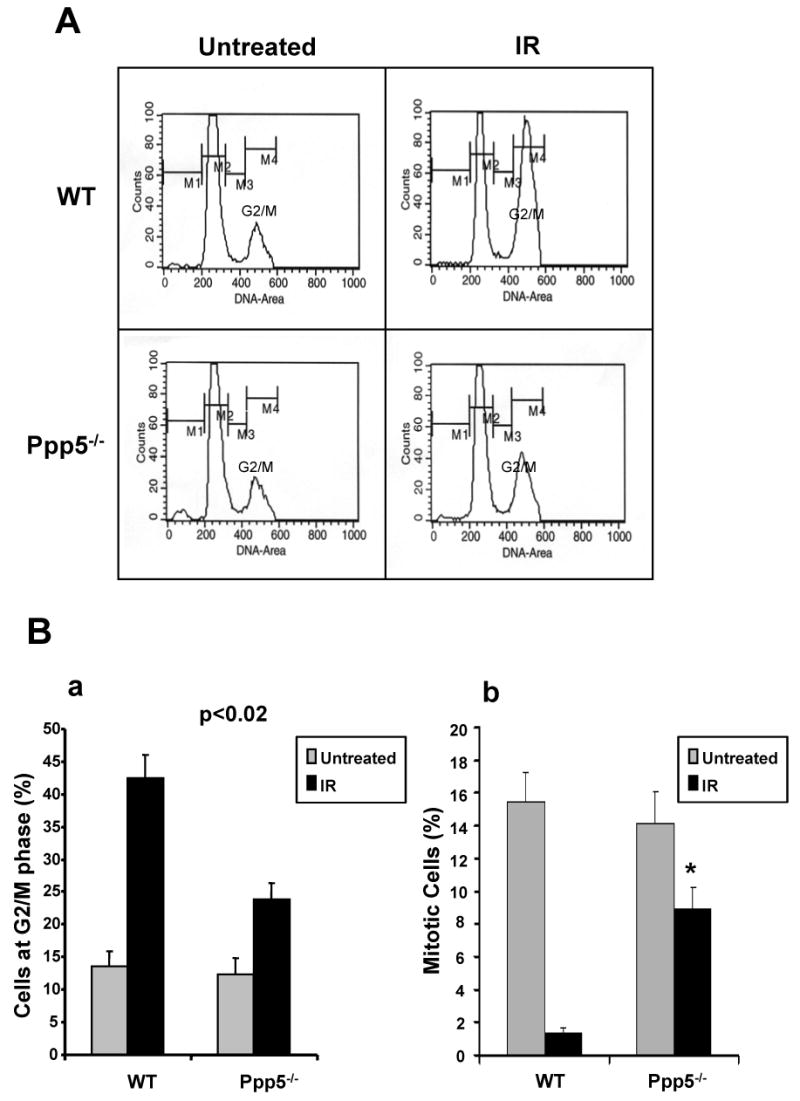
Ppp5-deficient MEF cells display a defect of ionizing radiation-induced G2/M checkpoint activation. (A) Cell-cycle profiles of Ppp5-deficient and wild-type cells with or without ionizing radiation exposure (3Gy). Cell-cycle distributions were determined by flow cytometry at 24 h post-IR treatment. (B-a) Statistical analysis of G2/M phase arrest in Ppp5-deficient and wild-type cells with or without IR exposure. The IR-induced G2/M arrest in Ppp5-deficient cells is significantly less than in wild-type cells. (B-b) Mitotic index confirmed that Ppp5-deficient cells displayed significantly greater fraction of mitotic cells after IR treatment.
In previous studies (12,14), Ppp5 was found in a complex with ATM. Ppp5 expression was also increased after the cells were exposed to the radiomimetic agent neocarzinostatin. To determine whether ablation of Ppp5 affects ATM activation induced by IR exposure, in vitro kinase assays were performed using the ATM-specific substrate Rad17 (15). A GST fusion protein containing a 185 amino acid C-terminal peptide of human Rad17 was incubated with ATM immunoprecipitates derived from wild-type and mutant Ppp5 MEF cells. ATM kinase activity was assessed by Western blot using a specific antibody against phosphorylated Rad17 at serine 635 (12,15). As expected, ATM kinase activity was greatly increased after IR treatment in wild-type MEF cells (Fig 3A-a). In contrast, ATM kinase activity was dramatically reduced in Ppp5-deficient cells after IR treatment, pointing to Ppp5 as a key regulator of ATM kinase activity.
Figure 3.
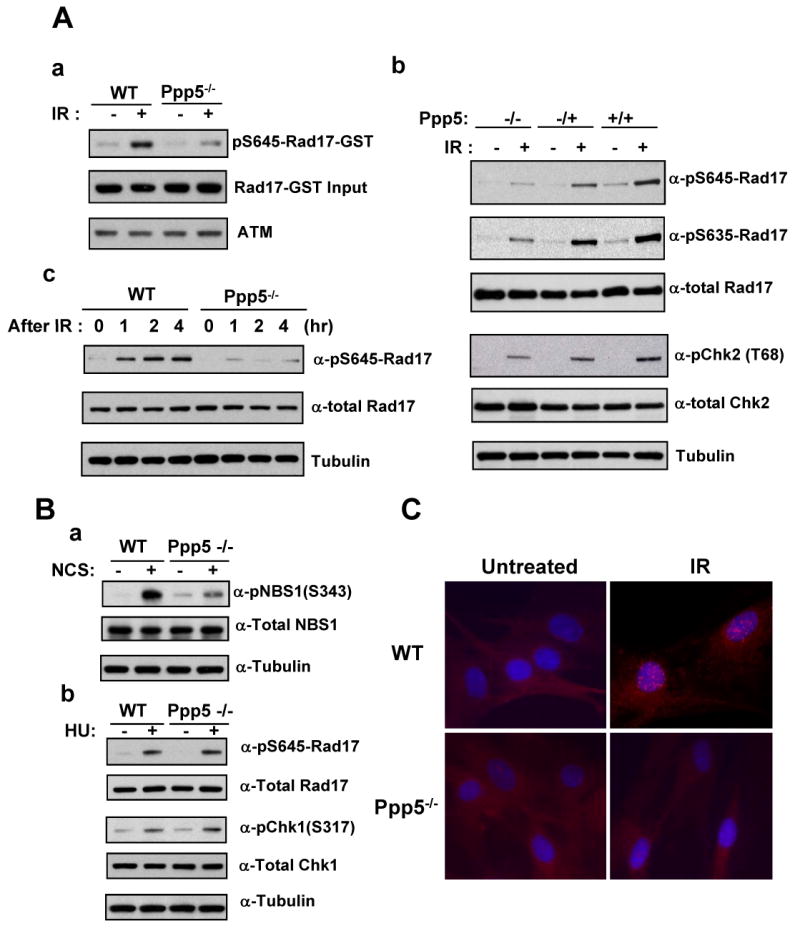
Ablation of Ppp5 decreased ATM kinase activity and IR- (A), NCS- and HU-induced (B) phosphorylation of Rad17, Chk2, NBS1 and Chk1. IR-induced formation of Rad17 nuclear foci is abolished in Ppp5-deficient MEF cells (C). (A-a) ATM kinase activity is reduced in Ppp5-deficient MEF cells. Wild type and Ppp5-deficient cells were exposed to IR (3Gy), followed by immunoprecipitation of ATM complexes and in vitro ATM kinase assay, as previously described (15). GST-hRad17 fusion protein was used as substrate, and detection of phosphorylation was performed with antibody specific to pS645-Rad17. (A-b) IR-induced phosphorylation of Rad17 and Chk2 is attenuated in Ppp5-deficient cells. Wild type (+/+), Ppp5 heterozygous (+/-) and homozygous (-/-) cells were exposed to 3 Gy ionizing radiation and harvested at 1 h post-IR. Levels of phosphorylated Rad17 (pS635-Rad17 and pS645-Rad17), phosphorylated Chk2 (pChk2-T68), and total Rad17 and Chk2 were determined by immunoblotting with specific antibodies. (A-c) Time course of Rad17 phosphorylation induced by IR exposure in wild type and Ppp5-deficient MEF cells. MEF cells were exposed to IR (3 Gy). Cellular extracts were collected at 0, 1, 2, and 4 h post-IR and were immunoblotted with specific antibodies against pS645-Rad17, total Rad17 or tubulin (used as internal loading control). (B) Western blot analysis of wild-type and Ppp5-deficient cells treated with HU (10mM, 20 hours) and NCS (100ng/ml, 2 hours). (B-a) phosphorylated NBS1 was reduced in Ppp5-deficient cells when compared to wild type controls after treating with NCS. (B-b) There is no significant difference in Rad17 and Chk1 phosphorylation between wild type and Ppp5-deficient MEF cell before and after HU treatments. (C) pS645-Rad17 immunofluorescent starining of wild type and Ppp5-deficient cells were exposed to IR (3Gy). At 3 h post-IR, cells were fixed and immuno-stained with a phospho-specific Rad17 antibody (a-pS645-Rad17). The Rad17 nuclear foci (in red) were examined under a fluorescence microscope. Nuclei were counter-stained with DAPI (in blue). IR-induced Rad17 nuclear foci appeared in wild-type MEF cells, but not in the Ppp5-deficient MEF cells.
To further investigate the ATM pathway in Ppp5-deficient cells, we measured phosphorylation of two ATM targets, Rad17 and Chk2, in intact cells. Rad17 is a checkpoint protein required for cell cycle arrest and DNA repair. This protein binds to chromatin prior to DNA damage and is phosphorylated by ATM post-damage (16). Phosphorylation of Rad17 is required for DNA-damage-induced cell cycle arrest, and is thought to be a critical early event during checkpoint signaling (15). The protein kinase Chk2 is also phosphorylated and activated in response to DNA damage by ionizing radiation, with both events dependent on ATM activity (17). Chk2-deficient embryonic stem cells fail to maintain radiation-induced arrest in the G2 phase of the cell cycle (18). Thus, phosphorylation levels of Rad17 and Chk2 are key indicators of endogenous ATM activity. As shown in Figure 3A-b, ionizing radiation induced phosphorylation of Rad17 at both Ser645 and Ser635 in wild-type MEF cells. Induction of phosphorylation was detected 1 h after IR exposure. Similarly, Chk2 phosphorylation was enhanced in wild-type MEF cells after irradiation. In contrast, IR-induced phosphorylation of both Rad17 and Chk2 was significantly reduced in Ppp5-deficient cells, even though levels of each protein remained normal. A time-course study further revealed that ATM activity remained dramatically reduced up to 4 hours post-IR (Fig. 3A-c). Furthermore, phosphorylated NBS1 was also significantly reduced in Ppp5-deficient cells when treating with a radio-mimic drug NCS (Fig. 3B-a). Consistent to normal cellular response to replication blocker HU, wild type and Ppp5-deficient MEF cells did not display a significant difference in the phosphorylation of Rad17 and Chk1 in response to HU treatment (Fig. 3B-b).
In addition to biochemical alterations, we also found that the biological function of Rad17 was defective in Ppp5-deficient MEF cells. Rad17 shares strong similarity with DNA replication factor C (RFC), and can form a complex with RFCs (19). The Rad17-RFC complex, along with another DNA-repair complex (Rad9-Rad1-Hus1), have both been implicated in the early phase of cell-cycle checkpoint control (20). They function as sensors for DNA damage and genome replication errors (21). Phosphorylation of Rad17 is required to form this complex (15), and a critical feature required for activity appears to be formation of Rad17-containing nuclear foci after IR (15,22). To determine whether Ppp5 activity is required for complex formation, we examined nuclear foci formation in Ppp5-deficient MEF cells after IR exposure. Wild type and Ppp5-deficient MEF cells were fixed and stained with a specific antibody against phospho-Rad17-Ser645 at 3 hours post-IR. As shown in Figure 3C, wild-type MEF cells displayed a typical punctuate pattern of nuclear staining after IR. In contrast, this nuclear staining pattern was almost completely missing in Ppp5-deficient MEF cells (Fig. 3C), suggesting that reduced phosphorylation of Rad17 in Ppp5-deficient cells may decrease nuclear foci (checkpoint complex) formation.
Taken as a whole, our data provide the first conclusive evidence that Ppp5 is an important regulator of ATM-mediated signaling. We have shown that Ppp5 is essential to the ability of ATM to phosphorylate key substrates, Rad17, Chk2 and NBS1, and that these phophorylation events are critical to check-point control of cell cycle arrest following DNA damage. One enigma from our initial characterization of Ppp5-deficient mice is that, despite significant alteration of ATM-mediated signaling, Ppp5 is not critical to mouse survival and fertility. In contrast, ATM-deficient mice display growth retardation, male and female infertility due to meiotic failure and abnormal chromosomal synapsis, defects in lymphocyte maturation, and extreme sensitivity to radiation. Moreover, most ATM-deficient mice die before 4 months of age from thymic lymphomas (23). Although we have not yet thoroughly investigated most of these parameters in the Ppp5-deficient mouse, it is clear that fertility appears to be unaffected and that lethality is much reduced compared to the ATM-deficient mouse. This suggests that ATM is involved in a broader checkpoint function than Ppp5. Our data demonstrates that Ppp5 is likely involved in regulating ATM kinase activity only in response to a subset of genotoxic stresses, such as IR. When we examined the HU-induced replication checkpoint activation in Ppp5-deficient and wild-type MEF cells, we found that the Rad17 and Chk1 phosphorylations were not significantly different between wild-type and Ppp5-deficient MEF cells, suggesting that Ppp5 was not involved in the intra-S-phase replication checkpoint. On the other hand, it was demonstrated that AT cells or ATM-deficient cells displayed IR-resistant DNA synthesis in S phase (2), suggesting that ATM was also associated with S-phase DNA replication checkpoint. Alternatively, other protein phosphatases may partially compensate for the loss of Ppp5 in the Ppp5-deficient cells. Together, these findings may help to explain the phenotypic difference between ATM-deficient and Ppp5-deficient mice. Given the high incidence of lymphomas in ATM null mice, it will be interesting to see whether Ppp5-dificient mice require a longer period of time to develop lymphomas or other tumors.
Also consistent with our hypothesis is the fact that Ppp5 appears to be a tightly regulated phosphatase. Several studies have shown extremely low basal phosphatase activity for Ppp5, most likely due to an auto-inhibitory function of the protein's TPR domain (24-26). It has been demonstrated that the TPR domain cooperates with the final 13 residues of the C-terminus to maintain the phosphatase in an inactive state. Removal of either the TPR domain or C-terminal 13 residues by limited proteolysis or deletion mutagenesis greatly increases the Ppp5 enzymatic activity (25,27-29). Interestingly, several reports now exist that both Hsp90 and fatty acids, such as arachidonic acid, can stimulate Ppp5 phosphatase activity by a direct interaction with the TPR domain (29,30). Although it is far from clear whether fatty acids are true physiological regulators of Ppp5, it is likely that other, as yet unknown, upstream factors will be found to regulate the phosphatase. Given the role uncovered here, it would not be surprising to find activation of Ppp5 either by radiation-induced signaling molecules, or via by-products of the DNA-damage process itself.
Clearly, the exact mechanisms that activate Ppp5 during DNA-damage stress or the direct targets of Ppp5 in the ATM survival mechanism remain unknown and will be the subject of much investigation. The Ppp5-deficient mice and cells we have generated may not only help to identify these missing factors, but may also contribute to the understanding of how genomic instability leads to tumorigenesis.
Acknowledgments
We wish to thank Dr. Shaolian Jing and Mr. William Carter of Indiana University Mouse Core for their superb assistance. We also thank Dr. Michael Chinkers for Ppp5 antibody. This study was supported in part by National Institute of Health grants DK73402 (W.S., E.S), DK70127 (E.S., W.S.), DK43867 (E.S.), and the Riley Children's Foundation (W.S.).
References
- 1.Shiloh Y, Lehmann AR. Nature cell biology. 2004;6(10):923–928. doi: 10.1038/ncb1004-923. [DOI] [PubMed] [Google Scholar]
- 2.Abraham RT. Genes Dev. 2001;15(17):2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 3.Pietenpol JA, Stewart ZA. Toxicology. 2002;181-182:475–481. doi: 10.1016/s0300-483x(02)00460-2. [DOI] [PubMed] [Google Scholar]
- 4.Kastan MB, Lim DS. Nature reviews. 2000;1(3):179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 5.Cohen PT. Trends in biochemical sciences. 1997;22(7):245–251. doi: 10.1016/s0968-0004(97)01060-8. [DOI] [PubMed] [Google Scholar]
- 6.Dean DA, Urban G, Aragon IV, Swingle M, Miller B, Rusconi S, Bueno M, Dean NM, Honkanen RE. BMC cell biology [electronic resource] 2001;2:6. doi: 10.1186/1471-2121-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ollendorff V, Donoghue DJ. J Biol Chem. 1997;272(51):32011–32018. doi: 10.1074/jbc.272.51.32011. [DOI] [PubMed] [Google Scholar]
- 8.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Embo J. 2001;20(21):6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi Y, Katoh H, Mori K, Negishi M. Curr Biol. 2002;12(15):1353–1358. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- 10.Gentile S, Darden T, Erxleben C, Romeo C, Russo A, Martin N, Rossie S, Armstrong DL. Proc Natl Acad Sci U S A. 2006;103(13):5202–5206. doi: 10.1073/pnas.0600080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. Nature cell biology. 2006;8(9):1011–1016. doi: 10.1038/ncb1465. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Bao S, Furumai R, Kucera KS, Ali A, Dean NM, Wang XF. Mol Cell Biol. 2005;25(22):9910–9919. doi: 10.1128/MCB.25.22.9910-9919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, Meek K, Cleaver JE, Chen DJ, Wabl M. Proc Natl Acad Sci U S A. 2004;101(5):1247–1252. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, Wang XF. Genes Dev. 2004;18(3):249–254. doi: 10.1101/gad.1176004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. Nature. 2001;411(6840):969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 16.Post SM, Tomkinson AE, Lee EY. Nucleic Acids Res. 2003;31(19):5568–5575. doi: 10.1093/nar/gkg765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Proc Natl Acad Sci U S A. 2000;97(19):10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. Science. 2000;287(5459):1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 19.Podust VN, Tiwari N, Ott R, Fanning E. The Journal of biological chemistry. 1998;273(21):12935–12942. doi: 10.1074/jbc.273.21.12935. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi M, Hirano A, Kumano T, Xiang SL, Mihara K, Haseda Y, Matsui O, Shimizu H, Yamamoto K. Genes Cells. 2004;9(4):291–303. doi: 10.1111/j.1356-9597.2004.00728.x. [DOI] [PubMed] [Google Scholar]
- 21.Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, Sancar A. Proc Natl Acad Sci U S A. 2003;100(4):1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou L, Cortez D, Elledge SJ. Genes Dev. 2002;16(2):198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Cell. 1996;86(1):159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 24.Chinkers M. Trends Endocrinol Metab. 2001;12(1):28–32. doi: 10.1016/s1043-2760(00)00335-0. [DOI] [PubMed] [Google Scholar]
- 25.Chen MX, Cohen PT. FEBS Lett. 1997;400(1):136–140. doi: 10.1016/s0014-5793(96)01427-5. [DOI] [PubMed] [Google Scholar]
- 26.Skinner J, Sinclair C, Romeo C, Armstrong D, Charbonneau H, Rossie S. J Biol Chem. 1997;272(36):22464–22471. doi: 10.1074/jbc.272.36.22464. [DOI] [PubMed] [Google Scholar]
- 27.Sinclair C, Borchers C, Parker C, Tomer K, Charbonneau H, Rossie S. The Journal of biological chemistry. 1999;274(33):23666–23672. doi: 10.1074/jbc.274.33.23666. [DOI] [PubMed] [Google Scholar]
- 28.Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M. Biochemistry. 2001;40(35):10485–10490. doi: 10.1021/bi010999i. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, Barford D. The EMBO journal. 2005;24(1):1–10. doi: 10.1038/sj.emboj.7600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramsey AJ, Chinkers M. Biochemistry. 2002;41(17):5625–5632. doi: 10.1021/bi016090h. [DOI] [PubMed] [Google Scholar]