


Abstract
Restriction endonucleases Ecl18kI and PspGI/catalytic domain of EcoRII recognize CCNGG and CCWGG sequences (W stands for A or T), respectively. The enzymes are structurally similar, interact identically with the palindromic CC:GG parts of their recognition sequences and flip the nucleotides at their centers. Specificity for the central nucleotides could be influenced by the strength/stability of the base pair to be disrupted and/or by direct interactions of the enzymes with the flipped bases. Here, we address the importance of these contributions. We demonstrate that wt Ecl18kI cleaves oligoduplexes containing canonical, mismatched and abasic sites in the central position of its target sequence CCNGG with equal efficiencies. In contrast, substitutions in the binding pocket for the extrahelical base alter the Ecl18kI preference for the target site: the W61Y mutant prefers only certain mismatched substrates, and the W61A variant cuts exclusively at abasic sites, suggesting that pocket interactions play a major role in base discrimination. PspGI and catalytic domain of EcoRII probe the stability of the central base pair and the identity of the flipped bases in the pockets. This ‘double check’ mechanism explains their extraordinary specificity for an A/T pair in the flipping position.
INTRODUCTION
Ecl18kI and EcoRII/PspGI restriction endonucleases recognize related nucleotide sequences 5′-CCNGG (N stands for A, T, G or C) and 5′-CCWGG (W stands for A or T), respectively. Ecl18kI (1) and PspGI (2) are single domain proteins. EcoRII shows modular organization and consists of three structural units: a catalytic core dimer, made from two copies of the C-terminal domain, and two regulatory N-terminal domains, that each bind a copy of the recognition sequence (3–5). The EcoRII catalytic unit (EcoRII-C) can be obtained by limited proteolysis and acts as a stand-alone restriction enzyme, which cuts at the 5′-CCWGG site (3,6). Ecl18kI, EcoRII-C and PspGI are evolutionarily related (7) and flip nucleotides at the centers of their pseudopalindromic target sequences CCNGG and CCWGG (1,8–10). Sequence and structural data indicate that conserved bases within the target sites are recognized analogously (5,7,11,12), but there are variations in the pockets that accommodate the flipped bases (1,2). In the co-crystal structure of Ecl18kI with DNA, the flipped bases are accommodated in pockets that are delineated by the side chains of R57 and the indole rings of W61 (1). These Ecl18kI residues coincide structurally with R222 and Y226 of EcoRII-C (1,4) and with E60 and F64 of PspGI [(2); Figure 1].
Figure 1.
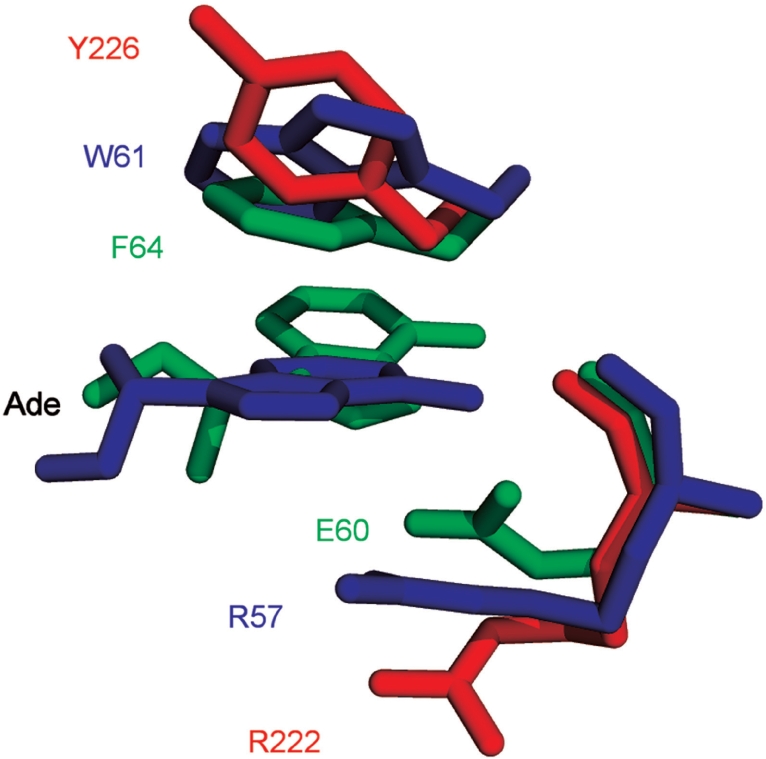
Binding pockets of Ecl18kI (blue), EcoRII-C (red) and PspGI (green). Coordinates for the Ecl18kI–DNA complex [2FQZ, (1)], the apo-form of EcoRII [1NA6, (4)] and the PspGI–DNA complex [3BM3, (2)] were used for the superposition.
Despite the similarity between Ecl18kI, EcoRII-C and PspGI, the specificities of these enzymes differ: Ecl18kI accepts both G/C and A/T base pairs at the center of the CCNGG sequence (13). In contrast, EcoRII-C (3,6) and PspGI (14) cut only when an A/T pair is present at the center of the target site. Why do the specificities for the flipped nucleotides differ? Some clues may be gained from a comparison with nucleotide flipping DNA modification and repair enzymes. Many of these make extensive contacts with an extrahelical base in the protein pocket (15–18). For example, uracil DNA glycosylase exploits every potential hydrogen bond donor and acceptor of the flipped uracil and makes a favorable edge–face aromatic interaction between a phenylalanine residue and the base ring (16). In addition, DNA repair enzymes, which flip out nucleotides to perform chemistry, often interrogate base pair stability while scanning DNA for the damaged site (19–21). A damaged or mismatched base pair is usually less stable than the canonical Watson–Crick base pair and therefore easier to flip (22,23). Not surprisingly, the N-methylpurine DNA glycosylase (24) and the mismatch-specific uracil DNA glycosylase (25,26) probe base pair stability searching for their target sites. By analogy, the specificity of the nucleotide flipping endonucleases could be due to direct interactions with the flipped bases, to the interrogation of base pair stability, or to a combination of both effects.
In order to distinguish these possibilities, we have tested the activity of the nucleotide flipping restriction endonucleases on oligoduplexes that contained canonical base pairs, mismatched bases or base analogs in the flipping position. The stability of the base pair in DNA depends on the hydrogen bonding and stacking interactions. Since these interactions are disrupted when nucleotides are extruded from the DNA helix, the base pair stability may affect Ecl18kI, EcoRII-C and PspGI function. The stability for base pairs obtained from statistical simulation follows the sequence: G/C > A/T > G/G > G/T = G/A > T/T = A/A > C/T ≥ A/C > C/C (27–31). Canonical Watson–Crick pairs G/C and A/T are most stable, while mismatches make less stable pairs. Our approach to characterize the interaction of the nucleotide flipping restriction endonucleases with their targets is similar to the strategy that has been used successfully for other classes of nucleotide flipping enzymes (19,20,24,25,32).
MATERIALS AND METHODS
Oligonucleotides
The 2-aminopurine and abasic site (1,3-propanediol spacer) containing oligodeoxynucleotides (Table 1) were purchased from Integrated DNA Technologies (Coralville, USA), other oligodeoxynucleotides were from Metabion (Martinsried, Germany). In order to assemble oligoduplexes (Table 1), appropriate oligodeoxynucleotides were mixed with complementary strands at equal molar ratio in the annealing buffer (33 mM Tris–acetate, pH 7.9 at 25°C, 66 mM potassium acetate), heated till boiling (or 85°C for modified oligoduplexes) and allowed to cool slowly over several hours to room temperature. A DNA labeling kit (Fermentas, Vilnius, Lithuania) was used for 5′-single or double strand labeling with [γ-33P]ATP (Hartmann Analytic, Braunschweig, Germany).
Table 1.
Oligoduplexes used in this study
| Oligoduplex | Sequencea | |
|---|---|---|
| Watson–Crick duplexes | A/T | 5′ CGCACGACTTCCTGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGACCTTCTCGTGCGTTG 5′ |
| G/C | 5′ CGCACGACTTCCCGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGGCCTTCTCGTGCGTTG 5′ | |
| Symmetrical mismatches | T/T | 5′ CGCACGACTTCCTGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGTCCTTCTCGTGCGTTG 5′ |
| A/A | 5′ CGCACGACTTCCAGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGACCTTCTCGTGCGTTG 5′ | |
| C/C | 5′ CGCACGACTTCCCGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGCCCTTCTCGTGCGTTG 5′ | |
| G/G | 5′ CGCACGACTTCCGGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGGCCTTCTCGTGCGTTG 5′ | |
| Asymmetric mismatches | G/T | 5′ CGCACGACTTCCTGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGGCCTTCTCGTGCGTTG 5′ |
| C/T | 5′ CGCACGACTTCCTGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGCCCTTCTCGTGCGTTG 5′ | |
| A/G | 5′ CGCACGACTTCCGGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGACCTTCTCGTGCGTTG 5′ | |
| A/C | 5′ CGCACGACTTCCCGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGGACCTTCTCGTGCGTTG 5′ | |
| 2-AP duplexes | 2/T | 5′ CGCACGCCTTCCTGGAAGCACACTA 3′ 3′ GCGTGCGGAAGG2CCTTCGTGTGAT 5′ |
| 2/A | 5′ CGCACGCCTTCC2GGAAGCACACTA 3′ 3′ GCGTGCGGAAGGACCTTCGTGTGAT 5′ | |
| 2/2 | 5′ CGCACGCCTTCC2GGAAGCACACTA 3′ 3′ GCGTGCGGAAGG2CCTTCGTGTGAT 5′ | |
| 2FSb | 5′ CGCACGCCTTCCTGGAAGCACACTA 3′ 3′ GCGTGCGGA2GGACCTTCGTGTGAT 5′ | |
| Abasic sites | −/A | 5′ CGCACGACTTCCAGGAAGAGCACGC 3′ 3′ GCGTGCTGAAGG-CCTTCTCGTGCGTTG 5′ |
| −/− | 5′ CGCACGACTTCC-GGAAGAGCACGC 3′ 3′ GCGTGCTGAAGG-CCTTCTCGTGCGTTG 5′ | |
| Non-specific | NSc | 5′ CGCACGACTTGTCACAAGAGCACGC 3′ 3′ GCGTGCTGAACAGTGTTCTCGTGCGTTG 5′ |
aEcl18kI/PspGI/EcoRII-C recognition site is in boldface; the central base pair of the recognition sequence is underlined; 2 stands for 2–aminopurine; a dash (−) marks the abasic site.
bOligoduplex 2FS contains 2-aminopurine introduced immediately adjacent to the target site.
cOligoduplex NS lacks the recognition sequence of Ecl18kI/PspGI/EcoRII-C.
Proteins
Wt Ecl18kI, the W61Y and W61A variants of Ecl18kI, PspGI and EcoRII-C were purified as described earlier (9). Their concentrations were determined from the absorption at 280 nm and refer to the dimers. Following extinction coefficients were calculated by ProtParam tool (http://www.expasy.ch/): PspGI—50 920 M−1 cm−1, EcoRII-C—43 240 M−1 cm−1, wt Ecl18kI—77 660 M−1 cm−1, W61Y—75 640 M−1 cm−1, W61A—72 660 M−1 cm−1.
Mutagenesis
The W61Y mutant was obtained similarly as described in ref. (9). Sequencing of the entire gene of the mutant confirmed that only the designed mutation had been introduced.
Gel mobility shift assay
33P-labeled oligoduplexes (Table 1) at 0.2 nM concentration were mixed in the binding buffer [(40 mM Tris–acetate (pH 8.3), 5 mM Ca(OAc)2, 0.1 mg/ml BSA, 10% (w/w) glycerol)] with increasing amounts of protein, incubated for 10 min at room temperature and reaction mixtures were analyzed on the nondenaturing 8% polyacrylamide gel (29:1, acrylamide/N,N′–methylenebisacrylamide) using 40 mM Tris–acetate (pH 8.3) supplemented with 5 mM calcium acetate as running buffer. Apparent Kd values were determined as described (5). Association constants Ka were calculated according to the equation Ka = 1/Kd.
Reactions with oligonucleotide duplexes
The Ecl18kI reactions were performed at 20°C by mixing manually, radiolabeled oligoduplexes (200 nM) with Ecl18kI (300 nM dimer) in the reaction buffer (33 mM Tris–acetate (pH 7.9 at 25°C), 66 mM potassium acetate, 10 mM magnesium acetate, 0.1 mg/ml BSA). Most of the EcoRII-C and PspGI reactions were performed in the same reaction buffer as above at 25°C and contained 200 nM of radiolabeled oligoduplexes and 1000 nM of enzyme (dimer). The samples were collected at timed intervals and quenched with loading dye solution (95% v/v formamide, 25 mM EDTA, 0.01% bromphenol blue). Some EcoRII-C reactions that were too fast to be measured by manual mixing were studied using a quench-flow device (KinTek, Austin, TX, USA): equal volumes of radiolabeled oligoduplexes (400 nM) and enzyme (2000 nM dimer) in the reaction buffer were mixed and quenched with 2.0 M HCl. The samples were neutralized by adding 3.5 M Tris and 3% SDS solution and mixed with loading dye solution. Separation of DNA hydrolysis products was performed by denaturing PAGE: the 20% polyacrylamide gel [acrylamide/N,N′–methylenebisacrylamide 29:1 (w/w)] in Tris–borate containing 8.5 M of urea was run at 30 V/cm. Radiolabeled DNA was detected and quantified by Cyclone Phosphor-Imager (Perkin-Elmer, Wellesley, MA, USA). When both oligoduplex strands were labeled, their cleavage rates could be independently monitored due to different strand lengths. In the case of DNA substrates containing 2-AP, cleavage of each DNA strand was monitored separately. Rate constants of DNA cleavage by PspGI and EcoRII-C were determined by fitting single exponentials to the time-courses of substrate depletion in the experiments. In the case of Ecl18kI, a biphasic decay of substrate was observed: half of substrate was cleaved rapidly and the remaining part much slower. Cleavage rate constants for Ecl18kI were determined by fitting a single exponential to the time–courses of substrate depletion of the fast phase.
Data analysis
The KYPLOT 2.0 software (33) was used for nonlinear regression analysis.
RESULTS
Strategy
In this study, we have measured the activity of nucleotide flipping restriction endonucleases on oligoduplexes that contained canonical base pairs, mismatched bases, base analogs or abasic sites in the flipping position. Altogether, 15 oligoduplexes with the CC(N/N)GG sequence (N stands for any base or an abasic site at the center of the target site) were used. Two of them were canonical oligoduplexes containing Watson–Crick base pairs A/T and G/C, while eight contained mismatches at the center (four symmetrical T/T, A/A, C/C, G/G and four mixed mismatches G/T, C/T, A/G, A/C). Together, these oligoduplexes represent all possible combinations of natural bases A, G, T and C at the center of the CC(N/N)GG sequence. Three additional oligoduplexes contained 2-AP at the center (Watson–Crick-like oligoduplex 2/T and the mismatched oligoduplexes 2/A and 2/2). The last two oligoduplexes, ‘−/A’ and ‘−/−’, carried, respectively, a single and a double abasic site generated by the introduction of the 1,3-propanediol spacer during the synthesis (Table 1). All substrates were tested with wt Ecl18kI, the Ecl18kI variants W61Y and W61A, and with EcoRII-C and PspGI.
Binding experiments
The affinity of Ecl18kI, its W61Y and W61A mutants, EcoRII-C and PspGI for all CC(N/N)GG sites was analyzed using a gel mobility shift assay (Figure 2). All DNA binding experiments were performed in the presence of Ca2+ ions, which do not support catalysis but are required for specific DNA binding (5,7,12). The apparent equilibrium association constant Ka values for DNA binding are presented in Figure 3 and Supplementary Table 1.
Figure 2.

Gel mobility shift analysis of the interactions between PspGI and oligoduplexes containing A/T (A), G/C (B) and G/G (C) base pairs at the center of CC(N/N)GG site. PspGI binding to nonspecific NS oligoduplex, which lacks the recognition site, is shown in (D). The binding reactions contained 33P-labeled oligoduplex (0.2 nM) and PspGI at concentrations indicated by each lane. Samples were analyzed by PAGE under nondenaturing conditions (see, Material and methods section).
Figure 3.

Binding of oligoduplexes containing different base pairs at the center of CC(N/N)GG sequences by wt Ecl18kI (A), W61Y (B) and W61A (C) mutants of Ecl18kI, EcoRII-C (D) and PspGI (E). Abbreviations below the bars specify the central base pair in the oligoduplex, 2 stands for 2-AP, a dash (−) marks the abasic site; oligoduplex NS lacks the recognition sequence of Ecl18kI/PspGI/EcoRII-C. WC stands for the Watson–Crick base pair; sMM, for the symmetrical mismatch; aMM, for the asymmetric mismatch; AB, abasic sites. Apparent Ka values determined for different DNA oligoduplexes by gel shift assay (see, Materials and methods section) are plotted in the diagrams.
Cleavage experiments
DNA cleavage experiments were performed in the buffer containing cofactor Mg2+ ions. Reactions were performed under the single turnover conditions. Enzyme was present in sufficient excess over substrate, so that all DNA was bound to the enzyme. Control experiments confirmed that further addition of enzyme did not alter the reaction rate (data not shown). Cleavage rate constants were determined for both strands of all oligoduplexes studied. In all cases, the rate constants for cutting both strands were equal, therefore only a single value is provided (Figure 4 and Supplementary Table 2). In some cases, digestion experiments were also performed with plasmid pEcoRII-2 [2 CC(A/T)GG and 8 CC(G/C)GG sites (6)] and dam−dcm− DNA from phage lambda (see Supplementary Figures 1 and 2).
Figure 4.

Cleavage of oligoduplexes containing different base pairs at the center of CC(N/N)GG sequences by wt Ecl18kI (A), W61Y (B) and W61A (C) mutants of Ecl18kI, EcoRII-C (D) and PspGI (E). The values of the cleavage rate constants for different DNA oligoduplexes are plotted in the diagrams. Rate constants are provided only for the cleavage of one strand in the oligoduplex, since cleavage rates of the second strand were similar. Abbreviations below and above the bars are the same as in Figure 3. DNA cleavage was performed under single-turnover conditions (see Materials and methods section). Reactions that were too slow in our experimental setup to measure the reaction rate are marked as (nd).
Wt Ecl18kI
Wt Ecl18kI binds all CC(N/N)GG oligoduplexes with the same affinity and cleaves them with similar rates in the range between 0.15 s−1 and 0.6 s−1 (Figures 3A and 4A). Thus, the enzyme tolerates any combination of the natural bases, 2-AP or even base deletion at the central position of the recognition sequence at the DNA binding and the cleavage steps.
Ecl18kI variant W61Y
This mutant was designed to make the base binding pocket of Ecl18kI more similar to the one in EcoRII-C. 2-AP fluorescence confirmed that it retains the ability to flip 2-AP if this adenine analog is placed at the center of the recognition sequence (Supplementary Figure 3). The mutant binds all mismatched and abasic oligoduplexes with the affinity similar to wt Ecl18kI (Figure 3A, B). However, the oligoduplexes with the Watson–Crick pairs A/T, G/C and 2/T in the central position of the CC(N/N)GG sequence, are smeared in the gel indicating that the protein–DNA complexes are unstable (data not shown). Quantitatively, the Ka values for W61Y mutant binding to the oligoduplexes with Watson–Crick paired bases at the center are 2- to 10-fold lower in comparison to the mismatched substrates, which is not observed with the wild-type enzyme.
W61Y mutant cleaves the duplexes with canonical Watson–Crick A/T and G/C pairs nearly 100-fold slower compared to the majority of the mismatched substrates (Figure 4B). At the same time, the cleavage rates of the mismatched oligoduplexes (except for the T-containing mismatches) and abasic oligoduplexes −/A and −/− approach those of wt Ecl18kI (Figure 4A and B). Surprisingly, the W61Y mutant cuts mismatched oligoduplex T/T even slower than the canonical A/T and G/C oligoduplexes. The cleavage rates of other T-containing oligoduplexes G/T, C/T and 2/T are also slow compared to their T-lacking equivalents (Figure 4B). Moreover, digestion experiments with the pEcoRII-2 plasmid suggest that the W61Y mutant preferentially cuts at CC(G/C)GG rather than at CC(A/T)GG sites (Supplementary Figure 1). Thus, a single amino acid exchange in the binding pocket alters the specificity of Ecl18kI.
Ecl18kI variant W61A
This mutant of Ecl18kI lacks one of the walls of the binding pocket (provided the pocket does not collapse) and has been shown to flip 2-AP at the center of the CC(N/N)GG sequence (9). Oligoduplexes with a Watson–Crick base pair in the center bind to this Ecl18kI variant 10- to 30-fold more weakly than those with a mismatch (Figure 3C). The W61A mutant does not cleave phage lambda DNA (Supplementary Figure 2), the oligoduplexes with canonical Watson–Crick base pairs or most duplexes with mismatches (Figure 4C). Residual activity (∼2000-fold lower compared to wt Ecl18kI) is observed only for the C/C, A/C, 2/A and 2/2 mismatched oligoduplexes. Surprisingly, the oligoduplex with the doubly abasic CC(−/−)GG site is a very good substrate (Figure 4A and C). Thus, the Ecl18kI variant W61A has the new specificity that differs markedly from those of wt Ecl18kI, EcoRII-C and PspGI.
EcoRII-C and PspGI
Unlike Ecl18kI, which recognizes the CCNGG site, EcoRII-C and PspGI are specific for the CCWGG sequence (14,34,35). Gel shift experiments indicate that EcoRII-C (Figure 3D) and PspGI (Figure 3E) have ∼10- and 100-fold lower affinity for the oligoduplex with a G/C base pair in the center in comparison to the A/T oligoduplex. Surprisingly, EcoRII-C and PspGI bind oligoduplexes containing the symmetrical C/C and G/G mismatches at the center of the recognition sequence with the same affinity as the canonical A/T oligoduplex (Figures 2, 3D and E). The binding affinity for other oligoduplexes is also similar to that of the cognate A/T substrate (Figure 3D and E). Thus, only the Watson–Crick G/C base pair at the center of the CC(N/N)GG sequence decreases the affinity to EcoRII-C and PspGI significantly. All other oligoduplexes including symmetrical mismatches C/C and G/G are bound as well as the canonical A/T oligoduplex.
EcoRII-C and PspGI cleave duplexes with a canonical A/T or symmetrical mismatched A/A or T/T pair at maximal rates. The enzymes digest the asymmetric mismatched substrates G/T, C/T, A/G and A/C poorly and the G/C, C/C and G/G oligoduplexes barely if at all (Figure 4D and E). Slow cleavage of the G/C oligoduplex correlates with the low binding affinity. In contrast, >1000-fold lower cleavage rate for the duplexes with G/G and C/C mismatches compared to the oligoduplex with the A/T match is remarkable, because all three duplexes are bound with similar affinity. The introduction of single and double abasic sites that plays no role at the DNA binding step (Figures 3 and 4), has major impact on the DNA cleavage. Indeed, a single abasic site slows down EcoRII-C and PspGI cleavage rates ∼30- and 25-fold, respectively. The introduction of two abasic sites reduces the cleavage rate 3000-fold for EcoRII-C and 500-fold for PspGI (Figure 4D and E). This effect is not observed for Ecl18kI, which does not require the central bases at the DNA binding or the cleavage step.
The specificities of EcoRII-C and PspGI are generally similar. For both enzymes, asymmetric mismatched oligoduplexes with G in the center are poorer substrates than their counterparts with C in this position. The effect is more pronounced for PspGI: the pocket of this enzyme selects against G so stringently that the G/G oligoduplex is cleavage resistant in our experimental conditions. PspGI is also more sensitive to the unnatural 2-AP base in the central position of the recognition sequence than EcoRII-C. Incorporation of this base in one or both strands (instead of a canonical A/T pair) slows down PspGI ∼10-fold and >100-fold, respectively (Figure 4E). In contrast, EcoRII-C cleaves these duplexes as efficiently as the duplex with the canonical A/T pair (Figure 4D).
EcoRII-C/PspGI versus Ecl18kI
The ratio of the specificity constants kchem × Ka is a useful index for comparing the relative reaction rates when alternative, competing substrates are present. Although kchem(A/T)/kchem(G/C) and Ka(A/T)/Ka(G/C) ratios differ between EcoRII-C and PspGI, their products are similar (Figure 5). They indicate a 3 × 106-fold kchem × Ka preference for CC(A/T)GG over CC(G/C)GG. This number is comparable to the ratio of specificity constants for substrates and nonsubstrates of orthodox restriction endonucleases like EcoRI and EcoRV (36,37).
Figure 5.

Preference for the CC(A/T)GG sequence by Ecl18kI, EcoRII-C and PspGI. The ratios of Ka (association constant), k (cleavage rate constant corresponding to kchem) and k × Ka (product of the cleavage rate constant and equilibrium association constant) for the CC(A/T)GG and CC(G/C)GG sequences for the Ecl18kI, EcoRII-C and PspGI. The same ratios are presented for EcoRV (37) and EcoRI (36) for the DNA sequences indicated above the diagram.
DISCUSSION
Wt Ecl18kI—no specificity
Our studies demonstrate that wt Ecl18kI binds all substrates with the same affinity and cleaves them at a similar rate (Figures 3A and 4A) indicating that phosphodiester bond cleavage is independent of the nature of the extrahelical base. The absence of a central base pair in the abasic oligoduplexes does not interfere with binding or catalysis. Thus, the extruded base is by itself not required for catalysis. It is likely that Ecl18kI has evolved a nonspecific pocket for the extrahelical base to pay off the energetic costs for extrusion of the base from the DNA stack.
Ecl18kI mutant W61Y—altered specificity
The conservative W61Y exchange in the pocket of Ecl18kI has surprisingly strong effects on its substrate preference. In contrast to wt Ecl18kI, the W61Y variant distinguishes Watson–Crick base pairs from other base combinations in the center of the target site already at the DNA binding step (Figure 3B), presumably due to the higher energetic cost for unstacking a Watson–Crick base pair.
The T-containing mismatched oligoduplexes bind tightly to Ecl18kI W61Y, but are cleaved very slowly (Figures 3B and 4B) suggesting that changes in the binding pocket imposes restrictions on the cleavage of substrates containing the T base at the central position. The specific effect of the T base on the cleavage implies that there might be a spatial position for this base that is compatible with binding, but not catalysis. Different conformations of the flipped out base have indeed been reported before for uracil DNA glycosylase and 8-oxoguanine DNA glycosylase I (18,38).
Ecl18kI mutant W61A—new specificity for the abasic CC(−/−)GG site
The binding phenotype of the Ecl18kI W61A variant is similar to that of the W61Y mutant (Figure 3B and C), but the discrimination against Watson–Crick pairs is more pronounced. Although the enzyme binds mismatched duplexes fairly well, it does not cleave them. The fluorescence of 2-AP in the duplex with the Watson–Crick 2/T pair increases strongly upon addition of the Ecl18kI W61A variant (more strongly than upon addition of the wild-type) (9), but the duplex is not cleaved by the mutant enzyme. Failure of the mutant protein to cleave this and similar substrates could be taken as a further evidence for a nonproductive binding mode of the flipped bases in the Ecl18kI pockets that does not lead to cleavage. This catalytically incompetent state may be related to the different conformation of the extrahelical base or to the unstable binding of the unstacked base in the protein pocket. It would explain the otherwise surprising finding that a complete elimination of the bases at the center of the target site results in the dramatic increase (nearly four orders of the magnitude!) of the mutant activity to a level comparable to that of wt Ecl18kI. It would also be consistent with the observation that abasic oligoduplexes are cleaved by wt Ecl18kI and the W61A mutant at the same position as the canonical oligoduplexes (before the first C; data not shown). In summary, the single W61A mutation in the binding pocket transforms Ecl18kI into a site-specific nuclease that cuts only at abasic CC(−/−)GG sites.
PspGI and EcoRII-C—inborn specificities
EcoRII-C and PspGI are specific for the CCWGG sites. According to both structural and 2-AP fluorescence data both flip central nucleotides in their recognition sequence [(2,9); D. Golovenko et al., unpublished data]. EcoRII-C and PspGI bind poorly to the CC(G/C)GG oligoduplex (Figure 3D and E), but surprisingly, duplexes with C/C and G/G mismatches are bound nearly as well as duplexes with canonical A/T or the mismatched T/T and A/A pairs in the center. Thus, the binding data alone suggest that the discrimination between the A/T and G/C base pairs by EcoRII-C and PspGI at the binding step may arise from the difference in the central base pair strength/stability. However, two lines of the experimental evidence argue that base pair strength is perhaps not the major factor in EcoRII-C and PspGI specificity. First, in vivo experiments suggest that PspGI is able to flip the C within the Watson–Crick base pair in the central position (8). Second, the chloroacetaldehyde assay for base flipping shows increased accessibility of the central C base in the PspGI complex (10). Moreover, cleavage experiments demonstrate that mismatched oligoduplexes G/G and C/C, which have a much weaker base pair at the center and therefore require less energy to disrupt it, are still poorly cleaved despite their high affinity to PspGI and EcoRII-C. Remarkably, the W61Y mutant of Ecl18kI displays a similar phenotype for the T-containing oligoduplexes. Low cleavage rates of the G/G and C/C oligoduplexes suggest that PspGI/EcoRII-C have developed an additional mechanism to discriminate between A/T and G/C base pairs within the binding pockets. In principle, one may assume that either the flipped C or G base is expelled from the EcoRII-C/PspGI binding pocket or/and that additional binding contacts are provided for the A and T in comparison to the G and C bases. Of note, substrates containing the asymmetric mismatches G/T, C/T, A/G and A/C at the center are cleaved faster by EcoRII-C/PspGI than the symmetrical oligoduplexes G/G and C/C (Figure 4D and E). Both strands of the asymmetric mismatched oligoduplexes are cleaved with similar rates (data not shown). Therefore, the binding of the T or A base in the binding pocket of one monomer should partially offset the deleterious effect of the G or C bases on the cleavage rate, which implies the cooperative interaction between monomers during catalysis. However, one cannot exclude that binding of the G and C bases in the pockets may be compromised at the same time. Furthermore, while wt Ecl18kI cleaves canonical and abasic oligoduplexes with the same rate, EcoRII-C and PspGI show much lower cleavage rates for abasic sites suggesting that interactions with the flipped out base are required in catalysis.
A double-check mechanism for nucleotide flipping restriction endonucleases
Apart from a comparison of binding constants and cleavage rates of Ecl18kI, EcoRII-C and PspGI, our data provide yet another argument for the combined influence of base pair strength and pocket specificity: the estimated stability difference for the G/C and A/T containing duplexes used in this study is 3.1 kcal/mol (http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/). Translation of this value into the kchem(A/T)/kchem(G/C) or Ka(A/T)/Ka(G/C) ratio should yield at maximum ∼200-fold difference in the specificity factors, which is at least three orders of magnitude smaller than the observed value (Figure 5). We therefore conclude again that base pair stability differences alone cannot explain the very stringent selection for A/T and against G/C pairs in the flipping position. We suggest that EcoRII-C and PspGI use a ‘double-check’ mechanism to distinguish A/T from G/C pairs. According to this mechanism, the enzymes ‘sense’ the strength/stability of a base pair (in the base binding step) and then ‘check’ the identity of the base in the binding pocket to assure that only correct substrates are cleaved.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
FUNDING
Lithuania State Science and Studies Foundation (T-14/07 and T-27/08); Polish Ministry of Science and Higher Education (grant No. 0295/B/PO1/2008/34). Funding for open access charge: Lithuania Science and Studies Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Saulius Klimasauskas for the KinTek access, to Giedre Tamulaitiene for help with figures and to Giedrius Sasnauskas and Honorata Czapinska for a critical reading of the article and comments. The authors also thank New England Biolabs for providing the recombinant PspGI strain and Monika Reuter (Humboldt University, Berlin) for sharing EcoRII constructs. M.B. acknowledges an EMBO/HHMI Young Investigator Award.
REFERENCES
- 1.Bochtler M, Szczepanowski RH, Tamulaitis G, Grazulis S, Czapinska H, Manakova E, Siksnys V. Nucleotide flips determine the specificity of the Ecl18kI restriction endonuclease. EMBO J. 2006;25:2219–2229. doi: 10.1038/sj.emboj.7601096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szczepanowski RH, Carpenter MA, Czapinska H, Zaremba M, Tamulaitis G, Siksnys V, Bhagwat AS, Bochtler M. Central base pair flipping and discrimination by PspGI. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mucke M, Grelle G, Behlke J, Kraft R, Kruger DH, Reuter M. EcoRII: a restriction enzyme evolving recombination functions? EMBO J. 2002;21:5262–5268. doi: 10.1093/emboj/cdf514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou XE, Wang Y, Reuter M, Mucke M, Kruger DH, Meehan EJ, Chen L. Crystal structure of type IIE restriction endonuclease EcoRII reveals an autoinhibition mechanism by a novel effector-binding fold. J. Mol. Biol. 2004;335:307–319. doi: 10.1016/j.jmb.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Tamulaitis G, Mucke M, Siksnys V. Biochemical and mutational analysis of EcoRII functional domains reveals evolutionary links between restriction enzymes. FEBS Lett. 2006;580:1665–1671. doi: 10.1016/j.febslet.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Tamulaitis G, Sasnauskas G, Mucke M, Siksnys V. Simultaneous binding of three recognition sites is necessary for a concerted plasmid DNA cleavage by EcoRII restriction endonuclease. J. Mol. Biol. 2006;358:406–419. doi: 10.1016/j.jmb.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 7.Tamulaitis G, Solonin AS, Siksnys V. Alternative arrangements of catalytic residues at the active sites of restriction enzymes. FEBS Lett. 2002;518:17–22. doi: 10.1016/s0014-5793(02)02621-2. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter M, Divvela P, Pingoud V, Bujnicki J, Bhagwat AS. Sequence-dependent enhancement of hydrolytic deamination of cytosines in DNA by the restriction enzyme PspGI. Nucleic Acids Res. 2006;34:3762–3770. doi: 10.1093/nar/gkl545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamulaitis G, Zaremba M, Szczepanowski RH, Bochtler M, Siksnys V. Nucleotide flipping by restriction enzymes analyzed by 2-aminopurine steady-state fluorescence. Nucleic Acids Res. 2007;35:4792–4799. doi: 10.1093/nar/gkm513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daujotyte D, Liutkeviciute Z, Tamulaitis G, Klimasauskas S. Chemical mapping of cytosines enzymatically flipped out of the DNA helix. Nucleic Acids Res. 2008;36:e57. doi: 10.1093/nar/gkn200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pingoud V, Kubareva E, Stengel G, Friedhoff P, Bujnicki JM, Urbanke C, Sudina A, Pingoud A. Evolutionary relationship between different subgroups of restriction endonucleases. J. Biol. Chem. 2002;277:14306–14314. doi: 10.1074/jbc.M111625200. [DOI] [PubMed] [Google Scholar]
- 12.Pingoud V, Conzelmann C, Kinzebach S, Sudina A, Metelev V, Kubareva E, Bujnicki JM, Lurz R, Luder G, Xu SY, et al. PspGI, a type II restriction endonuclease from the extreme thermophile Pyrococcus sp.: structural and functional studies to investigate an evolutionary relationship with several mesophilic restriction enzymes. J. Mol. Biol. 2003;329:913–929. doi: 10.1016/s0022-2836(03)00523-0. [DOI] [PubMed] [Google Scholar]
- 13.Den’mukhametov MM, Zakharova MV, Kravets AN, Pertsev AV, Sineva EV, Repik AV, Beletskaia IV, Gromova ES, Solonin AS. [Characteristics of a plasmid bearing a gene of a restriction modification type II system–the SsoII isoschizomer] Mol. Biol. 1997;31:831–838. [PubMed] [Google Scholar]
- 14.Morgan R, Xiao J.-P, Xu S.-Y. Characterization of an extremely thermostable restriction enzyme, PspGI, from a Pyrococcus strain and cloning of the PspGI restriction-modification system in Escherichia coli. Appl. Environ. Microbiol. 1998;64:3669–3673. doi: 10.1128/aem.64.10.3669-3673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 16.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl Acad. Sci. USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 18.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krosky DJ, Schwarz FP, Stivers JT. Linear free energy correlations for enzymatic base flipping: how do damaged base pairs facilitate specific recognition? Biochemistry. 2004;43:4188–4195. doi: 10.1021/bi036303y. [DOI] [PubMed] [Google Scholar]
- 20.Krosky DJ, Song F, Stivers JT. The origins of high-affinity enzyme binding to an extrahelical DNA base. Biochemistry. 2005;44:5949–5959. doi: 10.1021/bi050084u. [DOI] [PubMed] [Google Scholar]
- 21.Stivers JT. Extrahelical damaged base recognition by DNA glycosylase enzymes. Chemistry. 2007;14:786–793. doi: 10.1002/chem.200701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuxreiter M, Luo N, Jedlovszky P, Simon I, Osman R. Role of base flipping in specific recognition of damaged DNA by repair enzymes. J. Mol. Biol. 2002;323:823–834. doi: 10.1016/s0022-2836(02)00999-3. [DOI] [PubMed] [Google Scholar]
- 23.Cao C, Jiang YL, Krosky DJ, Stivers JT. The catalytic power of uracil DNA glycosylase in the opening of thymine base pairs. J. Am. Chem. Soc. 2006;128:13034–13035. doi: 10.1021/ja062978n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas T, Clos LJII, SantaLucia J., Jr, Mitra S, Roy R. Binding of specific DNA base-pair mismatches by N-methylpurine-DNA glycosylase and its implication in initial damage recognition. J. Mol. Biol. 2002;320:503–513. doi: 10.1016/S0022-2836(02)00519-3. [DOI] [PubMed] [Google Scholar]
- 25.Valinluck V, Liu P, Burdzy A, Ryu J, Sowers LC. Influence of local duplex stability and N6-methyladenine on uracil recognition by mismatch-specific uracil-DNA glycosylase (Mug) Chem. Res. Toxicol. 2002;15:1595–1601. doi: 10.1021/tx020062y. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Theruvathu JA, Darwanto A, Lao VV, Pascal T, Goddard W., III, Sowers LC. Mechanisms of base selection by the Escherichia coli mispaired uracil glycosylase. J. Biol. Chem. 2008;283:8829–8836. doi: 10.1074/jbc.M707174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peyret N, Seneviratne PA, Allawi HT, SantaLucia J., Jr Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 28.Allawi HT, SantaLucia J., Jr Thermodynamics of internal C.T mismatches in DNA. Nucleic Acids Res. 1998;26:2694–2701. doi: 10.1093/nar/26.11.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allawi HT, SantaLucia J., Jr Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 30.Allawi HT, SantaLucia J., Jr Nearest-neighbor thermodynamics of internal A.C mismatches in DNA: sequence dependence and pH effects. Biochemistry. 1998;37:9435–9444. doi: 10.1021/bi9803729. [DOI] [PubMed] [Google Scholar]
- 31.Tawa K, Knoll W. Mismatching base-pair dependence of the kinetics of DNA-DNA hybridization studied by surface plasmon fluorescence spectroscopy. Nucleic Acids Res. 2004;32:2372–2377. doi: 10.1093/nar/gkh572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klimasauskas S, Roberts RJ. M.HhaI binds tightly to substrates containing mismatches at the target base. Nucleic Acids Res. 1995;23:1388–1395. doi: 10.1093/nar/23.8.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshioka K. KyPlot—a user-oriented tool for statistical data analysis and visualization. CompStat. 2002;17:425–437. [Google Scholar]
- 34.Bigger CH, Murray K, Murray NE. Recognition sequence of a restriction enzyme. Nat. New Biol. 1973;244:7–10. doi: 10.1038/newbio244007a0. [DOI] [PubMed] [Google Scholar]
- 35.Boyer HW, Chow LT, Dugaiczyk A, Hedgpeth J, Goodman HM. DNA substrate site for the EcoRII restriction endonuclease and modification methylase. Nat. New Biol. 1973;244:40–43. doi: 10.1038/newbio244040a0. [DOI] [PubMed] [Google Scholar]
- 36.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the EcoRI endonuclease—DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 37.Engler LE, Welch KK, Jen-Jacobson L. Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol. 1997;269:82–101. doi: 10.1006/jmbi.1997.1027. [DOI] [PubMed] [Google Scholar]
- 38.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.