

Abstract
p27 mediates Cdk2 inhibition and is also found in cyclin D1-Cdk4 complexes. The present data support a role for p27 in the assembly of D-type cyclin-Cdk complexes and indicate that both cyclin D1-Cdk4-p27 assembly and kinase activation are regulated by p27 phosphorylation. Prior work showed that p27 can be phosphorylated by protein kinase B/Akt (PKB/Akt) at T157 and T198. Here we show that PKB activation and the appearance of p27pT157 and p27pT198 precede p27-cyclin D1-Cdk4 assembly in early G1. PI3K/PKB inhibition rapidly reduced p27pT157 and p27pT198 and dissociated cellular p27-cyclin D1-Cdk4. Mutant p27 allele products lacking phosphorylation at T157 and T198 bound poorly to cellular cyclin D1 and Cdk4. Cellular p27pT157 and p27pT198 coprecipitated with Cdk4 but were not detected in Cdk2 complexes. The addition of p27 to recombinant cyclin D1 and Cdk4 led to cyclin D1-Cdk4-p27 complex formation in vitro. p27 phosphorylation by PKB increased p27-cyclin D1-Cdk4 assembly in vitro but yielded inactive Cdk4. In contrast, Src pretreatment of p27 did not affect p27-cyclin D1-Cdk4 complex formation. However, Src treatment led to tyrosine phosphorylation of p27 and catalytic activation of assembled cyclin D1-Cdk4-p27 complexes. Thus, while PKB-dependent p27 phosphorylation appears to increase cyclin D1-Cdk4-p27 assembly or stabilize these complexes in vitro, cyclin D1-Cdk4-p27 activation requires the tyrosine phosphorylation of p27. Constitutive activation of PKB and Abl or Src family kinases in cancers would drive p27 phosphorylation, increase cyclin D1-Cdk4 assembly and activation, and reduce the cyclin E-Cdk2 inhibitory function of p27. Combined therapy with both Src and PI3K/PKB inhibitors may reverse this process.
Progression through the G1 phase of the cell division cycle is rate limiting for mammalian cell proliferation and is driven by the sequential formation and activation of D-type cyclin-Cdk4 and Cdk6 complexes, followed by cyclin E-Cdk2 activation (53). The inhibitor of Cdk4 proteins binds Cdk4 and Cdk6, leading to the loss of cyclin binding (54). The kinase inhibitor protein (KIP) family members, p21, p27, and p57, bind and inhibit cyclin E-bound Cdk2. p27 is expressed ubiquitously and importantly regulates G1 progression. While p27 was initially identified as a Cdk2 inhibitor (29, 48, 57), it also appears to promote the assembly of p27-cyclin D1-Cdk4 and Cdk6 complexes in vitro (32) and/or the stabilization of these complexes in cells (3, 12, 32). Mechanisms regulating the putative assembly function of p27 have remained largely unknown.
Cyclin D1 accumulation in early G1 results from mitogen-mediated increases in its transcription, translation, and protein stabilization (16, 43). Cyclin D1 is unstable in G0 (3, 46), and newly synthesized cellular cyclin D1 does not appreciably assemble into cyclin D1-Cdk4 complexes in quiescent cells (12). Cyclin D1-Cdk4 assembly is growth factor dependent since exogenously overexpressed cyclin D1 cannot form complexes with Cdk4 in serum-starved fibroblasts, but it does so readily in cells grown in complete medium (40). Cyclin D1-Cdk-KIP assembly is associated with cyclin D1 stabilization (2, 9, 32). In mouse embryonic fibroblasts lacking both p21 and p27, D-type cyclins are rapidly degraded, and cyclin D1-Cdk complexes, while present, are barely detectable. Reintroducing p27 or p21 into these mouse embryonic fibroblasts reversed the accelerated cyclin D1 proteolysis and led to the appearance of stable cyclin D-Cdk4 complexes (2, 9).
In addition to contributing to D-type cyclin stabilization, the assembly of p27-cyclin D-Cdk complexes would contribute to G1-to-S progression via at least two other mechanisms. First, cyclin D1, Cdk4, and Cdk6 lack nuclear localization signals. LaBaer et al. provided evidence that p21 and/or p27 could facilitate nuclear import of the assembled complexes (32). Second, a shift in the p27 binding equilibrium toward cyclin D-Cdks could potentiate cyclin E-Cdk2 activation. Posttranslational events that promote p27 binding to cyclin D-Cdks in early G1 may reduce p27 availability to cyclin E-Cdk2, contributing to cyclin E-Cdk2 activation (47). These events characterize the restriction point in late G1 when cell cycle progression becomes mitogen independent and resistant to G1 arrest by inhibitory cytokines, such as transforming growth factor β (TGF-β) (6, 33).
Prior work indicates that protein kinase B (PKB) can phosphorylate p27 and contributes to the regulation of cellular p27 (38, 55, 59). TGF-β-resistant human mammary epithelial cells (HMEC) showed elevated PKB activity, and transfection of constitutively active PKB conferred TGF-β resistance to sensitive cells (38) and increased p27-cyclin D1-Cdk4 complexes and activity (12). We and others showed that PKB phosphorylates p27 at threonine 157 (T157) (38, 55, 59), and PKB was also shown to phosphorylate p27 at T198 in vitro (19, 42). SGK1 also appears to phosphorylate p27 in a PDK1- and mTOR-dependent manner (24). While both Ras and PKB overexpression increase p21- and p27-bound cyclin D-Cdk complexes (12, 35, 50), the relationship between PKB action on p27 phosphorylation sites and the potential for p27 to promote cyclin D-Cdk assembly has not been elucidated.
In the present study, we show that cellular PKB activation and the appearance of p27pT157, p27pT198, and p27-cyclin D1-Cdk4 complexes have similar kinetics in early G1. PI3K/PKB inhibition causes p27-cyclin D1-Cdk4 dissociation. Cellular Cdk4-bound p27 is more phosphorylated at T157 and T198 than Cdk2-bound p27 in vivo, and transfected p27T157A and p27T198A show reduced binding to cellular cyclin D1 and Cdk4 compared to that of p27WT. PKB phosphorylates p27 in a T157- and T198-dependent manner and increases p27-cyclin D1-Cdk4 assembly, but this does not yield active Cdk4 complexes in vitro. Activation of the assembled p27-cyclin D1-Cdk4 complex appears to require the tyrosine phosphorylation of p27 that can be stimulated by Src treatment. These data support a model in which PKB-mediated p27 phosphorylation promotes the assembly of p27-cyclin D1-Cdk4 assembly in G1, while action of other kinases, including Src, would permit the tyrosine phosphorylation of p27 and cyclin D-Cdk activation.
MATERIALS AND METHODS
Cell culture.
MCF-7 cells were grown in improved Eagle's medium (8), and the WM35 melanoma line was cultured in RPMI 1640 medium as described previously (8, 13, 23). WM35 cells were arrested by serum starvation (0.1% fetal bovine serum) for 48 h. Cell cycle entry was stimulated by the addition of 5% fetal bovine serum, and cells were recovered at intervals for protein and cell cycle assays. The finite life span HMEC strain 184 was cultured as described previously (58) and synchronized in G0 by deprivation of epidermal growth factor (EGF) receptor signaling for 48 h (51). Cell cycle reentry was stimulated by the addition of complete medium containing EGF.
Plasmids, site-directed mutagenesis, and transfections.
Mammalian expression vectors YFPp27WT and YFPp27T157A were described previously (38). YFPT198A and YFPp27T157A/T198A were generated using the QuikChange site-directed mutagenesis kit (Stratagene). Mutations were confirmed by DNA sequencing. Five micrograms of each vector was transfected into asynchronous MCF-7 cells using Lipofectamine Plus (Gibco) according to the manufacturer's instructions. His-tagged p27WT, p27S10A, p27T157A, p27T198A, p27T157A/T198A, and p27S10A/T157A/T198A vectors were generated by site-directed mutagenesis of the pET28b His-p27WT. Mutations were confirmed by DNA sequencing.
Flow cytometry.
Cells were pulse labeled with 10 mM of bromodeoxyuridine (BrdU) for 2 hours and processed for flow cytometry as described previously (51).
Antibodies.
The anti-EGF receptor monoclonal antibody mAb225 was provided by Steve Wiley (University of Utah Medical Center, Salt Lake City, UT). Antibodies to phospho-PKB (Ser473), total PKB, phospho-mitogen-activated protein kinase (MAPK), and total MAPK were obtained from Cell Signaling; antibodies to Cdk4 (C-22), p27 (C-19), phospho-p27 (T187), phospho-p27 (S10), and Cdk2 (M-2) were from Santa Cruz Biotechnology; antibody to p27 was from Transduction Laboratories; antibodies to phospho-p27 (T157) and phospho-p27 (T198) were from R&D Systems; antibodies to cyclin D1 (DCS-6 and Ab-3) and Cdk4 (DCS-35 and Ab-4) were from Lab Vision (Neomarkers); and antibody to β-actin was obtained from Sigma. Cyclin E1 antibody (E12) was obtained from E. Harlow.
Immunoprecipitation and Western blotting.
Cell lysis, immunoprecipitation, and immunoblotting were performed as described previously (51). Immunoprecipitation and immunoblotting experiments used 200 μg and 50 μg of protein lysates, respectively, unless otherwise indicated. All proteins precipitated in immunoprecipitation-Western blotting have been shown not to bind to nonspecific antibody controls. Controls with antibody alone were run alongside all immunoprecipitates, and equal protein loading of all Western blots was verified by blotting for β-actin.
Recombinant proteins and purification.
Recombinant His-tagged p27WT, p27S10A, p27T157A, p27T198A, p27T157A/T198A, and p27S10A/T157A/T198A were transformed into Escherichia coli BL21(DE3) cells grown in Terrific Broth medium (Difco) to the mid-exponential phase and induced with 1 mM isopropyl-β-d-thiogalactopyranoside for 3 h at 37°C. Cells were harvested and lysed using the BugBuster protein extraction reagent (Novagen), following the manufacturer's protocol. The soluble fraction was then applied to a 5-ml HisTrap nickel-nitrilotriacetic acid column (Amersham), washed with 10 column volumes of buffer I (50 mM Tris, 233 mM NaCl, 20 mM imidazole, 5% glycerol, pH 8.3) and eluted with linear imidazole gradient in buffer II (50 mM Tris, 233 mM NaCl, 500 mM imidazole, 5% glycerol, pH 8.3). Fractions containing His-p27 proteins were pooled and concentrated to about 5 mg/ml.
Baculoviral supernatant for recombinant Flag-cyclin D1 and His-Cdk4 were kindly provided by Alan Diehl. Flag-cyclin D1 was harvested from infected Sf-9 cells, and protein was purified using anti-Flag M2 affinity gel (Sigma). Protein was eluted by competitive elution using Flag peptide (Sigma). Recombinant His-Cdk4 was also harvested from infected Sf-9 cells, and protein was purified using a 5-ml HisTrap nickel-nitrilotriacetic acid column as described above for recombinant His-p27. Cyclin D-Cdk4 assembly assays were carried out using either purified cyclin D1 and Cdk4 or crude baculovirus lysate (see below).
PKB kinase assays.
Recombinant active PKB was obtained from Upstate and Cell Signaling. Reaction conditions (time and PKB concentrations of 20 to 200 ng) were varied to determine the linear range of kinase assay conditions. For the quantitative kinase reactions, 100 ng of PKB (Upstate) or 25 ng of PKB (Cell Signaling) was incubated with 5 μg of recombinant His-p27 (His-p27WT, -S10A, -T157A, -T198A, -T157A/T198A, or -S10A/T157A/T198A) in 20 μl of kinase reaction buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 1 mM dithiothreitol, 100 μM ATP). Reactions were carried out at 30°C for 15 min in the presence of 10 μCi [γ-32P]ATP, products were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and incorporated radioactivity was quantified by phosphorimager.
To test the action of cellular PKB on p27 in vitro, activated PKB was immunoprecipitated with PKBpS473 antibody from increasing amounts of asynchronous WM239 cell lysate and reacted with His-p27 proteins as described above. The WM239 cell line has no PTEN and has high levels of activated cellular PKB (38). Amounts of immunoprecipitated PKB and substrate and reaction duration were adjusted to ensure that reactions were in the linear range. Immunoprecipitated PKB was reacted with 5 μg of recombinant p27-His as described above.
Assembly of cyclin D1, Cdk4, and p27 complexes.
To assay cyclin D1 and Cdk4 assembly, baculovirus-expressed human Flag-tagged cyclin D1 and His-tagged Cdk4 were purified as described above and then incubated in assembly buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 1 mM dithiothreitol) at 25°C for 20 min in the presence or absence of 100 ng His-p27. The reactions were then diluted with 500 ml of ice-cold Nonidet P-40 lysis buffer (0.5% NP-40, 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, and 0.02 mg each of aprotinin, leupeptin, and pepstatin per ml) (51) for immunoprecipitation with a cyclin D1 antibody (Ab-3) or a Cdk4 antibody (DCS-35), followed by immunoblotting for cyclin D1, Cdk4, and p27. Assembly reactions using crude baculovirus lysate as the source for cyclin D1 and Cdk4 yielded similar results to those carried out with purified proteins.
To assay the effect of PKB and/or Src on the assembly function of p27, His-p27 proteins were reacted with immunoprecipitated cellular PKB as described above or with recombinant Src using kinase assay conditions described previously (10). Reaction products were boiled to denature and precipitate PKB and/or Src, and heat-stable p27 was recovered from the supernatant following centrifugation and assayed for the ability to assemble purified recombinant cyclin D1 and Cdk4 as described above. To test the effect of Src on PKB-treated p27 in p27-cyclin D-Cdk4 assembly, p27 was treated with PKB as described above and boiled, and heat-soluble p27 was recovered from the supernatant and mixed with cyclin D1 and Cdk4 for 30 min under the assembly conditions described above. Src kinase was then added for a further 15 min prior to the addition of Cdk4 antibody, assembled complexes were resolved, and associated proteins were detected by immunoblotting.
To compare the abilities of cellular p27 isolated from different cyclin-Cdk complexes to promote the assembly of recombinant cyclin D1 and Cdk4 in vitro, immunoprecipitation of either cellular cyclin D1 or Cdk2 was performed using appropriate antibodies, and the heat-stable cellular p27 was released from the immunoprecipitates by boiling in assembly buffer. Recovered p27 was titrated to ensure that equivalent p27 amounts were added to reaction mixtures, and their abilities to assemble recombinant cyclin D1-Cdk4 were then assayed as described above.
Activity of the p27-cyclin D1-Cdk4 complex.
His-p27 was treated with PKB, Src, or both as described above and then isolated by boiling. The isolated p27 was then used in assembly assays with purified recombinant cyclin D1 and Cdk4 as described above. The assembled p27-cyclin D1-Cdk4 complexes were precipitated with anti-Cdk4 antibody, and cyclin D1-Cdk4 kinase activity was assayed using recombinant pRb as the substrate as described previously (51). Cdk4 immune complexes were washed and reacted with 3 μg recombinant pRb in kinase reaction buffer (50 mM HEPES, pH 7.5, 10 mM MgCl2, 5 mM MnCl2). Reactions were carried out at 30°C for 20 min in the presence of 10 μCi [γ-32P]ATP, products were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and incorporated radioactivity was quantified by a phosphorimager.
RESULTS
PKB activation and p27-cyclin D1-Cdk4 complexes increase in G1.
PKB activation is required for G1 cell cycle progression (37). To assay the temporal relationship between PKB activation and p27-cyclin D1-Cdk4 assembly, HMEC strain 184 cells were synchronized in G0 by EGF depletion and then released by repletion of EGF (Fig. 1A). In addition, the human melanoma line WM35 was serum starved, and then cell cycle reentry was stimulated by the addition of 5% serum (Fig. 1B). HMEC and WM35 cells were recovered at intervals for cell cycle distribution analysis by flow cytometry and for protein analysis. The percent S phase is indicated for each time point (Fig. 1A and B). The total PKB levels showed little variation in HMEC and a subtle increase in mid-G1 in WM35. Levels of activated Ser 473-phosphorylated PKB (PKBpS473) were minimal in G0, increased within 1 h of G0 release, and peaked at 3 h in both cell types. PKBpS473 remained elevated during G1 and declined as cells entered the S phase. The kinetics of PKB activation were similar in cells released from contact inhibition (data not shown).
FIG. 1.

PKB activation precedes cyclin D1-Cdk4-p27 assembly in G1. (A) HMEC 184 cells were G0 arrested by EGF deprivation and released by the addition of EGF at time zero. At intervals thereafter, cells were collected for BrdU labeling, flow cytometry, and protein analysis. Protein levels were detected by Western blotting. Left panel, cell cycle entry is indicated by S%; right panel, Cdk4 complexes were immunoprecipitated and resolved, and associated proteins were detected by immunoblotting. (B) WM35 cells were G0 arrested by serum deprivation and released by the addition of full serum at time zero. Cells were collected as described above. Cdk4 complexes were immunoprecipitated, and associated proteins were detected by immunoblotting (right panel). (C) p27 phosphorylated at S10, T198, T157, or T187 was detected using phospho-specific antibodies. “Ab only” indicates reaction of precipitating antibody with protein A-Sepharose without lysate to control for reactivity of immunoblotting antibody with heavy or light chain of the antibody used in immunoprecipitation (IP). FBS, fetal bovine serum; NR-IgG, normal rabbit immunoglobulin G.
p27 protein was maximal in G0 and declined with S phase entry as described previously (51). Levels of p27 phosphorylated at either S10, T157, T198, or T187 were detected using phospho-specific antibodies (Fig. 1C). In WM35 cells, p27S10 was maximally phosphorylated in G0 and fell rapidly in early G1, during the same time that PKB activation increased. While total p27 levels fell during this interval, the detection of p27pT198 increased over 1 to 3 h after release from quiescence and remained detectable for several hours thereafter. p27pT157 was minimal in G0 and rising in early G1 with a peak at 6 h. Both p27pT157 and p27pT198 decreased as cells moved into the S phase. As expected, the T187 phosphorylation of p27 was only minimally detected until late G1 (52, 60), coincident with peak cyclin E-Cdk2 activation (18).
p27-cyclin D1-Cdk4 complexes increased during early G1 despite the concurrent reduction of total p27 protein levels and was temporally associated with PKB activation and the appearance of both p27pT157 and p27pT198. The increase in p27-cyclin D1-Cdk4 complexes preceded the rise in total cyclin D1 levels in both lines, more notably in HMEC, where S phase entry was slower than in WM35 (Fig. 1A and B), consistent with the notion that Cdk binding stabilizes cyclin D1 (32, 46).
PI3K/PKB inhibition causes p27-cyclin D1-Cdk4 dissociation.
Sustained inhibition of the PI3K/PKB pathway with resultant G0 arrest has been shown to reduce cyclin D1 and increase p27 (38, 62). To test the effects of short-term PI3K inhibition on p27 complexes, p27 was immunoprecipitated from proliferating MCF-7 cells before and at intervals after the addition of 10 μM LY294002. While 6 h of PI3K inhibition did not affect levels of PKB, cyclin D1, Cdk4, Cdk2, cyclin E2, or p27, the drug rapidly reduced levels of PKBpS473, p27pT157, p27pT198, and p27-cyclin D1-Cdk4 complexes (Fig. 2A). The loss of Cdk4 from p27 complexes was slower than that observed for cyclin D1 at 6 h (Fig. 2A) but was appreciable within 6 h and fell further within 24 h of PI3K/PKB inhibition (Fig. 2D). p27 association with cyclin E1 and Cdk2 increased progressively during 6 h of LY294002 treatment. The loss of p27-bound cyclin D1 and Cdk4 following PI3K/PKB inactivation was notable within 6 h of PI3K/PKB inhibition and preceded G1 arrest. Cells took 24 h to undergo full G1 arrest (Fig. 2B).
FIG. 2.

PI3K inhibition dissociates cyclin D1/Cdk4. (A to B) Lysates were prepared from MCF-7 before and at intervals after the addition of 10 μM LY294002 (LY). (A) Left panel, cell cycle proteins were detected by Western blotting; right panel, p27 immune complexes were resolved, and associated proteins were analyzed by immunoblotting. (B) MCF-7 cells were pulse labeled with BrdU, and cell cycle profiles were analyzed by flow cytometry. Graphed data represent the average of three independent experiments for each time point. (C) Asynchronous MCF-7 cells were treated with 10 μM LY294002 at time zero and lysed 15, 30, 60, and 240 min later, and immunoblots prepared for total p27 using anti-phospho-p27 antibodies shown. (D) MCF-7 cells were treated with (+) or without (−) the MEK inhibitor U0126 (5 μM) or LY294002 (10 μM) for 24 h. Left panel, proteins detected by immunoblotting; right panel, p27 was immunoprecipitated, and associated proteins were detected by immunoblotting. “Ab only” indicates reaction of precipitating antibody with protein A-Sepharose without lysate to control for reactivity of immunoblotting antibody with heavy or light chain of the antibody used in immunoprecipitation (IP).
LY294002 inhibits PI3K and PKB within minutes. To test the effects of LY294002 on p27 over a more rapid time course, cells were recovered within 15 min and at intervals over a 4-hour period. p27pT157 and p27pT198 levels fell within 15 min of LY294002 addition, while total p27 was unchanged. It is noteworthy that p27pS10 and p27pT187 were unchanged over 6 h of PI3K/PKB inhibition (data shown are results for 0 to 4 h in Fig. 2C).
In contrast to the effects of LY294002, MEK inhibition by 5 μM U0126 for 24 h did not cause p27-cyclin D1-Cdk4 dissociation (Fig. 2D). Inhibition of PKB and MAPK is shown by diminished PKBpS473 and pMAPK, respectively (Fig. 2D).
p27S10A, p27T157A, and p27T198A show reduced phosphorylation by PKB in vitro.
PKB has been reported to phosphorylate p27 at T157, T198, and S10 in vitro. To test how phosphorylation at one site may influence phosphorylation at the others, recombinant His-tagged p27WT, p27S10A, p27T157A, p27T198A, p27T157D, p27T198D, p27T157A/T198A, and p27S10A/T157A/T198A (p27AAA) were each used as substrates in PKB kinase assays (Fig. 3A). For data shown, cellular PKBpS473 was immunoprecipitated from WM239 asynchronous cell lysates. All experiments were repeated using recombinant PKB from two different commercial sources, and effects on p27 phosphorylation were confirmed. While the loss of the potential to phosphorylate p27 at T157 reduced PKB-mediated phosphorylation by approximately 50%, mutation converting T198 to alanine reduced p27 phosphorylation by over 50%, suggesting that phosphorylation at T198 influences that at T157. As was observed for p27T198A, mutation converting S10 to alanine also reduced p27 phosphorylation by PKB by 80%, suggesting that the conformation of p27S10 influences the potential for p27 phosphorylation at other sites. While the PKB phosphorylation of p27T157A/T198A was similar to that of p27T198A, p27AAA was not phosphorylated by PKB in vitro compared to mock reactions lacking PKB. The p27T157D and p27T198D mutants showed the same loss of phosphorylation by PKB, as did their respective alanine mutants. Thus, they did not appear to function as phospho-mimetic in PKB kinase assays (not shown).
FIG. 3.
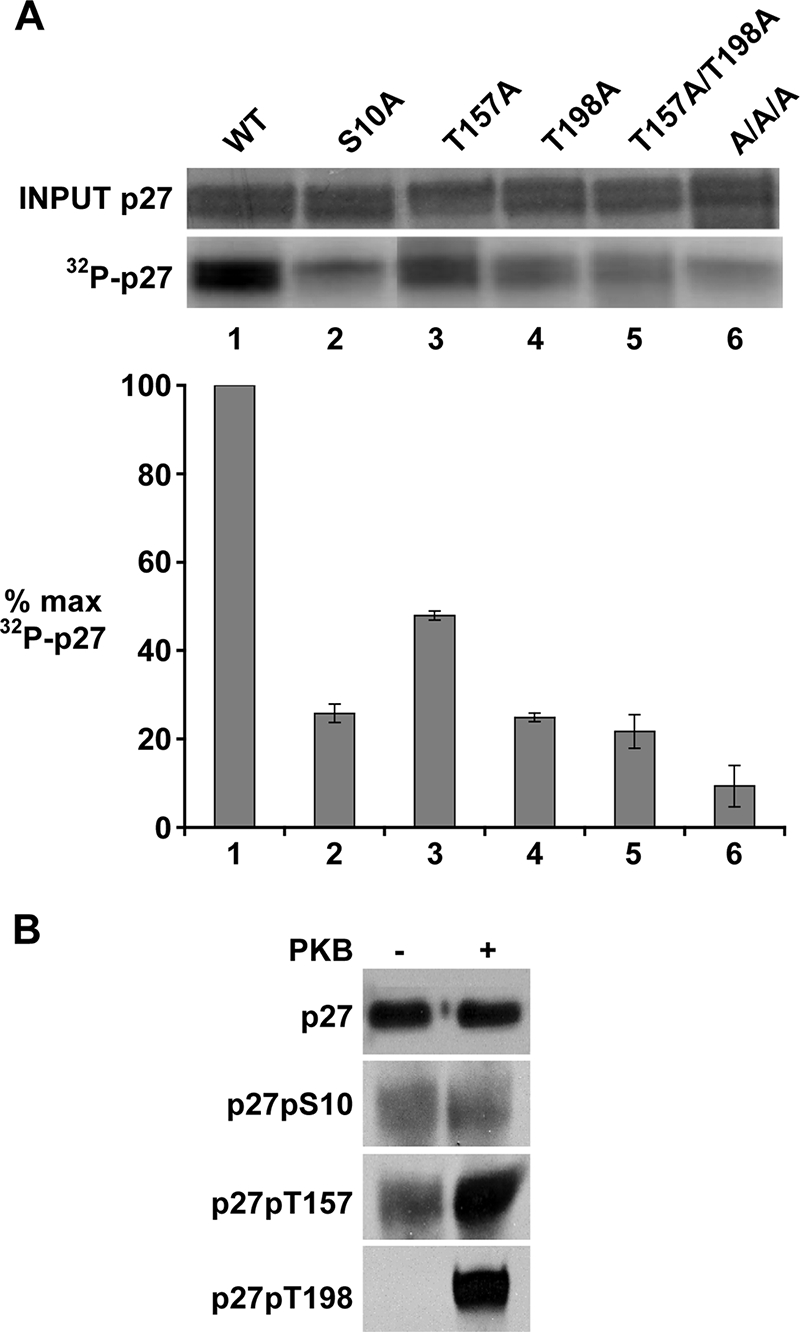
Phosphorylation of p27 by PKB in vitro. (A) PKB immunoprecipitated from asynchronous WM239 cells was incubated with His-p27 proteins in the presences of 10 μCi [γ-32P]ATP. The Coomassie blue stain of the input protein and the radioactivity of His-p27 bands are shown above the graph. The kinase reaction products shown were run on the same gel, and are all from the same exposure of the gel. The mean of four independent experiments is graphed. Error bars indicate the standard deviation. Kinase activity was plotted as a percentage of the maximum (% max), setting His-p27WT as 100% maximum kinase activity. (B) The PKB kinase reaction was carried out using His-p27WT as a substrate in the presence of cold ATP in the presence (+) or absence (−) of PKB. Phosphorylation of p27 at S10, T157, and T198 sites was detected by immunoblotting using phospho-specific antibodies.
The reduced action of recombinant PKB on p27S10A observed by us and others (19, 44) could indicate that this site is phosphorylated by PKB in vitro, or that the conformation of S10 is important for PKB action at other sites (e.g., T157 and T198). In WM35 cells, detection of p27pS10 is maximal in G0 and early G1 prior to PKB activation (Fig. 1B and C). To further test the effects of PKB on p27 phosphorylation at S10, T157, and T198, His-p27 was reacted with cellular PKBp473 in vitro and then immunoblotted and reacted with different anti-p27 phospho-antibodies (Fig. 3B). While PKB treatment increased p27 reactivity with antibodies raised against pT157 and pT198, levels of p27pS10 were not increased by PKB. Thus, while S10 phosphorylation may prime p27 for PKB phosphorylation, S10 does not appear to be a direct PKB site under our experimental conditions. This is in keeping with the observation that drug-mediated PKB inhibition also failed to reduce cellular p27pS10 levels (Fig. 2C).
Loss of phosphorylation at T157 and T198 impairs p27 binding to cyclin D1 and Cdk4 but not to Cdk2.
Prior work showed that the p27 phospho-isoforms present in cyclin D1-Cdk4 complexes differ from those in cyclin E-Cdk2-bound p27 (12). To further evaluate how p27 phosphorylation may differ in these respective cyclin-Cdk complexes, Cdk4 and Cdk2 were immunoprecipitated from asynchronous MCF-7 cells and associated cellular p27 was immunoblotted with phospho-specific antibodies (Fig. 4A). While more p27 was present in Cdk4 than in Cdk2 precipitates, the precipitates were titrated to allow a comparison of p27 phospho-isoforms in comparable amounts of total precipitated p27. Cellular p27pT157 and p27pT198 were detected only in Cdk4 precipitates and not in Cdk2 complexes.
FIG. 4.

The potential for T157 and T198 phosphorylation of p27 affects its association with cyclin D1 and Cdk4. (A) Cdk4 (IP Cdk4) and Cdk2 (IP Cdk2) were immunoprecipitated from asynchronous MCF-7 cells. p27 phosphorylated at T157 (p27pT157) and at T198 (p27pT198), total p27, total Cdk4, and total Cdk2 were detected by immunoblotting. (B to E) MCF-7 cells were transiently transfected with YFPp27 vectors, and cells were collected 12 h posttransfection. (B) Protein levels were detected by Western blotting. Cyclin D1 (C), Cdk4 (D), and Cdk2 (E) were immunoprecipitated, and associated YFPp27 was detected by immunoblotting. “Ab only” indicates reaction of precipitating antibody with protein A-Sepharose without lysate to control for reactivity of immunoblotting antibody with heavy or light chain of the antibody used in immunoprecipitation.
To test how the potential for T157 and T198 phosphorylation would affect p27 levels and p27-cyclin-Cdk complexes, YFPp27WT, YFPp27T157A, YFPp27T198A, or YFPp27T157A/T198A was transfected into MCF-7 cells. Total cyclin D1, Cdk4, Cdk2, and YFPp27 protein levels are shown (Fig. 4B). All p27 alleles caused G1 arrest within 48 h of transfection (not shown). At 12 h posttransfection, cells were not arrested, and while YFPp27WT and YFPp27T157A levels were similar, YFPp27T198A and YFPp27T157A/T198A protein levels were reduced compared to those of YFPp27WT. When differences in the expression levels of the p27 variants were taken into account, less YFPp27T157A, YFPp27T198A, and YFPp27T157A/T198A were detected in cyclin D1 and Cdk4 complexes compared with YFPp27WT (Fig. 4C and D). Steady-state binding to Cdk2 was similar for all YFPp27 variants compared with YFPp27WT (Fig. 4E). Since p27T198A and p27T157A/T198A levels were reduced compared to those of p27WT, the relative binding of these p27 mutants to Cdk2 was increased, in keeping with the observations of Kossatz et al. (30). Thus, the absence of phosphorylation at T157 and T198 reduces p27 association with cyclin D1 and Cdk4 in cells and may promote Cdk2 binding.
The ability of p27 to promote cyclin D-Cdk4 assembly is phosphorylation dependent and activated by PKB.
We next tested whether p27 phosphorylation by PKB could affect cyclin D1-Cdk4 assembly using recombinant proteins in vitro. Baculovirus-produced cyclin D1 and Cdk4 were mixed with or without the addition of His-p27WT. Their assembly was detected by immunoprecipitating cyclin D1 or Cdk4, followed by immunoblotting of associated proteins. While purified cyclin D1 and Cdk4 did not form detectable complexes when mixed alone, the addition of purified His-p27WT increased the amount of Cdk4 detected in cyclin D1 immunoprecipitates (Fig. 5A), consistent with prior observations (32). To test how PKB pretreatment may influence the effect of added p27 on Cdk4 binding to cyclin D1 in vitro, p27 was reacted with PKB in vitro and p27 was isolated from these reaction mixtures prior to addition to cyclin D1 and Cdk4 in vitro. The p27-PKB reaction mixture was boiled to heat denature the PKB. The heat-precipitated PKB was removed by centrifugation, the heat-stable p27 was recovered from the supernatant, and isolated p27 was then added to cyclin D1-Cdk4. Treatment of His-p27WT with PKB prior to the assembly reaction markedly increased the ability of p27 to mediate cyclin D1-Cdk4 assembly (Fig. 5A).
FIG. 5.

Cyclin D1-Cdk4 assembly by p27 in vitro is increased by PKB action on p27 and is phosphorylation dependent. (A) Recombinant cyclin D1 and Cdk4 were incubated without p27 (No p27), with His-p27WT, or with cellular p27 released by boiling from Cdk2 (K2-p27) and cyclin D1 (D1-p27) immune complexes from MCF-7 cells. Where indicated (+ or −), p27 was treated with PKB and boiled to remove active PKB prior to the addition of cyclin D1 and Cdk4 to p27 for the assembly assays. Cyclin D1 was then immunoprecipitated (IP Cyclin D1), and Cdk4 binding was detected by Western blotting. The bottom panel shows Western blots of the input proteins. (B) p27 was pretreated with (+) or without (−) CIP prior to assembly assays as described above. Input proteins are shown in the bottom panel. (C) Cyclin D1 and Cdk4 were incubated without p27, with His-p27WT, or with His-p27S10A/T157A/T198A (AAA). Where indicated (+ or −), p27 was incubated with PKB, and then p27 was isolated prior to the addition of cyclin D1 and Cdk4 for the assembly assays. Cdk4 was then immunoprecipitated (IP Cdk4), and Cdk4, cyclin D1, and His-p27 were detected by Western blotting. The input proteins are indicated.
The assembly activity of cellular p27 from either Cdk2 or cyclin D1 immunoprecipitates was similarly assayed. Cellular p27 present in immunoprecipitates of Cdk2 or cyclin D1 was released from these complexes by boiling, and the heat-stable cellular p27 was recovered. Cellular p27 isolated from Cdk2 was able to mediate the assembly of cyclin D1 and Cdk4 but did so less effectively than equivalent amounts of p27 isolated from cellular cyclin D1 complexes (Fig. 5A). When the p27 recovered from Cdk2 precipitates was pretreated with PKB, its ability to assemble p27-cyclin D1 complexes was increased (Fig. 5A). These data suggest that the PKB-dependent phosphorylation of p27 promotes its assembly function for cyclin D1 and Cdk4.
To further demonstrate the dependence of p27 assembly function on phosphorylation, p27 was treated with calf intestinal alkaline phosphatase (CIP) prior to cyclin D1-Cdk4 assembly assays. MCF-7 cells were transfected with Flag-tagged p27. Immunoprecipitated Flag-p27 was either treated with CIP or not prior to assembly reactions. p27 was recovered by boiling and reimmunoprecipitating p27 with anti-Flag antibody. CIP pretreatment markedly reduced the assembly function of p27 compared with that of the untreated p27 (Fig. 5B). Thus, the cyclin D1-Cdk4 assembly function of p27 is phosphorylation dependent.
While the PKB treatment of p27 increased the assembly of p27-cyclin D1-Cdk4 complexes in vitro, the p27S10A/T157A/T198A mutant protein showed minimal cyclin D1-Cdk4 assembly. Treatment with PKB did not increase the ability of this mutant p27 protein to promote cyclin D1-Cdk4 complexes (Fig. 5C). Taken together, these data indicate that site-specific phosphorylation of p27 is required to activate its cyclin D1-Cdk assembly function.
Cyclin D1-Cdk4-p27 assembled in vitro is inactive and requires tyrosine phosphorylation for catalytic activation.
Whether p27-cyclin D1-Cdk complexes can adopt an active conformation has been a matter of some controversy (1, 2, 9, 26, 32). We therefore tested whether p27-cyclin D1-Cdk4 assembled in vitro had catalytic activity toward pRb protein substrate. While treatment of His-p27WT with PKB prior to the assembly reaction increased cyclin D1-Cdk4 assembly (Fig. 6A, lane 3), these complexes were catalytically inactive and failed to phosphorylate pRb substrate (Fig. 6B, lane 3). Active cyclin D1-Cdk4 precipitates from asynchronous cell cultures served as a positive control for these Cdk4 assays.
FIG. 6.
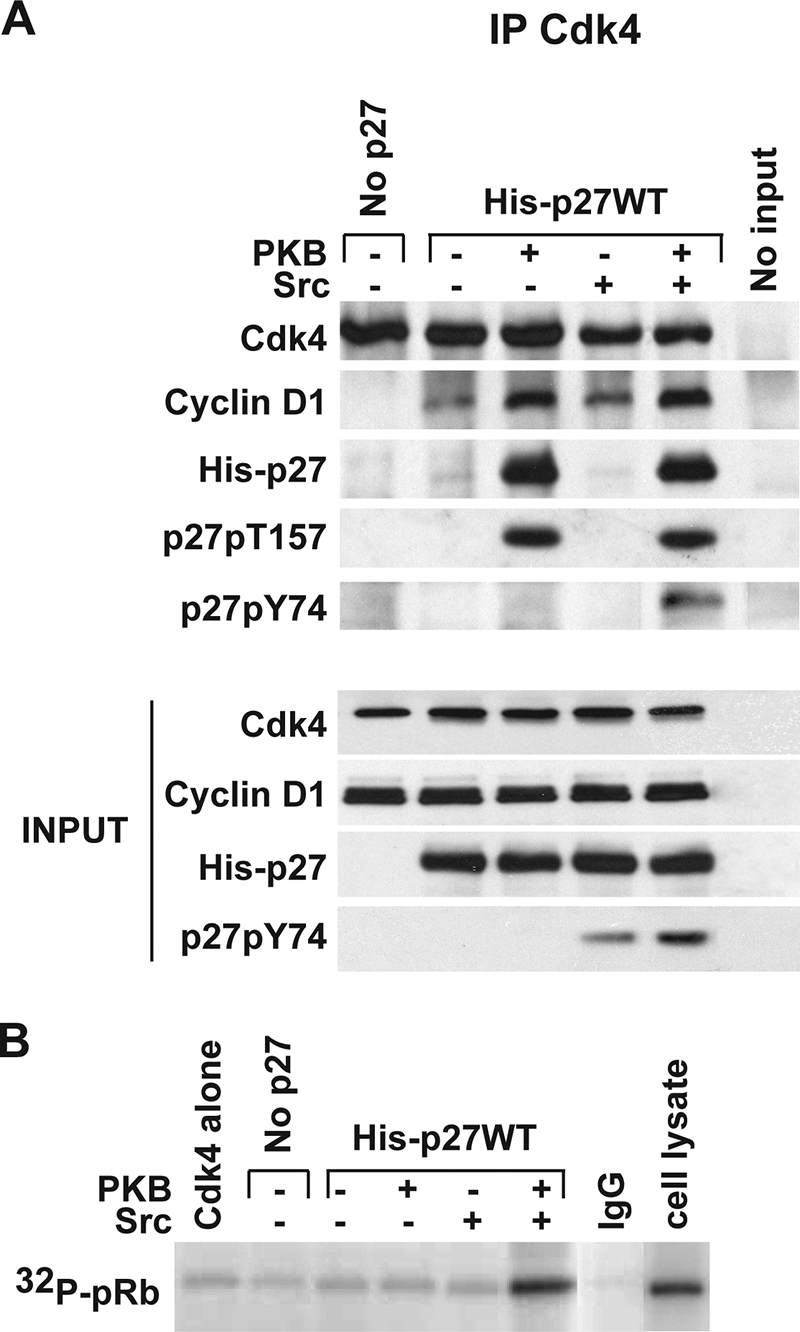
Activation of cyclin D1-Cdk4-p27 requires the tyrosine phosphorylation of p27. Cyclin D1 and Cdk4 were incubated without or with His-p27WT. Where indicated (+ or −), p27 was incubated with PKB and/or with Src prior to the addition to cyclin D1 and Cdk4 assembly reaction mixtures as shown in Fig. 5. (A) Assembly was detected by immunoprecipitation of Cdk4, followed by detection of associated cyclin D1 and p27, p27pT157, and p27pY74 by Western blotting. The input of cyclin D1, Cdk4, and p27 to reaction mixtures is indicated. (B) The activity of the complexes assembled as in panel A was measured using GST-pRb as a substrate in Cdk4 kinase assays. Active cyclin D1-Cdk4 precipitates from asynchronous WM35 cells served as a positive control, and nonspecific monoclonal immunoglobulin G immunoprecipitates (IgG) reacted with lysate served as a negative kinase reaction control.
We and others recently demonstrated that p27 is phosphorylated by Abl and Src family kinases at tyrosine residues (10, 21). p27 phosphorylation at Y88 prevents its association with the catalytic cleft of cyclin-bound Cdk2 (21), and Y74 phosphorylation of p27 appears to reduce p27-Cdk2 steady-state binding (10). We therefore tested whether Src treatment would influence p27-cyclin D1-Cdk4 assembly and/or the activation of this complex. In contrast to the effect of PKB, pretreatment of p27 with Src (without treatment with PKB) led to detection of tyrosine phosphorylation of p27 (Fig. 6A, bottom) but did not promote p27 binding to cyclin D1-Cdk4 over that observed with untreated p27 (Fig. 6A and B, lane 2 versus lane 4).
We then assayed the effect of Src on cyclin D1-Cdk4-p27 complexes. p27 was pretreated with PKB, isolated, and then mixed with cyclin D1 and Cdk4 under assembly conditions. Src treatment led to tyrosine phosphorylation of both input p27 and Cdk4-bound p27 (Fig. 6A, lane 5) and catalytic activation of p27-cyclin D1-Cdk4 toward pRb (Fig. 6B, lane 5). These data suggest that while phosphorylation of p27 by PKB promotes assembly of p27-cyclin D1-Cdk4, activation of the complex requires further phosphorylation of p27 at tyrosine sites.
DISCUSSION
Recent data indicate that p27 function is phosphorylation dependent. The Cdk2-inhibitory function of p27 is attenuated by tyrosine phosphorylation (10, 21). p27 also binds Cdk4 and cyclin D1, but whether p27-cyclin D1-Cdk has catalytic activity has been controversial (1, 2, 9, 26, 32). p27 was shown to facilitate in vitro assembly of D-type cyclin-Cdks (32), and double p21/p27 knockout cells show reduced cyclin D-Cdk complexes, but mechanisms regulating p27-cyclin D-Cdk assembly have been unclear. The observation that cyclin D1-bound p27 and cyclin E-bound p27 have different phospho-isoform patterns (12) provided early evidence that phosphorylation may regulate inhibitor and assembly functions of p27. Assembly of cyclin D1-Cdk4 is mitogen dependent. Despite their active synthesis in G0, newly formed cyclin D1 and Cdk4 do not assemble in G0 cells (12). Overexpressed cyclin D1 fails to activate Cdk4 in serum-starved cells, but does activate with serum repletion (40). Whether mitogen-dependent mechanisms regulate CIP/KIP phosphorylation to facilitate D-type cyclin-Cdk assembly has not been clear. The present work indicates that PKB-dependent phosphorylation of p27 at T157 and T198 appears to promote p27-cyclin D1-Cdk4 assembly, but further tyrosine phosphorylation of p27 is required for catalytic activation of these complexes.
Human p27 is phosphorylated by PKB at T157 (38, 55, 59) and at T198 in vitro and in cells (19, 20, 42). The present data provide evidence for coordinate regulation of T157 and T198 phosphorylation in cells. PKB activation, p27 phosphorylation at T157 and T198 and the appearance of p27-cyclin D1-Cdk4 show close temporal kinetics during G0-to-S progression. Moreover, the loss of the potential to phosphorylate p27 at T157 or T198 reduces p27 binding to cellular cyclin D1 and Cdk4. p27T198A levels were lower than those of either p27WT or p27T157A, consistent with findings that T198 phosphorylation stabilizes p27 (30, 36). However, the reduction in p27T198A binding to cyclin D1 was even greater than that predicted by the reduced p27T198A levels.
Mutation of T198 to alanine attenuated T157 phosphorylation by PKB in vitro. Phosphorylation at these two sites may be processive, with phosphorylation at T198 facilitating that at T157. Phosphorylation of p70S6K by PDK1 and MAPK shows a similar cooperativity (17). PKB and other AGC kinases, including SGK1 (24) and RSK1 (20; M. D. Larrea and J. M. Slingerland, unpublished data) may regulate cooperative p27 phosphorylation at T157 and T198. While p27T157 is not conserved in mice, p27T198 is (30) and may serve to regulate cyclin D-Cdk complexes as would murine ACG kinase action on p21. Coordinate T157 and T198 phosphorylation may permit fine-tuning of p27-cyclin D-Cdk assembly in humans.
p27pT157 and p27pT198 were detected in cellular Cdk4 but not in Cdk2. It is noteworthy that Kossatz et al. showed that murine p27T198A binds better to Cdk2 than p27WT (30), consistent with our finding of a relative increase in p27T198A binding to cellular Cdk2 compared to p27WT in human cells. p27 isolated from cellular Cdk2 complexes also assembled p27-cyclin D1-Cdk4 poorly in vitro compared to p27 from cyclin D1 complexes. p27 phosphorylation by PKB increased cyclin D1-Cdk4 assembly in vitro, while CIP-dephosphorylated p27 and the p27AAA mutant failed to do so. Taken together, these data suggest that PKB-dependent T157 and T198 phosphorylation may promote p27-cyclin D1-Cdk4 assembly. The shift of p27 from Cdk2 to cyclin D1-Cdk4 complexes observed in cells upon PKB activation may in part reflect increased p27 phosphorylation at T157 and T198. Since the p27T157 site is also conserved in p21 (35, 50, 64), PKB-mediated p21 phosphorylation may also promote cyclin D-Cdk assembly and would account for the increased cyclin D1-Cdk4-p21 observed in PKB-activated cells (35).
In WM35 and other cells, p27pS10 is maximal in G0 and falls rapidly in early G1 (25). While MAPK can phosphorylate S10 in vitro, MEK inhibition does not reduce p27pS10 in vivo (25). In Rat1 cells, S10 phosphorylation increases with G0 exit (49), and hKis and Mirk/dyrkB can phosphorylate p27 at S10 in early G1 (7, 14). We and others found that p27S10A is a poor substrate for PKB in vitro (19, 44). While S10 may be a phospho-site for PKB, PKB treatment of recombinant p27 did not increase p27 reactivity with anti-phospho-p27pS10 antibody under our experimental conditions. Cellular p27pS10 was maximal in G0 when PKB is inactive and fell in early G1 as PKB activation increased. In addition, PI3K/PKB inhibition rapidly attenuated cellular p27pT157 and p27pT198 but did not reduce p27pS10 levels. These data allow that S10 conformation and indeed prior S10 phosphorylation may facilitate subsequent phosphorylation at T157 and T198. S10 phosphorylation-dependent nuclear export of p27 would augment cytoplasmic p27 in early G1, when new synthesis of p27 may be rate limiting for assembly of newly synthesized cyclin D1-Cdk complexes. While the loss of S10 did not yield any apparent cell cycle defects in S10A knock-in to p27 null animals (4, 31), compensation for cell cycle defects is not uncommon in murine embryogenesis (39).
Prior work has shown that p27-bound cyclin D1-Cdk from proliferating cells can have catalytic activity (26, 32, 63), while other data indicated that p27-bound cyclin-D complexes are inactive (1, 2). The present data together with recent work of others provides a unifying model for these observations. We found p27-cyclin D1-Cdk4 complexes formed in vitro were catalytically inactive. Treatment of assembled p27-cyclin D1-Cdk4 with Src yielded tyrosine phosphorylation of p27 and Cdk4 activation. The James et al. group reported that p27 in inactive p27-cyclin D-Cdk complexes from contact-inhibited cells is not tyrosine phosphorylated, while p27 is tyrosine phosphorylated in cyclin D1-Cdk4 complexes from proliferating cells, and these have activity toward pRb (26). Tyrosine phosphorylation of p27 abrogates its inhibition of cyclin-Cdk2 (10, 21, 28). These data and the present findings suggest a model in which C-terminal p27 phosphorylation by AGC kinases may promote p27-cyclin D1-Cdk4 formation or stability, while the action of Abl or Src family kinases on p27 would be required for catalytic activation of the assembled complexes. While the crystal structure of cyclin D1-cdk4-p27 has not been solved, Y88 of p27 may also lie in the portion of p27 that occludes the ATP-binding pocket of Cdk4 as it does for Cdk2 and be released upon Y88 phosphorylation. Thus, multiple signaling kinases may cooperate to phosphorylate p27 to promote the assembly and activation of cyclin D1-Cdk4-p27 complexes and cellular proliferation.
p27 is frequently deregulated in human cancers through accelerated degradation or mislocalization in the cytoplasm (56). Increased cytoplasmic p27 is observed in many human cancers (11). p27pT157 and p27pT198 localize to the cytoplasm in certain human cancers (42, 59), and cytoplasmic p27 is uniformly observed in cancers with activated PKB (38, 55, 59). Constitutive mitogenic signaling would activate S10-dependent p27 export, increase cytoplasmic p27, and thus reduce cyclin E-Cdk2-dependent p27 proteolysis. In addition, T198 phosphorylation may further stabilize p27 (30, 36) in p27-cyclin D-Cdk complexes, protecting p27 from KPC-mediated degradation, a proteolytic mechanism that acts only on non-Cdk-bound p27 in the cytoplasm (22, 27). Stable cytoplasmic p27 may acquire novel functions to promote cell motility and tumor metastasis (5, 15, 34, 41, 45, 61). In the current model, oncogenic PKB and Src family activation would cooperate to regulate T157, T198, and tyrosine phosphorylation to stabilize cytoplasmic p27, promote cyclin D1-Cdk assembly and activation, and the acquisition of prooncogenic p27 action during tumorigenesis. These data provide a rationale for the combined use of PI3K/PKB and Src family inhibitors to restore p27 function in human cancers.
Acknowledgments
We thank Alan Diehl for providing baculovirus supernatant for recombinant cyclin D1 and Cdk4 production and members of the lab for helpful discussions during the course of this work.
This work was supported by a grant from the Doris Duke Charitable Foundation and NCI grant 1R01CA105118-01 to J.M.S. and by NIH/NCI predoctoral fellowship 5F31CA113284 to M.D.L. J.M.S. is supported by the Braman Family Breast Cancer Institute of UM/Sylvester Comprehensive Cancer Center.
Footnotes
Published ahead of print on 18 August 2008.
REFERENCES
- 1.Bagui, T. K., R. J. Jackson, D. Agrawal, and W. J. Pledger. 2000. Analysis of cyclin D3-cdk4 complexes in fibroblasts expressing and lacking p27kip1 and p21cip1. Mol. Cell. Biol. 208748-8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagui, T. K., S. Mohapatra, E. Haura, and W. J. Pledger. 2003. p27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol. Cell. Biol. 237285-7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates, S., D. Parry, L. Bonetta, K. Vousden, C. Dickson, and G. Peters. 1994. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene 91633-1640. [PubMed] [Google Scholar]
- 4.Besson, A., M. Gurian-West, X. Chen, K. S. Kelly-Spratt, C. J. Kemp, and J. M. Roberts. 2006. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2047-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besson, A., M. Gurian-West, A. Schmidt, A. Hall, and J. M. Roberts. 2004. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 18862-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blagosklonny, M. V., and A. B. Pardee. 2002. The restriction point of the cell cycle. Cell Cycle 1103-110. [PubMed] [Google Scholar]
- 7.Boehm, M., T. Yoshimoto, M. F. Crook, S. Nallamshetty, A. True, G. J. Nabel, and E. G. Nabel. 2002. A growth factor-dependent nuclear kinase phosphorylates p27Kip1 and regulates cell cycle progression. EMBO J. 213390-3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cariou, S., J. C. Donovan, W. M. Flanagan, A. Milic, N. Bhattacharya, and J. M. Slingerland. 2000. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc. Natl. Acad. Sci. USA 979042-9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng, M., P. Olivier, J. A. Diehl, M. Fero, M. F. Roussel, J. M. Roberts, and C. J. Sherr. 1999. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 181571-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu, I., J. Sun, A. Arnaout, H. Kahn, W. Hanna, S. Narod, P. Sun, C. K. Tan, L. Hengst, and J. Slingerland. 2007. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 128281-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu, I. M., L. Hengst, and J. M. Slingerland. 2008. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 8253-267. [DOI] [PubMed] [Google Scholar]
- 12.Ciarallo, S., V. Subramaniam, W. Hung, J. H. Lee, R. Kotchetkov, C. Sandhu, A. Milic, and J. M. Slingerland. 2002. Altered p27Kip1 phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor β-mediated G1 arrest. Mol. Cell. Biol. 222993-3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornil, I., D. Theodorescu, S. Man, M. Herlyn, J. Jambrosic, and B. Kerbel. 1991. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc. Natl. Acad. Sci. USA 886028-6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng, X., S. E. Mercer, S. Shah, D. Z. Ewton, and E. Friedman. 2004. The cyclin-dependent kinase inhibitor p27Kip1 is stabilized in G0 by Mirk/dyrk1B kinase. J. Biol. Chem. 27922498-22504. [DOI] [PubMed] [Google Scholar]
- 15.Denicourt, C., C. C. Saenz, B. Datnow, X. S. Cui, and S. F. Dowdy. 2007. Relocalized p27KiP1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res. 679238-9243. [DOI] [PubMed] [Google Scholar]
- 16.Diehl, J. A., F. Zindy, and C. J. Sherr. 1997. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 11957-972. [DOI] [PubMed] [Google Scholar]
- 17.Dufner, A., and G. Thomas. 1999. Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 253100-109. [DOI] [PubMed] [Google Scholar]
- 18.Florenes, V. A., N. Bhattacharya, M. R. Bani, Y. Ben-David, R. S. Kerbel, and J. M. Slingerland. 1996. TGF-beta mediated G1 arrest in a human melanoma cell line lacking p15INK4B: evidence for cooperation between p21Cip1/WAF1 and p27Kip1. Oncogene 132447-2457. [PubMed] [Google Scholar]
- 19.Fujita, N., S. Sato, K. Katayama, and T. Tsuruo. 2002. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 27728706-28713. [DOI] [PubMed] [Google Scholar]
- 20.Fujita, N., S. Sato, and T. Tsuruo. 2003. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 27849254-49260. [DOI] [PubMed] [Google Scholar]
- 21.Grimmler, M., Y. Wang, T. Mund, Z. Cilensek, E. M. Keidel, M. B. Waddell, H. Jakel, M. Kullmann, R. W. Kriwacki, and L. Hengst. 2007. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 128269-280. [DOI] [PubMed] [Google Scholar]
- 22.Hara, T., T. Kamura, K. Nakayama, K. Oshikawa, S. Hatakeyama, and K.-I. Nakayama. 2001. Degradation of p27Kip1 at the G0-G1 transition mediated by a Skp2-independent ubiquitination pathway. J. Biol. Chem. 27648937-48943. [DOI] [PubMed] [Google Scholar]
- 23.Herlyn, M. 1990. Human melanoma: development and progression. Cancer Metastasis Rev. 9101-112. [DOI] [PubMed] [Google Scholar]
- 24.Hong, F., M. D. Larrea, C. Doughty, D. J. Kwiatkowski, R. Squillace, and J. M. Slingerland. 2008. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol. Cell 30701-711. [DOI] [PubMed] [Google Scholar]
- 25.Ishida, N., M. Kitagawa, S. Hatakeyama, and K. Nakayama. 2000. Phosphorylation at serine 10, a major phosphorylation site of p27Kip1, increases its protein stability. J. Biol. Chem. 27525146-25154. [DOI] [PubMed] [Google Scholar]
- 26.James, M., A. Ray, D. Leznova, and S. W. Blain. 2008. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol. Cell. Biol. 28498-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamura, T., T. Hara, M. Matsumoto, N. Ishida, F. Okumura, S. Hatakeyama, M. Yoshida, K. Nakayama, and K. I. Nakayama. 2004. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat. Cell Biol. 61229-1235. [DOI] [PubMed] [Google Scholar]
- 28.Kardinal, C., M. Dangers, A. Kardinal, A. Koch, D. T. Brandt, T. Tamura, and K. Welte. 2006. Tyrosine phosphorylation modulates binding preference to cyclin-dependent kinases and subcellular localization of p27Kip1 in the acute promyelocytic leukemia cell line NB4. Blood 1071133-1140. [DOI] [PubMed] [Google Scholar]
- 29.Koff, A., M. Ohtsuki, K. Polyak, J. M. Roberts, and J. Massague. 1993. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science 260536-539. [DOI] [PubMed] [Google Scholar]
- 30.Kossatz, U., J. Vervoorts, I. Nickeleit, H. A. Sundberg, J. S. C. Arthur, M. P. Manns, and N. P. Malek. 2006. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 255159-5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotake, Y., K. Nakayama, N. Ishida, and K. I. Nakayama. 2005. Role of serine 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a serine 10 mutation. J. Biol. Chem. 2801095-1102. [DOI] [PubMed] [Google Scholar]
- 32.LaBaer, J., M. D. Garrett, L. F. Stevenson, J. M. Slingerland, C. Sandhu, H. S. Chou, A. Fattaey, and E. Harlow. 1997. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 11847-862. [DOI] [PubMed] [Google Scholar]
- 33.Laiho, M., J. A. DeCaprio, J. W. Ludlow, D. M. Livingston, and J. Massague. 1990. Growth inhibition by TGF-beta linked to suppression of retinoblastoma protein phosphorylation. Cell 62175-185. [DOI] [PubMed] [Google Scholar]
- 34.Lee, S., and D. M. Helfman. 2004. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J. Biol. Chem. 2791885-1891. [DOI] [PubMed] [Google Scholar]
- 35.Li, Y., D. Dowbenko, and L. A. Lasky. 2002. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 27711352-11361. [DOI] [PubMed] [Google Scholar]
- 36.Liang, J., S. H. Shao, Z. Xu, B. Hennessy, Z. Ding, M. Larrea, S. Kondo, D. J. Dumont, J. U. Gutterman, C. L. Walker, J. M. Slingerland, and G. B. Mills. 2007. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9218-224. [DOI] [PubMed] [Google Scholar]
- 37.Liang, J., and J. M. Slingerland. 2003. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2339-345. [PubMed] [Google Scholar]
- 38.Liang, J., J. Zubovitz, T. Petrocelli, R. Kotchetkov, M. K. Connor, K. Han, J. H. Lee, S. Ciarallo, C. Catzavelos, R. Beniston, E. Franssen, and J. M. Slingerland. 2002. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 81153-1160. [DOI] [PubMed] [Google Scholar]
- 39.Malumbres, M., and M. Barbacid. 2005. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30630-641. [DOI] [PubMed] [Google Scholar]
- 40.Matsushime, H., D. E. Quelle, S. A. Shurtleff, M. Shibuya, C. J. Sherr, and J. Y. Kato. 1994. D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 142066-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McAllister, S. S., M. Becker-Hapak, G. Pintucci, M. Pagano, and S. F. Dowdy. 2003. Novel p27kip1 C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol. Cell. Biol. 23216-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motti, M. L., C. De Marco, D. Califano, A. Fusco, and G. Viglietto. 2004. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle 31074-1080. [PubMed] [Google Scholar]
- 43.Muise-Helmericks, R. C., H. L. Grimes, A. Bellacosa, S. E. Malstrom, P. N. Tsichlis, and N. Rosen. 1998. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 27329864-29872. [DOI] [PubMed] [Google Scholar]
- 44.Nacusi, L. P., and R. J. Sheaff. 2006. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div. 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagahara, H., A. M. Vocero-Akbani, E. L. Synder, A. Ho, D. G. Latham, N. A. Lissy, M. Becker-Hapak, S. A. Ezhevsky, and S. F. Dowdy. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 41449-1452. [DOI] [PubMed] [Google Scholar]
- 46.Parry, D., S. Bates, D. J. Mann, and G. Peters. 1995. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. EMBO J. 14503-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polyak, K., J. Y. Kato, M. J. Solomon, C. J. Sherr, J. Massague, J. M. Roberts, and A. Koff. 1994. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 89-22. [DOI] [PubMed] [Google Scholar]
- 48.Polyak, K., M. H. Lee, H. Erdjument-Bromage, A. Koff, J. Roberts, P. Tempst, and J. Massagué. 1994. Cloning of p27KIP1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 7859-66. [DOI] [PubMed] [Google Scholar]
- 49.Rodier, G., A. Montagnoli, L. Di Marcotullio, P. Coulombe, G. F. Draetta, M. Pagano, and S. Meloche. 2001. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 206672-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rössig, L., A. S. Jadidi, C. Urbich, C. Badorff, A. M. Zeiher, and S. Dimmeler. 2001. Akt-dependent phosphorylation of p21Cip1 regulates PCNA binding and proliferation of endothelial cells. Mol. Cell. Biol. 215644-5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandhu, C., J. Garbe, N. Bhattacharya, J. Daksis, C. H. Pan, P. Yaswen, J. Koh, J. M. Slingerland, and M. R. Stampfer. 1997. Transforming growth factor β stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol. Cell. Biol. 172458-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheaff, R. J., M. Groudine, M. Gordon, J. M. Roberts, and B. E. Clurman. 1997. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 111464-1478. [DOI] [PubMed] [Google Scholar]
- 53.Sherr, C. J. 2000. The Pezcoller Lecture: cancer cell cycles revisited. Cancer Res. 603689-3695. [PubMed] [Google Scholar]
- 54.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 131501-1512. [DOI] [PubMed] [Google Scholar]
- 55.Shin, I., F. M. Yakes, F. Rojo, N. Y. Shin, A. V. Bakin, J. Baselga, and C. L. Arteaga. 2002. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat. Med. 81145-1152. [DOI] [PubMed] [Google Scholar]
- 56.Slingerland, J., and M. Pagano. 2000. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 18310-17. [DOI] [PubMed] [Google Scholar]
- 57.Slingerland, J. M., L. Hengst, C. H. Pan, D. Alexander, M. R. Stampfer, and S. I. Reed. 1994. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor β-arrested epithelial cells. Mol. Cell. Biol. 143683-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stampfer, M. 1985. Isolation and growth of human mammary epithelial cells. J. Tissue Cult. Methods 9107-115. [Google Scholar]
- 59.Viglietto, G., M. L. Motti, P. Bruni, R. M. Melillo, A. D'Alessio, D. Califano, F. Vinci, G. Chiappetta, P. Tsichlis, A. Bellacosa, A. Fusco, and M. Santoro. 2002. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27Kip1 by PKB/Akt-mediated phosphorylation in breast cancer. Nat. Med. 81136-1144. [DOI] [PubMed] [Google Scholar]
- 60.Vlach, J., S. Hennecke, and B. Amati. 1997. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 165334-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu, F. Y., S. E. Wang, M. E. Sanders, I. Shin, F. Rojo, J. Baselga, and C. L. Arteaga. 2006. Reduction of cytosolic p27Kip1 inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 662162-2172. [DOI] [PubMed] [Google Scholar]
- 62.Yakes, F. M., W. Chinratanalab, C. A. Ritter, W. King, S. Seelig, and C. L. Arteaga. 2002. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 624132-4141. [PubMed] [Google Scholar]
- 63.Zhang, H., G. Hannon, and D. Beach. 1994. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 81750-1758. [DOI] [PubMed] [Google Scholar]
- 64.Zhou, B. P., Y. Liao, W. Xia, B. Spohn, M. H. Lee, and M. C. Hung. 2001. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3245-252. [DOI] [PubMed] [Google Scholar]