

Abstract
The thiazolidinone 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (CFTRinh-172) inhibits cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel conductance with submicromolar affinity and blocks cholera toxin-induced intestinal fluid secretion. Fifty-eight CFTRinh-172 analogs were synthesized to identify CFTR inhibitors with improved water solubility, exploring modifications in its two phenyl rings, thiazolidinone core, and core-phenyl connectors. Greatest CFTR inhibition potency was found for 3-CF3 and polar group-substituted-phenyl rings, and a thiazolidinone core. Two compounds with ~1 μM CFTR inhibition potency and solubility >180 μM (>10-fold more than CFTRinh-172) were identified: Tetrazolo-172, containing 4-tetrazolophenyl in place of 4-carboxyphenyl, and Oxo-172, containing thiazolidinedione in place of the thiazolidinone core. These water soluble thiazolidinone analogs had low cellular toxicity. The improved water solubility of Tetrazolo- and Oxo-172 make them potential lead candidates for therapy of secretory diarrheas and polycystic kidney disease.
Keywords: CFTR, cystic fibrosis, diarrhea, SAR, CFTR inhibitors, CFTRinh-172
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel, which when mutated can produce the hereditary disease cystic fibrosis. CFTR inhibition is a potential strategy for therapy of secretory diarrheas1–6 and polycystic kidney disease,7–9 as well as for pharmacological creation of the cystic fibrosis phenotype in human and animal tissues.10 We previously discovered by high-throughput screening the thiazolidione CFTR inhibitor 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (CFTRinh-172), which blocked CFTR chloride conductance fully with IC50 < 0.5 μM.11 Patch-clamp analysis indicated a voltage-independent channel block mechanism with prolongation of mean channel closed time,12 involving interaction at arginine-347 located near the CFTR cytoplasmic surface.13 In vivo analysis indicated CFTRinh-172 efficacy in reducing cholera toxin-induced intestinal fluid secretion in rodent models.11, 14, 15 CFTRinh-172 was found to have low toxicity, renal excretion with minimal metabolism, and intestinal accumulation by enterohepatic recirculation.14 CFTRinh-172 has been used extensively to block CFTR chloride channel function in variety of cell culture, tissue and in vivo systems.16–23
Limitations of CFTRinh-172 include its low water solubility and oral bioavailability. CFTRinh-172 is a weak acid with a single negative charge at neutral pH, which precipitates in an acid environment. Also, the concentration of CFTRinh-172 in cytoplasm, where it acts, is likely reduced in a Nernstian manner compared with its extracellular concentration because of the interior-negative membrane potential of its target epithelial cells. We report here a structure-activity analysis of CFTRinh-172, with the goal of improving water solubility while maintaining CFTR inhibition potency. We systematically modified Rings A and C of CFTRinh-172, as well as the thiazolidinone core (Ring B) and the core-ring linkers (Fig. 1). CFTR analogs with >10-fold higher water solubility than CFTRinh-172 and only mildly reduced potency were synthesized and characterized.
Figure 1.
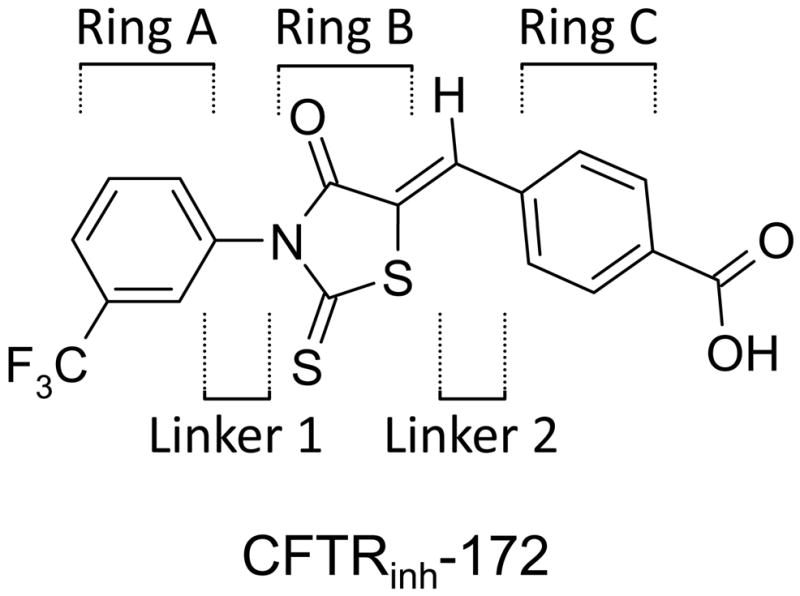
Chemical structure of CFTRinh-172 showing Rings A, B, C and Linkers 1 and 2.
Results
Chemistry
A total of 58 CFTRinh-172 analogs were assayed for CFTR inhibition activity, with the most potent compounds further characterized. Of the 58 compounds, several (compounds 5, 6, 9, 14, 15, 17, 20, 21, 25, 26, 27, 29, 33, 38) were reported previously as part of other studies from our lab.9, 11, 13, 14
Compounds with different substituents on Ring A were synthesized, keeping the remainder of the molecule the same. Commercially available substituted anilines 1 were reacted with carbon disulfide, followed by reaction with bromoacetate and acidic cyclization to give thiazolidinone intermediate 3 (Route 1, Scheme 1). The same reaction route was used for synthesis of analog containing a methylene group between Rings A and B (Route 1, Scheme 1). These intermediates upon Knoevenagel condensation with aromatic aldehydes in ethanol under reflux in presence of piperidine produced analogs 5–41, 47–49. TLC showed quantitative formation of products. This reaction generates a double bond that can produce E and Z isomers. Similar analogs are reported to exist predominantly as Z-isomers.24–29 It is presumed that the analogs synthesized here are mainly Z-isomers.
Scheme 1.

Reagents and conditions: (a) CS2, TEA, EtOAc, rt, 12 h; (b) BrCH2COOH, NaHCO3, rt, 2 h; (c) HCl, reflux, 2 h; (d) COCl2 or CSCl2, TEA, 10 °C, 2 h (e) Triphosgene, CHCl3, reflux, 3h; (); (f) HSCH2COOH, TEA, 4 h. (g) aldehyde, piperidine/ethanol, reflux, 2 h.; (h) SOCl2, cat. DMF, rt, 2 h; (i) DCC, N-hydroxy-succinimide, rt,; (j) HX-R, pyridine, 0°-rt, 2 h; (k) SCN-(3X2-4X3-Ph) or Sodium 4-SCN-furan-2-sulfonate (for 55), DBU, THF, rt, 2h. (l) LiBH4, pyridine, rt, 12 h.
Scheme 1 also shows an alternate isothiocyanate route (Route 2) for synthesis of the 2-thiaoxo-4-thiazolidinone ring intermediate 3. Route 2 was used for the synthesis of 13, 18, 20, 22, and 27. Isothiocyantes 2 were prepared by single step reaction of corresponding amino compounds 1 with thiophosgene and reacted with thioglycolic acid in presence of triethylamine to yield dithiaocarbamate intermediates. This intermediate upon in situ acidification (HCl) and reflux generated 2-thioxo-4-thiazolidinones 3. Route 2 was single-pot and increased overall yields compare to Route 1, though use of toxic compounds (phosgene or thiophosgene) was required.
Carboxylate modification to various esters and amides was achieved by three reaction routes as shown in Scheme 1. Amide 42 and ester 45 were synthesized by condensation of thiazolidinone intermediate 3 with 4-carbamoylbenzaldehyde and ethyl 4-formylbenzoate, respectively. Alternatively, the carboxyl in CFTRinh-172 was converted to the acid chloride using thionyl chloride, followed by reaction with equimolar amounts of amino compounds (ammonia, aminoethanol, ethylenediamine, acetoxymethanol) to yield 42–44 and 46. Reaction involving activation of carboxy function by DCC also generated these 42–44 and 46 amides (Scheme 1).
Thiazolidinedione 48 was synthesized by condensation of 2,4-thiazolidinedione intermediate 3 (R1, R3 = H, R2=CF3, Y =S, Z = O) with 4-carboxybenzaldehyde (Scheme 1). Route 2 was used for efficient synthesis of corresponding intermediate 3. For synthesis of compounds 50 and 51, maleimide intermediates 4 (R4 = Cl or H) were prepared by reaction of 3-trifluoromethylaniline with dichloromaleic anhydride (R4 = Cl) or maleic anhydride (R4 = H) in refluxing acetic anhydride (Scheme 2). Subsequent reaction with 4-aminobenzoic acid and 4-mercaptobenzoic acid produced compounds 50 and 51 (Scheme 2, dotted line indicate double bond in 50).30–33 Compound 52–55 were synthesized by reaction of aryl isothiocyanates with 3 in presence of base DBU at room temperature (Scheme 1) Reduction of the double bond in CFTRinh-172 using LiBH4 in pyridine24, 34 at room temperature gave 56 (Scheme 1).
Scheme 2.

Reagents and conditions: (a) Malic anhydride/dichloromalic anhydride, Ac2O, NaOAc, 80°, 2 h; (b) (4-COOH)-Ph-WH, TEA, THF, rt, 5 h.
Thiazole analogs 59 and 60 were synthesized by bromination of acetophenone 57 in acetic acid at 0 °C for 2 h, followed by reaction with substituted phenylthiourea in refluxing ethanol (Scheme 3).35, 36 Synthesis of thiadiazole compounds 64 and 65 was accomplished in three steps. Reaction of the acid hydrazide (prepared from benzoyl chloride 61 and hydrazine) with substituted phenylisothiocyanates gave thiosemicarbazide 62 and 63 in good yields. Treatment of 62 and 63 with sulfuric acid produced the 2-aminothiadiazoles 64 and 65 (Scheme 4).37
Scheme 3.

Reagents and conditions: (a) Br2, AcOH, 0°-rt, 2 h; (b) n-(4-carboxyphenyl)thiourea, AcOH, reflux, 4 h.
Scheme 4.

Reagents and conditions: (a) NH2NH2·xH2O, pyridine, rt, 2 h; (b) (4-COOH)-Ph-NCS, TEA, THF, rt, 12 h; (c) H2SO4, rt, 2 h.
Scheme 5 shows the synthesis of 1,2,3-triazoles. 1,3-Dipolar cycloaddition38, 39 of alkynes 66 and 67 with 4-azidobenzoic acid produced the 1,2,3-triazoles (compounds 68 and 69) in high yields. Single spot in TLC indicated that adducts were predominantly 4-regioisomers.
Scheme 5.

Reagents and conditions: (a) 1 mol-% CuSO4, 0.1 eq. sodium ascorbate, H2O/t-BuOH (1:1), rt, 24h.
Functional studies
CFTR inhibition potency for all compounds was measured by short-circuit current analysis, with representative original data and concentration-inhibition curves for compounds of greatest interest provided in Fig. 2. In the short-circuit current assay, CFTR was stimulated by the cAMP agonist forskolin, followed by compound additions at increasing concentration. Table 1 summarizes the effects of Ring A substitutions on CFTR inhibition potency. CF3 substitution at the 3-position, as in CFTRinh-172, gave greater inhibition potency than at the 2- or 4-positions (compounds 20 and 25).9 Substitution of 3-CF3 by 3-CH3 gave the less active analog 12. Compounds 8 and 11, in which Ring A contained 3-CF3 along with 4-F substituents, had intermediate activity. Similar substitutions by chloro at 2- or 4- positions greatly reduced activity (compounds 24 and 30).
Figure 2. Short-circuit current measurements of CFTR inhibition.

A CFTR-mediated apical membrane chloride current measured in FRT cells expressing human wildtype CFTR after permeabilization of the basolateral membrane in the presence of a chloride gradient (see Methods). CFTR was activated by 100 μM CPT-cAMP and indicated concentrations of inhibitors were added. B. Concentration-inhibition data for CFTRinh-172, Tetrazolo-172, Oxo-172, α-Me-172, and Pyridine-NO-172. See Table 3 for fitted IC50 values.
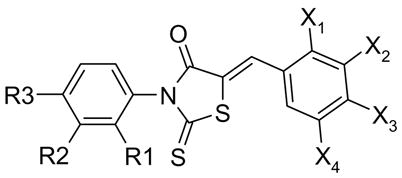
Table 1.
Variations in Rings A and C
 |
 |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R3 | R2 | R1 | X1 | X2 | X3 | X4 | IC50 (μM) |
| 5 CFTRinh-172 | H | CF3 | H | H | H | COOH | H | 0.4 |
| 6 Tetrazolo-172 | H | CF3 | H | H | H | Tetrazolo | H | 0.8 |
| 7 | H | CF3 | H | H | COOH | OH | H | 1.4 |
| 8 | F | CF3 | H | H | H | COOH | H | 1.5 |
| 9 | H | CF3 | H | H | COOH | H | H | 1.5 |
| 10 | H | CF3 | H | H | H | O-CH2-COOH | H | 2.6 |
| 11 | F | CF3 | H | H | COOH | H | H | 5 |
| 12 | H | CH3 | H | H | H | COOH | H | 5 |
| 13 | H | CH3 | CH3 | H | H | COOH | H | 7 |
| 14 | H | CF3 | H | H | Br | OH | Br | 7 |
| 15 | H | CF3 | H | OH | Br | OH | Br | 8 |
| 16 | H | CF3 | H | OH | H | COOH | H | |
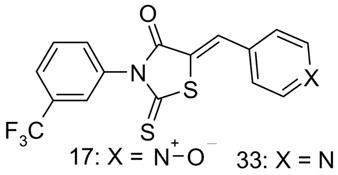
| 17 Pyridine-NO-172 | 9 | |||||||
| 18 α-Me-172 | H | CF3 | CH3 | H | H | COOH | H | 9 |
| 19 | H | CF3 | H | H | OH | OH | OH | 11 |
| 20 | H | H | CF3 | H | H | COOH | H | 12 |
| 21 | CF3 | H | H | H | COOH | H | H | 13 |
| 22 | H | H | CF3 | H | COOH | OH | H | 14 |
| 23 | Ring A: 3,5-di-CF3-Ph- | H | H | COOH | H | 14 | ||
| 24 | Cl | CF3 | H | H | H | COOH | H | 15 |
| 25 | CF3 | H | H | H | H | COOH | H | 15 |
| 26 | H | CF3 | H | COOH | H | H | H | 15 |
| 27 | H | H | CF3 | COOH | H | H | H | 18 |
| 28 | H | H | CF3 | H | COOH | H | H | 19 |
| 29 | CF3 | H | H | COOH | H | H | H | 20 |
| 30 | Ring A: 2-Cl-5-CF3-Ph- | H | H | COOH | H | 20 | ||
| 31 | H | CF3 | H | H | H | OH | H | 24 |
| 32 | CF3 | H | H | H | COOH | OH | H | 25 |
| 33 | -- | 28 | ||||||
| 34 | H | CF3 | H | OH | OH | OH | H | 28 |
| 35 | H | CF3 | H | H | H | SO3Na | H | 29 |
| 36 | H | CF3 | H | H | O-CH2-COOH | H | H | 30 |
| 37 | H | CF3 | H | SO3Na | H | SO3Na | H | 50 |
| 38 | H | CF3 | H | H | OH | OH | H | Inactive |
| 39 | H | CF3 | H | OH | Br | OMe | Br | Inactive |
| 40 | H | CF3 | H | OCH3 | H | Br | H | Inactive |
| 41 | H | CF3 | H | H | H | OMe | H | Inactive |
| 42 | H | CF3 | H | H | H | CONH2 | H | Inactive |
| 43 | H | CF3 | H | H | H | CONHC2H4OH | H | Inactive |
| 44 | H | CF3 | H | H | H | CONHC2H4NH2 | H | Inactive |
| 45 | H | CF3 | H | H | H | COOCH2CH3 | H | Inactive |
| 46 | H | CF3 | H | H | H | COOCH2OCOCH3 | H | Inactive |
To improve aqueous solubility, we first tried to disrupt the planar configuration of Rings A and B, which is predicted to reduce ring-ring stacking, by adding bulky ortho substituents to Ring A. Introduction of methyl at the 2-position in α-Me-172, 18, increased water solubility to 259 μM, but reduced inhibition potency considerably. Fig. 2 shows concentration-dependent inhibition of CFTR by CFTRinh-172 (A) and α-Me-172 (E) with IC50 0.4 and 8 μM, respectively. As determined by LC/MS, the solubility of α-Me-172 in saline was 259 μM, substantially greater than that of CFTRinh-172 (17 μM; Table 3). Addition of polar substituents such as hydroxy, SO3Na or COOH in Ring A, or removal of CF3, produced highly water-soluble though inactive compounds.
Table 3.
Inhibition potency, solubility, and toxicity of CFTR inhibitors.
| Compound | IC50 (μM) | Solubility in saline (μM) | % Cell viability | |
|---|---|---|---|---|
| 20 μM | 50 μM | |||
| CFTRinh-172 | 0.38 ± 0.04 | 17 | 86 | not soluble |
| Tetrazolo-172 | 0.76 ± 0.2 | 189 | 87 | 73 |
| Oxo-172 | 1.4 ± 0.2 | 420 | 93 | 91 |
| α-Me-172 | 8.2 ± 0.4 | 259 | 89 | 72 |
| Pyridine-NO-172 | 8.7 ± 0.7 | 264 | not determined | |
The thiazolidinone core, Ring B, was replaced by thiazolidinedione, maleimide, succinimide, thiazole, thiadiazole and triazole, while keeping Rings A and C and their substituents the same as in CFTRinh-172. These heterocycles were selected because of their resemblance to Ring B, their ease of synthesis, and prior knowledge of good bioavailability and metabolic and chemical stability.40, 41
The 2,4-thiazolidinedione 48 is a close analog of CFTRinh-172 in which the thioxo group is replaced by an oxo-group (referred to as Oxo-172). Replacement of 2-thioxo by 2-oxo increased solubility in saline by ~25-fold, while reducing CFTR inhibition potency 3.6-fold in short-circuit current assays (Figs. 2B, 2F; Table 2). Maleimide analog 50 had weak activity, though 2,5-pyrrolidinedione 51 had moderate activity with IC50 ~7 μM. The Ring B variation in 51 removed the double bond in CFTRinh-172, making it less reactive for Michael addition. The aminothiazole and aminothiadiazole analogs were inactive, however. Replacement of thiazolidinone by other heterocycles such as aminothiazoles (59, 60) gave weakly active compounds. Replacement of thiazolidinone by aminothiadiazole (64, 65), and 1,2,3-triazoles (68, 69) yielded inactive compounds.
Table 2.
Variations in Ring B and Linkers 1 and 2
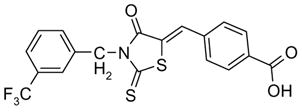 47 |
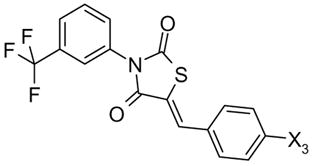 48,49 |
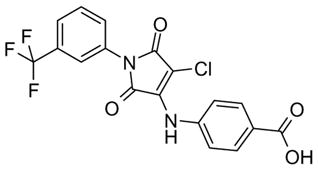 50 |
|
|---|---|---|---|
| Compound | X3 | IC50 (μM) | |
| 47 | Inactive | ||
| 48 Oxo-172 | COOH | 1.4 | |
| 49 | (2H)-Tetrazolo-5-yl | 17 | |
| 50 | 36 | ||
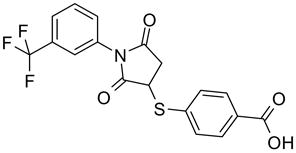
51 |
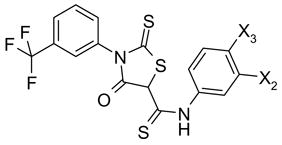
52-54 |
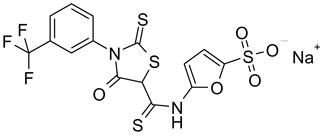
55 |
|
| Compound | X2 | X3 | IC50 (μM) |
| 51 | 7 | ||
| 52 | H | COOH | 9 |
| 53 | OH | COOH | 11 |
| 54 | H | SO3Na | 25 |
| 55 | 27 | ||
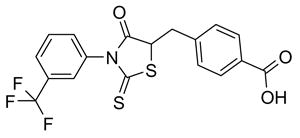
56 |
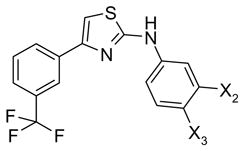
59, 60 |
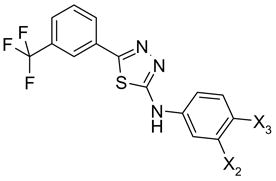
64, 65 |
|
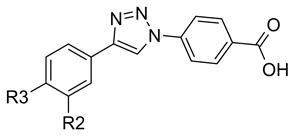
68, 69 |
|||
| Compound | X3/R3 | X2/R2 | IC50 (μM) |
| 56 | 27 | ||
| 59 | COOH | H | 28 |
| 60 | OH | COOH | 32 |
| 64 | COOH | H | Inactive |
| 65 | OH | COOH | Inactive |
| 68 | H | CF3 | Inactive |
| 69 | CF3 | H | Inactive |
C-ring substitutions were carried out in an attempt to increase compound solubility, and to convert compound net negative charge at physiological pH to neutral in order to increase compound accumulation in cytoplasm. Analogs were synthesized with different substitutions on Ring C, keeping Rings A and B, and both linkers the same as in CFTRinh-172. Substitutions were chosen to increase compound polarity and H-bond capacity, including carboxy, esters, amides, hydroxy, methoxy and sulfonate.
Monosubstituted compounds containing 4-COOH (CFTRinh-172) or 3-COOH, 9, showed greater CFTR inhibition potency than 2-COOH, 26. Esterification or amidation of 4-COOH in CFTRinh-172 gave inactive compounds 42–46. Compounds 31, 38, 19, and 34 containing mono, di or tri hydroxy functions at Ring C had low activity. Based on predicted pKa of >7.5, these compounds are expected to be neutral at physiological pH. Creation of a negative charge by addition of 3,4-dibromo electron withdrawing moieties, that lower the pKa of 4-OH, resulted in modest inhibition activity (compounds 14 and 15; Table 1), whereas modification of the ionizable 4-OH to 4-OMe gave the inactive compound 39. However, sulfonic acid derivatives 35 and 37, which carry at physiological pH single and double negative charges, respectively, were inactive.
Ring C was also replaced by heterocyclic-equivalent ring systems. Replacement of the phenyl by a pyridyl ring gave the inactive neutral compound 33; however, N-oxidation of pyridyl nitrogen gave the first, net-neutral thiazolidinone CFTR inhibitor, pyridine-NO-172, 17, with IC50 9 μM (Figs. 2D, 2F). This is a zwitterionic compound containing a positive charge on the ring nitrogen and negative charge on the oxygen. Addition of hetero atoms is expected to increase aqueous solubility by H-bonding and increased polarity. Pyridine-NO-172 had high aqueous solubility of 264 μM. Similarly, the polar analog 10 containing a 4-carboxymethoxy group had an IC50 of 2.6 μM (Table 1). Moving the carboxymethoxy group from the 4-position (as in 10) to the 3-postion (as in 36) substantially reduced CFTR inhibition.
As another approach to improve water solubility, the 4-COOH in ring C was replaced by tetrazolo-5-yl, an isoster of carboxy with delocalized negative charge at physiological pH. The tetrazolo substitution increased water solubility 11-fold (Table 3) with IC50 of 0.8 μM (Fig. 2F). As shown in Fig. 2C, Tetrazolo-172 showed slower inhibition kinetics than CFTRinh-172 or Oxo-172. Similarly replacement of carboxylate in 48 by tetrazolo in 49, gave lower inhibition potency (IC50 17 μM; Table 2).
CFTRinh-172 contains a single bond as a Linker 1 that directly connects lipophilic Ring A to heterocyclic Ring B. We examined the effect of introduction of a methylene group as a bridge between Rings A and B. Compound 47 was inactive, however, indicating little tolerance of Linker 1 modification.
Because the double bond in Linker 2 is a Michael electrophile, alternative linkers between Ring B and Ring C were investigated. First, reduction of double bond linker of CFTRinh-172 gave compound 56, which was inactive. The double bond reduction disturbed the rigid geometry of CFTRinh-172, which is presumed as Z-configuration, allowing free rotation of Rings B and C. Further, replacement of the double bond-methylidyne bridge by thioamide in 52 and 53, though highly water soluble, were inactive, as were analogs 54 and 55 containing sulfonic acid substituents (Scheme 1 and Table 2).
Preliminary cytotoxicity analysis was done for the principle compounds of interest, Tetrazolo-172, Oxo-172, α-Me-172 and Pyridine-NO-172, comparing with CFTRinh-172 (Table 3). Compounds were incubated with cell cultures for 48 h and cell viability determined by crystal violet staining. Compounds at 20 μM showed little cytotoxicity, with staining ~90% of that of control (vehicle-treated cultures). Crystal violet staining was reduced for Tetrazolo-172 and α-Me-172 at 50 μM. CFTRinh-172 was not studied at 50 μM because of its limited aqueous solubility.
Discussion
We synthesized and characterized fifty-eight CFTRinh-172 analogs with the goal to increase water solubility and retain CFTR inhibition potency. As summarized in Fig. 3, SAR analysis revealed that compounds containing 3-CF3 on Ring A, thiazolidinone core as Ring B, and 4-carboxy at Ring C had greatest CFTR inhibition potency. Polar ionizable substituents such as carboxy, hydroxy, tetrazolo, which created a negative charge on Ring C, gave greatest inhibition activity, whereas non-polar substituents were required on Ring A. The thiazolidinone ring (Ring B) could be replaced by thiazolidinedione, and the COOH group on Ring C by tetrazolo, yielding compounds with greatly increased water solubility compared to CFTRinh-172. Tetrazolo-172 and Oxo-172 are thus potential candidates for further development.
Figure 3.

Structure-activity analysis of thiazolidinone CFTR inhibitors.
Though the most active thiazolidinones contain a negative charge at neutral pH, compound Pyridine-NO-172, which has IC50 of 9 μM for CFTR inhibition, is zwitterionic and net neutral. Together with analysis of site-directed CFTR mutants,13 the existence of a moderately potent CFTR inhibitor with net neutral charge indicates that a negative charge is not necessary for CFTR inhibition by thiazolidinones. Though Pyridine-NO-172 is not a candidate for further development because of its predicted poor in vivo stability, it may be possible to identify other net neutral thiazolidinones with improved accumulation in cytoplasm compared to CFTRinh-172.
A recent study from our lab identified Tetrazolo-172 as the best thiazolidinone for inhibition of renal cyst growth in models of polycystic kidney disease.9 Prevention of cyst formation by Tetrazolo-172 in an MDCK cell model was substantially better than by CFTRinh-172. Tetrazolo-172 reduced cyst formation and expansion in an embryonic kidney organ culture model and in a mouse model of pkd1 gene deletion. Whether Tetrazolo-172 or other small-molecule CFTR inhibitors are effective in human polycystic kidney disease will require clinical trials. Our recent study also identified a cell permeable phenyl derivative of the glycine hydrazide-type CFTR inhibitor as effectively reducing cyst formation and growth in in vitro and mouse models of polycystic kidney disease.
In contrast to polycystic kidney disease, which is a life-long condition, therapy of enterotoxin-mediated secretory diarrheas such as cholera or Traveler’s diarrhea requires short-term therapy of days or less. Small-molecule CFTR inhibitors are predicted to reduce intestinal fluid secretion. Thiazolidinones, as ‘absorbable-type’ CFTR inhibitors that act from the cytoplasmic surface of CFTR, are taken up into enterocytes and enter the systemic circulation. Absorbable-type CFTR inhibitors are expected to resist potential washout, a theoretical concern of non-absorbable CFTR inhibitors in which rapid fluid transit through the intestine may dilute and wash out compounds that weakly associate with targets on the surface of the intestinal lumen. Our laboratory has developed a series of non-absorbable CFTR inhibitors that block the CFTR pore from its external surface,42, 43 including macromolecular conjugates that stick tightly to the intestinal surface.44 The clinical utility of absorbable vs. non-absorbable CFTR inhibitors in the treatment of secretory diarrheas will require human clinical trials.
Experimental Methods
Synthesis procedures
1H nuclear magnetic resonance spectra were obtained in CDCl3 or dimethyl sulfoxide (DMSO)-d6 using a 400-MHz Varian Spectrometer referenced to CDCl3 or DMSO. Mass spectrometry was done on a Waters LC/MS system (Alliance HT 2790+ZQ, HPLC: Waters model 2690, Milford, MA). Flash chromatography was done using EM silica gel (230–400 mesh), and thin-layer chromatography was performed on Merk silica gel 60 F254 plates (Darmstadt, Germany).
Intermediates were confirmed by mass spectrometry and 1H NMR. Full analytical data for representative and the main compounds, including their intermediates, are provided. Melting points are uncorrected. Weakly active and inactive compounds and their intermediates were characterized by mass spectral analysis (LC/MS). Purity was determined by TLC and HPLC. Compounds with purity > 95% were used for CFTR inhibition testing.
Synthesis of thiazolidinone intermediate 3
Typical procedure for Route 1:15 An equimolar amount of carbon disulfide was added dropwise to an ice-cold solution of aniline 1 and triethylamine in ethyl acetate over 30 min (Scheme 1). After stirring overnight, a yellow dithiocarbamate was isolated by filtration and reacted with an equimolar amount of aqueous bromoacetic acid solution (NaHCO3, pH 8–9). After 2 h, the solution was acidified (HCl), refluxed, and the resultant precipitate crystallized from ethanol to yield thiazolidinone intermediate 3. Typical procedure for Route 2: A solution of isothiocyante 2 (5 mmol, in THF) was added dropwise to a stirred aqueous solution of thioglycolic acid (0.347 g, 3.7 mmol) and triethylamine (1.38 ml, 10 mmol). After 30 min at 0 °C, the reaction mixture was further stirred at room temperature for 3 h. The reaction mixture was acidified (HCl), refluxed, and resultant precipitate crystallized from ethanol to yield thiazolidinone intermediate 3.
Isothiocyanates and isocyanates 2, if not available commercially, were prepared by reaction of respective amino compounds 1 with phosgene or thiophosgene, following known procedures.29
2-Thioxo-3-[3-(trifluoromethyl)phenyl]-4-thiazolidinone (intermediate 3; R1 = H, R2 = CF3, R3 = H, n = 0, Y & Z = S). mp 177–178 °C; 1H nmr (CDCl3):δ 7.72 (d, 1H, phenyl, J = 7.6 Hz), 7.64 (t, 1H, phenyl, J = 8.0 Hz), 7.48 (s, 1H, phenyl), 7.40 (d, 1H, phenyl, J = 8.0 Hz), d 4.18 (s, 2H, CH2); MS (ES+) (m/z): [M+1]+ calculated for C10H6F3NOS2, 278.29, found 277.93.
2-Thioxo-3-[2-methyl-3-(trifluoromethyl)phenyl]-4-thiazolidinone
MS (ES+) (m/z): [M+1]+ calculated for C11H8F3NOS2, 292.32, found 292.02.
Synthesis of compounds 5–41, 47, 48
4-[[4-Oxo-2-thioxo-3-[3-(trifluoromethyl)phenyl]-5-thiazolidinylidene]methyl]benzoic acid (5, CFTRinh-172).14
A mixture of 2-thioxo-3-(3-trifluoromethyl phenyl)-4-thiazolidinone 3 (55 mg, 0.2 mmol,15 4-carboxybenzaldehyde (30 mg, 0.2 mmol), and a drop of piperidine in absolute ethanol (0.5 ml) was refluxed for 2 h. Solvent was evaporated, and the residue was crystallized from ethanol and further purified by normal phase flash chromatography to yield 54 mg yellow powder (yield 67 %); mp 182–183 °C; 1H nmr (DMSO-d6): δ 13.20 (bs, 1H, COOH, D2O exchange), 8.07 (d, 2H, carboxyphenyl, J = 8.31 Hz), 7.80–8.00 (m, 5H, trifluoromethyl-phenyl and CH), 7.78 (d, 2H, carboxyphenyl, J = 8.2 Hz); HRMS (ESI−) (m/z): [M−1] − calculated for C18H9F3NO3S2, 407.9976, found 407.9976.
5-[[4-(2H-Tetrazol-5-yl)phenyl]methylene]-2-thioxo-3-[3-(trifluoromethyl)phenyl]-4-thiazolidinone (6, Tetrazolo-172).9
mp 216–219 °C; 1H nmr (DMSO-d6): δ11.92 (bs, 1H, tetrazolo-H, D2O exchange), 8.16 (d, 2H, phenyl, J = 8.3 Hz), 8.02 (s, 1H), 7.92 (s, 1H), 7.87-7.84 (m, 3H, phenyl or trifluoromethyl-phenyl and/or CH), 7.79-7.76 (m, 2H, trifluoromethyl-phenyl); MS (ES−) (m/z): [M−1]− calculated for C18H9F3N5OS2, 432.0201, found 432.0205.
5-(1-Oxido-4-pyridinyl)methylene)-2-thioxo-3-[3-(trifluoromethyl)phenyl]-4-thiazolidinone (17, Pyridine-NO-172)13
mp: 209–210 °C (decomp); MS (ES+) (m/z): [M+H]+ calculated for C16H9F3N2O2S2, 382.39, found 383.01.
4-[[4-Oxo-2-thioxo-3-[2-methyl-3-(trifluoromethyl)phenyl]-5-thiazolidinylidene] methyl] benzoic acid (18, α-Me-172)
mp 156–158 °C; 1H nmr (DMSO-d6): δ 12.74 (bs, 1H, COOH, D2O exchange), 8.05 (d, 2H, carboxyphenyl, J = 8.3 Hz), 7.90 (1H, s, =CH-), 7.85 (d, 1H, J = 7.8 Hz, trifluoromethylphenyl), 7.79 (d, 2H, J = 8.3, carboxyphenyl), 7.74 (d, 1H, J = 7.3 Hz, trifluoromethylphenyl), 7.58 (t, 1H, J = 7.8 Hz, trifluoromethylphenyl), 2.13 (s, 3H, CH3); HRMS (ESI−) (m/z): [M−1]− calculated for C19H11F3NO3S2, 422.0132, found 422.0140.
5-(4-Pyridinylmethylene)-2-thioxo-3-[3-(trifluoromethyl)phenyl]-4-thiazolidinone (33)
mp 186–188 °C; 1H nmr (DMSO-d6): δ8.72 (dd, 2H, J = 6.35, 2.93 Hz, pyridine), 7.92 (s, 1H, =CH-), 7.87 (d, 1H, J = 7.32 Hz, trifluoromethylphenyl), 7.81-7.75 (m, 3H, trifluoromethylphenyl), 7.59 (dd, 2H, J = 6.35, 2.93 Hz, pyridine); MS (ES+) (m/z): [M+1]+ calculated for C16H9F3N2OS2, 367.40, found 367.20.
4-[[4-Oxo-2-thioxo-3-[(3-(trifluoromethyl)phenyl)methyl]-5-thiazolidinylidene]methyl] benzoic acid (47)
1H nmr (DMSO-d6): δ 13.19 (s, 1H, COOH), 8.01 (d, 2H, J = 8.3 Hz, carboxyphenyl), 7.86 (s, 1H, trifluoromethylphenyl), 7.73-7.70 (m, 3H, carboxyphenyl and trifluoromethylphenyl), 7.63-7.51 (m, 3H, trifluoromethyl and =CH-), 5.29 (s, 2H, CH2); MS (ES+) (m/z): [M+1]+ calculated for C19H12F3NO3S2, 424.45, found 424.17.
4-[[3-[3-(trifluoromethyl)phenyl]-2,4-dioxo-5-thiazolidinylidene]methyl]benzoic acid (48, Oxo-172)
mp 168–170 °C; 1H nmr (DMSO-d6): δ 13.05 (bS, 1H, COOH, D2O exchange), 8.06-8.04 (d, 2H, carboxyphenyl, J = 8.3 Hz), 8.01 (s, 1H), 7.92 (s, 1H), 7.86-7.84 (m, 1H), 7.78-7.74 (m, 4H); HRMS (ESI−) (m/z): [M−1]− calculated for C18H9F3NO4S, 392.0204, found 392.0207.
4-[[2,5-dioxo-1-[3-(trifluoromethyl)phenyl]-3-pyrrolidinyl]thio]-benzoic acid (51)
MS (ES−) (m/z): [M−1] − calculated for C18H12F3NO4S, 394.35, found 394.18.
4-Oxo-[(3-trifluoromethyl)phenyl]-2-thioxo-N-[4-(carboxy)phenyl]-5-thiazolidinethio carboxamide (52)
MS (ES+) (m/z): [M+1]+ calculated for C18H11F3N2O3S3, 457.49, found 457.37.
4-Oxo-[(3-trifluoromethyl)phenyl]-2-thioxo-N-[(4-carboxy-3-hydroxy)phenyl]-5-thiazolidinethiocarboxamide (53)
MS (ES+) (m/z): [M+1]+ calculated for C18H11F3N2O4S3, 473.50, found 473.23.
4-[[4-Oxo-2-thioxo-3-[3-(trifluoromethyl)phenyl]-5-thiazolidinyl]methyl]benzoic acid (56).24, 34
To a stirred solution of 5 (102 mg, 0.25 mmole) in pyridine (1 ml), lithium borohydride (1 M solution in THF, 500 μl) was added dropwise over 30 min. The reaction mixture was refuxed (2 h), cooled, and added to an ice-cold solution of hydrochloric acid (4 N, 5 ml). The resulting material was refluxed for 30 min, filtered hot, and cooled. The precipitate was dried to yield 56. MS (ES+) (m/z): [M−1]− calculated for C18H12F3NO3S2, 410.42, found 410.13.
3-Trifluoromethyl benzoic acid hydrazide
Hydrazine hydrate (4 equivalents) was added to the stirred solution of 3-trifluoromethyl benzoyl chloride 61 in pyridine at 0 °C. The reaction mixture was added to the ice cold water and the collected precipitate was recrystallized from ethanol (yield 83 %). MS (ES+) (m/z): [M+1]+ calculated for C8H7F3N2O, 205.16, found 205.01.
3-Trifluoromethyl-2-[[(4-carboxyphenyl)amino]thioxomethyl]benzoic acid hydrazide (62)
A mixture of 3-trifluoromethyl benzoic acid hydrazide (1 g, 5 mmol, prepared as above) and appropriate carboxyphenylisothiocyanate (5 mmol) in THF was refluxed for 2 h. The solid obtained after solvent evaporation was purified by recrystallization from ethanol to give thiosemicarbazide 62 (yield 74 %). MS (ES+) (m/z): [M+1]+ calculated for C16H12F3N3O3S, 384.35, found 384.06.
4-[(5-(3-Trifluoromethylphenyl)-1,3,4-thiadiazol-2-yl)amino]-benzoic acid (64).37
Thiosemicarbazide 62 (2.6 mmol) was added slowly to 10 ml of concentrated sulfuric acid, stirred for 30 min at room temperature, and slowly dumped into a stirred ice-water mixture. The precipitated product was purified by flash chromatography to give 64 (yield 77%). 1H nmr (DMSO-d6): δ 10.98 (bs, 1H, COOH), 8.14-8.11 (m, 3H, trifluoromethylphenyl), 7.90 (d, 2H, J = 8.3 Hz, carboxyphenyl) 7.84-7.71 (m, 4H, (2H carboxyphenyl, 1H trifluoromethylphenyl, NH)); MS (ES+) (m/z): [M+1]+ calculated for C16H10F3N3O2S, 366.35, found 366.46.
4-Hydroxy-3-[[5-(3-trifluoromethyl)phenyl-1,3,4-thiadiazol-2-yl]amino]-benzoic acid (65)
Synthesized by same procedure as for 64. MS (ES+) (m/z): [M+1]+ calculated for C16H10F3N3O2S, 382.34, found 382.03.
4-[4-[3-(trifluoromethyl)phenyl]-1H-1,2,3-triazol-1-yl]-benzoic acid (68).38
1-Ethynyl-3-(trifluoromethyl)benzene (0.85 g, 5 mmol) and 4-azidobenzoic acid (0.815 g, 5 mmol) were suspended in a 1:1 mixture of water and tert-butyl alcohol (10 mL). A freshly prepared solution of sodium ascorbate (0.3 mmol, 300 μL of 1 M) was added, followed by copper (II) sulfate pentahydrate (7.5 mg, 0.03 mmol, in 100 μL of water). The mixture was stirred for 24 hr at room temperature. The reaction mixture was diluted with water, and the white precipitate was collected by filtration, washed and dried to give 1.53 g (92 %) of pure product as an off-white powder. mp >250 °C; MS (ES+) (m/z): [M+1]+ calculated for C16H10F3N3O2, 334.28, found 334.09.
Solubility
As reported,45 a saturated compound solution was prepared by addition of DMSO stock to phosphate buffered saline (final DMSO 2%) followed by sonication for 5 min at 25 °C and shaking at room temperature for 1 h. After centrifugation at 15 000 rpm for 1 h, the supernatant was analyzed by LC/MS with concentration determined from area under the curve, standardized against calibration data. Standard curve for each compound was obtained by plotting area under the curve from chromatograms against inhibitor concentration. The concentration range of the standard solutions was 1–15 μM (for 5) and 1–100 μM (other compounds). In all cases, standard curves prepared were linear with r > 0.998.
Short-circuit current measurements
FRT cells (stably expressing human wildtype CFTR) were cultured on Snapwell filters with 1 cm2 surface area (Corning-Costar) to resistance >1,000Ω.cm2 as described.11 Filters were mounted in an Easymount Chamber System (Physiologic Instruments, San Diego). For apical Cl− current measurements the basolateral hemichamber contained (in mM): 130 NaCl, 2.7 KCl, 1.5 KH2PO4, 1 CaCl2, 0.5 MgCl2, 10 Na-HEPES, 10 glucose (pH 7.3). The basolateral membrane was permeabilized with amphotericin B (250 μg/ml) for 30 min. In the apical solution 65 mM NaCl was replaced by sodium gluconate, and CaCl2 was increased to 2 mM. Solutions were bubbled with 95% O2/5% CO2 and maintained at 37 °C. Current was recorded using a DVC-1000 voltage-clamp (World Precision Instruments) using Ag/AgCl electrodes and 1 M KCl agar bridges.
Cytotoxicity
FRT cells in confluent monolayers were incubated with compounds for 2 days. Cells were washed 3 times, fixed (cytofix, 30 min) and stained with crystal violet (100 μl, 0.5%, 10 min) using standard procedures.46 Excess crystal violet was removed by washing and dye was extracted with Sorenson’s buffer (0.1 M sodium citrate, 50% ethanol, pH 4.2). Crystal violet was quantified by measurement of absorbance at 650 nm. Percentage crystal violet staining was determined from test wells measured 8 times, compared to blanks (wells not containing cells) and vehicle-treated cells.
Acknowledgments
Supported by grants DK72517, HL73854, EB00415, EY13574, DK35124 and DK43840 from the National Institutes of Health, and Drug Discovery and Research Development Program grants from the Cystic Fibrosis Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barrett KE, Keely SJ. Annu Rev Physiol. 2000;62:535. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- 2.Field M. J Clin Invest. 2003;111:931. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Science. 1994;266:107. doi: 10.1126/science.7524148. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, Naren AP. J Exp Med. 2005;202:975. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao MC. Annu Rev Physiol. 2004;66:385. doi: 10.1146/annurev.physiol.66.032902.134726. [DOI] [PubMed] [Google Scholar]
- 6.Thiagarajah JR, Verkman AS. Trends Pharmacol Sci. 2005;26:172. doi: 10.1016/j.tips.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Findlay IA, Sheppard DN. Kidney Int. 2004;66:1926. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- 8.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP. J Am Soc Nephrol. 2006;17:3424. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 9.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. J Am Soc Nephrol. 2008;19:1300. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiagarajah JR, Song Y, Haggie PM, Verkman AS. FASEB J. 2004;18:875. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- 11.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. J Clin Invest. 2002;110:1651. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. FEBS Lett. 2004;558:52. doi: 10.1016/S0014-5793(04)00011-0. [DOI] [PubMed] [Google Scholar]
- 13.Caci E, Caputo A, Hinzpeter A, Arous N, Fanen P, Sonawane N, Verkman AS, Ravazzolo R, Zegarra-Moran O, Galietta LJ. Biochem J. 2008;413:135. doi: 10.1042/BJ20080029. [DOI] [PubMed] [Google Scholar]
- 14.Sonawane ND, Muanprasat C, Nagatani R, Jr, Song Y, Verkman AS. J Pharm Sci. 2005;94:134. doi: 10.1002/jps.20228. [DOI] [PubMed] [Google Scholar]
- 15.Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. Gastroenterology. 2004;126:511. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Factor P, Mutlu GM, Chen L, Mohameed J, Akhmedov AT, Meng FJ, Jilling T, Lewis ER, Johnson MD, Xu A, Kass D, Martino JM, Bellmeyer A, Albazi JS, Emala C, Lee HT, Dobbs LG, Matalon S. Proc Natl Acad Sci U S A. 2007;104:4083. doi: 10.1073/pnas.0601117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Luo M, Zhang L, Ding W, Yan Z, Engelhardt JF. Am J Respir Cell Mol Biol. 2007;36:313. doi: 10.1165/rcmb.2006-0286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizumori M, Choi Y, Guth PH, Engel E, Kaunitz JD, Akiba Y. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1318. doi: 10.1152/ajpgi.00025.2008. [DOI] [PubMed] [Google Scholar]
- 19.Perez A, Issler AC, Cotton CU, Kelley TJ, Verkman AS, Davis PB. Am J Physiol Lung Cell Mol Physiol. 2007;292:L383. doi: 10.1152/ajplung.00403.2005. [DOI] [PubMed] [Google Scholar]
- 20.Pietrement C, Da Silva N, Silberstein C, James M, Marsolais M, Van Hoek A, Brown D, Pastor-Soler N, Ameen N, Laprade R, Ramesh V, Breton S. J Biol Chem. 2008;283:2986. doi: 10.1074/jbc.M704678200. [DOI] [PubMed] [Google Scholar]
- 21.Rakonczay Z, Jr, Hegyi P, Hasegawa M, Inoue M, You J, Jr, Iida A, Ignath I, Alton EW, Griesenbach U, Ovari G, Vag J, Da Paula AC, Crawford RM, Varga G, Amaral MD, Mehta A, Lonovics J, Argent BE, Gray MA. J Cell Physiol. 2008;214:442. doi: 10.1002/jcp.21220. [DOI] [PubMed] [Google Scholar]
- 22.Sakuma T, Gu X, Wang Z, Maeda S, Sugita M, Sagawa M, Osanai K, Toga H, Ware LB, Folkesson G, Matthay MA. Crit Care Med. 2006;34:676. doi: 10.1097/01.CCM.0000201403.70636.0F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sommer D, Bogdan R, Berger J, Peters DM, Morty RE, Clauss WG, Fronius M. Respir Physiol Neurobiol. 2007;158:97. doi: 10.1016/j.resp.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Cutshall NS, O’day C, Prezhdo M. Bioorg Med Chem Lett. 2005;15:3374. doi: 10.1016/j.bmcl.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Deubner R, Holzgrabe U. Magn Reson Chem. 2002;40:762. [Google Scholar]
- 26.Fresneau P, Cussac M, Morand JM, Szymonski B, Tranqui D, Leclerc G. J Med Chem. 1998;41:4706. doi: 10.1021/jm9801399. [DOI] [PubMed] [Google Scholar]
- 27.Bulletin CaP, Momose Y, Meguro K, Ikeda H, Hatanaka C, Oi S, Sohda T. Chem Pharm Bull. 1991;39:1440. doi: 10.1248/cpb.39.1440. [DOI] [PubMed] [Google Scholar]
- 28.Ohishi Y, Mukai T, Nagahara M, Yajima M, Kajikawa N, Miyahara K, Takano T. Chem Pharm Bull. 1990;38:1911. doi: 10.1248/cpb.38.1911. [DOI] [PubMed] [Google Scholar]
- 29.Sing WT, Lee CL, Yeo SL, Lim SP, Sim MM. Bioorg Med Chem Lett. 2001;11:91. doi: 10.1016/s0960-894x(00)00610-7. [DOI] [PubMed] [Google Scholar]
- 30.Badawy MA, Kadry AM, Abdel-Hady SA, Ibrahim YA. Sulfur Lett. 1988;8:43. [Google Scholar]
- 31.Pastor SD, Hessell ET, Odorisio PA, Spivack JD. J Heterocycl Chem. 1985;22:1195. [Google Scholar]
- 32.Mustafa A, Asker W, Khattab S, Zayed SMAD. J Org Chem. 1961;26:787. [Google Scholar]
- 33.Augustin M, Mueller W. J Prakt Chem. 1985;327:857. [Google Scholar]
- 34.Giles RG, Lewis NJ, Quick JK, Sasse MJ, Urquhart MWJ, Youssef L. Tetrahedron. 2000;56:4531. [Google Scholar]
- 35.Ikemoto N, Liu JC, Brands KMJ, McNamara JM, Reider PJ. Tetrahedron. 2003;59:1317. [Google Scholar]
- 36.Qiao Q, So SS, Goodnow RA. Org Lett. 2001;3:3655. doi: 10.1021/ol010175t. [DOI] [PubMed] [Google Scholar]
- 37.Oruc EE, Rollas S, Kandemirli F, Shvets N, Dimoglo AS. J Med Chem. 2004;47:6760. doi: 10.1021/jm0495632. [DOI] [PubMed] [Google Scholar]
- 38.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 39.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 40.Kempf DJ, Sham HL, Marsh KC, Flentge CA, Betebenner D, Green BE, McDonald E, Vasavanonda S, Saldivar A, Wideburg NE, Kati WM, Ruiz L, Zhao C, Fino L, Patterson J, Molla A, Plattner JJ, Norbeck DW. J Med Chem. 1998;41:602. doi: 10.1021/jm970636+. [DOI] [PubMed] [Google Scholar]
- 41.Zhang A, Xiong W, Hilbert JE, DeVita EK, Bidlack JM, Neumeyer JL. J Med Chem. 2004;47:1886. doi: 10.1021/jm049978n. [DOI] [PubMed] [Google Scholar]
- 42.Sonawane ND, Hu J, Muanprasat C, Verkman AS. FASEB J. 2006;20:130. doi: 10.1096/fj.05-4818fje. [DOI] [PubMed] [Google Scholar]
- 43.Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Chem Biol. 2008 doi: 10.1016/j.chembiol.2008.05.015. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Gastroenterology. 2007;132:1234. doi: 10.1053/j.gastro.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Fish PV, Allan GA, Bailey S, Blagg J, Butt R, Collis MG, Greiling D, James K, Kendall J, McElroy A, McCleverty D, Reed C, Webster R, Whitlock GA. J Med Chem. 2007;50:3442. doi: 10.1021/jm061010z. [DOI] [PubMed] [Google Scholar]
- 46.Flick DA, Gifford GE. J Immunol Methods. 1984;68:167. doi: 10.1016/0022-1759(84)90147-9. [DOI] [PubMed] [Google Scholar]