

Abstract
Megakaryoblastic leukemia 1 (MKL1) is a myocardin-related coactivator of the serum response factor (SRF) transcription factor, which has an integral role in differentiation, migration, and proliferation. Serum induces RhoA-dependent translocation of MKL1 from the cytoplasm to the nucleus and also causes a rapid increase in MKL1 phosphorylation. We have mapped a serum-inducible phosphorylation site and found, surprisingly, that its mutation causes constitutive localization to the nucleus, suggesting that phosphorylation of MKL1 inhibits its serum-induced nuclear localization. The key site, serine 454, resembles a mitogen-activated protein kinase phosphorylation site, and its modification was blocked by the MEK1 inhibitor U0126, implying that extracellular signal-regulated kinase 1/2 (ERK1/2) is the serum-inducible kinase that phosphorylates MKL1. Previous results indicated that G-actin binding to MKL1 promotes its nuclear export, and we found that MKL1 phosphorylation is required for its binding to actin, explaining its effect on localization. We propose a model in which serum induction initially stimulates MKL1 nuclear localization due to a decrease in G-actin levels, but MKL1 is then downregulated by nuclear export due to ERK1/2 phosphorylation.
Megakaryoblastic leukemia 1/2 proteins (MKL1/2, MRTF-A/B, MAL, BSAC), along with the related protein myocardin, are transcriptional coactivators of serum response factor (SRF) (7). SRF is a transcription factor that belongs to the MADS box family and binds to serum response elements (SRE) in the promoters of various immediate-early and muscle-specific genes (32, 33, 38). The core sequence of the SRE has the consensus sequence CC(AT)6GG and is called a CArG box (13, 17). Serum and growth factors stimulate SRF activity via two seemingly independent pathways, one that is dependent on the phosphorylation of ternary complex factors (TCFs) by a mitogen-activated protein kinase (MAPK) cascade and the other that is dependent on Rho signaling and actin dynamics (12, 40). The TCF proteins Elk-1, SAP-1, and Net make sequence-specific DNA contacts with Ets motifs adjoining the CArG boxes of some immediate-early genes, and phosphorylation of their transcriptional activation domains potentiates their ability to activate transcription (29). The second pathway that activates SRF involves the small GTPase RhoA, since the inhibition of RhoA blocks serum induction of TCF-independent SRE reporter genes and some SRF target genes, while activated RhoA can stimulate SRE reporter genes (12). RhoA activation causes stress fiber formation and the reduction of monomeric G-actin. The use of actin mutants and drugs that interfere with actin treadmilling suggests that SRE activation is controlled by the G-actin pool (21, 31).
Myocardin was originally identified as a strong coactivator for SRF in heart and smooth muscle cells (35). We and others have identified two myocardin-related SRF-specific coactivators, MKL1 and MKL2, that are expressed in a wide range of embryonic and adult tissues and that strongly activate SRE reporter genes when overexpressed (6, 8, 16, 18, 24, 28, 36). Experiments using dominant-negative MKL1 or inhibition of MKL1/2 expression with RNA interference have shown that these proteins are required for the induction of many immediate-early genes, such as SRF and vinculin, as well as for the serum induction of TCF-independent SRE reporter genes (unpublished results) (6, 18, 27). Miralles and colleagues showed that MKL1 is sequestered in the cytoplasm in serum-starved cells and accumulates in the nucleus upon serum induction and RhoA GTPase signaling (18). RhoA signaling causes the polymerization of actin and the formation of stress fibers (22). This leads to a reduction of monomeric G-actin. G-actin was found to bind directly to MKL1 through the RPEL domains at the N terminus of MKL1, suggesting that G-actin binding to MKL1 dictates its cytoplasmic localization (11, 18). In support of a model in which alterations in the cellular G-actin content regulate MKL1 localization, tension-mediated cytoskeletal changes in Drosophila melanogaster also resulted in nuclear accumulation of MKL1 in vivo (30).
In serum-starved cells, MKL1 constantly circulates between the nucleus and the cytoplasm. Live cell imaging demonstrated that the cytoplasmic steady-state localization of MKL1 in serum-starved cells is maintained by very efficient Crm1-dependent nuclear export (34). This rate of nuclear export is dependent upon MKL1 binding to actin and is reduced in serum-induced cells, resulting in the nuclear localization of MKL1 (34). Nuclear import of MKL1 may also be regulated, as it was dependent upon RhoA activity; however, changes in nuclear export appear to be the determining factor for MKL1 localization.
Besides changes in actin binding, serum also induces rapid phosphorylation of MKL1, yet the serum-induced phosphorylation site(s) has not been defined. In addition to induction by serum, brain-derived neurotrophic factor (BDNF) treatment of cortical neurons also induced phosphorylation of MKL1 in an extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent manner (14). It remained to be determined whether or how phosphorylation contributed to the regulation of MKL1 and the regulation of SRF transcriptional activity. In the case of the closely related MKL family member myocardin, phosphorylation by GSK3β has been reported to moderately repress its transcriptional activity (3).
In the current report, we have mapped the serum-induced site of phosphorylation of MKL1 to serine 454. During the course of this work, epidermal growth factor (EGF)-inducible phosphorylation sites were determined by using a proteomic approach that identified the same S454 site in MKL1 as that described here (20). We have found that rather than activating MKL1 following serum stimulation, MKL1 phosphorylation serves as a downregulatory mechanism that promotes nuclear export of MKL1 by increasing its binding to actin. This phosphorylation requires the ERK1/2 MAPK pathway and shows how cross-talk from this pathway can limit induction by the RhoA pathway.
MATERIALS AND METHODS
Cloning and plasmids.
The MKL1 coding region was expressed in p3x-FLAG-CMV-7.1, as described previously (6). The deletion mutants lack the region up to and including the amino acid in their names, e.g., the N-terminal deletion of N100 lacks amino acids 1 to 100, and the internal deletion, Δ444 to 500, lacks amino acids (aa) 444 to 500. Point mutations were introduced into the p3x-FLAG-MKL1 construct by using a QuikChange multisite-directed mutagenesis kit (Stratagene). For the Tet-off expression vectors, all constructs were individually recloned into the BamHI/SalI site of the pRevTRE expression vector (Clontech). The p5xSRE-luciferase reporter was from Stratagene. Green fluorescent protein (GFP)-actin was expressed with pcDNA3-EGFP-β-actin as described previously (4).
Cell culture, transfections, and luciferase assays.
HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum at 37°C in a 5% CO2 incubator. HeLa cells were transiently transfected on six-well plates, with the standard calcium phosphate DNA precipitation method (23). The MEF/3T3 Tet-off (TO3T3) cell line from Clontech was used to stably express the tetracycline-controlled transactivator. Cell lines with Flag-tagged MKL1, MKL1-STS/A, N100, and N100-STS/A were prepared by retroviral infection and selection with hygromycin (e.g., STS/A represents changes from three conserved serine and threonine residues, S449, S454, and T450, to alanine). The viruses expressing Flag-tagged MKL1, MKL1-STS/A, N100, and N100-STS/A were generated by transfection of 293 cells with pRevTRE MKL1, MKL1-STS/A, N100, and N100-STS/A and a packaging site-defective Moloney murine leukemia virus construct. Stable TO3T3 cell lines were maintained in DMEM with 10% fetal bovine serum (FBS), 50 μg/ml hygromycin (InvivoGen), 100 μg/ml G418 (InvivoGen), and 1 μg/ml tetracycline (Sigma).
TO3T3 cell lines were transiently transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions in 24-well plates with reporter plasmids, as indicated in the figure legends, including a Renilla luciferase simian virus 40 (SV40) promoter reporter as an internal control. On the next day, cells were serum starved for another 24 h before being stimulated with 20% tetracycline-free FBS for 3 h. Luciferase assays were performed using a dual luciferase system (Promega). The firefly luciferase activities were normalized to the Renilla luciferase activities to compensate for variability in transfection efficiencies. All experiments were performed with duplicate plates of cells for each time point.
Immunoprecipitation and immunoblots.
For immunoprecipitation assays, cells from six-well plates were rinsed once in ice-cold phosphate-buffered saline (PBS) and then lysed in 500 μl of immunoprecipitation buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, and a protease inhibitor cocktail [catalog no. P8340; Sigma]). The lysates were rotated for 2 h at 4°C and cleared by centrifugation at 13,000 × g for 10 min at 4°C. Flag-MKL1 (3×), GFP-actin, or endogenous actin was immunoprecipitated with 1 μl of anti-Flag antibody M2 (Sigma), GFP antibody (Invitrogen), or actin antibody (Sigma) overnight. Recombinant protein G-Sepharose (Zymed) (100 μl of a 50% slurry in immunoprecipitation buffer) was added, and then the mixture was incubated for 3 h. The protein G beads were washed three times with immunoprecipitation buffer, and the proteins were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For dephosphorylation, MKL1 immunoprecipitates were incubated at 37°C for 30 min with 1 μl of calf intestinal alkaline phosphatase (CIP) in 50 μl of 1× CIP buffer as provided by the manufacturer (Boehringer Mannheim Biochemicals) prior to SDS-PAGE. The proteins were then immunoblotted with anti-Flag (Sigma) or anti-GFP (Invitrogen) antibodies at 1:1,000 dilution. A commercial source (Quality Controlled Biochemicals) was used for peptide and antibody synthesis and antibody purification of the phospho-S454-specific MKL antibody. The successful antibody was raised against the conserved MKL2 sequence (SPLPIpSPSPSE, where pS indicates phosphoserine) and cross-reacted with MKL1. The corresponding MKL1 antigen did not provide specific sera. The antiserum indicates to total MKL1 was described previously (6). Immunoblots were visualized using fluorescently labeled secondary antibodies and detected with a Li-Cor Odyssey infrared imaging system.
RNA analyses.
For reverse transcription-PCR (RT-PCR) experiments, total RNA was made from stable MEF/3T3 cell lines using Trizol as described by the manufacturer (Invitrogen). cDNA was synthesized from the RNA (1 μg) with ImProm-II reverse transcriptase (Promega) and random hexamer primers. Specific cDNAs were quantified by SYBR Green-based real-time PCR, using an ABI PRISM 7000 sequence detection system (Applied Biosystems). The signals for vinculin and SRF cDNAs were normalized to that for 18S rRNA. The sequences of the primers are available upon request.
Immunofluorescence.
HeLa cells were fixed with 3.5% paraformaldehyde in PBS for 10 min, which was followed by extraction in 0.2% Triton X-100 in PBS for 7 min. Anti-Flag antibody and anti-mouse secondary antibody coupled to Alexa Fluor 488 were diluted 1:500. Cells were washed four times in PBS after antibody incubations and prior to mounting. Images were obtained with a Nikon Diaphot 300 epifluorescent microscope and processed as PICT files, using Adobe Photoshop CS software. MKL1 localization was scored as predominantly nuclear, predominantly cytoplasmic, or both in 100 to 200 cells.
RESULTS
MKL1 is subject to serum-inducible phosphorylation.
Treatment of quiescent HeLa cells with serum results in a change in the electrophoretic mobility of MKL1, in which the entire pool of MKL1 shifts to a slower-migrating form by 30 min of treatment (Fig. 1A). The shifted form persists for 90 min, and the original mobility is restored afterwards. This change in mobility is due to increased phosphorylation, since treatment of the shifted MKL1 protein from serum-induced cells with alkaline phosphatase changed MKL1 to the faster migrating form, while there was little or no effect on MKL1 from serum-starved cells (Fig. 1B). This result is similar to that obtained by Miralles et al. (18).
FIG. 1.

MKL1 is subject to serum-inducible phosphorylation at aa 444 to 500. (A) Quiescent HeLa cells transfected with Flag-tagged MKL1 were stimulated as indicated with serum prior to lysis and immunoblotted with anti-Flag antibodies. (B) Lysates from quiescent and serum-stimulated HeLa cells expressing Flag-MKL1 were treated with (+) or without (−) calf intestinal alkaline phosphatase and immunoblotted as above. MKL1 electrophoretic mobility was examined as described in the legend to panel A. (C) The MKL1 derivatives used for mapping of phosphorylation sites are shown. Conserved domains, RPEL domain; B, basic domain; Q, glutamine-rich domain; SAP domain; LZ, leucine zipper-like domain. (D) HeLa cells expressing Flag-MKL1 or its derivatives were left unstimulated or stimulated for 30 min with serum and analyzed by immunoblotting.
To assess the importance of MKL1 phosphorylation relative to its regulation, we mapped serum-inducible phosphorylation sites by mutagenesis of Flag-tagged MKL1 and transfection into HeLa cells. We first used different deletion derivatives to gain insight into the sites of phosphorylation, using the shift in gel mobility as an indication of phosphorylation (Fig. 1C). While there was still inducible phosphorylation following serum stimulation with the MKL1 mutant lacking aa 301 to 380, there was consistently no induction with the internal deletion of aa 381 to 506 (Fig. 1D). Similarly, an MKL1 mutant that lacks aa 444 to 630 was not phosphorylated upon serum stimulation. We next divided the internal deletion of aa 444 to 630 into two regions consisting of aa 444 to 500 and 500 to 630. Deletion of aa 500 to 630 had no effect and exhibited a change in mobility following serum stimulation. In contrast, deletion of aa 444 to 500 was sufficient to abolish the serum-inducible shift (Fig. 1D). These data indicate that the site(s) of phosphorylation is in the region of aa 444 to 500.
Identification of MKL1 phosphorylation sites.
In addition to other conserved regions in MKL1, the sequence within aa 444 to 500 is conserved in different species and among the MKL family members MKL1, MKL2, and myocardin (Fig. 2A). We mutated together three conserved serine and threonine residues (S449, S454, and T450) to alanine (STS/A) and noted that these point mutations abolished the serum-inducible phosphorylation of MKL1 (Fig. 2B). Among the single aa mutations, S454A completely blocked a change in mobility, while the S449A and T450A mutations reduced but did not abolish the serum-induced mobility change (Fig. 2C and D). We next replaced S449, T450, and S454 with glutamates to mimic phosphorylation (STS/E). As a result, we observed a change in the mobility of the STS/E mutant in serum-starved HeLa cells compared to that in the MKL1 wild type (wt). There was no further increase in mobility following serum stimulation (Fig. 2E). The change in mobility suggests the similarity between the glutamate mutant (STS/E) and constitutively phosphorylated MKL1.
FIG. 2.

Identification of MKL1 phosphorylation sites. (A) Conserved sequence motifs found between aa 444 and 500 in the MKL/myocardin protein family. (B to E) HeLa cells expressing MKL1 and the indicated mutants were stimulated with serum for 30 min, lysed, and immunoblotted with anti-Flag antibody. The STS/A and STS/E mutants have changes of S449/T450/S454 to alanines and glutamates, respectively.
Phosphorylation of MKL1 at S454.
To further substantiate the identification of S454 as a key site for serum-inducible phosphorylation of MKL1, we raised a phospho-specific antibody capable of recognizing MKL1 only when it was phosphorylated at S454. We immunoblotted cell lysates from the TO3T3 cells overexpressing MKL1 or the phosphorylation site mutants (Fig. 2). Consistent with the results described above, the antibody recognized MKL1 phosphorylated at S454 after 30 min of serum stimulation (Fig. 3A). The antibody did not react with the MKL1 STS/A or S454A mutant but did react with the S449A and T450A mutants, showing the antibody's specificity (Fig. 3A and B). The mutations of S449 and T450 had only moderate effects on the signal with the phospho-specific antibody (Fig. 3B). This may be due to effects of the recognition of the S454 site or effects of the phosphorylation of the site. We also tested phosphorylation of endogenous MKL1 in TO3T3 cells (Fig. 3C). Phosphorylation was most strongly induced at 30 min and a decreased level was maintained until at least 120 min after serum treatment. Endogenous MKL1 was not seen in the other blots (Fig. 3A and B) because it runs slightly above transfected MKL1 (due to differences at the N terminus) and because endogenous MKL1 is expressed much more weakly than the transiently transfected MKL1 variants (Fig. 3A and B). The above-described results demonstrate that MKL1 S454 is required for serum-inducible phosphorylation and that MKL1 is phosphorylated on this site. S449 and T450 may be phosphorylated, but they were not required for phosphorylation at S454. A large-scale detection of EGF-inducible phosphorylation sites by mass spectrometry also identified the S449, T450, and S454 sites on MKL1 in addition to serine 6 (http://www.phosida.com/) (20). Deletion of aa 1 to 100 did not affect the serum-inducible mobility shift (data not shown), showing that serine 6 is not required for this shift.
FIG. 3.
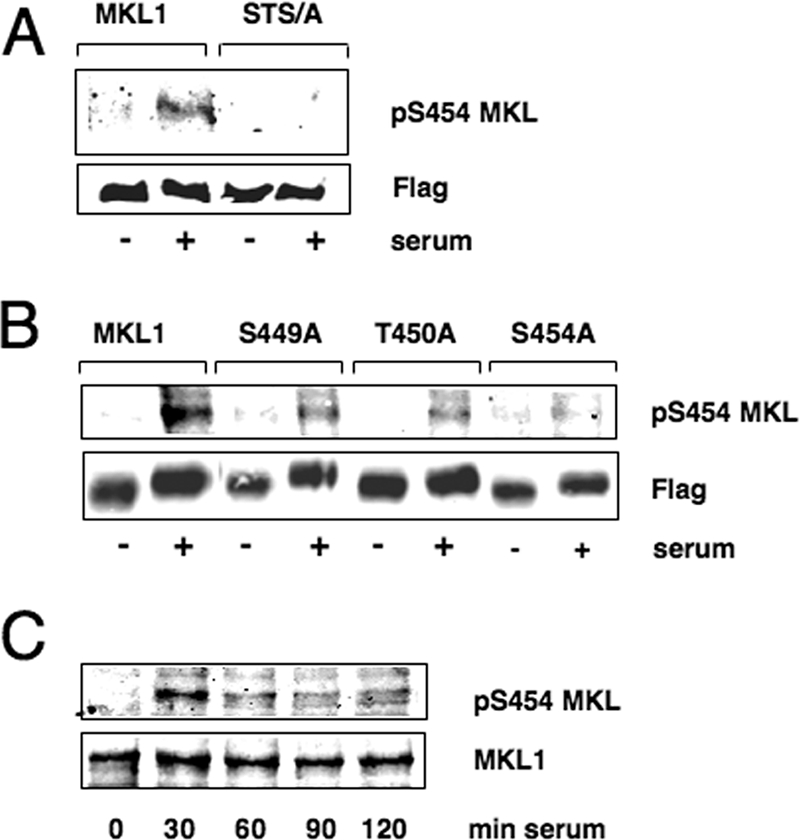
Phosphorylation of MKL1 at serine 454. (A and B) Serum-starved TO3T3 cells transiently transfected with MKL1 and the indicated mutants in the p3x-FLAG-CMV-7.1 vector (2 μg/35-mm dish) were treated with serum for 30 min and immunoblotted with antisera that recognize phospho-S454 or Flag-MKL1. (C) Serum-starved TO3T3 cells were incubated with serum for the times indicated and detected with anti-phospho-S454 and anti-MKL1 antibodies.
The MKL1 phosphorylation site mutant is constitutively localized to the nucleus.
Since the subcellular localization of MKL1 is regulated by serum (18), we tested whether mutations of the phosphorylation sites affected the serum induction of its movement from the cytoplasm to the nucleus. We transfected HeLa cells with constructs encoding epitope-tagged MKL1 and the MKL1 phosphorylation site mutant (MKL1-STS/A) and followed the localization through immunofluorescence with anti-FLAG antibodies. Wild-type MKL1 exhibited predominantly cytosolic staining in serum-starved cells and accumulated in the nucleus after serum stimulation for 30 min (Fig. 4A and B). In contrast, the STS/A mutant showed predominantly nuclear localization of the protein, even in serum-starved cells. Serum stimulation caused no further increase in nuclear accumulation of MKL1-STS/A (Fig. 4A and B). We similarly found that deletion of the region, such as Δ444 to 630 and Δ444 to 500, led to constitutive nuclear localization of MKL1 (data not shown). Among the single mutants, only the S454A mutant exhibited nuclear localization in serum-starved cells. The S449A and T450A mutants behaved like wt MKL1, suggesting that the S454 phosphorylation site is required for localization in the cytoplasm in serum-starved cells (Fig. 4C).
FIG. 4.

The MKL1 phosphorylation site mutant is constitutively localized to the nucleus. (A) HeLa cells were transfected with Flag-MKL1 or the phosphorylation site mutant (STS/A), serum starved, and treated with (+) or without (−) serum for 30 min. The cells were analyzed by immunofluorescence with anti-Flag antibodies. (B) Subcellular localization shown in panel A was scored as predominantly cytoplasmic (C), nuclear (N), or both (C/N). Statistical analyses were carried out for three independent experiments, with 100 to 200 cells per condition; error bars indicate standard errors of the means. (C) Quantitation of immunofluorescence in HeLa cells transiently transfected with MKL1 and the indicated mutants as described in the legend to panel A. (D) Mutants in putative nuclear export sequences were analyzed for cellular localization as described in the legend to panel B.
In these experiments, we have used a form of MKL1 that starts at the first start codon, is 931 aa long, and contains two RPEL motifs at the N terminus. However, full-length MKL1 appears to start at a noncanonical leucine codon, adding 92 amino acids and an additional RPEL motif (18). We determined whether this additional sequence would affect the localization of MKL1 or the phosphorylation site mutants. We transfected this full-length version of MKL1 with an N-terminal Flag tag into HeLa cells and found localization similar to that shown in Fig. 4A. In addition, we made the phosphorylation site STS/A mutant in the full-length form of MKL1 and similarly found that it was constitutively nuclear (data not shown). Thus, the effect of the phosphorylation site mutation on localization, as shown in Fig. 4A, is not dependent upon the lack of the first RPEL motif.
The localization of MKL1 is regulated predominantly at the level of nuclear export (34), as seen most simply by its nuclear localization in the presence of the nuclear export inhibitor leptomycin B (34) (data not shown). However, it is not known whether a nuclear export signal (NES) contributes to nucleocytoplasmic shuttling of MKL1. To address this question, we looked for an NES in MKL1 to determine whether it is critical for nuclear export. The NetNES server (5, 15) predicted two putative nuclear export signals in MKL1 at aa 272 and aa 555. The aa 272 site is in the glutamine-rich domain and is well conserved among the MKL family members. We found that mutation of L272 to alanine was sufficient to cause nuclear localization of MKL1, consistent with impaired nuclear export, while mutation of L555 had no effect (Fig. 4D). This result suggests that other regions of MKL1, such as the phosphorylation sites and the N-terminal actin binding domain, may regulate the accessibility of the NES around aa 272.
MKL1 phosphorylation is regulated by the ERK1/2 MAPK pathway.
Since S454 is followed by a proline, it fits the consensus sequence for a MAPK phosphorylation site (2). We determined whether ERK1/2 might mediate phosphorylation at this specific site by using the MEK1 inhibitor UO126 to prevent the activation of ERK1/2 (9). We found that UO126 completely abolished serum-inducible phosphorylation (Fig. 5A). We then determined whether U0126 also affected the kinetics of serum-induced localization of MKL1. Wild-type MKL1 accumulates in the nucleus within 30 min and then gradually exits until it is nearly entirely cytoplasmic at 6 h (Fig. 5B). In contrast, U0126 inhibition of the ERK1/2 pathway did not affect the initial nuclear localization of MKL1 but blocked the subsequent movement back to the cytoplasm for as long as 8 h following serum stimulation (Fig. 5C).
FIG. 5.

MKL1 phosphorylation is regulated by the ERK1/2 MAPK pathway. (A) HeLa cells were transfected with Flag-MKL1, serum-starved, preincubated with or without the MEK1 inhibitor UO126 (10 μM) for 30 min, and then stimulated with serum for 30 min. Lysates were immunoblotted with anti-Flag antibodies. (B) For immunofluorescence analysis, HeLa cells were transfected with Flag-MKL1, serum starved, and then serum stimulated for the indicated times and quantitated for cellular localization as shown in Fig. 4. (C) As in panel B, except that cells were pretreated for 30 min with 10 μM U0126. Localization of FLAG-MKL1 was scored in 100 to 200 cells; N, nuclear; C, cytoplasmic; C/N, both nuclear and cytoplasmic. Three independent experiments were performed. Error bars, standard errors of the means. (D) Left panel, quiescent HeLa cells transfected with wt MKL1 or the phosphorylation site mutant (STS/A) were incubated with (+) or without (−) TPA (100 ng/ml) for 30 min and immunoblotted as described in the legend to panel A. (D) Right panel, untransfected (lane 1) or wt MKL1-transfected (lanes 2 and 3) cells were treated with or without TPA and immunoblotted using the anti-phospho-S454 MKL or anti-Flag antibodies as indicated. (E) Time course of MKL1 localization in HeLa cells preincubated with TPA for 30 min and stimulated with serum was determined by immunostaining as described in the legends to panels B and C. (F) HeLa cells were transfected with the STS/E MKL1 mutant, where the phosphorylation sites were changed to glutamates and analyzed for cellular localization, as described above.
Since UO126 blocked serum-induced phosphorylation of MKL1, we sought to induce phosphorylation of MKL1 by selectively activating the MAPK pathway. We used the phorbol ester 12-O-tetradecanoyl-13-acetate (TPA), which activates protein kinase C and leads to activation of ERK1/2 (26). We confirmed that TPA induced ERK1/2 phosphorylation in HeLa cells, using phospho-ERK1/2-specific sera (data not shown). TPA also induced a shift in the mobility of MKL1 and was not able to modify the STS/A mutant (Fig. 5D, left panel). Furthermore, the phospho-specific pS454 antibody also recognized MKL1 phosphorylated in response to TPA (Fig. 5D, right panel); together, these findings demonstrate that TPA induces phosphorylation at S454.
Interestingly, prior TPA treatment blocked MKL1 localization to the nucleus in serum-induced cells at all time points tested (Fig. 5E). Nuclear import still functions in the presence of TPA, since TPA-treated cells exhibited nuclear localization of MKL1 in the presence of leptomycin B, an inhibitor of nuclear export (data not shown). Next, we studied the localization of the phosphomimetic STS/E mutant with the phosphorylation sites mutated to glutamate instead of alanine. In contrast to the localization of the STS/A mutant, the STS/E mutant was localized to the cytoplasm in serum-starved cells and was nuclear following serum stimulation for 30 min. However, the phosphomimetic mutant was rapidly redistributed to the cytoplasm much more quickly than wt MKL1 (compare Fig. 5B and F). While the initial import of the STS/E mutant differs from that found with the TPA-induced phosphorylation of MKL1, the rapid movement back to the cytoplasm further shows that MKL1 phosphorylation promotes cytoplasmic localization. The difference between the STS/E mutant response and that of the TPA suggests that the STS/E mutant may not be a perfect phosphomimetic mutant. To show that the TPA block of MKL1 nuclear localization was specific to its induction of MKL1 phosphorylation rather than a general effect of TPA on signaling pathways, we determined whether TPA would block serum-induced nuclear localization of the STS/E mutant. This mutant was inducibly localized to the nucleus at 30 min (Fig. 5F and 6B), and we expected that, in contrast to the effect on wt MKL1 (Fig. 5E and 6A), TPA would not affect this localization, since it cannot induce phosphorylation of the STS/E mutant. Indeed, TPA had no effect on serum-induced localization of the STS/E mutant (Fig. 6B). This suggests that the TPA-increased cytoplasmic localization of MKL1 (Fig. 5E and 6A) is due to a specific effect on its phosphorylation rather than to a more indirect effect through other cellular proteins. Together, these findings suggest that the ERK1/2 kinase mediates the phosphorylation of MKL1 and thereby facilitates nuclear export.
FIG. 6.
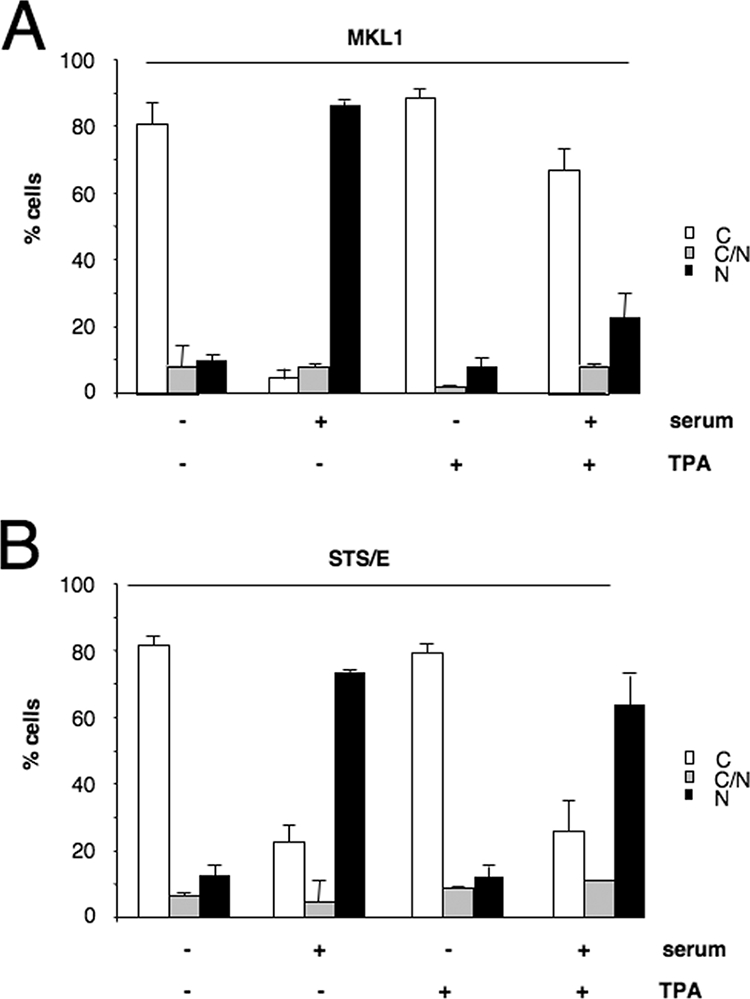
TPA treatment inhibits serum-induced MKL1 nuclear localization. (A) HeLa cells were transfected with Flag-tagged MKL1, serum-starved, and treated with (+) or without (−) TPA for 30 min, followed by serum induction for 30 min as indicated. The cellular localization of MKL1 was determined by immunofluorescence with anti-Flag antibodies as shown in Fig. 4. (B) As described in the panel A legend above, except that the STS/E phosphomimetic MKL1 mutant was used. C, cytoplasmic; N, nuclear; C/N, both cytoplasmic and nuclear.
Effect of MKL1 phosphorylation sites on SRF activity.
To assess the importance of the phosphorylation sites for the regulation of SRF target genes, we used a relatively weak Tet-off-regulated promoter to express MKL1, since strong expression of MKL1 under the control of a cytomegalovirus (CMV) promoter results in the activation of SRE-containing reporter genes in both serum-starved and -induced cells (6; data not shown). We tested the effect of the transient transfection of the Tet-off MKL1 constructs expressing wt MKL1; an STS/A mutant of the three phosphorylation sites, N100, which lacks the N-terminal 100 amino acids of MKL1 containing the RPEL motifs that bind actin; and the N100-STS/A double mutant. Each of the mutant proteins was expressed at similar levels as shown by immunoblotting (data not shown). As expected, without actin binding, N100 was localized to the nucleus in serum-starved and -induced cells (data not shown). Expression of a low amount of wt MKL1-expressing plasmid with a 5xSRE-luciferase reporter gene led to an increase in the basal luciferase levels in serum-starved cells, but there was also a clear serum induction of luciferase activity (Fig. 7A). Expression of both the STS/A and the N100 mutants led to an increase in activity in serum-starved cells and little further activation by serum, consistent with these two mutants being constitutively active due to their nuclear localization. The N100-STS/A double mutant gave results similar to those of the N100 and STS/A mutants individually, suggesting that the phosphorylation sites are not required for MKL1 function in the absence of its actin binding domain and that their mutation also does not further increase activity. This latter result shows that the phosphorylation sites are not required for MKL1 activity when MKL1 is nuclear.
FIG. 7.

The nonphosphorylatable mutant is constitutively active. (A) The indicated MKL1 expression vectors in pRevTRE (5 ng) or vector alone (Vec) were transiently transfected into TO3T3 cells with an SRE-dependent luciferase reporter gene (p5xSRE; 100 ng), together with a Renilla luciferase internal control (pRL-SV40P; 50 ng). Two days after cells were transfected, and 3 days after doxycycline was removed, the cells were treated with or without serum (20%) for 3 h. Luciferase assays were performed for firefly luciferase and normalized to the Renilla luciferase control. Data are represented as standard errors of the means of three independent experiments, each with triplicates. STS/A, phosphorylation site mutant; N100, deletion of aa 1 to 100; N100-STS/A, double mutant. (B) TO3T3 cells stably expressing the indicated mutants were transfected with the p5xSRE and pRL-SV40P reporters as described in the legend to panel A. Inset, immunoblot with anti-Flag antibodies of the indicated stably transfected TO3T3 cells. (C) RNA was isolated from the stably expressing MKL1 variant cells shown in panel B that were serum starved and treated with (+) or without (−) serum for 2 h. The abundance of vinculin mRNA was analyzed by quantitative RT-PCR, normalized to 18S rRNA levels, and represented as the means ± standard deviations of three independent experiments. (D) TO3T3 cells were serum starved and treated with TPA or serum, as indicated, or with TPA for 30 min, followed by serum for the indicated times. RNA was then isolated and analyzed for vinculin expression as described above.
We also established cell lines stably expressing the Tet-off MKL1 expression constructs as an alternative means to express the proteins at lower levels in the cells. The wt and mutant proteins were expressed at similar levels (Fig. 7B). The expression levels were about twice that of endogenous MKL1, as determined using anti-MKL1 sera in immunoblots of cell lysates (data not shown). The subcellular localization of MKL1 and its mutants in the stable cell lines is similar to that found following transient transfections, as determined by immunofluorescence microscopy, i.e., the STS/A, N100, and double mutants were constitutively nuclear, while wt MKL1 moved from the cytoplasm to the nucleus upon serum stimulation (data not shown). Transfection of the 5xSRE reporter gene yielded results similar to those of cells transiently transfected with the MKL1 constructs. Luciferase expression was constitutively activated in the STS/A, the N100, and the double mutant cell lines (Fig. 7B). There was a modest serum-induced luciferase expression in the N100 mutant cells; however, this change was not statistically significant compared to that of serum-starved cells with wt MKL1 (Student's t test, P > 0.05) (Fig. 7A and B). The differences between luciferase activities in the serum-starved cells of the STS/A, N100, and STS/A-N100 double mutants and that of the wt MKL1 cells were all statistically significant (P < 0.03).
The stable cell lines also allowed us to examine the effect of the MKL1 mutant expression on endogenous MKL1-dependent target genes. We examined the serum-inducible expression of vinculin, whose expression we previously found to be inhibited by dominant-negative MKL1 (6, 27). We have also found that knockdowns of MKL1 and MKL2 with short hairpin RNA strongly reduced serum-induced vinculin expression (unpublished results). We measured vinculin mRNA by quantitative real-time PCR (Fig. 7C). Surprisingly, there was little increase in the N100 and STS/A mutant cell lines on basal, serum-starved vinculin expression, in contrast to the result with the SRE reporter genes. Compared to the expression of wt MKL1, there was a twofold increase in serum-induced vinculin expression with the STS/A mutant and a fivefold increase with the N100 mutant, demonstrating higher activity under these conditions (Fig. 7C). Similar results were found when we tested another MKL1 target gene, the SRF gene (data not shown). These results indicate that while the STS/A and MKL1-N100 mutants are constitutively active when assayed with reporter genes, the mutations are not sufficient to activate the endogenous target genes, suggesting that there must be an additional step(s) required for activation of the endogenous genes (see Discussion).
Since TPA treatment blocked serum-induced MKL1 nuclear localization (Fig. 5E and Fig. 6), we determined whether this blockage also corresponded with the inhibition of MKL1 target gene expression. TPA alone did not affect vinculin expression but was sufficient to block serum-induced vinculin expression (Fig. 7D). These results show that prior activation of the ERK1/2 pathway can serve to block activation of the RhoA-MKL-SRF pathway by phosphorylation and nuclear exclusion of MKL1.
Phosphorylation sites are required for actin binding.
In a recent study, Vartiainen and colleagues showed that G-actin binding is necessary for rapid nuclear export of MKL1 (34). The N-terminal RPEL motifs of MKL1 are required for actin binding (11). Since the N100 and STS/A mutants had similar effects on MKL1 localization and activity (Fig. 7A and B and data not shown), we determined whether they affected actin binding in similar ways. Lowered actin binding could also explain why localization of the STS/A mutant is nuclear. We tested the association of GFP-actin with MKL1 by immunoprecipitation of MKL1 and immunoblotting for GFP-actin (Fig. 8A). Indeed, binding between MKL1 and GFP-actin was markedly reduced with the nonphosphorylatable STS/A mutant. As expected, actin binding to the N100 mutant, lacking the RPEL motifs, was reduced. We also assessed the properties of the single phosphorylation site mutants in regard to actin binding. While the S449A and T450A mutants were able to bind to actin, the S454A mutant did not (Fig. 8A). To exclude the possibility that the reason for the different actin binding properties might reside in the constitutively nuclear localization of the N100, STS/A, and S454A mutants, we performed actin binding studies with the NES mutants. Although the L272A mutant is constitutively localized to the nucleus (Fig. 4D), it bound actin, as did wt MKL1 and the L555A mutant (Fig. 8A).
FIG. 8.

The phosphorylation site mutant has a weaker affinity for actin. (A) HeLa cells were cotransfected with Flag-tagged MKL1 or the indicated MKL1 mutants together with GFP-actin expression vector (2 μg each). Immunoprecipitates with anti-Flag antibody were immunoblotted with anti-GFP or anti-Flag antibodies. A portion of the cell lysate (1/30) was also directly immunoblotted with anti-GFP antibodies (bottom panel). (B) HeLa cells were transfected with GFP-actin and Flag-MKL1 (lanes 1 and 2) or neither (lane 3), serum-starved, and treated with TPA (100 ng/ml) for 30 min (lane 2). Immunoprecipitates with anti-GFP antibodies were immunoblotted with anti-Flag or anti-GFP antibodies. A portion of the cell lysate (1/30) was also directly immunoblotted with anti-Flag antibodies (bottom panel). (C) The model for serum regulation of MKL1: serum induction results in the activation of the RhoA- and Ras/MEK/ERK pathways. RhoA activation stimulates MKL1 nuclear localization due to the formation of actin stress fibers and a decrease in G-actin levels, while nuclear export is stimulated due to ERK1/2 phosphorylation of MKL1 and increased G-actin binding. Export of actin-bound MKL1 reduces the amount of MKL1 available to bind to SRF and activate transcription (see Discussion for details).
Since a mutation of the phosphorylation sites reduced actin binding, we sought to determine whether inducing phosphorylation of the sites would increase actin binding. We treated the cells with TPA, which induces phosphorylation of S454 (Fig. 5D). Coimmunoprecipitation of HeLa cells transfected with GFP-actin and Flag-MKL1 revealed increased binding of MKL1 and actin after TPA treatment, while there was no effect on MKL1 or GFP-actin expression levels (Fig. 8B). Together, these results show that MKL1 phosphorylation regulates its binding to actin. Given the effect of actin on MKL1 nuclear export, increased actin binding to phosphorylated MKL1 would increase its export and cytoplasmic localization.
DISCUSSION
MKL1/2 are critical for RhoA activation of SRF and its target genes (6, 18, 28). One mechanism for MKL1 regulation is the movement from the cytoplasm to the nucleus upon serum stimulation (18). This is accomplished by a reduction in the G-actin pool due to its incorporation into stress fibers (18). Since G-actin binds directly to MKL1's N-terminal RPEL motifs, the reduction of G-actin levels leads to its dissociation from MKL1 and reduced nuclear export of MKL1 (11, 18, 34). At the same time serum induces MKL1 movement from the cytoplasm to the nucleus, it is phosphorylated. We suggest that this phosphorylation at S454 by the ERK1/2 pathway provides an inhibitory mechanism that promotes nuclear export of MKL1. In the model shown in Fig. 8C, serum induction activates both the RhoA and the ERK1/2 pathways. RhoA then induces stress fibers and a reduction in G-actin levels. MKL1 can actively shuttle into the nucleus but is normally quickly exported due to its association with actin. Reduced G-actin results in nuclear accumulation of MKL1 (34). We have found that MKL1 becomes phosphorylated by the ERK1/2 pathway. This phosphorylation is required for nuclear export because the sites are required for optimal binding to actin, such that MKL1 is relocalized out of the nucleus. As such, the ERK1/2 pathway limits activation by the RhoA-MKL1 pathway. Aspects of this model are discussed below.
Phosphorylation of MKL1 at serine 454.
We have identified three residues in MKL1, S449, T450, and S454, whose mutations affect the serum-induced electrophoretic mobility shift. Mutation of S454 completely blocked the phosphorylation-induced mobility shift of MKL1, while mutation of the other sites reduced the mobility only partially. These sites cluster in a previously unrecognized conserved region between the SAP and leucine zipper domains. The S454 site in particular is conserved in MKL1, MKL2, and myocardin, suggesting that all of these family members may be regulated by phosphorylation. A previous study by Badorff et al. demonstrated that GSK3β phosphorylates myocardin on eight amino acids, among which is the site that is homologous to the MKL1 S454 site (3). We found, however, that inhibition of GSK3β in cells with lithium chloride did not affect serum-inducible phosphorylation of MKL1 (data not shown). The S449, T450, and S454 sites in MKL1 were also identified in a proteomic screen for EGF-inducible phosphorylation sites, suggesting that they may be targets for multiple growth factors (20). Additional phosphoacceptor sites may exist that do not contribute to the serum-induced mobility shift, although only one other site, serine 6, was identified in the EGF-induced cells (20). We have focused on S454 since it was able to abolish the serum-inducible mobility shift. The phospho-specific antibody raised against phospho-S454 showed that this site is indeed inducibly phosphorylated in response to serum. The effect of the S454 mutation on MKL1 cellular localization and activity further shows the significance of this site.
The S454 site resembles a MAPK phosphorylation site, and we found that the MEK1 inhibitor UO126 inhibited phosphorylation, suggesting the involvement of the ERK1/2 kinase. In support of this possibility, activation of the ERK1/2 pathway by the protein kinase C activator TPA induced phosphorylation of wt MKL1 but not the phosphorylation site mutant. Kalita et al. found that, besides serum induction, treatment of cortical neurons with BDNF also induced MKL1 phosphorylation in an MEK1-dependent manner (14). They also found that ERK2 could phosphorylate MKL1 in vitro, though the site(s) of phosphorylation was not identified. We also found that ERK2 phosphorylated MKL1 in vitro; however, phosphorylation was not affected by the S454 mutation (data not shown). There are 23 other (S/T)P sequences in MKL1, indicating that there are many potential ERK2 sites. These sites may be phosphorylated in vitro, even though there must be more specificity in vivo. Nevertheless, the blocking of MKL1 phosphorylation by the MEK1 inhibitor U0126 suggests that this pathway is required for serum-induced phosphorylation. Kalita et al. found that inhibiting the ERK1/2 pathway with U0126 blocked MKL1 activation of the SRE reporter genes in cortical neurons, but they did not demonstrate that this was due to phosphorylation of MKL1 (14). While this inhibition is opposite to the effect we observed for MKL1 localization, it may also be due to the different cell type used, since MKL1 was constitutively nuclear in cortical neurons (14). Alternatively, U0126 may affect other processes required for SRF and MKL1 activity.
Nuclear import/export of MKL1.
One of the most interesting aspects of this study is the observation that the nonphosphorylatable mutant (S454A) is constitutively localized to the nucleus. This implicates an important role for phosphorylation in the regulation of MKL1 movement in the cell. This regulation appears to be at the level of nuclear export. Treatment of cells with the nuclear export inhibitor leptomycin B resulted in rapid nuclear localization of MKL1 in serum-starved cells, in which all the cytoplasmic MKL1 was trapped in the nucleus within 4 min (34). Since the S454A mutant localized to the nucleus in serum-starved cells, the simplest explanation is that the rate of nuclear export was reduced. Further support for the fact that the lack of phosphorylation promotes nuclear localization is that the MEK inhibitor UO126 impaired MKL1 phosphorylation and resulted in persistent nuclear localization of MKL1 for up to 8 h following serum stimulation, whereas MKL1 is normally redistributed to the cytoplasm in untreated cells within 4 h. In addition, the phosphomimetic MKL1 mutant with glutamate substitutions was rapidly exported out of the nucleus after 1 h of serum treatment. To show that phosphorylation is consistent with cytoplasmic localization, we induced ERK1/2 activity and MKL1 phosphorylation with TPA. In contrast to the loss of phosphorylation found with U0126, TPA treatment restricted MKL1 to the cytoplasm.
These results show that nuclear localization can be regulated by two regions of MKL1, the phosphorylation site at aa 454 and the N-terminal RPEL motifs that bind actin (11, 18). Deletion or mutation of the RPEL domain or S454 leads to nuclear localization (18; unpublished results). These effects may be related, as we found that mutation of the phosphorylation sites reduced actin binding to MKL1, similar to deletion of the N-terminal domain. These results are consistent with a role for actin in promoting nuclear export. This could occur through an NES in MKL1, actin, or an unknown cofactor. We searched MKL1 for a putative NES and found that the mutation of one at aa 272 abolished its activity and caused nuclear localization of MKL1. This NES sequence is within MKL1's glutamine-rich region, and deletion of this domain was previously shown to cause nuclear localization in serum-starved cells (18). We propose that actin binding to MKL1 allows the utilization of this NES sequence due to conformational changes in the protein.
Importantly, constitutive localization of MKL1 to the nucleus is not sufficient to reduce actin binding, as shown by the L272A mutant. This mutant is constitutively localized to the nucleus but binds to actin, indicating that the nuclear localization of the phosphorylation site mutant cannot explain its inability to bind actin.
It is interesting that the N-terminal 204 amino acids of MKL1 were sufficient to mediate serum-induced cytoplasm-to-nucleus translocation when fused to pyruvate kinase or GFP (11). This translocation is independent of MKL1 phosphorylation or its NES. It is possible that pyruvate kinase or GFP provides a cryptic NES for the fusion proteins that are blocked by the free (non-actin-bound) RPEL motifs. Alternatively, in the context of the full-length protein, the NES at aa 272 may be more critical for export than the N-terminal fragments due to the conformation of the protein. In addition, the effect of phosphorylation would be seen only in the context of folding of the entire protein, where phosphorylation at S454 could affect binding to actin by the N-terminal RPEL motifs.
Nuclear import of MKL1 appears to be constitutive, as shown by the leptomycin B experiment. However, inhibition of RhoA or sequestration of actin monomers with latrunculin B blocked the nuclear import of MKL1, suggesting that these pathways are required for nuclear import (18). These two treatments strongly affect the cytoskeleton such that they may cause indirect effects on nuclear import, and a serum-induced change in import rates has not been found. Nevertheless, some of our results suggest that there may be additional levels of regulation of localization. In particular, MKL1 is cytoplasmic in serum-starved cells, even though it is not phosphorylated. One possibility is that the G-actin levels are high in this state, such that they bind MKL1 sufficiently to induce its nuclear export. After serum stimulation, phosphorylation of MKL1 would shift the equilibrium of MKL1-actin binding such that less actin is required for binding to MKL1 and nuclear export. Additional experiments will be required to address whether this or other mechanisms are involved.
Regulation of SRF activity by phosphorylation of MKL1.
The nuclear localization of the phosphorylation site STS/A mutant predicts that it will be active. Indeed, in transient transfections of SRE reporter genes, the STS/A mutation caused constitutive activation in serum-starved and -induced cells. Similar results were found with the N100 mutant that lacks the actin binding domain and is also constitutively nuclear. We were surprised to find a different result when we looked at endogenous MKL1 and SRF target genes. While the vinculin and SRF genes had higher serum induction in cells expressing the STS/A and N100 mutants, the basal expression in serum-starved cells was not activated, even though the MKL1 mutants were nuclear. This result likely reflects the possibility that an additional step(s) is required for the induction of the target genes tested, vinculin and SRF.
The discrepancy between the activation of a transiently transfected template and a chromosomally located SRF-controlled reporter gene was analyzed in a previous study by Alberts et al. (1). They similarly found that a transiently transfected SRE reporter gene could be induced by activated RhoA, while a chromosomally integrated SRE reporter gene required an additional signal. This signal was mediated by RacI and resulted in global histone 4 (H4) hyperacetylation. However, this mechanism may not explain our findings. Similar to the method described by Alberts et al., we used trichostatin A, a histone deacetylase inhibitor, to induce H4 hyperacetylation, but this treatment did not affect vinculin expression in serum-starved cells containing the N100 or the MKL1-STS/A mutants (unpublished results), suggesting that other steps are required for serum-induced vinculin expression.
Vartiainen et al. also found that leptomycin B treatment did not activate MKL1 target genes, even though it caused MKL1 nuclear localization (34). They proposed that even though MKL1 was nuclear, it was still inhibited by actin binding. That cannot be the case here, since the N100 mutant lacks the actin binding domain. It is possible that the differences between the apparent results we see with SRE reporter genes and those with endogenous target genes are due to copy numbers of the genes or to the effect of the chromosomal location of the endogenous genes. The chromosomal location could be important for certain regulatory mechanisms. It should be noted that EGF induced histone H3 phosphorylation at serine 10 (25). This or other chromatin modifications might be required for the induction of endogenous immediate-early genes. Alternatively, additional steps may be required for activation of the endogenous genes. These steps could be other modifications to MKL1, additional SRF or MKL1 cofactors, or the action of other transcription factors on the target gene promoters. This will be an important question to resolve.
Cross-talk of MAPK and RhoA pathways.
It is interesting to note that SRF lies at the nexus of two major signaling pathways, the RhoA and MAPK pathways. Many SRF binding sites contain flanking p62TCF sites. p62TCF is encoded by a family of Ets-related factors, Elk-1, SAP-1, and Net, and can bind only adjacent to the SRE if SRF is bound. The MAPKs can phosphorylate p62TCF in a conserved transcriptional activation domain and activate its function (29). In contrast, the RhoA pathway does not require the flanking TCF site and is dependent upon MKL family members (6, 18, 19). Two classes of SRF target genes can be distinguished by their sensitivities to these two pathways, partially determined by the existence of a TCF site flanking the SRE (10, 19).
p62TCF binds to the SRE due to its binding to SRF, as well as to DNA. The binding sites of p62TCF and MKL1 on SRF are overlapping such that binding is mutually exclusive (18, 39). In smooth muscle cells, myocardin binding to SRF on target genes can lead to cell differentiation. However, platelet-derived-growth factor can stimulate proliferation and block differentiation by activating ERK1/2 and p62TCF. This activation of p62TCF displaces myocardin, a more potent transcriptional coactivator, and represses target gene expression (37). As the S454 phosphorylation site is conserved in myocardin, it is possible that an additional mechanism for myocardin downregulation is its phosphorylation by ERK1/2.
In general, MAPK and RhoA activation pathways of SREs have been considered separate pathways. Our results here suggest cross-talk between the pathways, where the activation of ERK1/2 promotes nuclear export and downregulation of MKL1. In this way, ERK1/2 signaling would increase the p62TCF-SRF association at the expense of MKL1-SRF binding. In fact, prior treatment with TPA, which leads to ERK1/2 activation, blocked serum-induced activation of MKL1. This provides an additional mechanism whereby proliferative signals would block MKL1 activation of target genes that may lead more to differentiation rather than proliferation.
Acknowledgments
The work was funded by grant CA050329 from the National Cancer Institute to R.P. S.M. was funded by a fellowship of the German Research Foundation (DFG). T.C.L. was funded by NIH predoctoral fellowship 1F31GM082027-01.
Footnotes
Published ahead of print on 11 August 2008.
REFERENCES
- 1.Alberts, A. S., O. Geneste, and R. Treisman. 1998. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell 92475-487. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez, E., I. C. Northwood, F. A. Gonzalez, D. A. Latour, A. Seth, C. Abate, T. Curran, and R. J. Davis. 1991. Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J. Biol. Chem. 26615277-15285. [PubMed] [Google Scholar]
- 3.Badorff, C., F. H. Seeger, A. M. Zeiher, and S. Dimmeler. 2005. Glycogen synthase kinase 3beta inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation. Circ. Res. 97645-654. [DOI] [PubMed] [Google Scholar]
- 4.Ballestrem, C., B. Wehrle-Haller, and B. A. Imhof. 1998. Actin dynamics in living mammalian cells. J. Cell Sci. 1111649-1658. [DOI] [PubMed] [Google Scholar]
- 5.Bogerd, H. P., R. A. Fridell, R. E. Benson, J. Hua, and B. R. Cullen. 1996. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol. Cell. Biol. 164207-4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cen, B., A. Selvaraj, R. C. Burgess, J. K. Hitzler, Z. Ma, S. W. Morris, and R. Prywes. 2003. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol. Cell. Biol. 236597-6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cen, B., A. Selvaraj, and R. Prywes. 2004. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J. Cell. Biochem. 9374-82. [DOI] [PubMed] [Google Scholar]
- 8.Du, K. L., M. Chen, J. Li, J. J. Lepore, P. Mericko, and M. S. Parmacek. 2004. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J. Biol. Chem. 27917578-17586. [DOI] [PubMed] [Google Scholar]
- 9.Favata, M. F., K. Y. Horiuchi, E. J. Manos, A. J. Daulerio, D. A. Stradley, W. S. Feeser, D. E. Van Dyk, W. J. Pitts, R. A. Earl, F. Hobbs, R. A. Copeland, R. L. Magolda, P. A. Scherle, and J. M. Trzaskos. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 27318623-18632. [DOI] [PubMed] [Google Scholar]
- 10.Gineitis, D., and R. Treisman. 2001. Differential usage of signal transduction pathways defines two types of serum response factor target gene. J. Biol. Chem. 27624531-24539. [DOI] [PubMed] [Google Scholar]
- 11.Guettler, S., M. K. Vartiainen, F. Miralles, B. Larijani, and R. Treisman. 2008. RPEL motifs link the serum response factor cofactor MAL but not myocardin to rho signaling via actin binding. Mol. Cell. Biol. 28732-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill, C. S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 811159-1170. [DOI] [PubMed] [Google Scholar]
- 13.Johansen, F. E., and R. Prywes. 1995. Serum response factor: transcriptional regulation of genes induced by growth factors and differentiation. Biochim. Biophys. Acta 12421-10. [DOI] [PubMed] [Google Scholar]
- 14.Kalita, K., G. Kharebava, J. J. Zheng, and M. Hetman. 2006. Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J. Neurosci. 2610020-10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.la Cour, T., L. Kiemer, A. Molgaard, R. Gupta, K. Skriver, and S. Brunak. 2004. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17527-536. [DOI] [PubMed] [Google Scholar]
- 16.Ma, Z., S. W. Morris, V. Valentine, M. Li, J. A. Herbrick, X. Cui, D. Bouman, Y. Li, P. K. Mehta, D. Nizetic, Y. Kaneko, G. C. Chan, L. C. Chan, J. Squire, S. W. Scherer, and J. K. Hitzler. 2001. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 28220-221. [DOI] [PubMed] [Google Scholar]
- 17.Minty, A., and L. Kedes. 1986. Upstream regions of the human cardiac actin gene that modulate its transcription in muscle cells: presence of an evolutionarily conserved repeated motif. Mol. Cell. Biol. 62125-2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miralles, F., G. Posern, A. I. Zaromytidou, and R. Treisman. 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113329-342. [DOI] [PubMed] [Google Scholar]
- 19.Murai, K., and R. Treisman. 2002. Interaction of serum response factor (SRF) with the Elk-1 B box inhibits RhoA-actin signaling to SRF and potentiates transcriptional activation by Elk-1. Mol. Cell. Biol. 227083-7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsen, J. V., B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen, and M. Mann. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127635-648. [DOI] [PubMed] [Google Scholar]
- 21.Posern, G., F. Miralles, S. Guettler, and R. Treisman. 2004. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J. 233973-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridley, A. J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70389-399. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook, J., E. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 24.Sasazuki, T., T. Sawada, S. Sakon, T. Kitamura, T. Kishi, T. Okazaki, M. Katano, M. Tanaka, M. Watanabe, H. Yagita, K. Okumura, and H. Nakano. 2002. Identification of a novel transcriptional activator, BSAC, by a functional cloning to inhibit tumor necrosis factor-induced cell death. J. Biol. Chem. 27728853-28860. [DOI] [PubMed] [Google Scholar]
- 25.Sassone-Corsi, P., C. A. Mizzen, P. Cheung, C. Crosio, L. Monaco, S. Jacquot, A. Hanauer, and C. D. Allis. 1999. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285886-891. [DOI] [PubMed] [Google Scholar]
- 26.Schonwasser, D. C., R. M. Marais, C. J. Marshall, and P. J. Parker. 1998. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 18790-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selvaraj, A., and R. Prywes. 2004. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol. Biol. 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selvaraj, A., and R. Prywes. 2003. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J. Biol. Chem. 27841977-41987. [DOI] [PubMed] [Google Scholar]
- 29.Shaw, P. E., and J. Saxton. 2003. Ternary complex factors: prime nuclear targets for mitogen-activated protein kinases. Int. J. Biochem. Cell Biol. 351210-1226. [DOI] [PubMed] [Google Scholar]
- 30.Somogyi, K., and P. Rorth. 2004. Evidence for tension-based regulation of Drosophila MAL and SRF during invasive cell migration. Dev. Cell 785-93. [DOI] [PubMed] [Google Scholar]
- 31.Sotiropoulos, A., D. Gineitis, J. Copeland, and R. Treisman. 1999. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98159-169. [DOI] [PubMed] [Google Scholar]
- 32.Takeda, S., D. L. North, M. M. Lakich, S. D. Russell, and R. G. Whalen. 1992. A possible regulatory role for conserved promoter motifs in an adult-specific muscle myosin gene from mouse. J. Biol. Chem. 26716957-16967. [PubMed] [Google Scholar]
- 33.Treisman, R. 1986. Identification of a protein-binding site that mediates transcriptional response of the c-fos gene to serum factors. Cell 46567-574. [DOI] [PubMed] [Google Scholar]
- 34.Vartiainen, M. K., S. Guettler, B. Larijani, and R. Treisman. 2007. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 3161749-1752. [DOI] [PubMed] [Google Scholar]
- 35.Wang, D., P. S. Chang, Z. Wang, L. Sutherland, J. A. Richardson, E. Small, P. A. Krieg, and E. N. Olson. 2001. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105851-862. [DOI] [PubMed] [Google Scholar]
- 36.Wang, D. Z., S. Li, D. Hockemeyer, L. Sutherland, Z. Wang, G. Schratt, J. A. Richardson, A. Nordheim, and E. N. Olson. 2002. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 9914855-14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, Z., D. Z. Wang, D. Hockemeyer, J. McAnally, A. Nordheim, and E. N. Olson. 2004. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428185-189. [DOI] [PubMed] [Google Scholar]
- 38.Williams, G. T., and L. F. Lau. 1993. Activation of the inducible orphan receptor gene nur77 by serum growth factors: dissociation of immediate-early and delayed-early responses. Mol. Cell. Biol. 136124-6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaromytidou, A. I., F. Miralles, and R. Treisman. 2006. MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA-binding domain. Mol. Cell. Biol. 264134-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zinck, R., R. A. Hipskind, V. Pingoud, and A. Nordheim. 1993. c-fos transcriptional activation and repression correlate temporally with the phosphorylation status of TCF. EMBO J. 122377-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]