

Abstract
There is a growing appreciation of the role that epigenetic alterations can play in oncogenesis. However, given the large number of genetic anomalies present in most cancers, it has been difficult to evaluate the extent to which epigenetic changes contribute to cancer. SNF5 (INI1/SMARCB1/BAF47) is a tumor suppressor that regulates the epigenome as a core member of the SWI/SNF chromatin remodeling complex. While the SWI/SNF complex displays potent tumor suppressor activity, it is unknown whether this activity is exerted genetically via maintenance of genome integrity or epigenetically via transcriptional regulation. Here we show that Snf5-deficient primary cells do not show altered sensitivity to DNA damaging agents, defects in γ-H2AX induction, or an abrogated DNA damage checkpoint. Further, the aggressive malignancies that arise following SNF5 loss are diploid and genomically stable. Remarkably, we demonstrate that most human SNF5-deficient cancers lack genomic amplifications/deletions and, aside from SNF5 loss, are indistinguishable from normal cells on single-nucleotide polymorphism arrays. Finally, we show that epigenetically based changes in transcription that occur following SNF5 loss correlate with the tumor phenotype. Collectively, our results provide novel insight into the mechanisms of oncogenesis by demonstrating that disruption of a chromatin remodeling complex can largely, if not completely, substitute for genomic instability in the genesis of aggressive cancer.
Many safeguards are in place to prevent a cell from undergoing oncogenic transformation. For example, an average of 14 separate cancer-promoting mutations are required for formation of breast or colon cancer (70). The mechanisms that underlie cancer formation are complex, however, since the low rate of mutation in normal human cells should essentially preclude the independent acquisition of so many mutations. Genome instability may explain this apparent paradox, since early mutations that compromise DNA repair can accelerate the acquisition of subsequent mutations (3). Consistent with this, both DNA repair defects and chromosomal instability are extremely common among highly malignant cancers. However, the cause-and-effect relationships between oncogenesis and genomic instability are not clear, and genetic mutation is not the only mechanism by which stable alteration of gene expression can occur.
It has become increasingly clear that epigenetic alterations, those changes in gene expression brought about by altered chromatin context rather than DNA sequence mutations, can also contribute to oncogenic transformation. For example, hypermethylation of DNA in CpG-rich promoter regions can silence tumor suppressor genes. Similarly, expression levels of histone methyltransferases, histone acetyltransferases, and histone deacetylases are frequently altered in cancer cells, as are global levels of histone acetylation and methylation (20, 30, 67). Recently nucleosome positioning was also implicated in oncogenesis, with the demonstration that heritable changes in nucleosome occupancy culminate in epigenetic silencing of tumor suppressor genes (36). The degree to which epigenetic changes can substitute for genetic instability is unclear, particularly during the genesis of highly aggressive cancers. Indeed, while diploidy is relatively common in early benign tumors and is found occasionally among low-grade malignant tumors, it is quite rare among highly malignant cancers.
SWI/SNF complexes represent a novel link between epigenetics and tumor suppression. These complexes utilize the energy of ATP hydrolysis to alter nucleosome positions and remodel chromatin. This activity contributes to the transcriptional regulation of numerous target genes by regulating access of the transcriptional machinery to target DNA sequences. Human SWI/SNF complexes are evolutionarily conserved and are comprised of a SWI2/SNF2 family ATPase (either BRG1 or BRM), common core subunits (SNF5, BAF155, and BAF170), and four to eight subunits that vary by cellular lineage (55). Transcriptional regulation by SWI/SNF has been implicated in the control of proliferation and differentiation in multiple tissues (16). Specific biallelic inactivation of SNF5 is present in the large majority of malignant rhabdoid tumors (MRTs), and inherited mutation of SNF5 also forms the basis of a familial cancer predisposition syndrome (8, 9, 58). MRTs are highly aggressive pediatric cancers that occur in kidney, brain (where they are referred to as atypical teratoid/rhabdoid tumors), and soft tissues (“extrarenal rhabdoid tumors”) (64). These tumors are poorly differentiated, nearly always locally invasive, and frequently metastatic. As a result, the large majority of children die of their disease, most within 1 year of diagnosis. Mutation of SNF5 has also recently been implicated in the genesis of epithelioid sarcomas and in familial schwannomatosis (26, 39). Mouse models have confirmed that Snf5 is a tumor suppressor, since heterozygous mice are tumor prone and biallelic conditional inactivation leads to rapid cancer formation with 100% penetrance (23, 32, 53, 54). The 11-week median cancer onset following inactivation of Snf5 in mice is remarkably rapid for the inactivation of a single gene and indicates a critical role for Snf5 in preventing cancer. In comparison, p53 loss leads to cancer at 20 weeks, p19Arf loss at 38 weeks, and p16Ink4a loss at 60 weeks (17, 25, 57). In addition to SNF5, the BRG1 ATPase subunit also has tumor suppressor activity, since specific mutations or loss of expression of BRG1 have been identified in lung, pancreatic, breast, and prostate cancer cell lines and primary tumors and mice haploinsufficient for Brg1 are predisposed to a low rate of breast tumors (11, 15, 38, 50, 69). Additionally, loss of the BAF180 subunit has been implicated in human breast cancer, while mice deficient in the Brm ATPase subunit have defective growth regulation (11, 51, 72).
While members of the SWI/SNF complex display potent tumor suppressor activity, the underlying mechanism is unclear. It is unknown whether this activity is exerted genetically via maintenance of genome integrity or epigenetically via transcriptional regulation. Since other SWI2/SNF2 family ATP-dependent chromatin remodelers, such as INO80, SWR1, and RAD54, have been linked to DNA repair and maintenance of genomic integrity (19, 40, 46, 59, 63, 71), a role for the Swi/Snf complex in these activities has been explored. Several studies have suggested that the Swi/Snf complex is involved in DNA repair (12, 24, 33, 47).
Given that SNF5 is mutated in human cancers and in a cancer predisposition syndrome, that conditional inactivation of Snf5 leads to the rapid onset of highly malignant cancers in all mice, and that SNF5 is a core member of a complex that contributes to regulation of chromatin structure, we sought to evaluate whether its tumor suppressor activity arises from a role in DNA repair and maintenance of genome stability or whether it arises from epigenetic activity. Based upon preliminary results that did not reveal defective DNA repair in the absence of SNF5, we hypothesized that its tumor suppressor activity is mediated epigenetically. Here we have used conditional genetic models to specifically inactivate Snf5 and confine our observations to the time period immediately after cells have lost the Snf5 protein but before secondary effects associated with impaired growth occurred. We show that loss of Snf5 does not affect the response to DNA damage or the completion of DNA repair. Snf5-deficient primary mouse embryonic fibroblasts (MEFs) are not hypersensitive to DNA damaging agents, induce γH2AX damage foci with normal kinetics, and have an intact DNA damage checkpoint. We further demonstrate that both mouse and human SNF5-deficient tumors are typically diploid and use high-density single-nucleotide polymorphism (SNP) arrays to reveal that, unlike other pediatric CNS tumors, human MRTs generally lack genomic deletions, amplifications, or microsatellite instability. Lastly we show that epigenetically driven changes in the expression of the key target cyclin D1 correlate strongly with the tumor phenotype. These results demonstrate that disruption of a chromatin remodeling complex can eliminate the selective pressure for genomic instability to occur in the genesis of aggressive cancer. These findings have important therapeutic implications since, unlike genetic mutations, epigenetic alterations are potentially reversible. Given the lack of confounding genomic alterations, Snf5-deficient cancer may constitute an ideal model with which to study epigenetic contributions to oncogenic transformation.
MATERIALS AND METHODS
Snf5 inactivation.
Primary MEFs were harvested from E13.5 embryos. Cre was introduced into cells via adenoviral or retroviral infection. Excision of Snf5 was achieved with adeno-Cre (Ad5CMV-Cre; University of Iowa Gene Transfer Vector Core) at a multiplicity of infection of 150. For some experiments, pBabe-puror-Cre retroviral supernatant was used instead to infect MEFs two times at 4-h intervals. Cells were stably selected in medium containing puromycin (2.5 μg/ml) 48 h after infection.
Immunoblots.
For fractionation experiments, chromatin-bound and soluble protein fractions were biochemically separated from whole-cell lysates by a 5-min incubation in KLB buffer on ice (150 mM NaCl, 50 mM Tris, 4 mM EDTA, 0.1% NP-40, 0.1% Triton) followed by a 3-min centrifugation at 5,000 × g. The soluble supernatant was removed, and the chromatin pellet was treated with DNase I (Worthington Biochemical Corporation) for 30 min at 37°C. For Western blots, cells were harvested, washed with 1× phosphate-buffered saline (PBS), and lysed in RIPA buffer. Protein concentrations were determined using a Bradford reagent (Bio-Rad). After the samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the proteins were transferred to a polyvinylidene difluoride membrane (Millipore) and blocked in PBS-Tween containing 5% milk. Antibodies included anti-SNF5/BAF47 (BD Biosciences and Bethyl Laboratories); anti-Brg1, -p21, -c-jun, and -histone H1 (Santa Cruz Biotechnology); anti-phosphorylated Chk1 (Bethyl Laboratories); anti-phosphorylated ATM and anti-phosphorylated p53 (Cell Signaling); and anti-phosphorylated H2AX and anti-phosphorylated histone H3 (Upstate). Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch). Proteins were visualized using enhanced chemiluminescence.
Immunofluorescence.
Cells were incubated on ice in prepermeabilization buffer (0.5% Triton X-100, 20 mM HEPES [pH 7.9], 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose) fixed with 2% paraformaldehyde for 20 min and 100% methanol for 10 min, and blocked with 4% bovine serum albumin in PBS. Primary and secondary antibody incubations were done in 4% bovine serum albumin in PBS. Antibodies and dilutions used in this study include anti-Snf5, 1:500 (Sigma-Aldrich); anti-phosphorylated H2AX (serine 139), 1:10,000 (Upstate); anti-Brg1, 1:200 (Santa Cruz Biotechnology); anti-histone H3 (trimethylated on lysine 9), 1:500 (Upstate); anti-heterochromatin protein 1β (HP1β), 1:7,500 (Chemicon); Alexa Fluor 488 goat anti-rabbit immunoglobulin G, 1:1,000 (Molecular Probes); and Cy3 goat anti-mouse antibody, 1:500 (Jackson Immunoresearch). DNA was stained with 1 mg/ml Hoechst 33342 dye (Invitrogen).
Drug sensitivity assays.
Primary MEFs were seeded into a 96-well plate at a concentration of 1 × 104 cells/well 56 h after Adeno-Cre transduction and treated with cisplatin, etoposide, (NovaPlus), or UV radiation (StrataLinker; Stratagene Corp, La Jolla, CA) at the appropriate concentrations/amounts. Proliferation was measured 24 h after cisplatin treatment, 48 h after etoposide treatment, and 72 h after UV exposure using the WST-1 reagent (Roche) and a microplate spectrophotometer (Bio-Rad Benchmark Plus).
Cell cycle analysis.
Primary MEFs were fixed by the dropwise addition of cold 70% ethanol. The cells were then permeabilized with 0.25% Triton and incubated with phosphorylated histone H3 (serine 10) antibody (Santa Cruz Biotechnology), followed by incubation with fluorescein isothiocyanate-conjugated secondary antibody (Jackson Immunoresearch). Flow cytometric analysis was performed using a CantoII (Becton Dickinson) machine, and data were analyzed using the FlowJO Software program (Tree Star).
SNP analysis.
Snap-frozen tumor samples were obtained from brain tumor resections collected at Children's Hospital Boston, Children's Hospital of Philadelphia, and St. Jude's Research Hospital for Children under approval from their respective Institutional Review Boards. Matched normal blood samples were collected through the Children's Hospital Boston under approval from the Institutional Review Board. The rhabdoid tumor cell lines TM87, 2004, and A204 were obtained from American Type Culture Collection and maintained in culture under conditions as previously published. All genomic DNA was prepared using the Puregene DNA extraction kit (Gentra Systems, Minneapolis, MN) according to the manufacturer's instructions.
Genomic DNA from 16 rhabdoid tumors and 16 medulloblastomas was applied to Affymetrix GeneChip Mapping 250K SNP arrays using the 96-well plate protocol per the manufacturer's protocols. An additional three primary rhabdoid tumor samples and the three rhabdoid tumor cell lines (A204, TM87, and 2004) were applied to the Affymetrix 6.0 SNP microarray. Plates were scanned on the GeneChip Scanner 3000 (Hewlett-Packard, Palo Alto, CA), and the Affymetrix Genotyping Tools 2.0 software program was used to examine the SNP hybridization patterns and to make SNP calls of all loci in each samples. Probe-level signal intensities were normalized to a baseline array with median intensity using invariant set normalization (34). All samples passed a minimum of 85% positive SNP calls. Genomic Identification of Significant Targets in Cancer (GISTIC) analysis was performed as previously described (4). Copy number gains and losses were also catalogued by direct visualization of the data using the SNP viewer module on GenePattern software (freely available at www.broad.mit.edu/software/GenePattern). For visualization, gains were defined as >2.8 copies and losses defined as <1.2 copies spanning multiple consecutive SNPs.
Expression array.
Snap-frozen primary tumor samples were obtained from Children's Hospital Boston as described above under approval from the Institutional Review Board. Total RNA was isolated using RNA Trizol per the manufacturer's protocol. RNA was applied to Affymetrix U133A2 arrays, and CEL files were preprocessed using the robust multichip average algorithm (27). Arrays were visualized with both the GenePattern and GeneSpring GX 7.3.1 (Agilent, Santa Clara, CA) software programs.
RESULTS
Intact DNA repair in the absence of Snf5.
Phosphorylation and recruitment of the histone variant H2AX to damaged sites is an immediate event following DNA damage, occurring within minutes (48, 56). To investigate whether Snf5 colocalized with γH2AX at sites of DNA damage, we exposed wild-type (WT) primary mouse embryonic fibroblasts (MEFs) to ionizing radiation (IR). At 1 h post-IR, although we identified clear induction of γH2AX foci at sites of DNA damage, Snf5 did not colocalize with these foci (Fig. 1A). We next examined whether the distribution of Snf5 or the Swi/Snf ATPase Brg1 was altered following IR. Immunofluorescence of Brg1 and Snf5 revealed no change in the pattern of either protein (Fig. 1B). Since chromatin remodeling complexes such as ISWI have been shown to be specifically involved in aiding progression of the DNA replication machinery through heterochromatin (10, 14), we investigated whether Swi/Snf subunits relocalized to heterochromatic foci after DNA damage to facilitate access to damaged heterochromatin. Snf5 and Brg1 do not relocalize to heterochromatic foci, demarcated by histone H3 trimethylated on lysine 9 (H3K9me3) and heterochromatin protein 1, in response to IR (Fig. 1B). We also evaluated whether Snf5 or Brg1 showed increased expression or altered affinity for chromatin following DNA damage. Following exposure of cells to UV, IR, or doxorubicin, the chromatin-bound and soluble protein fractions were separated and analyzed by Western blotting. Neither Brg1 nor Snf5 expression was altered in response to UV, IR, or doxorubicin, and neither became more tightly associated with chromatin (Fig. 1C and D).
FIG. 1.

Snf5 neither colocalizes with γH2AX foci nor relocalizes after DNA damage. (A) Snf5 does not colocalize with γH2AX foci after IR. NIH 3T3 cells were fixed 1 h after IR (10 Gy), double stained for Snf5 and γH2AX (phospho-Ser 139), and counterstained with Hoechst 33342. (B) DNA damage has no effect on the nuclear localization of Swi/Snf subunits. Unirradiated and irradiated (10 Gy) NIH 3T3 cells were fixed 1 h after IR, double stained for either Brg1 and H3K9me3 or Snf5 and heterochromatin protein 1β (HP1b). The exclusion of Swi/Snf subunits from heterochromatic foci (marked by H3K9Me3 and HP1β staining) observed in unirradiated cells is unaltered following IR. (C) DNA damage does not affect Snf5 expression in WT cells. The Snf5 protein level was analyzed by Western blotting 1 h after UV treatment (30 J/m2), 1 h after IR (2.5 Gy), and 16 h after treatment with 1.3 μM doxorubicin and compared to untreated controls. (D) The affinity of Snf5 for chromatin is unchanged following DNA damage. Chromatin-bound (C) and soluble (S) protein fractions were biochemically separated from whole-cell lysates using a detergent buffer containing 0.1% Triton and 0.1% NP-40 1 h after damage. The distribution of Snf5, histone H1 (chromatin marker), and c-jun (soluble nuclear marker) was analyzed by Western blotting. DNA damage conditions are as described for panel C.
Although Snf5 does not colocalize with γH2AX at damage foci, we sought to determine whether loss of Snf5 would impair the kinetics of the DNA damage response. Snf5-conditional MEFs were exposed to Cre recombinase to ablate Snf5 (Fig. 2A). While cells that lack the Snf5 protein are initially viable, they begin to lose proliferative capacity 5 days following exposure to Cre and the majority succumb to cell death by 7 days (28). Since lack of proliferation would be scored as defective DNA repair in some assays, we monitored viability and performed these experiments soon after Snf5 protein loss in order to avoid confounding effects due to the secondary arrest phenotype. We thus used immunoblotting to monitor Snf5 expression and performed all experiments during the 3-day window when Snf5 was no longer detectable but cells remained viable and proliferative. Primary WT or Snf5Flox/− MEFs were infected with pBabe-Cre, selected in puromycin, and examined for their ability to induce γH2AX expression following IR. Recruitment of γH2AX to sites of DNA damage was not impaired in Snf5-deficient cells (Fig. 2B). In addition, the absence of Snf5 had no discernible effect upon the kinetics of γH2AX induction, as assessed by both Western blotting for γH2AX and quantification of γH2AX foci by immunofluorescence (Fig. 2B, C, and D). Further, loss of the Snf5 protein did not result in any increase of γH2AX expression, suggesting that Snf5 loss does not lead to damage (Fig. 2C and D). In both WT and Snf5-deficient cells, γH2AX levels peaked at 1 h post-IR and decreased over time, falling to the levels of unirradiated cells by 12 h. Together these results suggest that Snf5 does not participate in the initial cellular response to DNA damage nor does Snf5 loss affect the kinetics of double-strand break (DSB) repair.
FIG. 2.

Snf5-deficient cells display normal induction of γH2AX following DNA damage. (A) The Snf5 protein is efficiently eliminated in Snf5Flox/− (Snf5Fl/−) cells exposed to Cre recombinase. WT and Snf5Fl/− primary MEFs were infected with pBabe-puror (+) or with pBabe-Cre-puror. Two days after transduction, cells were selected in 2.5 μg/ml puromycin, and at day 4, they were harvested for Western analysis or plated for experimental assays. (B) Snf5-deficient cells are able to induce H2AX phosphorylation following DNA damage. WT and Snf5-deficient cells were exposed to 2.5 Gy IR and fixed on slides after 1 h. Immunofluorescence of phosphorylated H2AX (serine 139) was compared to that of unirradiated controls. Hoechst 3342 was used as a counterstain. (C and D) Snf5-deficient cells display normal kinetics of repair. WT and Snf5-deficient cells were either untreated (no IR) or exposed to 5 Gy IR and fixed on slides for immunostaining or harvested for protein analysis at 1, 3, and 6 h after damage. For panel C, cells were immunostained for H2AX (serine 139) and a minimum of 100 cells were counted for each time point. (D) Western blot analysis of H2AX (serine 139) expression. H2AX expression peaked 1 h after damage and decreased over time, indicating that the cells have repaired the damage.
Other DNA damaging agents can also be used to test for deficiencies in DSB repair and other DNA repair pathways. Alkylated, abasic, or UV-damaged DNA is repaired by the nucleotide excision repair and base excision repair pathways, while drug-induced DSBs are repaired by nonhomologous end joining. To investigate whether Snf5 loss specifically affected these repair pathways, we exposed WT and Snf5-deficient cells to UV (cross-links), cisplatin (alklyation), and etoposide (DSBs). Unlike the loss of other mammalian chromatin remodelers, such as components of the INO80 complex (71), loss of Snf5 had no effect upon the proliferative response to any of the tested DNA damaging agents (Fig. 3).
FIG. 3.

Snf5-deficient cells respond appropriately to DNA damaging agents. Snf5-deficient MEFs do not display increased sensitivity to cisplatin, etoposide, or UV. WT and Snf5Flox/− MEFs were transduced with Adeno-Cre and treated 56 h later with cisplatin, etoposide, or UV at doses indicated. (A) Cellular proliferation was measured by absorbance of the WST-1 reagent at 24 h (cisplatin), 48 h (etoposide), or 72 h (UV) after plating in order to optimize detection of an impaired response to each agent. Immortalized ataxia-telangiectasia patient-derived fibroblasts (ATM-/-) (75) were included as a positive control for sensitivity to etoposide-induced DSBs, and immortalized Rad18-deficient MEFs (61) were included as a positive control for sensitivity to UV-induced cross-links and cisplatin-induced alkylation. Data are presented as the ratio of the absorbance of treated cells to untreated cells of each genotype and are the means ± standard errors for at least three independent experiments using MEFs isolated from different embryos. Snf5 Fl/Fl, Snf5Flox/Flox. (B) Western blot of Snf5 protein levels at the beginning of the assay, 56 h after Adeno-Cre infection (start), i.e., the time point when DNA damaging agents were added to the cells, and at 24, 48, and 72 h after plating, i.e., the time points when cellular proliferation was measured after DNA damage.
Intact DNA damage checkpoint in Snf5-deficient cells.
The normal induction of γH2AX observed in Snf5-deficient cells does not exclude a DNA damage checkpoint defect. Therefore, we investigated whether Snf5 contributes to DNA damage checkpoints. Activation of the ATM (ataxia-telangectasia mutated) and ATR (ataxia-telangectasia-related) kinases are critical for initiation of the DNA damage checkpoint. To investigate whether Snf5-deficient cells displayed defects in initiating the DNA damage signaling response, we exposed WT and Snf5-deficient cells to IR and immunoblotted for activated ATM (phospho-serine 1981). Snf5-deficient cells displayed normal activation of ATM (Fig. 4A). Following their activation in response to DNA damage, the ATM and ATR kinases in turn phosphorylate and activate the Chk1 kinase and p53. We found that phosphorylation of Chk1 (serine 317) and p53 (serine 18) was intact in Snf5-deficient MEFs following exposure to IR (Fig. 4A). Finally, we found that Snf5 loss did not impair the ability of p53 to induce p21 (Fig. 4A).
FIG. 4.

The G2 checkpoint is intact in Snf5-deficient cells. (A) DNA damage signaling is appropriately initiated in Snf5-deficient cells. WT and Snf5-deficient cells were immunoblotted for ATM (serine 1981), p53 (serine 18), phosphorylated Chk1 (pChk1) (serine 317), p21, and Snf5 expression in the absence (−) or presence (+) of 2.5 Gy IR. Snf5Fl/Fl, Snf5Flox/Flox. (B and C) The mitotic index of Snf5-deficient cells appropriately decreases following DNA damage and recovers by 6 h. The percentage of pH3-positive cells for unirradiated (−IR) or irradiated (+IR) WT or Snf5-deficient cells was quantified by fluorescence-activated cell sorting and is noted in each panel. Data represented in panel C are presented as means for at least three independent experiments ± standard errors.
Since the results indicated that initiation of the DNA damage response was intact, we next tested whether Snf5-deficient cells could appropriately activate the G2 checkpoint in order to prevent mitotic entry following exposure to IR. The mitotic index is easily monitored by flow cytometric analysis of phosphorylated histone H3 (pH3), a marker of mitotic cells. Snf5-deficient cells were able to appropriately prevent entry into mitosis following IR-induced DNA damage, indicating that the G2 checkpoint was intact (Fig. 4B). Both WT and Snf5-deficient cells reentered the cell cycle following completion of repair, as indicated by the increase in pH3-positive cells at 6 h post-IR (Fig. 4C). To further investigate mitosis in the Snf5-deficient cells, we fixed and immunostained WT and Snf5-deficient MEFs for microtubules (DM1α) and for chromosomes (4′,6′-diamidino-2-phenylindole). The Snf5-deficient cells displayed normal mitoses, and importantly, they displayed normal spindle assembly and normal anaphase (see Fig. S1 in the supplemental material).
SNF5-deficient cancers are diploid and genomically stable.
Since defects either in the DNA damage response or in DNA repair frequently result in genome instability, we evaluated the DNA content of Snf5-deficient tumor cells. MRT is an aggressive and highly lethal cancer of early childhood that is poorly responsive to both chemotherapy and radiation. Intriguingly, studies of small series of MRTs or MRT cell lines have suggested that these cancers are sometimes diploid by karyotype or comparative genomic hybridization (CGH) analysis (5, 18, 52, 68). Notably, we previously detected a small increase in polyploidy in primary MEFs following the inactivation of Snf5 (28). Since polyploidy has been proposed to be a precursor to both aneuploidy and oncogenesis, we sought to determine whether this effect contributed to oncogenesis in the absence of Snf5. To investigate whether Snf5-deficient tumors were prone to chromosomal aberrations, we first performed chromosome counts on six immunopurified Snf5-deficient murine T-cell lymphomas and on a cell line derived from a seventh lymphoma. In all cases, including the cell line, the predominant populations were diploid and lacked visible breakage, translocations, and dicentric chromosomes, suggesting a lack of genomic instability (see Table S1 in the supplemental material). In order to definitively examine the requirement for SNF5 in the maintenance of genomic stability, we next evaluated the genome of human MRTs at high resolution by utilizing high-density SNP arrays, since they allow for detailed assessment of much smaller amplified or deleted regions than current array CGH technology. To identify specific chromosomal aberrations contributing to MRT onset, we subjected the DNA from 16 primary human rhabdoid tumors to genome-wide SNP analysis using the 250K Affymetrix SNP array. DNA from 16 primary medulloblastomas (another pediatric brain cancer) and nine normal peripheral blood samples were included as controls. The SNP array data revealed a striking lack of genomic instability in the MRTs. While the medulloblastomas, as is typical of aggressive cancers, displayed numerous amplifications and deletions, MRTs displayed no recurrent amplifications or deletions other than at the SNF5 locus itself (chromosome 22) (Fig. 5A). Indeed, out of 16 tumors, only 2 had other abnormalities that were clearly distinct from copy number variation present in normal blood samples, 1 with a partial deletion of chromosome 1 and the other with a partial deletion of chromosome 19. While deletions at the SNF5 locus were common (12 of 16 samples), not all tumors displayed copy number loss, consistent with the fact that some rhabdoid tumors lose SNF5 function via point mutations and others lack coding region/promoter mutations and may be silenced epigenetically (7). Recently we were able to utilize the new Affymetrix genome-wide human SNP array 6.0, containing 900,000 SNPs and an additional 940,000 copy number probes, to analyze the genomic profile of three additional MRTs. Consistent with the prior 250K array data, the higher-resolution array showed remarkably normal results, with only one sample displaying two deletions (Fig. 5B). Notably, even established human MRT cell lines that have been extensively passaged lacked widespread copy number alterations, with the 2004 cell line lacking any chromosomal alterations or large deletions/amplifications (Fig. 5B).
FIG. 5.

High-density SNP array analysis of SNF5-deficient MRTs. (A) DNA from 16 primary MRTs, 16 medulloblastomas, and 9 normal blood samples was analyzed on the Affymetrix 250K SNP array. A closeup view of the SNF5 locus on chromosome 22q11 is shown in the bottom panel. Red indicates regions of genomic amplifications, while blue indicates regions of genomic deletions. (B) DNA from an additional three primary MRTs, three MRT cell lines, eight medulloblastomas, and seven normal blood samples was analyzed on the Affymetrix 6.0 SNP array containing 940,000 SNPs. The SNF5 locus on chromosome 22q11 is shown in the bottom panel. (C) GISTIC was utilized to search for small deletions in the rhabdoid tumors. Deletion of SNF5 itself at 22q11 is the only significant finding (q < 10−24). Other changes are not significant and are of a magnitude consistent with normal copy number variation.
We next used GISTIC (4) to search for small deletions or amplifications that could be cooperating with SNF5 loss in oncogenesis. The only genomic change identified with a minimum false discovery rate (q value) of less than 0.05 was deletion of SNF5 itself (q = 10−24), suggesting either that MRTs lack recurrent deletions/amplifications that cooperate with SNF5 loss in oncogenesis or that cooperating deletions/amplifications are rare (Fig. 5C). Collectively, these data demonstrate that despite their highly malignant nature, MRTs are diploid and lack genomic instability.
Given the lack of genomic alterations, we sought to evaluate whether epigenetically induced changes in gene expression may underlie oncogenesis following Snf5 loss. Snf5 regulates the transcription of cyclin D1 by directly binding to its promoter (74). Cyclin D1 promotes cell cycle progression, and its overexpression can contribute to oncogenic transformation (29, 37). Previously, examination of six human MRT samples found that all expressed cyclin D1 at high levels (21, 74). In our tumors, there were no copy number alterations of the cyclin D1 locus detected by the SNP array analysis. In order to determine whether increased expression correlates with the rhabdoid tumor phenotype, we compared cyclin D1 transcript levels in MRTs, medulloblastomas, and normal cerebellum. Cyclin D1 was upregulated in all MRTs, while it was expressed at high levels in only a few medulloblastoma cases (P < 10−13) (Fig. 6). This effect was specific, since other cyclins were not similarly upregulated, and likely a critical step in tumorigenesis since cyclin D1 ablation prevents proliferation of human MRT cell lines and tumor formation in Snf5+/− mice (1, 62). We searched for additional oncogenes that displayed specific gene expression changes in MRTs and found that expression of c-Myc was elevated in 10 out of 11 (91%) MRTs compared to 24 out of 56 (42%) medulloblastomas (Fig. 6). c-myc is a classic oncogene that is frequently activated by either genomic amplification or chromosomal translocation. The SNP results rule out amplification, and while SNP analysis is insensitive to balanced translocations, we are not aware of such a translocation ever being found in MRTs. In contrast, the SWI/SNF complex binds directly to the c-myc promoter and is required for its transcriptional repression during differentiation (43, 44). Taken together, these results suggest that epigenetically based transcriptional changes are essential for the tumor suppressor activity of SNF5.
FIG. 6.
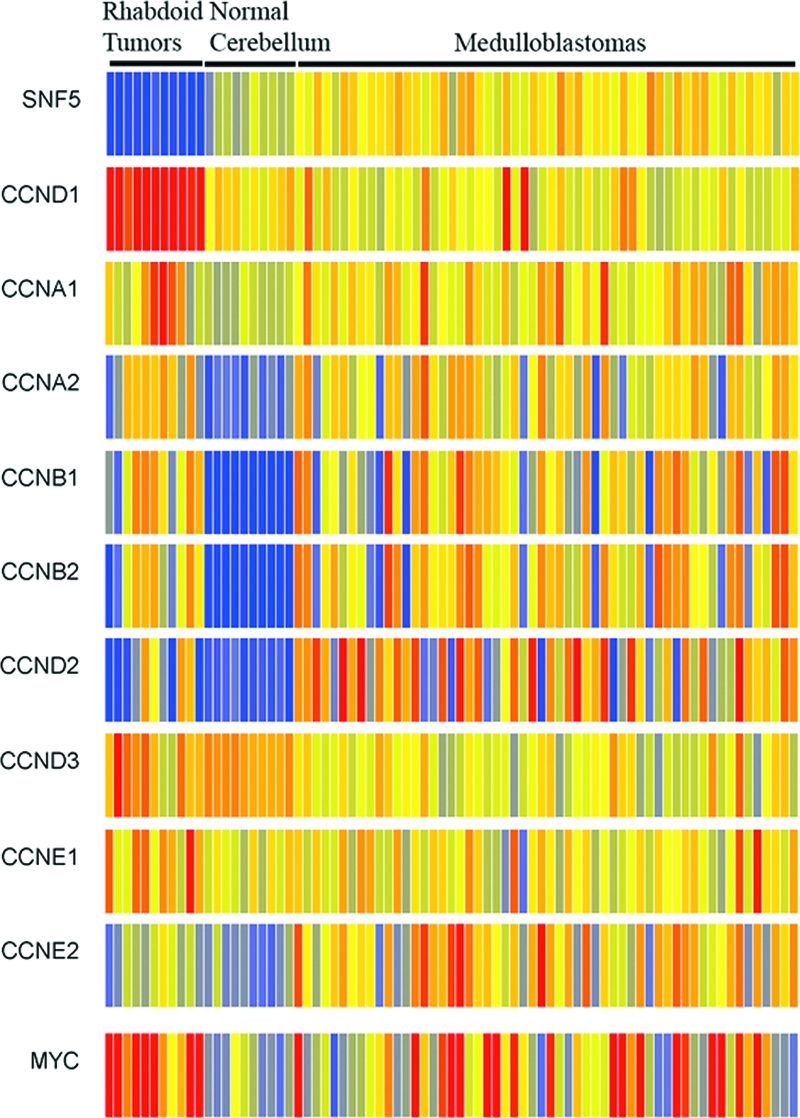
Cyclin D1 overexpression significantly correlates with the MRT phenotype. Affymetrix U133A2 microarray data from a panel of medulloblastomas, normal cerebellum, and MRTs were visualized using GeneSpring GX 7.3.1 software. Cyclin D1 (CCND1) overexpression significantly correlated with the MRT class (P = 1.74 × 10−14), in contrast to the expression of other cyclins associated with cell proliferation. Elevated expression of c-myc is evident in 91% of MRT cases compared to 42% of medulloblastomas. Red and blue indicates comparatively high- and low-level gene expression, respectively.
DISCUSSION
Genomic instability caused by defects in DNA repair and the DNA damage response is a common feature of many tumor cells. Epigenetic changes can also contribute to oncogenic transformation. However, the extent to which epigenetic changes can substitute for chromosomal instability is unclear, since highly malignant cancers are rarely diploid. Inactivation of SNF5 leads to aggressive tumors in children, and the median cancer onset following Snf5 loss in mouse models is remarkably short for inactivation of a single tumor suppressor. We hypothesized that the tumor suppressor activity of SNF5 arose not from a role in DNA repair or maintenance of genome integrity but rather from its role in the epigenetic control of gene expression.
Here we have demonstrated that loss of a gene involved in chromatin remodeling leads to aggressive cancers that lack other genomic aberrations. We show that Snf5 does not play a direct role in the DNA repair process or in the DNA damage response. Snf5 does not colocalize with DNA damage-induced γH2AX foci, its loss does not impair formation or resolution of γH2AX foci, and its loss does not confer sensitivity to DNA damaging agents that cause cross-links or DSBs. Furthermore, the DNA damage response is also intact, as indicated by normal phosphorylation of ATM, Chk1, and p53 and normal induction of p21, culminating in a G2 arrest following IR. Thus, we conclude that the mechanism of tumor suppression by SNF5 does not involve regulating the accessibility of damaged DNA to repair factors or initiating a proper DNA damage response.
Mammalian cells contain more than 20 SWI2/SNF2-related genes, and some of these, including INO80, SWR1, and RAD54, have been implicated in the execution of chromatin remodeling associated with DNA repair. Our results are in contrast to those in previous studies for which increased sensitivity to DNA damage in cells with conditionally inactivated Snf5 or overexpressed dominant-negative Brg1 was reported (33, 47). The reason for this discrepancy is not clear, although it may lie in the toxicity of these events. Both Snf5 inactivation and overexpression of dominant-negative BRG1 result in impaired proliferation after 5 days, making it difficult to interpret cellular responses to other stimuli. Once cellular viability begins to be impaired, cells may display reduced proliferation compared to normal cells when exposed to toxic agents (65). We performed experiments early after Snf5 protein loss but before secondary effects upon growth had occurred (28, 33) in order to better distinguish sensitivity to DNA damaging agents from the lethal effects of Snf5 deletion. In addition to impaired proliferation, it is conceivable that overexpression of a dominant-negative version of the BRG1 subunit disrupts stoichiometry of the SWI/SNF complex and could cause off-target interactions that affect DNA repair. While we found no direct role for Snf5 in DNA repair nor any alteration of the DNA damage response up to 4 days after Snf5 deletion, it remains possible that some of the 1,400-plus genes whose transcription is affected by Snf5 loss (28) may secondarily affect DNA repair at later time points. Additionally, while we are able to quantitatively remove the Snf5 protein as assessed by immunoblotting, it is conceivable that a small amount of protein remains bound to chromatin and obscures our ability to detect defects in DNA repair. We feel that the latter scenario is unlikely due to the absence of our ability to detect altered localization of Snf5 in response to IR, the absence of colocalization with γH2AX, the absence of abnormalities in numerous assays for DNA repair defects, and ultimately the fact that SNF5-deficient tumors are diploid and lack instability. While we have found no evidence of impaired DNA repair processes in Snf5-deficient cells or recurrent genomic amplifications/deletions in MRTs, it is important to note that we cannot rule out the possibility that pro-oncogenic point mutations exist in SNF5-deficient tumors. Doing this will ultimately require genome-wide single molecule DNA sequencing.
The intact DNA repair and damage response pathways in Snf5-deficient cells may underlie the surprising lack of genomic aberrations detected by chromosome counts of Snf5-deficient murine tumors and SNP arrays of human MRTs. Thus, we conclude that genomic instability does not account for the early onset and rapid development of SNF5-deficient tumors. Indeed, our data suggest that the epigenetic role of SNF5 in contributing to transcriptional regulation via nucleosome remodeling is the major, and perhaps sole, mechanism by which SNF5 acts as a tumor suppressor. Emerging evidence has implicated the chromatin remodeling activity of SWI/SNF in controlling the balance between cell proliferation and differentiation during development. Via the incorporation of lineage-restricted subunits, the SWI/SNF complex serves highly specific roles in fate determination in many tissues, processes known to go awry during transformation (2, 16, 34, 35, 73). Loss of Snf5 disrupts developmental progression, alters the expression of numerous target genes, and has been implicated in known tumorigenic events that select for proliferation over differentiation, including specific upregulation of cyclin D1, p16INK4a silencing, and derepression of E2F targets (6, 13, 22, 28, 31, 45, 66, 74). Furthermore, we and others have previously shown that Snf5 loss alters the expression of numerous target genes (22, 28, 66). Thus, deregulation of transcriptional activities of the SWI/SNF complex due to SNF5 loss may be equivalent to multiple genetic changes, which undergo similar selective processes during tumorigenesis. In this model, SNF5 loss may result in “epigenetic instability” due to impaired nucleosome positioning or mobility. Since malpositioned nucleosomes presumably occur at numerous targets, oncogenesis and clonal selection may be dependent upon an individual cell being left with a specific combination of nucleosomes positioned both in silencing positions over tumor suppressor genes and in activating positions away from the promoters of genes that stimulate proliferation and survival. Oncogenic clonal selection, based upon the epigenetic state rather than the genetic state of a cell, may then explain the emergence of malignant cells from the otherwise widespread death caused by SNF5 loss in normal cells.
The epigenetic mechanisms acting in SNF5-deficient cancers likely are relevant to many other tumor types. A small percentage of numerous types of cancers, including acute myeloid leukemia, hepatocellular carcinoma, colorectal cancer, and others, do not exhibit detectable chromosomal or microsatellite instability (41, 49, 60). Furthermore, while other pediatric solid tumors, such as Wilms' tumor, rhabdomyosarcoma, and Ewing's sarcoma, often have genomic anomalies, 56%, 35%, and 28% of these cancers, respectively, exhibit normal diploid genomes at the resolution of CGH (Progenetix CGH database [http://www.progenetix.de/∼pgscripts/progenetix/]). In addition, SNP arrays of acute lymphoblastic leukemias recently revealed a low instance of genomic aberrations in MLL-rearranged B-cell lineage acute lymphoblastic leukemia, with an average of one deletion per case. This is noteworthy, since MLL possesses histone methyltransferase activity, directly interacts with SNF5, and is recruited by SNF5 to cooperatively regulate tumor suppressor loci (31, 41, 42), raising the possibility of a shared epigenetic mechanism between SNF5-mutant and MLL-mutant cancers. Collectively, these findings provide novel insight into the fundamental mechanisms required for oncogenic transformation and raise the intriguing possibility that epigenetic mechanisms can be sufficient to eliminate the need for genome instability during cancer formation.
Unlike genetic mutations, epigenetic changes are potentially reversible, and this may have important therapeutic relevance. Early attempts at developing drugs capable of altering the epigenetic state of cancer cells have met with some success, since the DNA methylation inhibitors decitabine and 5-azacytidine have both been FDA approved for the treatment of myelodysplastic syndrome and the histone deacetylase inhibitor vorinostat has been FDA approved for cutaneous T-cell lymphoma. Since the large majority of cancers that arise following SNF5 loss lack genomic aberrations, these tumors may constitute a useful model both which to investigate the mechanisms by which epigenetic changes contribute to oncogenesis and also with which to test therapeutic interventions aimed at reversing the epigenetic state of cancer.
Supplementary Material
Acknowledgments
We thank Tom Curran for providing tumor samples and Yosef Shiloh, Satoshi Tateishi, and Alan D'Andrea for providing the positive-control cell lines for the DNA damage sensitivity assays. We also thank Neil J. Ganem for assistance with the preparation and imaging of mitotic cells, David Pellman and Alan D'Andrea for critical reading of the manuscript, and Rameen Beroukhim for helpful discussions.
The work was supported by PHS awards F32CA123776 (to C.G.S.), R01CA46274 (to J.A.B.), R01CA109467 (to S.L.P.), and R01CA113794 (to C.W.M.R). C.W.M.R. gratefully acknowledges support from the Garrett B. Smith Foundation, the Claudia Adams Barr Foundation, the Kate Lambert Rhabdoid Tumor Research Fund, and the Murphy Family Research Fund. C.G.S. gratefully acknowledges support from Hope Street Kids.
We declare no competing financial interests.
Footnotes
Published ahead of print on 18 August 2008.
REFERENCES
- 1.Alarcon-Vargas, D., Z. Zhang, B. Agarwal, K. Challagulla, S. Mani, and G. V. Kalpana. 2006. Targeting cyclin D1, a downstream effector of INI1/hSNF5, in rhabdoid tumors. Oncogene 25722-734. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong, J. A., J. J. Bieker, and B. M. Emerson. 1998. A SWI/SNF-related chromatin remodeling complex, E-RC1, is required for tissue-specific transcriptional regulation by EKLF in vitro. Cell 9593-104. [DOI] [PubMed] [Google Scholar]
- 3.Bartkova, J., Z. Horejsi, K. Koed, A. Kramer, F. Tort, K. Zieger, P. Guldberg, M. Sehested, J. M. Nesland, C. Lukas, T. Orntoft, J. Lukas, and J. Bartek. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434864-870. [DOI] [PubMed] [Google Scholar]
- 4.Beroukhim, R., G. Getz, L. Nghiemphu, J. Barretina, T. Hsueh, D. Linhart, I. Vivanco, J. C. Lee, J. H. Huang, S. Alexander, J. Du, T. Kau, R. K. Thomas, K. Shah, H. Soto, S. Perner, J. Prensner, R. M. Debiasi, F. Demichelis, C. Hatton, M. A. Rubin, L. A. Garraway, S. F. Nelson, L. Liau, P. S. Mischel, T. F. Cloughesy, M. Meyerson, T. A. Golub, E. S. Lander, I. K. Mellinghoff, and W. R. Sellers. 2007. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. USA 10420007-20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berrak, S. G., M. M. Ozek, C. Canpolat, A. Dagcinar, A. Sav, A. El-Naggar, and L. A. Langford. 2002. Association between DNA content and tumor suppressor gene expression and aggressiveness of atypical teratoid/rhabdoid tumors. Childs Nerv. Syst. 18485-491. [DOI] [PubMed] [Google Scholar]
- 6.Betz, B. L., M. W. Strobeck, D. N. Reisman, E. S. Knudsen, and B. E. Weissman. 2002. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 215193-5203. [DOI] [PubMed] [Google Scholar]
- 7.Biegel, J. A. 2006. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg. Focus 20E11. [DOI] [PubMed] [Google Scholar]
- 8.Biegel, J. A., B. Fogelgren, L. M. Wainwright, J. Y. Zhou, H. Bevan, and L. B. Rorke. 2000. Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 2831-37. [DOI] [PubMed] [Google Scholar]
- 9.Biegel, J. A., J. Y. Zhou, L. B. Rorke, C. Stenstrom, L. M. Wainwright, and B. Fogelgren. 1999. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 5974-79. [PubMed] [Google Scholar]
- 10.Bozhenok, L., P. A. Wade, and P. Varga-Weisz. 2002. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 212231-2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bultman, S. J., J. I. Herschkowitz, V. Godfrey, T. C. Gebuhr, M. Yaniv, C. M. Perou, and T. Magnuson. 2008. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 27460-468. [DOI] [PubMed] [Google Scholar]
- 12.Chai, B., J. Huang, B. R. Cairns, and B. C. Laurent. 2005. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 191656-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chai, J., A. L. Charboneau, B. L. Betz, and B. E. Weissman. 2005. Loss of the hSNF5 gene concomitantly inactivates p21CIP/WAF1 and p16INK4a activity associated with replicative senescence in A204 rhabdoid tumor cells. Cancer Res. 6510192-10198. [DOI] [PubMed] [Google Scholar]
- 14.Collins, N., R. A. Poot, I. Kukimoto, C. Garcia-Jimenez, G. Dellaire, and P. D. Varga-Weisz. 2002. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 32627-632. [DOI] [PubMed] [Google Scholar]
- 15.Decristofaro, M. F., B. L. Betz, C. J. Rorie, D. N. Reisman, W. Wang, and B. E. Weissman. 2001. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol. 186136-145. [DOI] [PubMed] [Google Scholar]
- 16.de la Serna, I. L., Y. Ohkawa, and A. N. Imbalzano. 2006. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat. Rev. Genet. 7461-473. [DOI] [PubMed] [Google Scholar]
- 17.Donehower, L. A., M. Harvey, B. L. Slagle, M. J. McArthur, C. A. Montgomery, Jr., J. S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356215-221. [DOI] [PubMed] [Google Scholar]
- 18.Douglass, E. C., M. Valentine, S. T. Rowe, D. M. Parham, J. A. Wilimas, J. M. Sanders, and P. J. Houghton. 1990. Malignant rhabdoid tumor: a highly malignant childhood tumor with minimal karyotypic changes. Genes Chromosomes Cancer 2210-216. [DOI] [PubMed] [Google Scholar]
- 19.Essers, J., R. W. Hendriks, S. M. Swagemakers, C. Troelstra, J. de Wit, D. Bootsma, J. H. Hoeijmakers, and R. Kanaar. 1997. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 89195-204. [DOI] [PubMed] [Google Scholar]
- 20.Fraga, M. F., E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero, K. Petrie, N. G. Iyer, A. Perez-Rosado, E. Calvo, J. A. Lopez, A. Cano, M. J. Calasanz, D. Colomer, M. A. Piris, N. Ahn, A. Imhof, C. Caldas, T. Jenuwein, and M. Esteller. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37391-400. [DOI] [PubMed] [Google Scholar]
- 21.Fujisawa, H., K. Misaki, Y. Takabatake, M. Hasegawa, and J. Yamashita. 2005. Cyclin D1 is overexpressed in atypical teratoid/rhabdoid tumor with hSNF5/INI1 gene inactivation. J. Neurooncol. 73117-124. [DOI] [PubMed] [Google Scholar]
- 22.Gresh, L., B. Bourachot, A. Reimann, B. Guigas, L. Fiette, S. Garbay, C. Muchardt, L. Hue, M. Pontoglio, M. Yaniv, and A. Klochendler-Yeivin. 2005. The SWI/SNF chromatin-remodeling complex subunit SNF5 is essential for hepatocyte differentiation. EMBO J. 243313-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guidi, C. J., A. T. Sands, B. P. Zambrowicz, T. K. Turner, D. A. Demers, W. Webster, T. W. Smith, A. N. Imbalzano, and S. N. Jones. 2001. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 213598-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara, R., and A. Sancar. 2002. The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol. 226779-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harvey, M., M. J. McArthur, C. A. Montgomery, Jr., J. S. Butel, A. Bradley, and L. A. Donehower. 1993. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 5225-229. [DOI] [PubMed] [Google Scholar]
- 26.Hulsebos, T. J., A. S. Plomp, R. A. Wolterman, E. C. Robanus-Maandag, F. Baas, and P. Wesseling. 2007. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 80805-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irizarry, R. A., B. Hobbs, F. Collin, Y. D. Beazer-Barclay, K. J. Antonellis, U. Scherf, and T. P. Speed. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4249-264. [DOI] [PubMed] [Google Scholar]
- 28.Isakoff, M. S., C. G. Sansam, P. Tamayo, A. Subramanian, J. A. Evans, C. M. Fillmore, X. Wang, J. A. Biegel, S. L. Pomeroy, J. P. Mesirov, and C. W. Roberts. 2005. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl. Acad. Sci. USA 10217745-17750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang, W., S. M. Kahn, P. Zhou, Y. J. Zhang, A. M. Cacace, A. S. Infante, S. Doi, R. M. Santella, and I. B. Weinstein. 1993. Overexpression of cyclin D1 in rat fibroblasts causes abnormalities in growth control, cell cycle progression and gene expression. Oncogene 83447-3457. [PubMed] [Google Scholar]
- 30.Jones, P. A., and S. B. Baylin. 2007. The epigenomics of cancer. Cell 128683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kia, S. K., M. M. Gorski, S. Giannakopoulos, and C. P. Verrijzer. 2008. SWI/SNF mediates Polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 283457-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klochendler-Yeivin, A., L. Fiette, J. Barra, C. Muchardt, C. Babinet, and M. Yaniv. 2000. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 1500-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klochendler-Yeivin, A., E. Picarsky, and M. Yaniv. 2006. Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol. Cell. Biol. 262661-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lessard, J., J. I. Wu, J. A. Ranish, M. Wan, M. M. Winslow, B. T. Staahl, H. Wu, R. Aebersold, I. A. Graef, and G. R. Crabtree. 2007. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55201-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lickert, H., J. K. Takeuchi, I. Von Both, J. R. Walls, F. McAuliffe, S. L. Adamson, R. M. Henkelman, J. L. Wrana, J. Rossant, and B. G. Bruneau. 2004. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 432107-112. [DOI] [PubMed] [Google Scholar]
- 36.Lin, J. C., S. Jeong, G. Liang, D. Takai, M. Fatemi, Y. C. Tsai, G. Egger, E. N. Gal-Yam, and P. A. Jones. 2007. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell 12432-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lovec, H., A. Sewing, F. C. Lucibello, R. Muller, and T. Moroy. 1994. Oncogenic activity of cyclin D1 revealed through cooperation with Ha-ras: link between cell cycle control and malignant transformation. Oncogene 9323-326. [PubMed] [Google Scholar]
- 38.Medina, P. P., O. A. Romero, T. Kohno, L. M. Montuenga, R. Pio, J. Yokota, and M. Sanchez-Cespedes. 2008. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum. Mutat. 29617-622. [DOI] [PubMed] [Google Scholar]
- 39.Modena, P., E. Lualdi, F. Facchinetti, L. Galli, M. R. Teixeira, S. Pilotti, and G. Sozzi. 2005. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 654012-4019. [DOI] [PubMed] [Google Scholar]
- 40.Morrison, A. J., J. Highland, N. J. Krogan, A. Arbel-Eden, J. F. Greenblatt, J. E. Haber, and X. Shen. 2004. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119767-775. [DOI] [PubMed] [Google Scholar]
- 41.Mrozek, K., G. Marcucci, P. Paschka, S. P. Whitman, and C. D. Bloomfield. 2007. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 109431-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullighan, C. G., S. Goorha, I. Radtke, C. B. Miller, E. Coustan-Smith, J. D. Dalton, K. Girtman, S. Mathew, J. Ma, S. B. Pounds, X. Su, C. H. Pui, M. V. Relling, W. E. Evans, S. A. Shurtleff, and J. R. Downing. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446758-764. [DOI] [PubMed] [Google Scholar]
- 43.Nagl, N. G., Jr., X. Wang, A. Patsialou, M. Van Scoy, and E. Moran. 2007. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 26752-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagl, N. G., Jr., D. R. Zweitzig, B. Thimmapaya, G. R. Beck, Jr., and E. Moran. 2006. The c-myc gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 661289-1293. [DOI] [PubMed] [Google Scholar]
- 45.Oruetxebarria, I., F. Venturini, T. Kekarainen, A. Houweling, L. M. Zuijderduijn, A. Mohd-Sarip, R. G. Vries, R. C. Hoeben, and C. P. Verrijzer. 2004. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J. Biol. Chem. 2793807-3816. [DOI] [PubMed] [Google Scholar]
- 46.Papamichos-Chronakis, M., J. E. Krebs, and C. L. Peterson. 2006. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 202437-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park, J. H., E. J. Park, H. S. Lee, S. J. Kim, S. K. Hur, A. N. Imbalzano, and J. Kwon. 2006. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 253986-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pilch, D. R., O. A. Sedelnikova, C. Redon, A. Celeste, A. Nussenzweig, and W. M. Bonner. 2003. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol. 81123-129. [DOI] [PubMed] [Google Scholar]
- 49.Pineau, P., S. Ezzikouri, A. Marchio, M. Benazzouz, E. Cordina, R. Afifi, L. Elkihal, M. T. Khalfallah, H. Mestiri, S. Tebbal, S. Berkane, N. Debzi, H. Triki, A. Dejean, F. Iguer, O. Bahri, A. E. Essaid El Feydi, and S. Benjelloun. 2007. Genomic stability prevails in North-African hepatocellular carcinomas. Dig. Liver Dis. 39671-677. [DOI] [PubMed] [Google Scholar]
- 50.Reisman, D. N., J. Sciarrotta, W. Wang, W. K. Funkhouser, and B. E. Weissman. 2003. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 63560-566. [PubMed] [Google Scholar]
- 51.Reyes, J. C., J. Barra, C. Muchardt, A. Camus, C. Babinet, and M. Yaniv. 1998. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). EMBO J. 176979-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rickert, C. H., and W. Paulus. 2004. Chromosomal imbalances detected by comparative genomic hybridisation in atypical teratoid/rhabdoid tumours. Childs Nerv. Syst. 20221-224. [DOI] [PubMed] [Google Scholar]
- 53.Roberts, C. W., S. A. Galusha, M. E. McMenamin, C. D. Fletcher, and S. H. Orkin. 2000. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 9713796-13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts, C. W., M. M. Leroux, M. D. Fleming, and S. H. Orkin. 2002. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2415-425. [DOI] [PubMed] [Google Scholar]
- 55.Roberts, C. W., and S. H. Orkin. 2004. The SWI/SNF complex—chromatin and cancer. Nat. Rev. Cancer 4133-142. [DOI] [PubMed] [Google Scholar]
- 56.Rogakou, E. P., D. R. Pilch, A. H. Orr, V. S. Ivanova, and W. M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 2735858-5868. [DOI] [PubMed] [Google Scholar]
- 57.Serrano, M., H. Lee, L. Chin, C. Cordon-Cardo, D. Beach, and R. A. DePinho. 1996. Role of the INK4a locus in tumor suppression and cell mortality. Cell 8527-37. [DOI] [PubMed] [Google Scholar]
- 58.Sevenet, N., E. Sheridan, D. Amram, P. Schneider, R. Handgretinger, and O. Delattre. 1999. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 651342-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaked, H., N. Avivi-Ragolsky, and A. A. Levy. 2006. Involvement of the Arabidopsis SWI2/SNF2 chromatin remodeling gene family in DNA damage response and recombination. Genetics 173985-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang, R., C. R. Changchien, M. C. Wu, C. W. Fan, K. W. Liu, J. S. Chen, H. T. Chien, and L. L. Hsieh. 2004. Colorectal cancer without high microsatellite instability and chromosomal instability—an alternative genetic pathway to human colorectal cancer. Carcinogenesis 25841-846. [DOI] [PubMed] [Google Scholar]
- 61.Tateishi, S., H. Niwa, J. Miyazaki, S. Fujimoto, H. Inoue, and M. Yamaizumi. 2003. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol. Cell. Biol. 23474-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsikitis, M., Z. Zhang, W. Edelman, D. Zagzag, and G. V. Kalpana. 2005. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc. Natl. Acad. Sci. USA 10212129-12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsukuda, T., A. B. Fleming, J. A. Nickoloff, and M. A. Osley. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438379-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Versteege, I., N. Sevenet, J. Lange, M. F. Rousseau-Merck, P. Ambros, R. Handgretinger, A. Aurias, and O. Delattre. 1998. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394203-206. [DOI] [PubMed] [Google Scholar]
- 65.Vreeswijk, M. P., B. E. Westland, M. T. Hess, H. Naegeli, H. Vrieling, A. A. van Zeeland, and L. H. Mullenders. 1998. Impairment of nucleotide excision repair by apoptosis in UV-irradiated mouse cells. Cancer Res. 581978-1985. [PubMed] [Google Scholar]
- 66.Vries, R. G., V. Bezrookove, L. M. Zuijderduijn, S. K. Kia, A. Houweling, I. Oruetxebarria, A. K. Raap, and C. P. Verrijzer. 2005. Cancer-associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes Dev. 19665-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, G. G., C. D. Allis, and P. Chi. 2007. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol. Med. 13363-372. [DOI] [PubMed] [Google Scholar]
- 68.Wharton, S. B., C. Wardle, J. W. Ironside, W. H. Wallace, J. A. Royds, and D. W. Hammond. 2003. Comparative genomic hybridization and pathological findings in atypical teratoid/rhabdoid tumour of the central nervous system. Neuropathol. Appl. Neurobiol. 29254-261. [DOI] [PubMed] [Google Scholar]
- 69.Wong, A. K., F. Shanahan, Y. Chen, L. Lian, P. Ha, K. Hendricks, S. Ghaffari, D. Iliev, B. Penn, A. M. Woodland, R. Smith, G. Salada, A. Carillo, K. Laity, J. Gupte, B. Swedlund, S. V. Tavtigian, D. H. Teng, and E. Lees. 2000. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 606171-6177. [PubMed] [Google Scholar]
- 70.Wood, L. D., D. W. Parsons, S. Jones, J. Lin, T. Sjoblom, R. J. Leary, D. Shen, S. M. Boca, T. Barber, J. Ptak, N. Silliman, S. Szabo, Z. Dezso, V. Ustyanksky, T. Nikolskaya, Y. Nikolsky, R. Karchin, P. A. Wilson, J. S. Kaminker, Z. Zhang, R. Croshaw, J. Willis, D. Dawson, M. Shipitsin, J. K. Willson, S. Sukumar, K. Polyak, B. H. Park, C. L. Pethiyagoda, P. V. Pant, D. G. Ballinger, A. B. Sparks, J. Hartigan, D. R. Smith, E. Suh, N. Papadopoulos, P. Buckhaults, S. D. Markowitz, G. Parmigiani, K. W. Kinzler, V. E. Velculescu, and B. Vogelstein. 2007. The genomic landscapes of human breast and colorectal cancers. Science 3181108-1113. [DOI] [PubMed] [Google Scholar]
- 71.Wu, S., Y. Shi, P. Mulligan, F. Gay, J. Landry, H. Liu, J. Lu, H. H. Qi, W. Wang, J. A. Nickoloff, C. Wu, and Y. Shi. 2007. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat. Struct. Mol. Biol. 141165-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia, W., S. Nagase, A. G. Montia, S. M. Kalachikov, M. Keniry, T. Su, L. Memeo, H. Hibshoosh, and R. Parsons. 2008. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 681667-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan, Z., Z. Wang, L. Sharova, A. A. Sharov, C. Ling, Y. Piao, K. Aiba, R. Matoba, W. Wang, and M. S. Ko. 2008. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 261155-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang, Z. K., K. P. Davies, J. Allen, L. Zhu, R. G. Pestell, D. Zagzag, and G. V. Kalpana. 2002. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol. 225975-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ziv, Y., N. G. Jaspers, S. Etkin, T. Danieli, L. Trakhtenbrot, A. Amiel, Y. Ravia, and Y. Shiloh. 1989. Cellular and molecular characteristics of an immortalized ataxia-telangiectasia (group AB) cell line. Cancer Res. 492495-2501. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.