

Abstract
Laforin, encoded by the EPM2A gene, by sequence is a member of the dual specificity protein phosphatase family. Mutations in the EPM2A gene account for around half of the cases of Lafora disease, an autosomal recessive neurodegenerative disorder, characterized by progressive myoclonus epilepsy. The hallmark of the disease is the presence of Lafora bodies, which contain polyglucosan, a poorly branched form of glycogen, in neurons, muscle and other tissues. Glycogen metabolizing enzymes were analyzed in a transgenic mouse over-expressing a dominant negative form of laforin that accumulates Lafora bodies in several tissues. Skeletal muscle glycogen was increased two-fold as was the total glycogen synthase protein. However, the −/+ glucose-6-P activity of glycogen synthase was decreased from 0.29 to 0.16. Branching enzyme activity was increased by 30%. Glycogen phosphorylase activity was unchanged. In whole brain, no differences in glycogen synthase or branching enzyme activities were found. Although there were significant differences in enzyme activities in muscle, the results do not support the hypothesis that Lafora body formation is caused by a major change in the balance between glycogen elongation and branching activities.
Lafora disease is an autosomal recessive, progressive myoclonus epilepsy (OMIM #254780), with onset typically in teenagers followed by decline and death usually within ten years [1–3]. The disease is characterized by the presence of Lafora bodies, periodic acid-Schiff positive structures containing an abnormal form of glycogen, the branched polymer of glucose that serves as a stored form of glucose in many tissues. Although Lafora body formation in neurons is believed to account for the symptoms of the disease, the bodies are present in other tissues including liver, muscle and skin [4–7].
Glycogen synthesis (Fig. 1) is mediated by glycogen synthase, which catalyzes formation of the predominant α-1,4-glycosidic linkage of the polymer and branching enzyme, which introduces the α-1,6-glycosidic branchpoints [8]. Degradation occurs in the cytosol through the action of glycogen phosphorylase and the debranching enzyme or alternatively in the lysosome via the activity of an α-glycosidase (acid maltase or GAA). In Lafora disease, a less branched form of glycogen, also called polyglucosan, accumulates and is associated with the Lafora bodies.
Figure 1.
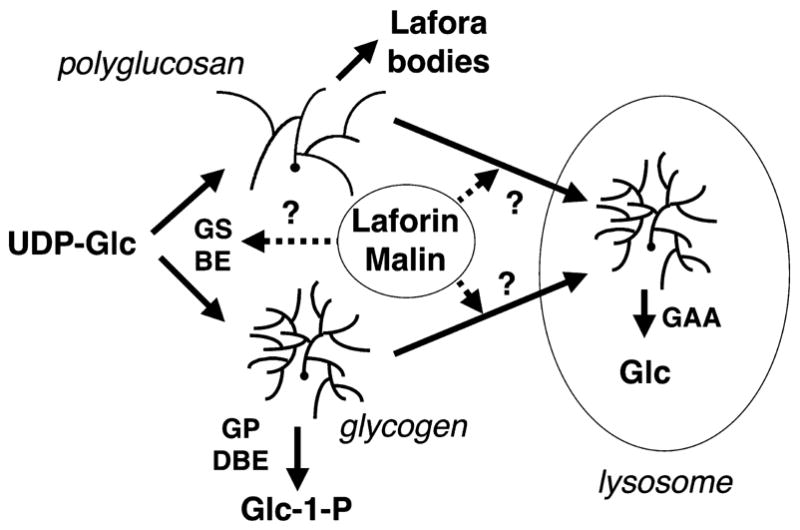
Glycogen metabolism. Normal glycogen metabolism is shown in the lower part of the figure, with normal activities of glycogen synthase (GS) and branching enzyme (BE) leading to a regularly branched polysaccharide. Its degradation is mediated in the cytosol by glycogen phosphorylase (GP) and debranching enzyme (DBE) or in the lysosome by α-glucosidase (GAA). In Lafora disease, poorly branched glycogen or polyglucosan is formed. Possible sites of action of laforin and malin are indicated. One hypothesis is that they control the balance between branching and elongating activities. A second idea is that laforin in some way monitors the branching state of glycogen, enabling its effective removal, presumably via the lysosome.
Mutations in two genes, EPM2A and EPM2B, account for approximately 90% of Lafora cases [1, 3]. EPM2A, encodes a protein, laforin, that belongs to the dual specificity protein phosphatase family [9]. Laforin also contains a functional polysaccharide binding domain [10–12] that binds preferentially to polyglucosan over glycogen [13]. Some forty mutations have been identified throughout all four exons of the EPM2A gene (http://projects.tcag.ca/lafora/)[9, 12, 14–17]. Most mutations cause a loss of phosphatase activity in recombinant laforin [11, 18]. However, one disease mutation, W32G, in the polysaccharide binding domain blocks glycogen binding [10, 11, 19] without completely destroying phosphatase activity [10, 19]. Therefore, binding to polysaccharide is required for normal laforin function in vivo and one might speculate that laforin is somehow involved in glycogen metabolism.
There are other possible links to glycogen metabolism. The EPM2B gene (also called NHLRC1) encodes malin, a 395 residue protein that contains an NH2-terminal RING finger domain[20] characteristic of E3 ubiquitin ligases [21]. Gentry et al. [22] reported that malin interacts with laforin and catalyzes its polyubiquitination and in cultured cells leads to the degradation of laforin. Loss of function mutations in EPM2B should thus stabilize laforin making it hard to understand how recessive mutations in either EPM2A or EPM2B result in the disease. Lohi et al. [23] found that malin interacts also with glycogen synthase and propose that the laforin-malin-glycogen synthase complex is targeted for degradation. In the same study, it was also reported that laforin could dephosphorylate the NH2-terminal inhibitory phosphorylation site of the protein kinase GSK-3 which should result in glycogen synthase inactivation. Another connection to glycogen came from the yeast two hybrid screen described by Fernandez-Sanchez et al. [18] which identified the type 1 protein serine/threonine phosphatase regulatory subunit, PTG. PTG, also called R5, binds glycogen and is implicated in the control of glycogen metabolism [24]. PTG has been proposed to act as a scaffold, binding also to glycogen synthase, phosphorylase and phosphorylase kinase [24]. Fernandez-Sanchez et al. [18] found that a disease-associated mutation of laforin, G240S, has no effect on either phosphatase activity or glycogen binding but impairs interaction with PTG.
Two mouse models of Lafora disease have been developed [13, 25]. Disruption of the mouse Epm2a gene resulted in viable homozygous null mice that accumulated structures similar to Lafora bodies in neurons and other tissues [25]. Lafora bodies began to appear in the brain by two months at which time neurons began to degenerate and die but not by typical apoptotic mechanisms. Behavioral abnormalities were detected at 4 months and by 9 months the animals had myoclonic seizures, ataxia and epileptiform electroencephelogram activity. The second mouse model utilized transgenic over-expression of a dominant negative form of laforin generated by mutating the catalytic Cys266 to Ser [13]. The mice developed Lafora bodies in muscle, liver and neurons and, by immunogold electron microscopy, laforin was shown to be in proximity of the polyglucosan deposits.
Despite these significant advances, the cause of Lafora disease and the reason for the accumulation of Lafora bodies is not understood (Fig. 1). There are two prevailing hypotheses in the field. One idea is that the defect in laforin blocks the breakdown or removal of excess and/or aberrant glycogen molecules [11, 13]. Some support for this hypothesis comes from the preferential binding of laforin to poorly branched glycogen [11, 13, 19], so that laforin may act in quality control of the synthetic process and initiate removal of defective molecules. The second is that there may be faulty glycogen synthesis, leading to the abnormal polysaccharide structure and reduced solubility. In this regard, it has been proposed that an imbalance between branching enzyme and glycogen synthase activities may cause the abnormal branching of the polyglucosan. In support of this idea, the GSL30 transgenic mouse that over-expresses constitutively active glycogen synthase in muscle hyper-accumulates glycogen that is less branched [26] and contains structures that resemble Lafora bodies [27]. One fundamental question yet to be answered is whether the activities of the glycogen metabolizing enzymes are affected in tissues that form Lafora bodies. To address this critical question, we analyzed several enzymes involved in glycogen metabolism in tissue from the transgenic mice over-expressing dominant negative laforin [13]. Although there were some statistically significant differences, there was no evidence for a gross imbalance between elongation and branching activities in the transgenic mice.
Experimental procedures
Genetically modified mouse models
Mice over-expressing mutant laforin, described in the Introduction, had gene expression driven by the β-actin promoter [13].
Generation of anti-laforin antibodies
Recombinant mouse laforin was produced in E. coli and purified as previously described [19]. The anti-laforin antibody was raised in rabbits using recombinant laforin protein as antigen by Cocalico Biologicals, Inc. The antibodies were affinity purified using recombinant laforin coupled with Affigel-15 (Bio-Rad). After dialysis against 100 mM MOPS pH 7.5 overnight at 4ºC, laforin (5 mg) was incubated with Affigel-15 overnight at 4ºC. Then 0.1 M ethanolamine-HCl pH 8 was added to block any unreacted sites. The resin was packed into a column and washed with PBS (0.27 mM KCl, 0.15 mM KH2PO4, 14 mM NaCl and 0.81 mM Na2HPO4, pH 7.5), 100 mM MOPS pH 7.5 and 100 mM glycine HCl pH 2.4/150 mM NaCl. Serum was passed through the column three times and the column was washed with PBS. Affinity purified antibody was eluted with 100 mM glycine HCl pH 2.4, 150 mM NaCl and immediately neutralized with Tris HCl pH 8.0. The antibody was dialyzed against PBS overnight, glycerol was added to 25% (v/v) and the antibody was stored at −80ºC.
Preparation of mouse tissues
Mouse tissues were harvested as rapidly as possible and frozen immediately in liquid nitrogen. Tissues were later powdered by a tissue pulverizer and stored at −80ºC. Samples of frozen tissues were homogenized with a Tissue Tearer (Biospec Products Inc.) in 10 or 30 volumes of a buffer containing 50 mM Tris-HCl pH 7.8, 2mM EGTA, 10 mM EDTA, 100 mM NaF, 0.5 mM PMSF, 2 mM benzamidine, 50 mM β-mercaptoethanol, 10 μg/ml leupeptin and 37 μg/ml TLCK. Total tissue homogenates were used for determination of enzyme activities. Protein concentration was measured by the method of Bradford [28] using bovine serum albumin as standard.
Western Blotting
Four volumes of SDS loading buffer was added to one volume of whole homogenate and boiled. Loading buffer was 62.5 mM Tris-HCl, pH 6.8, 50% (v/v) glycerol, 2.5% (w/v) SDS, 5% (v/v) β-mercaptoethanol, and 1.25% (w/v) bromophenol blue. Typically 30 μg protein was loaded per lane and separated by SDS-PAGE with 10% acrylamide. The protein was transferred to nitrocellulose and incubated with the indicated first antibody. Horseradish peroxidase-conjugated secondary antibody (Sigma) and Enhanced Chemiluminescence (Amersham) were used for detection. Autoradiograms were quantitated by densitometric scanning of films by using Kodak 1D Image Analysis Software (Kodak).
Enzyme activity assays
Glycogen synthase or phosphorylase activities were measured by monitoring the incorporation of radioactivity from UDP-[U-14C]glucose by the method of Thomas [29] or [U-14C]glucose-1-phosphate by the method of Gilboe [30], respectively, into glycogen as previously described [31]. Activity ratio is defined as the enzymatic activity measured in the absence of allosteric effector divided by that determined in the presence of 7.2 mM glucose-6-phosphate for glycogen synthase or 3 mM AMP for phosphorylase, under standard conditions. The activity ratio provides a kinetic index of the relative phosphorylation state of the enzymes. For glycogen synthase, a low value is indicative of a highly phosphorylated enzyme. Branching enzyme activity was determined as previously described [26].
Measurement of glycogen
Glycogen content in tissue was determined in samples of frozen tissue (~30 mg) by measuring amyloglucosidase-released glucose from glycogen by the method of Bergmeyer [32] as previously described by Suzuki et al. [33].
Other Materials and Methods
Antibodies against total mouse GSK-3α/β were from Biosource International. Anti-phospho-GSK3α/β (Ser21/Ser9) antibody and anti-phospho-GS (site 3a) antibodies were from Cell Signaling Technology. Anti-phospho-GSK3α/β (Tyr 279/Tyr216) antibody (mouse) was from Upstate Biotechnology. Results are presented as means ± standard error of the mean. Unpaired Student’s t-test was used to determine statistical significance using the Statview package (Abacus Concept, Inc.). A value of p<0.05 was considered significant.
Results and discussion
To evaluate whether there were gross impairments of glycogen metabolism in a mouse model of Lafora disease, we took advantage of the Cys266Ser laforin over-expressing mice [13]. These animals form Lafora bodies in both muscle and neurons. Using antibodies raised to recombinant laforin, we could identify an immunoreactive species of Mr ~40,000 in muscle extracts from wild-type mice (Fig. 2). We failed to detect laforin in brain and liver extracts, presumably because the levels were too low. Using the same antibodies to analyze muscle from the transgenic overexpressing mice, we estimated an approximately 150-fold over-expression of the mutant laforin protein with little change in the endogenous, wild-type laforin (Fig. 2). This result is in keeping with a more than 100-fold elevation in the Epm2a transcript level [13]. The muscle of the transgenic animals contained approximately twice as much glycogen as wild-type (Fig. 3), with a 30% increase in glycogen synthase protein based on Western analysis (Fig. 3). Consistent with this result, the glycogen synthase activity measured in the presence of glucose-6-P, which is an indicator of total synthase protein, was increased by ~50%. The −/+glucose-6-P activity ratio, however, was paradoxically decreased in the glycogen synthase from the transgenic muscle, suggestive of increased phosphorylation of the enzyme. This was reflected in an increase in the phosphorylation of site 3a, one of the key phosphorylation sites, as judged by a phospho-specific antibody (Fig. 3). If the signal from the phospho-specific antibody was normalized by the intensity for total synthase, there was an increase of 22% (p=0.014) in the tissue from the transgenic animals. Branching enzyme activity was increased by 30% in the transgenic animals whereas glycogen phosphorylase, whether measured in the presence or absence of AMP, was unchanged (Fig. 3). The muscle of the transgenic mouse contains significant levels of the mutant laforin, which will be able to bind glycogen, and has the potential to interfere with enzyme assays. We therefore performed controls in which recombinant mouse C265S laforin was added back to muscle extracts from wild type mice at a level, ~70 μg/ml, comparable to the laforin level in the transgenic animals. We observed no effect on glycogen synthase assays, with or without glucose-6-P, or on branching enzyme assays (data not shown).
Figure 2.
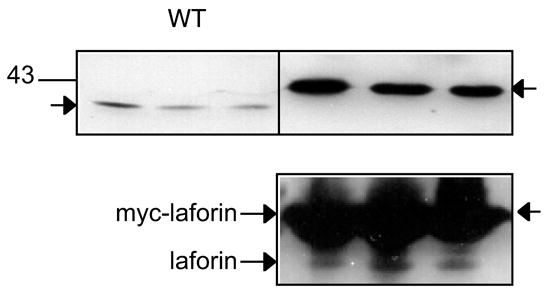
Laforin expression in skeletal muscle of wild-type, transgenic mice overexpressing myc-Cys266Ser laforin. Upper Panel: Wild type laforin runs as a species of ~40,000. The transgenic Cys266Ser dominant negative laforin also has a myc tag leading to an apparent Mr of ~43,000. Note that the loading for the wild-type extract was 30 μg of protein but 1 μg for the transgenic animals, so that the endogenous laforin is not visible. Lower Panel: Equal loadings (30 μg protein) of extracts of muscle from wild-type and transgenic mice.
Figure 3.
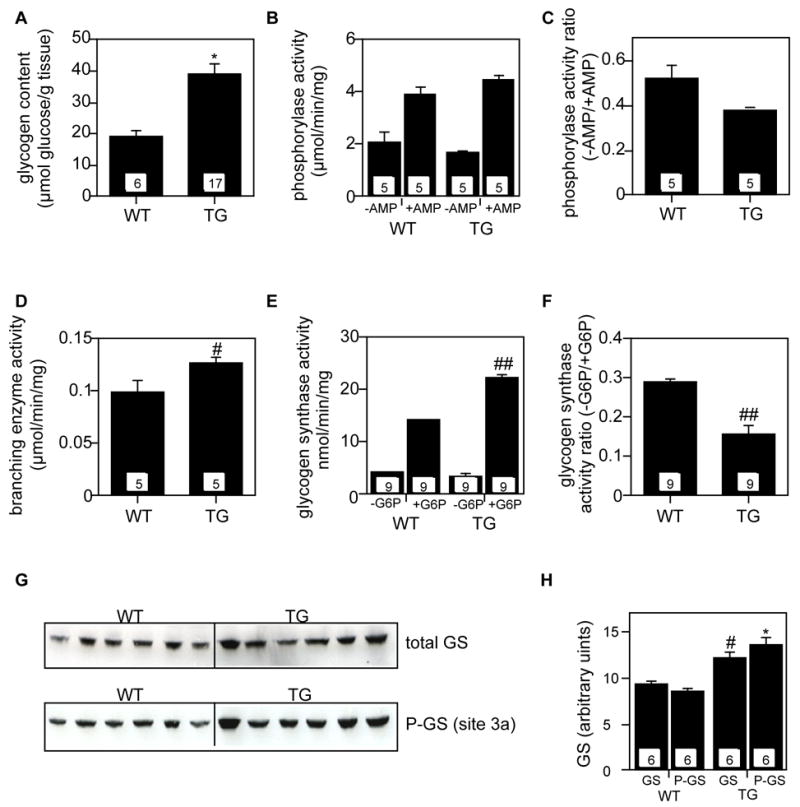
Activities of glycogen metabolizing enzymes in skeletal muscle of transgenic mice overexpressing myc-Cys266Ser laforin. “WT”, wild type. “TG”, myc-Cys266Ser laforin transgenic. Measurements were as described under Experimental Procedures. The numbers within the bars indicate the number of animals analyzed. *p<0.005 vs WT. #p<0.05 vs WT. ##p<0.0001 vs WT. A. Glycogen content measurement. B. Phosphorylase activity, measured in the presence or absence of AMP. C. Phosphorylase −/+ AMP activity ratio. D. Branching enzyme measurement. E. Glycogen synthase activity, measured in the presence or the absence of glucose-6-phosphate (G6P). F. Glycogen synthase −/+glucose-6-phosphate activity. G. Western blotting analysis to detect total glycogen synthase (upper panel) and glycogen synthase phosphorylation at site 3a (lower panel). The loading was 30 μg protein to a 10% SDS-PAGE. H. Quantitation of the data in Panel G.
We additionally analyzed GSK-3 phosphorylation in muscle using phospho-specific antibodies against the inactivating NH2-terminal phosphorylation site (Ser21 for GSK-3α and Ser9 for GSK-3β) or the activating phosphorylation site in the T-loop (Tyr279 for GSK-3α and Tyr216 for GSK-3β). Ser phosphorylation was scarcely detected in GSK-3α and in GSK-3β was unchanged in the transgenic over-expressors (Fig. 4). There was a 40% decrease in Tyr phosphorylation of GSK-3α in the transgenic mice (Fig. 4). Tyr phosphorylation of GSK-3β, though detectable, was too faint to quantitate. No major differences between genotype were apparent (Fig. 4).
Figure 4.
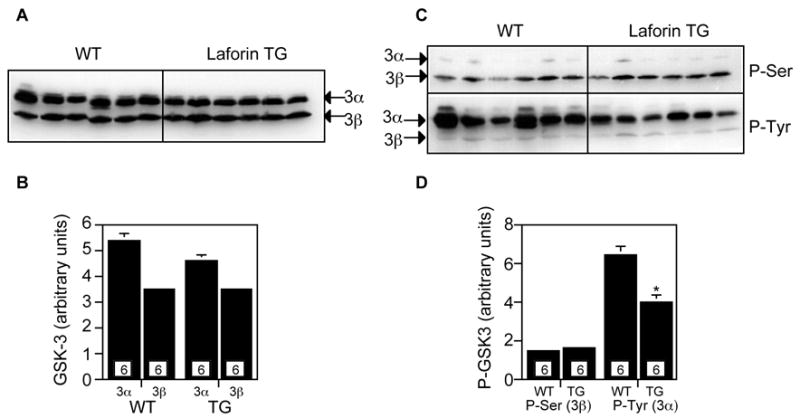
Analysis of glycogen synthase kinase-3α/β (GSK3α/β) in skeletal muscle of transgenic mice overexpressing myc-Cys266Ser laforin. Loadings were 45 μg protein 10% SDS-PAGE gels. Numbers within the bars indicate the number of mice analyzed. A. Total GSK3α/β protein levels. B. Quantitation of the data shown in panel A. C. Ser21(GSK3α) and Ser9 (GSK3β) phosphorylation as detected by phospho-specific antibodies (upper). Tyr279 (GSK3α) and Tyr 216 (GSK3β) phosphorylation as detected by phospho-specific antibodies (lower). D. Quantitation of the data in panel C. *p<0.005 vs WT.
The elevated glycogen content in the transgenic mouse muscle is consistent with the increase in glycogen synthase protein. The activation state of the glycogen synthase, however, was reduced and one would have to infer that local metabolite conditions, including the glucose-6-P concentration, were able to sustain increased glycogen synthesis [34]. The measurements of bulk enzyme activity, though, do not give much mechanistic insight into the cause of Lafora body formation. The increased glycogen synthase protein would be consistent with the proposal that laforin participates in glycogen synthase degradation but not that it activates GSK-3 by NH2-terminal dephosphorylation [23]. Branching enzyme activity is also increased in the transgenic muscle so that the ratio of glycogen synthase to branching activities is little changed. Therefore, we have no evidence that Lafora bodies in muscle in this mouse model are caused by an imbalance in these activities.
Muscle glycogen metabolism is especially well-understood and, since the muscle of the transgenic mice developed Lafora bodies, we reasoned that any gross perturbations in glycogen metabolism caused by laforin downregulation should be evident in muscle. For this reason, we first analyzed muscle. We also measured branching enzyme and glycogen synthase activities in whole brain and found that there were no changes in the transgenic mice (Fig. 5). We did not analyze brain glycogen because it is very difficult to preserve post-mortem without specialized strategies, such as in situ microwaving [35]. The negative results with enzyme activity measurements are difficult to interpret since most brain glycogen is present in the glial cells and not the neurons [35]. Therefore, a substantial derangement of neuronal glycogen metabolism could be easily masked by the contribution of the glia. Glycogen phosphorylase is predominantly found in the glia with only occasional neurons displaying immunoreactivity to phosphorylase antibodies [36]. In contrast, by immunocytochemistry, glycogen synthase was found in both neurons and glial cells, and if anything, was more prominent in neurons [37].
Figure 5.
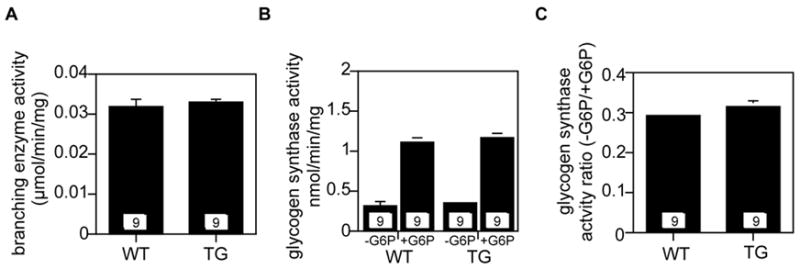
Glycogen synthase and branching enzyme activities in brain of transgenic mice overexpressing myc-Cys266Ser laforin. The numbers within the bars indicate the number of animals analyzed. “WT”, wild type. “TG”, transgenic mice overexpressing myc-Cys266Ser laforin. A. Branching enzyme activity. B. Glycogen synthase activity in the presence and absence of glucose-6-P (G6P). C. Glycogen synthase −/+ G6P activity ratio.
Conclusion
The first conclusion from the present study is that genetic manipulation of laforin function does result in altered glycogen metabolism in an organ, skeletal muscle, that produces Lafora bodies. However, the mechanism remains unclear and the changes in glycogen and glycogen synthase activity do not fit with GSK-3 dephosphorylation by laforin. Loss of laforin activity should lead to decreased GSK-3 activity and increased glycogen synthase −/+ glucose-6-P activity ratio. Also, no alterations in GSK-3 Ser phosphorylation were detected in the transgenic tissue. The reason for these apparent discrepancies is not clear at this time. It has to be acknowledged that Worby et al. [38] did not observe GSK-3 dephosphorylation by laforin.
Although some glycogen metabolizing enzymes were affected in the transgenic mice, our data do not make a case for a radical perturbation of the balance between glycogen elongating and branching, an observation that future models of Lafora body formation will need to take into account. Perhaps laforin function has more to do with monitoring glycogen branching and/or managing its disposal. However, a very recent observation is that laforin can remove covalently attached phosphate from amylopectin [38]. Glycogen has also long been known to contain trace amounts of phosphate [39] although whether it serves a specific function is not really known. Obviously, more work is needed to understand laforin function and its relation to Lafora disease.
Acknowledgments
Supported by NIH grants DK27221 (P.J.R.), DK36569 (A.D.R.), an American Heart Association Fellowship (W.W.), the Canadian Institutes of Health Research (B.A.M), and the Sigrid Juselius and Emil Aaltonen Foundations (H.L.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chan EM, Andrade DM, Franceschetti S, Minassian B. Progressive myoclonus epilepsies: EPM1, EPM2A, EPM2B. Adv Neurol. 2005;95:47–57. [PubMed] [Google Scholar]
- 2.Delgado-Escueta AV, Ganesh S, Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Gen. 2001;106:129–138. doi: 10.1002/ajmg.1575. [DOI] [PubMed] [Google Scholar]
- 3.Ganesh S, Puri R, Singh S, Mittal S, Dubey D. Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J Hum Genet. 2006;51:1–8. doi: 10.1007/s10038-005-0321-1. [DOI] [PubMed] [Google Scholar]
- 4.Cavanagh JB. Corpora-amylacea and the family of polyglucosan diseases. Brain Research - Brain Research Reviews. 1999;29:265–295. doi: 10.1016/s0165-0173(99)00003-x. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura RN, Ishak KG, Reddick R, Porter R, James S, Barranger JA. Lafora disease: diagnosis by liver biopsy. Ann Neurol. 1980;8:409–415. doi: 10.1002/ana.410080412. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter S, Karpati G. Ultrastructural findings in Lafora disease. Ann Neurol. 1981;10:63–64. doi: 10.1002/ana.410100116. [DOI] [PubMed] [Google Scholar]
- 7.DiMauro S, Lamperti C. Muscle glycogenoses. Muscle & Nerve. 2001;24:984–999. doi: 10.1002/mus.1103. [DOI] [PubMed] [Google Scholar]
- 8.Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–120. doi: 10.2174/1566524024605761. [DOI] [PubMed] [Google Scholar]
- 9.Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, Jardim L, Satishchandra P, Andermann E, Snead OC, 3rd, Lopes-Cendes I, Tsui LC, Delgado-Escueta AV, Rouleau GA, Scherer SW. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nature Genetics. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Stuckey JA, Wishart MJ, Dixon JE. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem. 2002;277:2377–2380. doi: 10.1074/jbc.C100686200. [DOI] [PubMed] [Google Scholar]
- 11.Ganesh S, Tsurutani N, Suzuki T, Hoshii Y, Ishihara T, Delgado-Escueta AV, Yamakawa K. The carbohydrate-binding domain of Lafora disease protein targets Lafora polyglucosan bodies. Biochem Biophys Res Commun. 2004;313:1101–1109. doi: 10.1016/j.bbrc.2003.12.043. [DOI] [PubMed] [Google Scholar]
- 12.Minassian BA, Ianzano L, Meloche M, Andermann E, Rouleau GA, Delgado-Escueta AV, Scherer SW. Mutation spectrum and predicted function of laforin in Lafora's progressive myoclonus epilepsy. Neurology. 2000;55:341–346. doi: 10.1212/wnl.55.3.341. [DOI] [PubMed] [Google Scholar]
- 13.Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P, Scherer SW, Minassian BA. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet. 2004;13:1117–1129. doi: 10.1093/hmg/ddh130. [DOI] [PubMed] [Google Scholar]
- 14.Ianzano L, Young EJ, Zhao XC, Chan EM, Rodriguez MT, Torrado MV, Scherer SW, Minassian BA. Loss of function of the cytoplasmic isoform of the protein laforin (EPM2A) causes Lafora progressive myoclonus epilepsy. Hum Mutat. 2004;23:170–176. doi: 10.1002/humu.10306. [DOI] [PubMed] [Google Scholar]
- 15.Minassian BA, Ianzano L, Delgado-Escueta AV, Scherer SW. Identification of new and common mutations in the EPM2A gene in Lafora disease. Neurology. 2000;54:488–490. doi: 10.1212/wnl.54.2.488. [DOI] [PubMed] [Google Scholar]
- 16.Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, Augustijn PB, Tassinari CA, Malafosse RM, Topcu M, Grid D, Dravet C, Berkovic SF, de Cordoba SR. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum Mol Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Garre P, Sanz Y, Rodriguez De Cordoba SR, Serratosa JM. Mutational spectrum of the EPM2A gene in progressive myoclonus epilepsy of Lafora: high degree of allelic heterogeneity and prevalence of deletions. Eur J Hum Genet. 2000;8:946–954. doi: 10.1038/sj.ejhg.5200571. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Sanchez ME, Criado-Garcia O, Heath KE, Garcia-Fojeda B, Medrano-Fernandez I, Gomez-Garre P, Sanz P, Serratosa JM, Rodriguez de Cordoba S. Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum Mol Genet. 2003;12:3161–3171. doi: 10.1093/hmg/ddg340. [DOI] [PubMed] [Google Scholar]
- 19.Wang W, Roach PJ. Glycogen and related polysaccharides inhibit the laforin dual-specificity protein phosphatase. Biochem Biophys Res Commun. 2004;325:726–730. doi: 10.1016/j.bbrc.2004.10.083. [DOI] [PubMed] [Google Scholar]
- 20.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, Bohlega S, Andermann E, Rouleau GA, Delgado-Escueta AV, Minassian BA, Scherer SW. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 21.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 22.Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohi H, Ianzano L, Zhao XC, Chan EM, Turnbull J, Scherer SW, Ackerley CA, Minassian BA. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005;14:2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- 24.Printen JA, Brady MJ, Saltiel AR. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science. 1997;275:1475–1478. doi: 10.1126/science.275.5305.1475. [DOI] [PubMed] [Google Scholar]
- 25.Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K, Agarwala KL, Hasegawa Y, Bai DS, Ishihara T, Hashikawa T, Itohara S, Cornford EM, Niki H, Yamakawa K. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11:1251–1262. doi: 10.1093/hmg/11.11.1251. [DOI] [PubMed] [Google Scholar]
- 26.Pederson BA, Csitkovits AG, Simon R, Schroeder JM, Wang W, Skurat AV, Roach PJ. Overexpression of glycogen synthase in mouse muscle results in less branched glycogen. Biochem Biophys Res Commun. 2003;305:826–830. doi: 10.1016/s0006-291x(03)00862-3. [DOI] [PubMed] [Google Scholar]
- 27.Raben N, Danon M, Lu N, Lee E, Shliselfeld L, Skurat AV, Roach PJ, Lawrence JC, Jr, Musumeci O, Shanske S, DiMauro S, Plotz P. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739–1745. doi: 10.1212/wnl.56.12.1739. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Thomas JA, Schlender KK, Larner J. A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal Biochem. 1968;25:486–499. doi: 10.1016/0003-2697(68)90127-9. [DOI] [PubMed] [Google Scholar]
- 30.Gilboe DP, Larson KL, Nuttall FQ. Radioactive method for the assay of glycogen phosphorylases. Anal Biochem. 1972;47:20–27. doi: 10.1016/0003-2697(72)90274-6. [DOI] [PubMed] [Google Scholar]
- 31.Pederson BA, Chen H, Schroeder JM, Shou W, DePaoli-Roach AA, Roach PJ. Abnormal cardiac development in the absence of heart glycogen. Mol Cell Biol. 2004;24:7179–7187. doi: 10.1128/MCB.24.16.7179-7187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergmeyer HU, Berndt E, Schmidt F, Stork H. In: Methods of enzymatic analysis. Bergmeyer HU, editor. Academic Press; New York: 1974. pp. 1196–1201. [Google Scholar]
- 33.Suzuki Y, Lanner C, Kim JH, Vilardo PG, Zhang H, Yang J, Cooper LD, Steele M, Kennedy A, Bock CB, Scrimgeour A, Lawrence JC, Jr, DePaoli-Roach AA. Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol. 2001;21:2683–2694. doi: 10.1128/MCB.21.8.2683-2694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roach PJ, Larner J. Rabbit skeletal muscle glycogen synthase. II. Enzyme phosphorylation state and effector concentrations as interacting control parameters. J Biol Chem. 1976;251:1920–1925. [PubMed] [Google Scholar]
- 35.Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer-Guglielmi B, Fleckenstein B, Jung G, Hamprecht B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J Neurochem. 2003;85:73–81. doi: 10.1046/j.1471-4159.2003.01644.x. [DOI] [PubMed] [Google Scholar]
- 37.Inoue N, Matsukado Y, Goto S, Miyamoto E. Localization of glycogen synthase in brain. J Neurochem. 1988;50:400–405. doi: 10.1111/j.1471-4159.1988.tb02926.x. [DOI] [PubMed] [Google Scholar]
- 38.Worby CA, Gentry MS, Dixon JE. Laforin: A dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006 doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lomako J, Lomako WM, Kirkman BR, Whelan WJ. The role of phosphate in muscle glycogen. Biofactors. 1994;4:167–171. [PubMed] [Google Scholar]