

Abstract
Avian Pancreatic Polypeptide is a 36 residue protein that exhibits a tertiary fold. Results of previous experimental and computational studies indicate that the structure of aPP is stabilized more by non-bonded interactions than by the hydrophobic effect. Aromatic residues are known to participate in a variety of long range non-bonded interactions, with both backbone atoms and the atoms of other side-chains, which could be responsible, in part, for the stability of both the local secondary structure and the tertiary fold. The effect of these aromatic interactions on the stability of aPP was calculated using BHandHLYP/cc-pVTZ. Aromatic residues were shown to participate in multiple hydrogen bonded and weakly polar interactions in the secondary structure. The energies of the weakly polar interactions are comparable with those of hydrogen bonds. Aromatic residues were also shown to participate in multiple weakly polar interactions across the tertiary fold, again with energies similar to those of hydrogen bonds.
Keywords: Aromatic residues, Avian Pancreatic Polypeptide, Density functional theory, Tertiary fold, Weakly polar interactions
Introduction
Avian Pancreatic Polypeptide (aPP), is a short protein with a tertiary fold formed by the packing of a polyproline-II helix (PPII) on an α-helix [1] (Figure 1). Its relatively small size makes it ideal for computational study and several investigators have used it as a model to study protein folding and for prediction of structure [2-6]. Vibrational and electronic circular dichroism spectroscopies demonstrated that, when the fragments of aPP representing the PPII helix and the α-helix, aPP(1-11)-NH2 and Ac-aPP(12-36), respectively, were placed in solution in equal concentrations, they refolded into the conformation of the full structure [7].
Fig. 1.
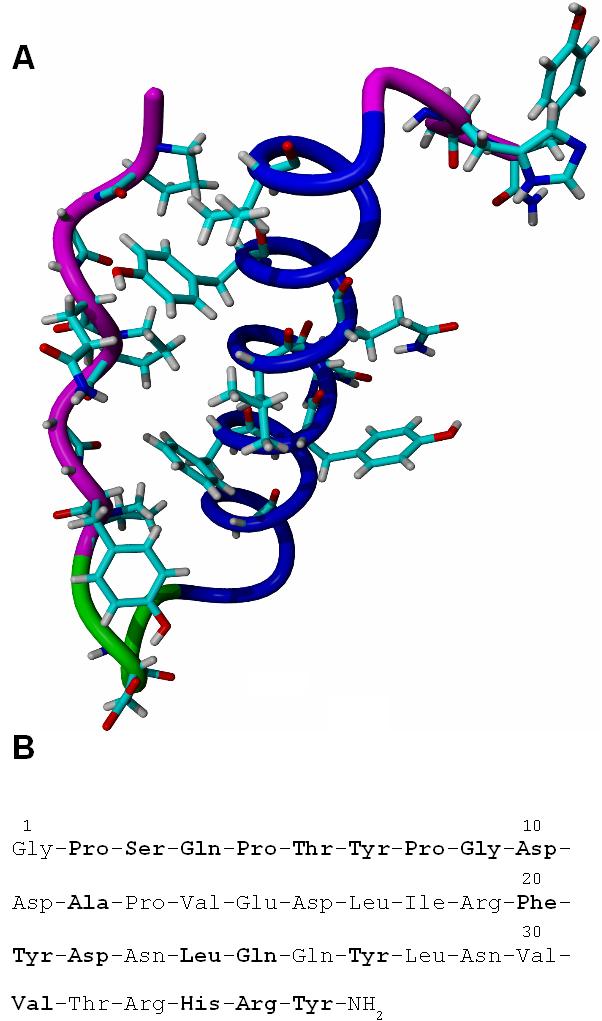
A. Backbone structure of aPP with interacting side chains and backbone atoms displayed. The N-terminal PPII helix is magenta, the turn structure is green and the α-helix is dark blue. B. The primary structure of aPP, interacting residues are in boldface.
A molecular dynamics (MD) simulation of the conformation of aPP by Zhang and associates [8] showed that the structure of aPP appears to be stabilized more by electrostatic interactions than by the hydrophobic effect. These stabilizing interactions are most likely to be weakly polar non-bonded interactions [9]. Aromatic side-chains, in particular, are known to participate in a variety of weakly polar interactions including those with other side-chains, such as those of Pro, Lys, Arg and aliphatic residues [10-13]. Aromatic sides-chains can also participate in weakly polar interactions with the peptide backbone [14].
The goal of this investigation was to determine the effect of aromatic residues on the structural stability of aPP by calculating the strength of the non-bonded interactions in which they participate, using DFT methods.
Methods
Initial structure
The structure of aPP (code 2BF9) used in this study was obtained from the Protein Data Bank [15].
Determination of non-bonded interactions
A “weakly polar” interaction was assigned when any backbone Hα, H, or O atoms, or any side chain atoms were within 6.0 Å of the centroid of an aromatic ring [16]. A mixed interaction was assigned when the criteria for a weakly polar interaction are satisfied and an atom of the aromatic residue participated in at least one hydrogen bond. A hydrogen bonded only interaction was assigned when the aromatic residue had an atom participating in at least one hydrogen bond but the ring was not close enough for a weakly polar interaction with another atom. Further classification of interactions was based on whether residues stabilized either local and/or secondary structure or the interaction between the α-helix and the PPII helix.
Interaction energy calculations
Intramolecular interaction energy calculations were performed at the BHandHLYP/cc-pVTZ level of theory because it closely approximates high-level results for weakly polar interactions [14,17]. The BHandHLYP functional used in this study was expressed as in ref. 14:
| (1) |
where and ESX are the exact HF and local Slater exchange, respectively, and is the Lee-Yang-Parr correlation functional.
To calculate intermolecular interaction energy between the PPII - and α-helices, an indirect approach was used. The aromatic side-chains of Phe20 and Tyr27 were removed and it was assumed that the interaction energy between the secondary structural elements is additive and thus, it can be calculated as follows:
| (2) |
where ΔEi(Phe20∨Tyr27) is the energy of the ith pairwise, tertiary structure-stabilizing interaction which involves Phe20 or Tyr27 (Table 2) and ΔEint(FM) is the interaction energy between the fragmented and mutated structures which were generated as described below. Firstly, aPP was fragmented into Gly-Pro-Ser-Gln-Pro-Thr-Tyr-Pro-Gly-Asp-NHMe and N-Ac-Val-Glu-Asp-Leu-Ile-Arg-Phe-Tyr-Asn-Asp-Leu-Gln-Gln-Tyr-Leu-Asn-Val-Val-NHMe. Secondly, residues which had side-chains far from the interacting surfaces of the fragments were replaced with Ala. Finally, Phe20 and Tyr27 were replaced with Gly, resulting in two fragments, Gly-Pro-Ala-Gln-Pro-Ala-Tyr-Pro-Gly-Asp-NHMe (PPIIfrag) and N-Ac-Ala-Ala-Asp-Leu-Ala-Ala-Gly-Ala-Ala-Asp-Leu-Ala-Gln-Gly-Leu-Ala-Val-Val-NHMe (αfrag) as shown in Figure 2. The positions of the introduced hydrogens were then optimized at the HF/3-21G level of theory and the interaction energies were calculated with the BHandHLYP/6-31+G** method. For interactions involving non-adjacent residues, the Boys and Bernardi basis set superposition error (BSSE) correction was used [18]. The peptide bond (PB) between Pro-Xaa, where Xaa was Ser3, Thr6, or Gly9 was not broken so as to preserve the electronic structure of the backbone. The energies of the interaction of the aromatic side-chains of Phe20 and Tyr27, respectively, with the peptide bonds of Thr6Tyr7 (PB6) and of Ser3Gln4 (PB3) were counted twice. Therefore the energies of interaction of the side-chains with the overlap structure, represented by CH3-NH-C(=O)-CH3, were subtracted from the total interaction energy. For interactions involving adjacent residues, a rotation method previously described [14] was used to correct the BSSE.
Table II.
Energies (kcal · mol-1) of non-bonded interactions which stabilize tertiary structure.
| Interactions | ΔEint |
|---|---|
| Pro8Gly9-Ala12 | -8.71 |
| Phe20-Pro5Thr6 | -6.05 |
| Phe20-Tyr7Pro8 | -5.25 |
| Tyr27-Pro2Ser3 | -7.43 |
| Tyr27-Gln4Pro5 | -5.53 |
| Phe20-PB6a | 3.42 |
| Tyr27-PB3a | 2.22 |
| Total | -27.33 |
For doubly counted peptide bonds (PB), see methods.
Fig. 2.

Structure of the PPIIfrag and αfrag. The color scheme is the same as Figure 1 except that the Gly residues which replace residues Phe20 and Tyr27 are red.
Program packages
The Jaguar V5.5 release 11 and V6.0 release 11 (Schrödinger LLC, Portland, OR), and Gaussian 03 (Revision C.01) program packages were used for all calculations. YASARA (http://www.yasara.org) was used for visualization and the preparation of figures.
Results and Discussion
Tyr7, Phe20, Tyr21, Tyr27 and Tyr36 participated in all three categories of non-bonded interactions (Table 1). Residues Phe20, Tyr21 and Tyr27 appear to have a role in stabilizing the α-helix, whereas Tyr7 and Tyr36 appear to participate in stabilizing local structure.
Table I.
Energies (kcal · mol-1) of non-bonded interactions which stabilize secondary and/or local structure.
|
Weakly polar interactions |
Mixed interactions |
H-bonded interactions |
|||
|---|---|---|---|---|---|
| Interacting residues | ΔEint | Interacting residues | ΔEint | Interacting residues | ΔEint |
| Tyr7-Pro8Gly9 | -14.17 | Tyr7-Asp10 | -24.76 | Phe20-Leu17 | -18.16 |
| Tyr21-Asp22 | -12.74 | Phe20-Leu24 | -13.00 | ||
| Tyr36-His34Arg35 | -2.33 | Tyr21-Gln25 | -13.11 | ||
| Tyr27-Val31 | -13.07 | ||||
The energies of all three categories of interaction which stabilize secondary and local structure are of the same order of magnitude. Furthermore, the energies of the weakly polar interactions between Phe20 and Pro5Thr6 and between Tyr27 and Pro2Ser3, were similar to those calculated for similar interactions which ranged from -4.35 to -4.80 kcal · mol-1 in a model helix [14]. Previously [19], it was found that the MD simulations estimate of the aromatic-backbone amide interaction energy is between -0.5 and -2.22 kcal · mol-1 whereas the quantum chemical calculation here shows that the energy of the interaction between Tyr27 and Pro2Ser3 is -7.43 kcal · mol-1. Thus, while the MD simulations are appropriate for estimation of geometries, the present level of theory is the minimum needed for determination of interaction energies. The weakly polar interactions of the aromatic side-chains of residues Phe20 and Tyr27 stabilize the tertiary fold of aPP.
Additionally, a hydrogen bond and three non-canonical CH..O hydrogen bonds [20] between Pro8Gly9 and Ala12 (Figure 3) contribute to the fold stability (Table 2).
Fig. 3.

Hydrogen bonds of residues Pro8Gly9 and Ala12. The hydrogen bond is indicated with a dashed line, and the three non-canonical CH..O hydrogen bonds are indicated with solid lines.
The energies of individual interactions across the tertiary fold are comparable with the lower energies of strong hydrogen bonds (-4 to -15 kcal · mol-1) [21]. They are also comparable with the energies, about -5.0 kcal · mol-1, found for other structures including the core of rubredoxin [22]. The interaction energies of the doubly counted peptide bonds, PB3 and PB6, respectively, were -2.22 kcal · mol-1 and -3.42 kcal · mol-1. After correction the total energy of the interactions which stabilize the tertiary fold formed by the α-helix and the PPII-helix is -27.33 kcal · mol-1. When Phe20 and Tyr27 were removed from the α-helical fragment, the interaction energy between PPIIfrag and αfrag, ΔEint(FM), was repulsive (20.88 kcal · mol-1). According to eq. 2, the interaction energy that stabilizes the tertiary fold is -6.45 kcal · mol-1.
The functional groups and distances of all interactions are given in Table 3. With few exceptions, they are between 2 and 5 Å, in close agreement with distances observed in a PDB search of proteins with weakly polar interactions [16], though Burley and Petsko [9] stated that distances of weakly polar interactions can be as long as 9 Å. Findings here are also in agreement with the conclusion following a previous clustering analysis [10] that Ar-Pro interactions can constrain local conformations in proteins.
Table III.
Distances (Å) of non-bonded interactionsa.
| Interacting residues | Functional group 1 | Functional group 2 | Distance | Type |
|---|---|---|---|---|
| Interactions which stabilize local and/or secondary structure | ||||
| Tyr7-Pro8Gly9 | Ar | Hδ of Pro8 | 3.58 | Weakly Polar |
| Tyr7-Pro8Gly9 | Ar | N-H of Gly9 | 2.99 | Weakly Polar |
| Tyr7-Asp10 | Ar-OH | Oδ | 1.87 | Mixed |
| Tyr7-Asp10 | Ar | PB9b | 4.38 | Mixed |
| Phe20-Leu17 | N-H of Tyr21 | C=O of Leu17 | 2.05 | H-bonded |
| Phe20-Leu17 | N-H of Phe20 | C=O of Asp16 | 2.19 | H-bonded |
| Phe20-Leu24 | Ar | Hβ | 3.67 | Mixed |
| Phe20-Leu24 | C=O | N-H | 1.95 | Mixed |
| Tyr21-Asp22 | Ar | PB21 | 4.06 | Weakly Polar |
| Tyr21-Gln25 | Ar | Hβ | 4.56 | Mixed |
| Tyr21-Gln25 | C=O | N-H | 2.11 | Mixed |
| Tyr27-Val31 | Ar | Hβ | 5.03 | Mixed |
| Tyr27-Val31 | C=O | N-H | 2.10 | Mixed |
| Tyr36-His34Arg35 | Ar | Ring of His34 | 4.85 | Weakly Polar |
| Tyr36-His34Arg35 | Ar | PB34 | 5.00 | Weakly Polar |
| Interactions which stabilize the tertiary structure | ||||
| Pro8Gly9-Ala12 | C=O of Pro8 | Hβ | 2.78 | Mixed |
| Pro8Gly9-Ala12 | C=O of Gly9 | Hβ | 2.98 | Mixed |
| Pro8Gly9-Ala12 | C=O of Gly9 | Hβ2 | 2.99 | Mixed |
| Pro8Gly9-Ala12 | C=O of Gly9 | N-H | 2.35 | Mixed |
| Phe20-Pro5Thr6 | Ar | Hβ of Pro5 | 4.03 | Weakly Polar |
| Phe20-Pro5Thr6 | Ar | PB6 | 5.65 | Weakly Polar |
| Phe20-Tyr7Pro8 | Ar | Ar-Hδ | 3.21 | Weakly Polar |
| Phe20-Tyr7Pro8 | Ar | Hδ | 3.59 | Weakly Polar |
| Tyr27-Pro2Ser3 | Ar-Hδ | Prolyl ring | 4.11 | Weakly Polar |
| Tyr27-Pro2Ser3 | Ar | C=O of Pro2 | 4.68 | Weakly Polar |
| Tyr27-Pro2Ser3 | Ar | PB3 | 4.72 | Weakly Polar |
| Tyr27-Gln4Pro5 | Ar | Hα of Gln4 | 3.15 | Weakly Polar |
| Tyr27-Gln4Pro5 | Ar | Hδ of Pro5 | 3.71 | Weakly Polar |
Functional groups 1 and 2, respectively, refer to left and right interacting residues, unless otherwise stated.
PB, peptide bond.
Conclusions
The aromatic residues Phe20 and Tyr27 contribute significantly to the tertiary fold stability of aPP through weakly polar interactions. Aromatic residues can significantly stabilize proteins through non-bonded interactions. They influence the stability of both secondary and tertiary structure by weakly polar interactions as strong as hydrogen bonds.
Acknowledgements
This work was supported by the NIH-INBRE grant (P20 RR016469) and the Carpenter Endowed Chair in Biochemistry, Creighton University.
References
- 1.Blundell TL, Pitts JE, Tickle IJ, Wood SP, Wu C-W. Proc Natl Acad Sci. 1981;78:4175–4179. doi: 10.1073/pnas.78.7.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Covell DG, Jernigan RL. Biochemistry. 1990;29:3287–3294. doi: 10.1021/bi00465a020. [DOI] [PubMed] [Google Scholar]
- 3.Sun S. Protein Sci. 1993;2:762–785. doi: 10.1002/pro.5560020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liwo A, Pincus MR, Wawak RJ, Rackovsky S, Scheraga HA. Protein Sci. 1993;2:1715–1731. doi: 10.1002/pro.5560021016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yue K, Dill KA. Protein Sci. 1996;5:254–261. doi: 10.1002/pro.5560050209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derreumaux P. J Chem Phys. 1998;109:1567–1574. [Google Scholar]
- 7.Copps J, Murphy RF, Lovas S. Biopolymers. 2006;83:32–38. doi: 10.1002/bip.20524. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Wong CF, Thacher T, Rabitz H. Proteins: Structure, Function, and Genetics. 1995;23:218–232. doi: 10.1002/prot.340230211. [DOI] [PubMed] [Google Scholar]
- 9.Burley SK, Petsko GA. Adv Protein Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 10.Tóth G, Murphy RF, Lovas S. Protein Engineering. 2001;14:543–547. doi: 10.1093/protein/14.8.543. [DOI] [PubMed] [Google Scholar]
- 11.Meng HY, Thomas KM, Lee AE, Zondlo NJ. Biopolymers. 2006;84:192–204. doi: 10.1002/bip.20382. [DOI] [PubMed] [Google Scholar]
- 12.Gallivan JP, Dougherty DA. Proc Natl Acad Sci. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macias AT, Mackerell AD., Jr. J Comput Chem. 2005;26:1452–1463. doi: 10.1002/jcc.20281. [DOI] [PubMed] [Google Scholar]
- 14.Palermo NY, Csontos J, Owen MC, Murphy RF, Lovas S. J Comput Chem. 2007;28:1208–1214. doi: 10.1002/jcc.20578. [DOI] [PubMed] [Google Scholar]
- 15.Berman HM, Westbrook J, Feng Z, Gilliand G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tóth G, Watts CR, Murphy RF, Lovas S. Proteins. 2001;43:373–381. doi: 10.1002/prot.1050. [DOI] [PubMed] [Google Scholar]
- 17.Csontos J, Palermo NY, Murphy RF, Lovas S. J Comput Chem. doi: 10.1002/jcc.20898. submitted.
- 18.Boys SF, Bernardi F. Mol Phys. 1970;19:553–566. [Google Scholar]
- 19.Tóth G, Murphy RF, Lovas S. J Am Chem Soc. 2001;123:11782–11790. doi: 10.1021/ja011245u. [DOI] [PubMed] [Google Scholar]
- 20.Baures PW, Beatty AM, Dhanasekaran M, Helfrich BA, Perez-Segarra W, Desper J. J Am Chem Soc. 2002;124:11315–11323. doi: 10.1021/ja0257366. [DOI] [PubMed] [Google Scholar]
- 21.Desiraju GR, Steiner T. The weak hydrogen bond. Oxford University Press; Oxford: 1999. ch. 4; pp. 13–14. [Google Scholar]
- 22.Vondrasek J, Bendova L, Klusak V, Hobza P. J Am Chem Soc. 2005;127:2615–2619. doi: 10.1021/ja044607h. [DOI] [PubMed] [Google Scholar]