

Abstract
Studies in vascular smooth muscle cells suggest that, angiotenisn II (Ang II)-mediated cellular response requires transactivation of epidermal growth factor receptor (EGF-R), and involves tyrosine phosphorylation of caveolin-1. Here we demonstrate that, exposure of WB rat liver cells to Ang II does not cause transactivation of EGF-R, but did rapidly activate p42/p44 mitogen-activated protein (MAP) kinases suggesting that it activates MAP kinases independent of EGF-R transactivation. We observed that the phospho-specific anti-caveolin-1 antibody detected a tyrosine phosphorylated, 75 kDa protein in Ang II-treated cells which we identified as glucose regulated protein-75 (GRP-75). Phosphoamino acid analysis showed that Ang II induced its phosphorylation at tyrosine, serine and threonine residues and was localized to the cytoplasm. The ability of Ang-II to induce GRP-75 phosphorylation suggests that it may play a role in the protection of cytoplasmic proteins from the damaging effect of oxidative stress known to be produced during Ang-II induced signaling.
Keywords: Angiotensin II, GRP-75, Phosphorylation, Signal transduction, Oxidative stress, Mitochondria, Chaperone, EGF-R transactivation
INTRODUCTION
Angiotensin II (Ang II) is a peptide hormone that plays a central role in the regulation of blood pressure and sodium homeostasis. It is also known to have a number of blood pressure independent actions including induction of gene expression and promotion of cell growth in cardiac fibroblasts, myocytes, and vascular smooth muscle cells (VSMCs) [1, 2]. Recent studies have demonstrated that components of renin-angiotensin system are also involved in growth promoting and remodeling effects in other cell types. For example, Ang II has been shown to have proliferative/fibrotic response in human lung fibroblasts [3], kidney epithelial cells [4], hepatic stellate cells [5] and hepatocytes [6]. Angiotensin II produces its action by binding to high affinity G-protein coupled receptors, of which there are two subtypes, Ang II type 1 and type 2 (AT1 and AT2) receptors [1, 7]. The AT1 receptors (AT1-R) regulate most physiological responses to Ang II and are principal receptors involved in pathophysiological signaling [1]. The AT1-R mediates a number of different signaling pathways through activation of the Gq/11 family of heterotrimeric G proteins. This results in the activation in phospholipase C-catalyzed hydrolysis of phosphatidyl-inositol 4,5-bisphosphate, generating inositol triphosphate and diacylglycerol [2]. These molecules mobilize calcium and activate protein kinase C. In addition, Ang II activates tyrosine kinases such a Src kinases, focal adhesion kinase and Janus kinase and serine-threonine kinases such as p42/44 kinases, and p38 kinases [1, 8, 9]. These signaling intermediates modulate the activity of cellular substrates including structural proteins, enzymes and transcription factors to cause distinct cellular responses.
Previous studies in VSMCs have shown that, the transactivation of epidermal growth factor receptor (EGF-R), which serves as a scaffold for various signaling molecules, plays an essential role in eliciting Ang II mediated tyrosine kinase signaling pathways [10–14]. The transactivation of EGF-R by Ang II is demonstrated to occur through a metalloproteinase activity and requires Ca2+, c-Src, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived reactive oxygen species (ROS), leading to activation of p42/44 MAP kinases and Akt/protein kinase B (PKB) [14–18]. Caveolin-1, the major structural protein (21 kDa) of caveolae [19] appears to play a central role in Ang II-induced transactivation of EGF-R in VSM cells [13, 14, 18] and hepatic clone 9 cells [20] and an important event in this process is the phosphorylation of caveolin-1 at Y14 [13, 14]. Based on co-precipitation and co-localization studies, it was postulated that caveolin-1 interacts with EGF-R to maintain the receptor in the inactivated state in unstimulated VSMCs; however, following Ang II stimulation, caveolin-1 tyrosine phosphorylation leads to the release and subsequent activation of EGF-R [14]. The presence of a consensus caveolin-1 binding site in AT1-R [14, 21], the demonstration of an interaction between AT1-R and EGF-R [20], and AT1-R and caveolin-1 [21] further suggests the importance of caveolin-1 in Ang II induced signaling. Interestingly, another study demonstrated that interaction between AT1-R and caveolin-1 is important for trafficking of the AT1-R to the cell surface, rather than acting as a scaffold for AT1-R at the cell surface during signaling [22].
The requirement for EGF-R transactivation in Ang II-mediated signaling appears to differ between cell types. In rat aortic VSMCs, EGF-R transactivation was shown to be crucial for Ang II mediated p42/p44 MAP kinases [10, 14, 16]; however, in human embryonic kidney (HEK) cells transfected with AT1-R [23] and preglomular smooth muscle cells expressing endogenous AT1-R [24], MAP kinase activation by Ang II has been shown to occur independent of EGF-R transactivation. Since involvement of caveolin-1 in AT1-R signaling has not been well studied in non-vascular smooth muscle cell types, we initiated studies to determine if exposure of WB cells (a continuously passaged rat liver cell line), which expresses endogenous AT1-R, to Ang II, causes tyrosine phosphorylation of caveolin-1 and transactivation of EGF-R. WB cells are epithelial cells that were originally isolated from the liver of adult rats [25, 26] and they have been used by numerous investigators to study Ang II-induced signal transduction pathways [25–34].
In this study, we demonstrate in WB rat liver cells that Ang II does not induce caveolin-1 phosphorylation or transactivation of EGF-R. Despite the lack of EGF-R transactivation, Ang II induced activation of p42/p44 MAP kinases in these cells. While studying the effect of Ang II on caveolin-1 phosphorylation, we observed that the phospho-specific anti-caveolin-1 antibody used in the study cross-reacted with a 75 kDa protein. Enhanced cross-reactivity of the phospho-specific anti-caveolin-1 antibody with the 75 kDa protein in multiple experiments suggested that Ang II may regulate its phosphorylation. Therefore, we performed studies to characterize and establish the identity of this protein. We demonstrate that Ang II rapidly induced phosphorylation of the 75 kDa protein at tyrosine, serine and threonine residue(s). Phosphorylated 75 kDa protein was localized to the cytoplasm in both unstimulated and Ang II stimulated cells. Using mass spectrometry, we identified it as GRP-75, a member of heat shock protein (HSP) family. Although known to be present in mitochondria, phosphorylation of GRP-75 in the cytoplasm did not cause its translocation to the mitochondria. Exposure of cells to pervanadate, a stress inducing agent, mimicked the actions of Ang II, suggesting that GRP-75 phosphorylation occurs in cells under the conditions of oxidative stress. Since GRP-75 is a stress response protein, its phosphorylation by Ang II suggests that, through interactions, it may protect cytoplasmic proteins from the damaging effects of H2O2 or superoxide, known to be produced during Ang II-induced signal transduction [2].
EXPERIMENTAL PROCEDURES
Materials
Cell culture reagents were purchased from Invitrogen. Richter’s improved minimal essential medium was obtained from Irvine Scientific Co; EXP3174 was provided by Merck Sharp & Dohme Research laboratories; Interleukin-6 (IL-6) and Epidermal Growth Factor (EGF) were obtained from R&D systems; Angiotensin II was obtained from Sigma. Nitrocellulose membrane was from Amersham; phospho-specific anti-caveolin-1 antibody was obtained from Cell Signaling Technology; phospho-specific anti-Stat3 antibody was from Biosource International; anti-Stat3 antibody antibody was from Santa Cruz Biotechnology; anti-phosphotyrosine antibody was from Upstate Biotechnology; anti-GRP-75 antibody was from Oxford Biomedical Research; goat anti-rabbit IgG and rabbit anti-mouse IgG were from Bio-rad; all other chemicals were either from Sigma or Fisher Scientific.
Cell Culture
WB rat liver cells were kindly provided by Dr. H. Shelton Earp (University of North Carolina, Chappell Hill, NC) and maintained at 37 °C in Richter’s improved minimal essential medium containing 10% fetal bovine serum and 0.1 μM insulin in a humidified 5% CO2 incubator as previously described [33]. Cells (passages 20–30) were grown for 12–24 hours and serum starved for 12 hours before the addition of Ang II or cytokines.
Immunoprecipitation and Western Blots
Serum starved cells were treated with Ang II or other agents for the indicated times and washed with phosphate buffered saline. Cells were scrapped in lysis bufer [10 mM Tris-Cl pH 7.4, 150 mM NaCl, 15% glycerol, 1% triton X-100, 1 mM sodium orthovanadate (tyrosine phosphatase inhibitor), 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM NaF (serine/threinine phosphatase inhibitor), and 1 mM phenylmethylsulfonyl fluoride (PMSF). Equal amounts of proteins were immunoprecipitated with indicated antibodies and protein A/G agarose. Immunocomplexes were collected by centrifugation, washed three times with imminoprecipiation buffer (lysis buffer), proteins resolved by 8% polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and were incubated with their respective primary antibodies. Immunoreactive bands were visualized using a chemiluminescence Western blotting system according to the manufacturers’ instructions (Amersham).
Cell Fractionation
All cell fractionation studies were carried out at 4 °C. Cells were harvested by scraping into ice-cold PBS, washed once with ice-cold PBS, resuspended in 0.8 ml of hypotonic buffer (10 mM Hepes, pH 7.9, 10 mM KCl, 3 mM MgCl2, 1 mM dithiothreitol, 1 mM PMSF, 1 mM sodium ortho vanadate, 1 mM NaF), per cell pellet derived from two 100-mm culture. Using a loose fitting Dounce homogenizer, cells were gently broken to fractionate into cytoplasmic and nuclear fractions as described earlier [35]. Nuclei were removed from cell homogenate by low speed centrifugation (1000 rpm for 4 min) in an eppendorf centrifuge. The crude post nuclear cytoplasm fraction was further subjected to 15000 x g in an eppendorf tube for 15 min. The pellet represented the membrane fraction (mitochondria, endoplasmic reticulum and plasma membrane). The supernatant represented the cytoplasmic fraction. Mitochondria were isolated using the mitochondria isolation kit as described by the manufacturer (Pierce).
Mass Spectrometry and Sequence Analysis
The 75 kDa protein was purified from Ang II-treated total cell lysates (150 mg) by immunoprecipitation with phospho-specific anti-caveolin-1 antibody. The immunoprecipitated protein was concentrated and separated on an 8% SDS-polyacrylamide gel, and stained with coomassie blue. The stained gel bands were rinsed in water, cut into ~1 mm pieces, dehydrated with 0.2 M NH4HCO3/50% acetonitile for 30 min and dried in a Speed-Vac. The gel pieces were then rehydrated in O.1 M NH4HCO3 containing 0.5–1 μg modified trypsin (Promega) and digested for 20 h at 37 °C. The supernatant was removed to a clean microfuge tube, the gel fragments extracted with aqueous 50% methanol/1% formic acid for 30 min, and combined with the initial extract. This was evaporated to ~20 μl and desalted on a C18ZipTip (Millipore) as recommended by the vendor. Peptides were eluted from the ZipTip with 2–5 μl aqueous solution of 50% methanol and 1% formic acid. One μl was mixed with matrix (alpha-cyano-4-hydroxycinnamic acid) and spotted on a MALDI target plate for analysis by Applied Biosystems Voyager DE-STR MALDI-TOF mass spectrometer. Peptide masses detected were sent to PROWL (Rockefeller University) or Protein prospector (University of California-San Francisco) for protein data base searches for possible matches. The search was done for all species (in the NCBI data base) for a molecular weight range of 5 kDa to 250 kDa (wide open search). Sequence comparison was performed using the BLAST search program and search was done with NCBInr data base.
In Vivo [32P] Labeling, Immunoprecipitation and Phosphoamino Acid Analysis
WB cells were serum starved for 12 hours in phosphate free medium, labeled with carrier free [32P] orthophosphoric acid (500 μCi/ml) for 3 h, and either left untreated or treated with Ang II for 10 min. Cell lysates were prepared and immunoprecipitation were done as previously described [36]. 200 μg of total cell extract were immunoprecipitated with phospho-specific anti-caveolin-1 antibody (cross-reactive to the 75 kDa protein), and immune complexes collected by the addition of protein A/G agarose. Samples after wash were dissolved in SDS-sample buffer and loaded on to the SDS – PAGE, transferred to polyvinylidene difluoride membrane, and subjected to autoradiography for 6 h to localize 32P-radiolabeled GRP-75. Phosphoamino acid analysis was performed as previously described [36]. The GRP-75 band was excised and the membrane suspended in 200 μl of 6 N HCl, and hydrolyzed at 110 °C, for 1h, dried, dissolved in 5 μl of thin layer electrophoresis buffer (formic acid:acetic acid:H2O-50:156:1794) (pH 1.9), containing phospho amino acid standards, spotted onto TLC plates and resolved electrophoretically in the pH 1.9 buffer, using the Hunter Thin Layer Electrophoresis system (HTLE) system. Following 1st dimension electrophoresis, the plates were dried and subjected to 2nd dimension electrophoresis in a solvent system of acetic acid:pyridine:H2O (100:10:1890) (pH 3.5) using the HTLE system. The plates were dried, sprayed with ninhydrin, and exposed to X-ray film.
RESULTS
Angiotensin II Activates p42/p44 MAP kinases Independent of EGF-R Transactivation
Previous reports have demonstrated in VSMCs that Ang II induces activation of p42/44 MAP kinases through transactivation of EGF-R and this occurs within 3 minutes following exposure to Ang II [10, 14, 16, 37]. One of the major Ang II-induced tyrosine phosphorylation sites on EGF-R was identified as Y1068 [37]. To determine if Ang II induces transactivation of EGF-R in WB cells, lysates prepared from cells untreated or treated with Ang II (3 min) or EGF (3 min) were run on a SDS-polyacrylamide gel and immunoblotted with phospho-specific (Y1068) anti-EGF-R antibody. Figure 1A demonstrates that the phospho-specific EGF-R antibody does not detect phosphorylated EGF-R in cells treated with Ang II (lane 2). However, it detected the phosphoryated EGF-R in cells treated with EGF (positive control). This suggests that in WB cells, Ang II does not cause transactivation of EGF-R. Reprobing the blot of Fig. 1A with anti-EGF-R antibody showed equal amount of EGF-R in all lanes (Fig. 1B). A detailed experiment involving exposure time from 1 min to 30 min with Ang II, also failed to show EGF-R transactivation (data not shown). To determine if Ang II activates p42/p44 MAP kinases, the samples of Fig. 1A were immunoblotted with phospho-specific anti-MAP kinase antibody. Figure 1C demonstrates that Ang II and EGF, both activate p42/44 MAP kinases. Reprobing the blot of Fig. 1C with anti-MAP kinase antibody showed similar amount of protein in all lanes (Fig. 1D) These results show that in WB cells, Ang II activates p42/p44 MAP kinases independent of transactivation of EGF-R.
FIG. 1. Angiotensin II activates p42/p44 MAP kinases independent of EGF-R transactivation; phosphospecific anti-caveolin-1 antibody cross-reacts with a 75 kDa protein.
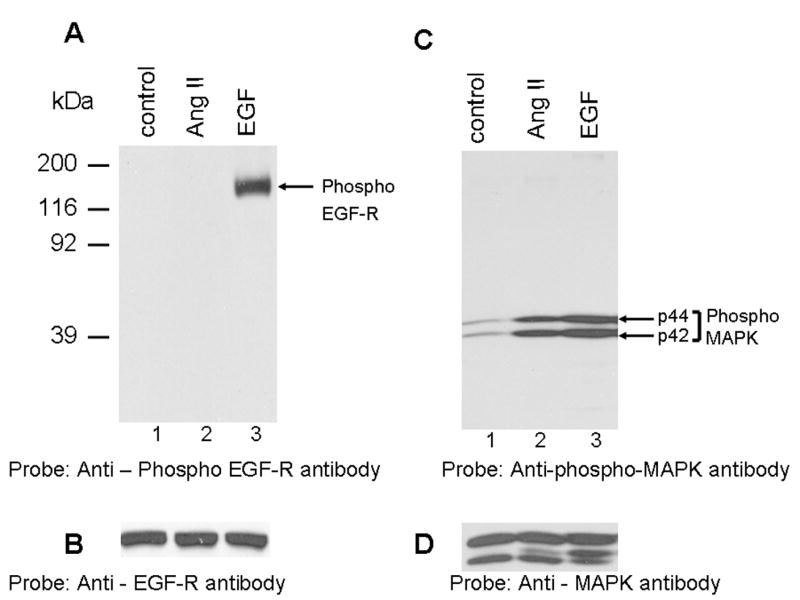
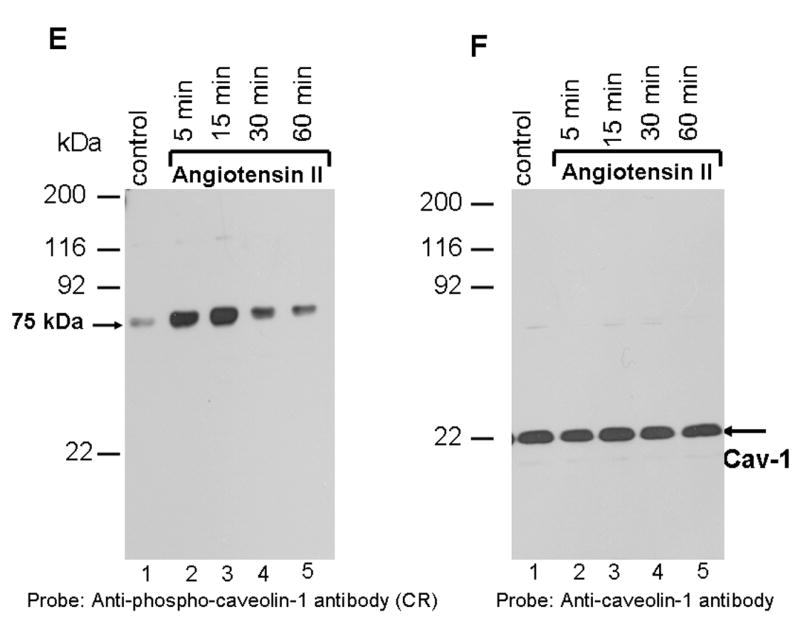
A, shows that Ang II does not transactivate EGF-R. Lysates were prepared from cells untreated (control), or treated with Ang II (100 nM, 3 min), or EGF (100 ng/ml; 3 min). Equal amount of proteins were run on a SDS-polyacrylamide gel and immunoblotted with phospho-specific EGF-R antibody specific for Y1068. B, the blot in A was stripped and reprobed with EGF-R antibody. C, shows that Ang II and EGF activate p42/p44 MAP kinases. The samples representing A were run on a SDS-polyacrylamide gel and immunoblotted with phospho-specific p42/p44 anti-MAP kinase antibody. D, The blot in C was stripped and reprbed with p42/p44 anti-MAP kinase antibody. E, shows that Ang II does not induce tyrosine phosphorylation of caveolin-1; anti-phospho caveolin-1 antibody cross reacts with a 75 kDa protein. Serum starved cells were left untreated (control) (lane 1), or treated with Ang II (100 nM) for different times (lanes 2–5), and total extracts were prepared, run on a 8% SDS-polyacrylamide gel, and immunoblotted with phospho-specific anti-caveolin-1 antibody. F, The blot in E was stripped and reprobed with anti-caveolin-1 antibody. These blots are representative of four independent experiments. The position of the 75 kDa protein and p42/p44 MAP kinases are shown in an arrows. CR, cross-reactive.
Angiotensin II Does Not Induce Tyrosine Phosphorylation of 21 kDa Caveolin-1 Protein; Phospho-specific Anti-Caveolin-1 Antibody Cross-Reacts with a 75 kDa Protein
The lack of EGF-R transactivation by Ang II suggested that in WB cells, Ang II may not induce tyrosine phosphorylation of caveolin-1. Previous reports have demonstrated that exposure of VSMCs to Ang II induces tyrosine phosphorylation (Y14) of caveolin-1 and this was implicated in the transactivation of EGF-R [14]. According to this report, Ang II-mediated tyrosine phosphorylation of caveolin-1 was observed within 1 min, peaked at 15 min, and gradually decreased to basal levels by 60 min. To determine the effect of Ang II on caveolin-1 tyrosine phosphorylation in WB cells, total cell lysates were prepared from cells treated with Ang II for varying periods of time. Equal amounts of proteins were immunoblotted with the phospho-specific anti-caveolin-1 antibody. This antibody is specific to caveolin-1 protein phosphorylated at tyrosine residue 14. Figure 1E demonstrates that phospho-specific anti-caveolin-1 antibody did not detect phosphorylated caveolin-1 (21 kDa) in WB rat liver cells suggesting that in these cells Ang II does not induce its tyrosine phosphorylation. This antibody, when used in immunoblots containing cell lysates treated with hydrogen peroxide (positive control), detected the 21 kDa caveolin-1 protein (data not shown), suggesting that it has the ability to detect phospho-caveolin-1 protein. Interestingly, we observed that the phospho-specific anti-caveolin-1 antibody prominently detected a 75 kDa protein following Ang II stimulation with activation observed as early as 5 min (lane 2), peaked at 15 min (lane 3) and decreased to almost basal levels at 60 min (lane 5). The interaction between phospho-specific anti-caveolin-1 antibody with the 75 kDa protein appears to be specific as pre-incubation of phospho-specific anti-caveolin-1 antibody with phospho-caveolin-1 peptide (immunogen) abrogated its ability to cross-react with the 75 kDa protein(s); however, when the antibody was incubated with unphosphorylated caveolin-1 peptide, it still retained the cross-reactivity with the 75 kDa protein (data not shown). To determine if caveolin-1 is expressed in WB cells, we stripped the blot of Fig. 1E and probed with caveolin-1 specific antibody. Figure 1F demonstrates that anti-caveolin-1 antibody detected the 21 kDa caveolin-1 protein in all lanes, suggesting that WB cells express caveolin-1 protein and that equal amounts of protein was present in individual samples (loading control). These results show that Ang II does not induce phosphorylation of caveolin-1 and that the phospho-specific anti-caveoilin-1 antibody specifically cross-reacts with a 75 kDa protein.
The 75 kDa Protein Detected by Phospho-Anti-Caveolin-1 Antibody is Phosphorylated at Tyrosine and Serine Residues in Ang II-Treated Cells
Specific recognition of the 75 kDa protein by the phospho-specific (tyrosine) anti-caveolin-1 antibody in Ang II treated cells suggested that it may be phosphorylated at tyrosine residues. To determine this, proteins from untreated and Ang II-treated cells were immunoprecipitated with anti-phosphotyrosine antibody, and samples were run on a gel and immunoblotted with phosphospecific anti-caveolin-1 antibody cross-reactive to the 75 kDa protein. Figure 2A demonstrates that the phospho-specific anti-caveolin antibody recognized the 75 kDa protein in the anti-phosphotyrosine antibody immunoprecipitates with enhanced cross-reactivity in Ang II-treated cells. Reprobing the blot of Fig. 2A with anti-caveolin-1 antibody did not detect a band corresponding to 21 kDa caveolin-1, confirming that Ang II does not induce caveolin-1 tyrosine phosphorylation (data not shown). In a reciprocal experiment, we immunoprecipitated the samples with phospho-specific anti-caveolin-1 antibody and the precipitated proteins were immunoblotted with anti-phosphotyrosine antibody. We again observed increased cross-reactivity of the 75 kDa protein by anti-phosphotyrosine antibody in anti-phospho-caveolin-1 antibody immunoprecipotates obtained from lysates of Ang II-treated cells (Fig. 2B). Reprobing the blot of Fig. 2B with anti-caveolin-1 antibody again did not detect caveolin-1 protein, which rules out the possibility that the 75 kDa protein is complexed with caveolin-1 protein (data not shown). These results establish that the 75 kDa protein is phosphorylated at tyrosine residue(s) in WB cells following Ang II stimulation and that the anti-phospho-caveolin-1 antibody directly recognizes the 75 kDa protein.
FIG. 2. The 75 kDa protein detected by phospho-specific anti-caveolin-1 antibody is tyrosine and serine phosphorylated in Ang II-treated cells.
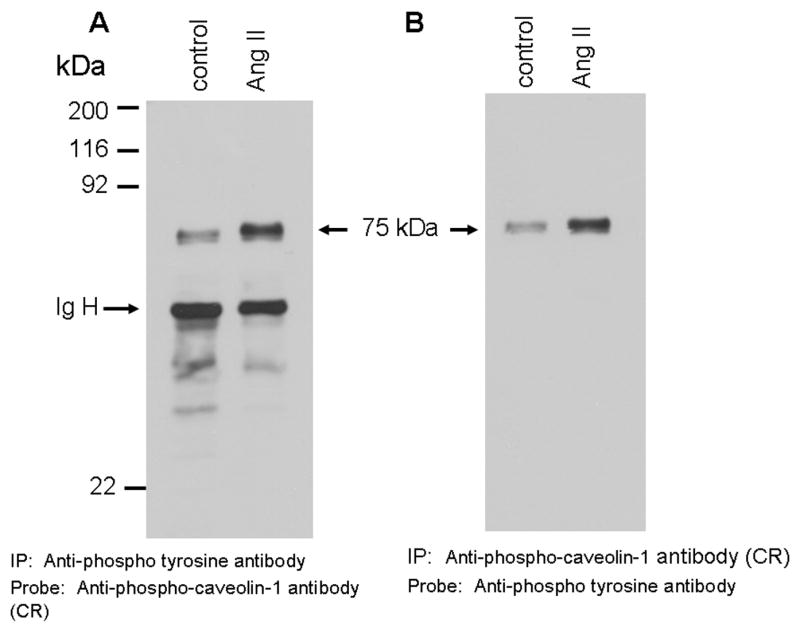
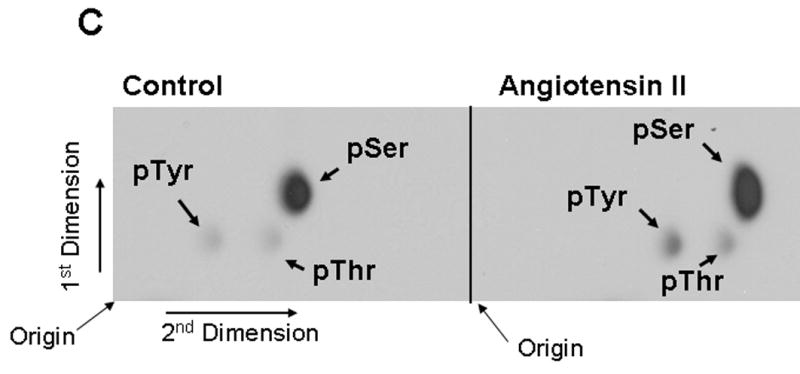
A and B show that Ang II-induces tyrosine phosphorylation of the 75 kDa protein. A, Serum starved cells were left untreated (control) or treated with Ang II (100 nM) for 15 min. Total cell lysates prepared and immunoprecipitated (IP) with anti-phospho-tyrosine antibody, immunocomplexes were run on a 8% SDS-polyacrylamide gel and immunoblotted with phosphospecific anti-caveolin-1 antibody. B, Lasates representing A were immunoprecipitated (IP) with phosphospecific anti-caveolin-1 antibody, immunocomplexes were analyzed by immunoblotting with anti-phospho-tyrosine antibody. C, Phosphoamino acid analysis of the 75 kDa protein following Angiotensin II treatment. Following in vivo labeling with [32P] as described in methods, cells were exposed to Ang II (100 nM) for 15 min. Five hundred μg of the total protein were immunoprecipitated with phospho-specific anti-caveolin-1 antibody (cross-reactive to the 75 kDa protein), run on a SDS-polyacrylamide gel, transferred to PVDF membrane, and exposed to X-ray film. The 75 kDa band from control and Ang II treated lanes were excised and subjected to phosphoamino acid analysis as described in the methods section. These blots are representative of four independent experiments. These blots are representative of four independent experiments. Ig H, Immunoglobulin heavy chain. CR, cross-reactive.
We then addressed if beside tyrosine, Ang II also induces phosphorylation of the 75 kDa protein at serine/threonine residues. For this, we performed in vivo [32P] labeling of proteins in presence of Ang II, followed by immunoprecipitation with anti-phospho-caveolin-1 antibody cross-reactive to the 75 kDa protein, and phosphoamino acid analysis. Figure 2C demonstrates that the 75 kDa protein is constitutively phosphorylated at only serine residues in untreated control; however, Ang II stimulated phosphorylation of not only tyrosine, but also serine residues. Phosphorylation at serine was more prominently observed compared to at tyrosine. Cerenkov counting of the radioactive spot showed a two fold increase in tyrosine phosphorylation, a four fold increase in serine phosphorylation, and 0.5-fold increase in threonine phosphorylation, following Ang II treatment. These data demonstrate that Ang II induces phosphorylation of the 75 kDa protein at tyrosine, serine and threonine residues.
Angiotensin II-Induced Phosphorylation of the 75 kDa Protein is Concentration Dependent and Mediated by AT1 Receptor
We performed experiments to determine if Ang II-mediated phosphorylation of the 75 kDa protein is concentration dependent. For this, cells were left untreated or treated with different concentration of Ang II, lysates prepared and immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to the 75 kDa protein. Figure 3A demonstrates that Ang II induces phosphorylation of the 75 kDa protein in a concentration dependent manner. Phosphorylation was observed at low concentration as low as 1 nM Ang II. As a loading control, we stripped and reprobed the blot in Fig. 3A with anti-Stat3 antibody. We used Stat3 as loading control in these and subsequent experiments because its levels remained unchanged upon Ang II addition (unpublished data), and additionally, we have also used it as loading control in our previous report [35]. Figure 3B demonstrates that all lanes contained equal amount of proteins. Pretreatment of cells with EXP3174, a selective non-peptide AT1-R antagonist completely blocked Ang II-induced phosphorylation of the 75 kDa protein (Fig. 3C). Reprobing the blot of Fig. 3C with anti-Stat3 antibody showed equal amount of protein in all lanes (Fig. 3D). These results demonstrate that the phosphorylation of the 75 kDa protein by Ang II is mediated by AT1-R.
FIG. 3. Angiotensin II-induced tyrosine phosphorylation of the 75 kDa protein is concentration dependent and blocked by the AT1 receptor antagonist, EXP3174.
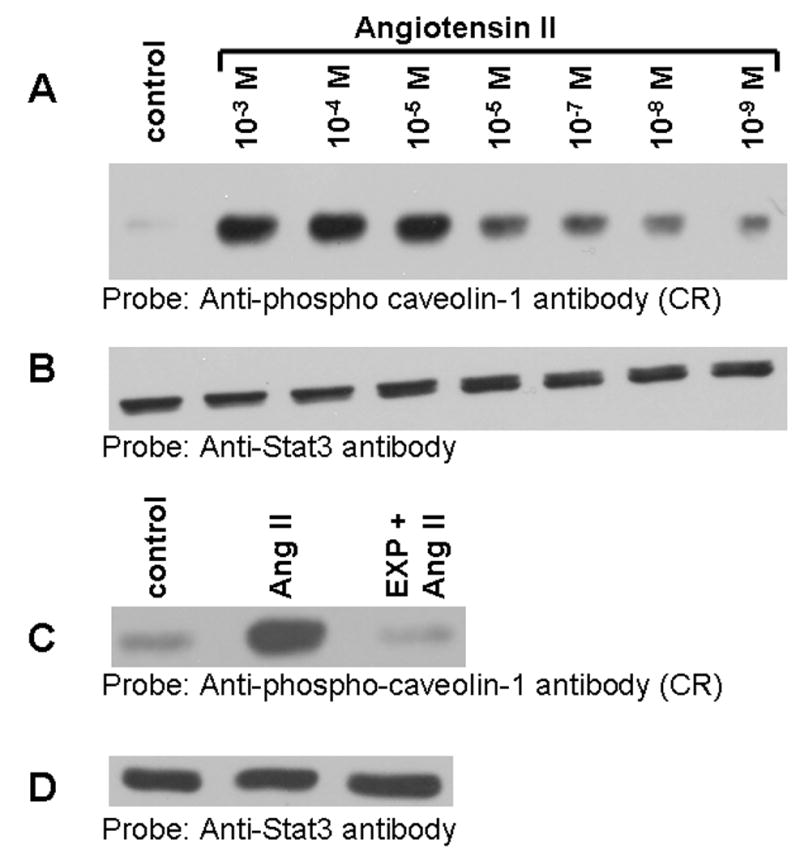
A, Serum starved cells were left untreated, or treated for 15 min with different concentrations with Ang II. Lysates were prepared and equal amount of proteins were run on a 8% SDS-polyacrylamide gel and immunoblotted with phosphospecific anti-caveolin-1 antibody cross-reactive to the 75 kDa protein. B, the blot in A was stripped and reprobed with anti-Stat3 antibody (loading control). C, shows that the EXP3174 pretreatment blocks Ang II-induced tyrosine phosphorylation of the 75 kDa protein. Cells were left untreated (control), or treated with Ang II for 15 min (10 nM), or first pre-treated with EXP3174 (1000 nM) for 15 min and then with Ang II (10 nM) for 15 min. Lysates were prepared, run on a 8% SDS-polyacrylamide gel, and immunoblotted with phosphospecific anti-caveolin-1 antibody. D, the blot in C was stripped and reprobed with anti-Stat3 antibody (loading control). These blots are representative of four independent experiments.
Identification of the 75 kDa Protein as GRP-75
The ability of the phospho-specific anti-caveolin-1 antibody to immunoprecipitate the 75 kDa protein suggested that this antibody could be used to purify the protein and establish its identity by mass spectrometry. For this, lysates from Ang II-treated cells were immunoprecipitated with phospho-specific anti-caveolin-1 antibody and the proteins electrophoresed on an 8% SDS-polyacrylamide gel and then stained with Coomassie blue. The 75 kDa protein band was excised from the gel, digested with trypsin and analyzed by mass spectrometry. The masses detected for 13 peptides derived from the 75 kDa protein matched (20 ppm accuracy window) perfectly with the rat GRP-75 sequence (Accession number P48721) (Fig. 4). The rat GRP-75 has a total of 679 amino acids [38]. Put together, the 13 peptides represented sequences anywhere between amino acids 86-653, and this covered ~30 % of the GRP-75 sequence (Fig. 4). In mass spectrometry based identification of proteins, peptide coverage of 30% or greater is considered excellent for a pure protein present in large abundance 1. The search was done for all species (in the NCBI data base) for a molecular mass range of 5-250 kDa, and the best match was found with rat GRP-75.
FIG. 4. Identification of the 75 kDa protein phosphorylated by Ang II as GRP-75.
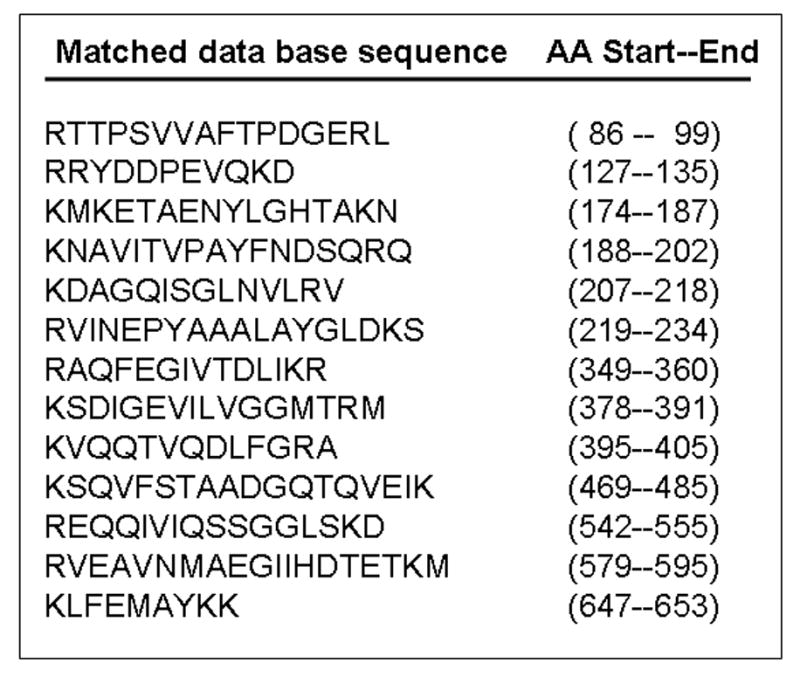
The 75 kDa protein was purified and subjected to mass spectrometry for protein identification. The sequences of the 13 peptides representing various regions of GRP-75 from the data base which matched with the peptides derived from the 75 kDa protein are indicated. The identity was established in three separate experiments. AA, amino acid.
GRP-75 is Tyrosine Phosphorylated in Cells Stimulated with Ang II
We considered the possibility that GRP-75 may undergo phosphorylation in cells treated with Ang II. To determine this, lysates from unstimulated and cells stimulated with Ang II (15 min) were immunoprecipitated with commercially available anti-GRP-75 antibody, protein complexes separated on a SDS-polyacrylamide gel, and immunoblotted with anti-phospho-tyrosine antibody. Figure 5A demonstrates that GRP-75 undergoes tyrosine phosphorylation in Ang II-treated cells. As a control for the immunoprecipitation, we stripped the blot of Fig. 5A and reprobed the blot with anti-GRP-75 antibody. Fig. 5B demonstrates that both lanes contain similar amount of GRP-75 protein. To verify whether the immunoprecipitated GRP-75 protein from Ang II treated cells can be detected with anti-phospho-caveolin-1 antibody; we stripped the blot of Fig. 5A and reprobed it with anti-phospho-caveolin-1 antibody. We observed that the anti-phospho-caveolin-1 antibody again detected the 75 kDa protein with enhanced cross-reactivity in Ang II treated cells (Fig. 5C). This confirms that the 75 kDa protein recognized by anti-phosphotyrosine antibody and anti-phospho-caveolin-1 antibody in anti-GRP-75 antibody immunoprecipitates is the same protein.
FIG. 5. GRP-75, a stress response protein undergoes phosphorylation in cells treated with Ang II and it is localized to the cytoplasm.
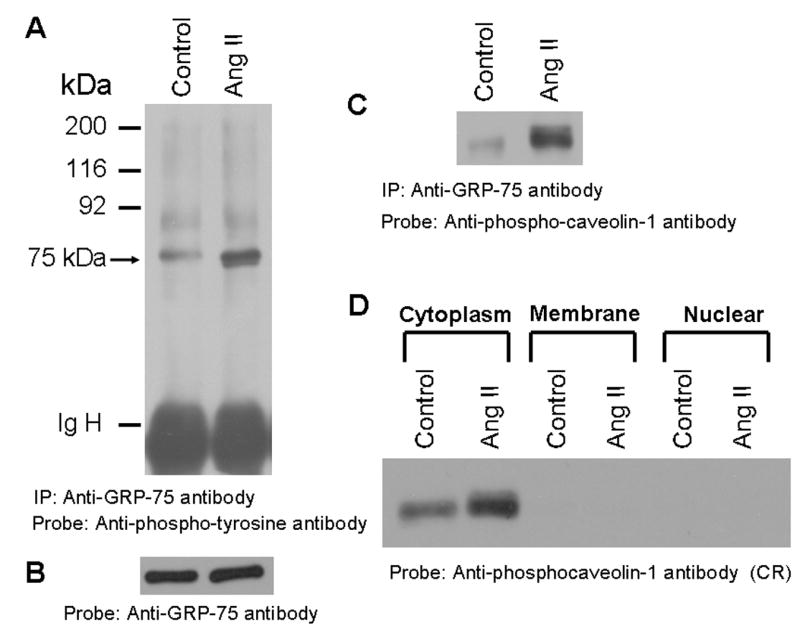
A, Phospho-tyrosine blot of immunoprecipitated GRP-75. Serum straved cells were left untreated, or treated with Ang II for 15 min (100 nM). Cell lysates were prepared and immunoprecipitated (IP) with anti-GRP-75 antibody, immunocomplexes were run on an 8% SDS-polyacrylamide gel and probed with anti-phosphotyrosine antibody. B, the blot in A was stripped and reprobed with anti-GRP-75 antibody. C, the blot representing A was stripped and reprobed with anti-phospho-caveolin-1 antibody. D, shows that the 75 kDa protein is localized to the cytoplasm. Serum starved cells were untreated or treated with Ang II for 15 min (100 nM) and lysed, and then membrane, cytoplasm and nuclear fractions were prepared. 15 μg of each sample were run on a 8% SDS-polyacrylamide gel and immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to the 75 kDa protein (phosphorylated GRP-75). These blots are representative of three independent experiments. IgH, Immunoglobulin heavy chain.
The Phosphorylated GRP-75 is Localized to the Cytoplasm in Both Unstimulated and Ang II-Stimulated Cells
GRP-75 is a highly conserved member of HSP family [38, 39]. It has been demonstrated to be predominantly localized to mitochondria; however, is also detected in other cellular compartments including endoplasmic reticulum, cytoplasmic vesicles and cytoplasm [40, 41]. We therefore determined the cellular localization of phosphorylated GRP-75 in WB cells using anti-phospho-caveolin-1 antibody cross reactive to phosphorylated GRP-75. We isolated total membrane (endoplasmic reticulum, plasma membrane and mitochondria), cytoplasm and nuclear fractions from un-stimulated and Ang II-stimulated cells, proteins immunoblotted with phospho-specific anti-caveolin-1 antibody. Figure 5D demonstrates that phosphorylated GRP-75 is localized to cytoplasm and completely absent in membrane and nuclear fractions. In control experiments, we confirmed the purity of cell fractionated samples by probing the immunoblots with anti-Stat3, anti-transforming growth factor-β receptor and anti-E2F antibodies specific respectively to cytoplasm, membrane and nuclear fractions (data not shown). These results demonstrate that phosphorylated 75 kDa protein is exclusively localized to the cytoplasm.
GRP-75 Undergoes Tyrosine Phosphorylation in Cells Treated with Pervanadate
Since GRP-75 is a stress response protein, we determined if exposure of cells to pervanadate, a known inducer of oxidative stress, stimulates GRP-75 phosphorylation in WB cells. Cells were treated with pervanadate for 15 min and lysate were immunoblotted with the phospho-specific anti-caveolin-1 antibody. Figure 6A (lane 5) demonstrates that pervanadate potently induced tyrosine phosphorylation of GRP-75; however, IL-6 (lane 3) and EGF (lane 4) did not elicit its tyrosine phosphorylation. Densitometry scanning of the blot confirmed that the levels of GRP-75 phosphorylation in EGF and IL-6 treated samples were similar to the untreated control levels (data not shown). Reprobing the blot of Fig. 6A with anti-Stat3 antibody showed equal amounts of protein in all lanes (Fig. 6B). Exposure of cells to heat shock (42 °C) for 30 min also failed to induce GRP-75 phosphorylation (data not shown). These results suggest that GRP-75 phosphorylation occurs under the conditions of oxidative stress.
FIG. 6. Pervanadate, an agent which induces oxidative stress, stimulates tyrosine phosphorylation of GRP-75; diphenylene iodonium (DPI), an inhibitor of NADH/NADPH oxidase suppresses Ang II-medaited action.
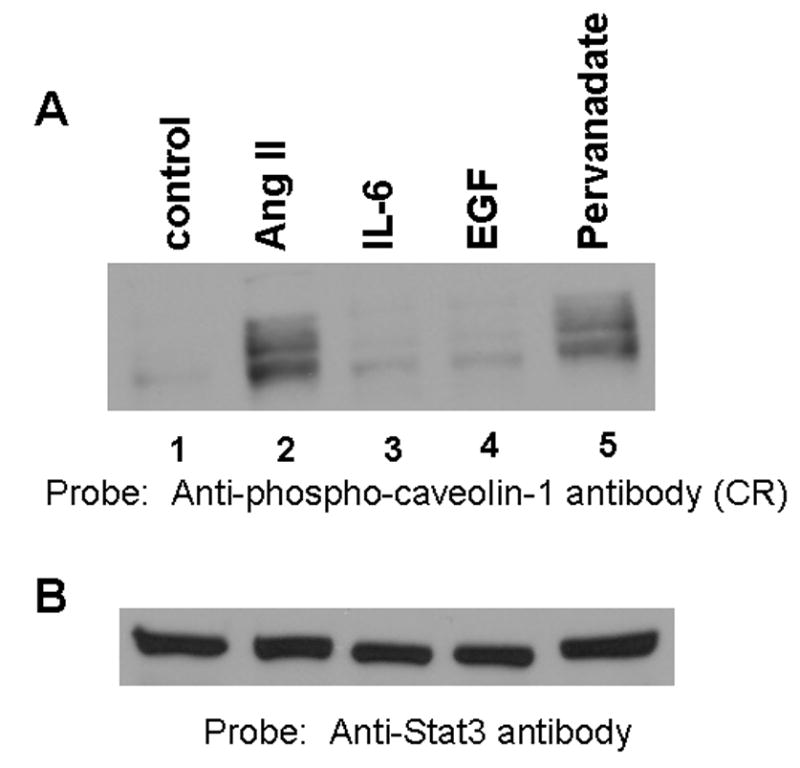
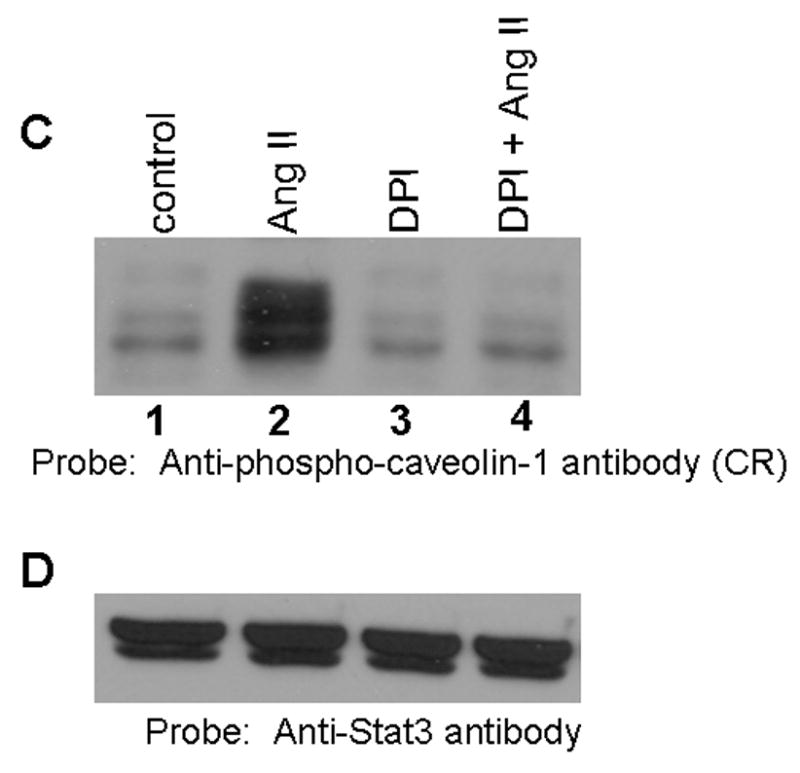
A, Serum starved cells were left untreated (control), or treated with Ang II (100 nM) for 15 min, or interleukin-6 (IL-6; 20 ng/ml) for 15 min, or EGF (100 ng/ml) for 5 min, or pervanadate (3 mM H2O2 together with 1 mM vanadate) for 15 min. Cell lysates were prepared and proteins run on an 8% SDS-polyacrylamide gel and immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to the 75 kDa protein (phopshorylated GRP-75). B, the blot in A was stripped and reprobed with anti-Stat3 antibody (loading control). C, Effect of DPI on Ang II-mediated GRP-75 tyrosine phosphorylation. Cells were left untreated, or treated with Ang II alone (100 nM) for 15 min, or DPI alone (10 μM) for 60 min, or first with DPI (10 μM) for 45 min followed by Ang II (100 nM) for 15 min. Cell lysates were prepared and equal amounts of protein were immunoblotted with anti-phospho-caveolin-1 antibody cross reactive to phosphorylated GRP-75. D, the blot in C was stripped and reprobed with anti-stat3 antibody. These blots are representative of four independent experiments.
Previous reports in VSM cells showed that Ang II induces the production of reactive oxygen species (ROS) in VSM cells, and this was mediated via NADH/NADPH oxidase (14). We hypothesized that Ang II mediated phosphorylation of GRP-75 may also occur via NADH/NADPH oxidase and generation of ROS. To address this, we tested the ability of Ang II to induce tyrosine phosphorylation of GRP-75 in presence diphenylene iodonium (DPI) an inhibitor of NADH/NADPH oxidase. Cells were left untreated, treated with DPI, or first pretreated with DPI followed by Ang II and immunoblotted with anti-phospho-caveolin-1 antibody cross reactive to phosphorylated GRP-75. Figure 6 C demonstrates that the pretreatment of cells with DPI completely inhibited Ang II-mediated increase in GRP-75 tyrosine phosphorylation (lanes 4). Reprobing the blot with anti-stat3 antibody showed similar amount of protein in all lanes (Fig. 6D). These results demonstrates that Ang II-mediated tyrosine phosphorylation of GRP-75 occurs via activation of NADH/NADPH oxidase and implicates ROS in this process. Since Ang II activates p42/p44 MAP kinases in these cells, we determined if PD98059, an inhibitor of MAP kinase kinase 1 would block Ang II-mediated increase in GRP-75 tyrosine phosphorylation. We observed that the GRP-75 tyrosine phosphorylation was insensitive to treatment with PD98059, suggesting that p42/p44 MAP kinases are not involved in Ang II-mediated effects.
Sequence Comparison Between Phospho-Caveolin-1 Peptide and GRP-75 Shows a Homology Region for Recognition by Phospho-Caveolin-1 Antibody
GRP-75 and caveolin-1 are unrelated proteins and therefore, the cross-reactivity of phospho-caveolin-1 antibody with GRP-75 is an intriguing observation. To explain the cross-reactivity and to identify a potential homology region, we compared the caveolin-1 phospho-peptide sequence (EGHLY*TVPIR; aa 10–19; where Y* represents phosphoyrosine), which was used as the immunogen, with GRP-75 amino acid sequence. Amino acid sequence comparison showed the presence of a common G-LY motif between phospho-caveolin-1 peptide and GRP-75 sequence (aa 158–161) (Fig. 7). It is likely that the tyrosine (Y161) in the G-LY of GRP-75 undergoes phosphorylation following treatment with Ang II, and this site then would be recognized by phospho-specific anti-caveolin-1 antibody.
FIG. 7. Amino acid sequence comparison of caveolin-1 phospho-peptide (immunogen) with GRP-75 shows a homogy region, G-LY.
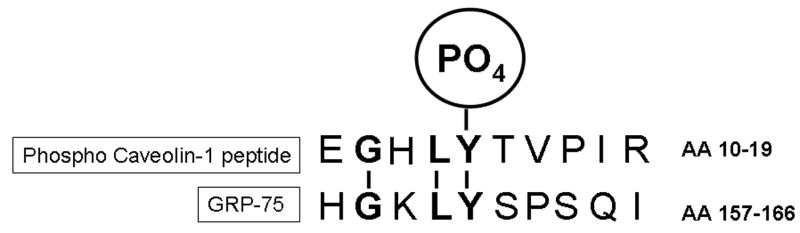
The caveolin-1 phospho-peptide (aa 10–19) was used as immunogen. The Y14 of caveolin-1 is the site of tyrosine phosphorylation by several agonists (13, 14, 19). It is likely that the Y in the GHLY motif in GRP-75 also undergoes phosphorylation in cells treated with Ang II, and this site therefore, would be recognized by phospho-specific anti-caveolin-1 antibody.
Angiotensin II Induces Tyrosine Phosphorylation of GRP-75 at Two Different Sites
In a previous report, we demonstrated that exposure of CCL39 lung fibroblasts to α-thrombin induces phosphorylation of GRP-75, and the potential site of tyrosine phosphorylation was identified as Y331 [35]. This was based on an observation that phospho-specific anti-Stat3 antibody raised against a phospho-peptide of Stat3 (SAAPY*LKTKF, aa 701–710; where Y represents phosphotyrosine) specifically cross-reacted with GRP-75 from α-thrombin-treated cells [35]. Comparison of the Stat3 phospho-peptide sequence with GRP-75 identified a tri-peptide sequence PYL common between these two sequences, providing an explanation for the cross-reactivity of phospho-specific anti-Stat3 antibody with GRP-75 [35]. Since the homology region identified in this study between phospho-caveolin-1 peptide and GRP-75 (sequence G-LY; aa 158–161) is different from the homology region identified in our previous report [35] between phospho-Stat3 peptide and GRP-75 (sequence PYL; 330–332), we hypothesized that Ang II, beside phosphorylation at Y161, may also induce phosphorylation at Y331. To determine this, we immunoprecipitated the lysates from untreated and Ang II-treated cells with phospho-specific anti-Stat3 antibody cross-reactive to phosphorylated GRP-75 [35], immunocomplexes collected, and immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to phosphorylated GRP-75. Figure 8A demonstrates that the phospho-specific anti-Stat3 antibody immunoprecipitates contained a 75 kDa protein cross-reactive to phospho-specific anti-caveolin-1 antibody. In a reciprocal experiment, we immunoprecipitated the control and Ang II-treated cell lysates with phospho-specific anti-caveolin-1 antibody and immunocomplexes immunoblotted with phospho-specific anti-Stat3 antibody cross-reactive to phosphorylated GRP-75. Figure 8B demonstrates that phospho-specific anti-caveolin-1 immunoprecipitates contained the 75 kDa protein crossreactive to phospho-specific anti-Stat3 antibody. These results collectively show that exposure of WB cells to Ang II induces tyrosine phosphorylation of GRP-75 at both Y161 and Y331 sites.
FIG. 8. Angiotensin II induces phosphorylation of GRP-75 at two different sites (Y331 and Y161).
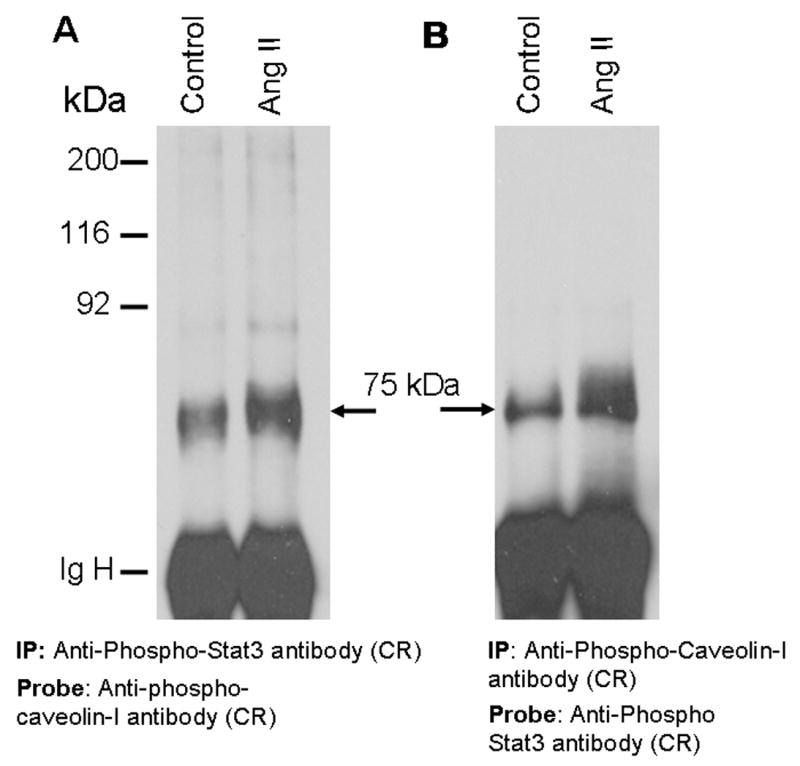
In these experiments two different antibodies [phospho-specific anti-Stat3 antibody (35), and phospho-specific anti-caveolin-1 antibody], both cross-reactive to phosphorylated GRP-75 were used. A, Anti-phospho-caveolin-1 antibody immunoblot of samples immunoprecipitated with phospho-specific anti-Stat3 antibody. Serum starved cells were left untreated, or treated with Ang II for 15 min (100 nM). Cell lysates were prepared and immunoprecipitated (IP) with phospho-specific anti-Stat3 antibody, immunocomplexes were run on an 8% SDS-polyacrylamide gel and probed with phospho-specific anti-caveolin-1 antibody. B, The samples representing A were immunoprecipitated with phospho-specific anti-caveolin-1 antibody, immunocomplexes were run on an 8% SDS-polyacrylamide gel and probed with phospho-specific anti-Stat3 antibody. These blots are representative of three independent experiments. IgH, Immunoglobulin heavy chain. CR= cross reactive.
GRP-75 Phosphorylation and Mitochondrial Translocation are Independent Events
Since GRP-75 is has been localized to both cytoplasm and mitochondria in many cell types [40–42], in a time course study, we examined if phosphorylation of GRP-75 has any relationship to its translocation to mitochondria. To determine this, we prepared cytoplasmic fraction and mitochondrial fraction from untreated and Ang II-treated (15 min and 30) cells. Equal amounts of proteins were run on a SDS-gel and immunoblotted with GRP-75 antibody. Figure 9A demonstrates that following Ang II stimulation, GRP-75 levels remained unchanged in cytoplasm and mitochondrial compartments. To determine if mitochondria contain phosphorylated GRP-75, untreated and Ang II-treated samples representing cytoplasmic and mitochondrial fractions were immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to phosphorylated GRP-75. Figure 9B demonstrates that phosphorylated GRP-75 is observed in the cytoplasmic fraction but not in mitochondria. These results demonstrate that translocation and phosphorylation of GRP-75 are not linked events. Furthermore, the result of Fig. 9B also confirms the observation in Fig. 5C that phosphorylated GRP-75 is exclusively localized to cytoplasm. As previously reported by other investigators [40], mitochondrial fraction contained higher levels of GRP-75 (Fig. 9A).
FIG. 9. GRP-75 phosphorylation in the cytoplasm does not cause its translocation to mitochondria.
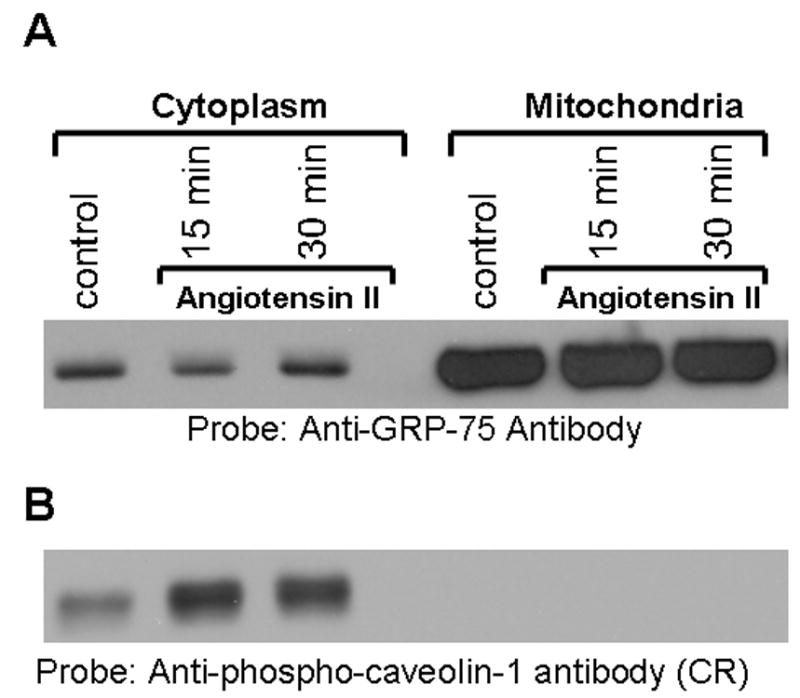
A, Serum starved cells were left untreated (control), or treated with Ang II (100 nM) for either 15 min or 30 min. Mitochondria and cytoplasmic fractions were isolated as described in the methods section. Mitochondrial proteins were solubilized and equal amounts of protein from cytoplasm and mitochondrial fraction were run on a 8% SDS-polyacrylamide gel, and immunoblotted with anti-GRP-75 antibody. B, the samples representing A was immunoblotted with phospho-specific anti-caveolin-1 antibody cross-reactive to phosphorylated GRP-75. These blots are representative of three independent experiments. CR= cross reactive.
DISCUSSION
Accumulating evidence suggests that mitogenic responses to AT1-R activation occur through transactivation of EGF-R [10–14], and tyrosine phosphorylation of caveolin-1 is an important step in this process. This pathway has been well characterized in the cardiovascular cell types; however, little information is available about Ang II effects on EGF-R transactivation and caveolin-1 phosphorylation in non vascular cells. In this study, we demonstrate in WB cells expressing endogenous AT1-R that, Ang II induces p42/44 MAP kinases independent of EGF-R transactivation. Consistent with the lack of EGF-R transactivation, Ang II also failed to induce caveolin-1 tyrosine phosphorylation. We identified the 75 kDa protein cross-reactive to the phospho-specific anti-caveolin-1 antibody as GRP-75, a member of the heat shock/stress response protein family. Angiotensin II induced phosphorylation of GRP-75 in a concentration dependent fashion, and this was mediated via AT1-R. Phosphorylation of GRP-75 was observed on serine, tyrosine and threonine residues; however, phosphorylation at serine was more prominent than at tyrosine, and phosphorylation at threonine was minimal. Phosphorylated GRP-75 was localized to the cytoplasm in both unstimulated and Ang II stimulated cells. Exposure of cells to pervanadate, which is a known inducer of oxidative stress, stimulated phosphorylation of the GRP-75; however, compared to control, its phosphorylation levels were similar with IL-6 or EGF treatment. This suggests that oxidative stress/generation of reactive oxygen species (ROS) is likely to be required for GRP-75 phosphorylation. In support of this, we observed that treatment of cells with DPI, an inhibitor of NADH/NADPH oxidase (a known inducer of super oxide production), abrogated Ang II-induced GRP-75 tyrosine phosphorylation. This is the first report of the ability of Ang II to induce phosphorylation of GRP-75 at tyrosine, serine and threonine residues and suggest that through its chaperone properties, it may protect cytoplasmic proteins from unfolding and degradation caused by oxidative stress, known to be produced during Ang II-induced signaling [2].
Previous reports have shown that cell types exhibiting Ang II signaling through EGF-R activation include VSMCs, cardiac myocytes and fibroblasts, COS 7 cells, C9 hepatic cells, glomerular mesengial cells, prostrate stromal cells, anterior pituitary cells, and breast cancer cells [23]. However, the EGF-R transactivation does not appear to be a universal process and has a minor or no role in HEK 293 cells [23] and pre-glomerular smooth muscle cells [24]. Our results in WB rat liver cells indicate that EGF-R transactivation is not an intermediate event in Ang II mediated signal transduction pathways. Out of the 5 autophosphorylation sites on the EGF-R [43, 44], Ang II has been shown to phosphorylate Y 1068 and Y 1173 of EGF-R in a redox sensitive manner [37]. In our experiments, initially we used phospho specific anti-EGF-R antibody directed against Y1068; yet, we did not observe any EGF-R phosphorylation. However, this antibody detected the EGF-R phosphorylation when cells were stimulated with EGF. Phospho-specific EGF-R antibody specific to Y1173 also failed to detect EGF-R from Ang II treated cells (data not shown). Although the reason for the lack of transactivation of EGF-R by Ang II in WB cells remains to be elucidated, it may be related to the lack of induction of matrix metalloproteinases required for the activation of HB-EGF-R, as has been previously shown in HEK 293 cells [23]. The inability of Ang II to cause trasactivation of EGF-R in WB cells further establishes that individual actions of Ang II on EGF-R transactivation occur in a cell type specific manner.
We used mass spectrometry to identify the 75 kDa protein which cross-reacted to phospho-specific anticaveolin-1 antibody, as GRP-75. It is a member of heat shock/stress response protein family [40–42] which function as molecular chaperones that have translocase and foldase activities [45]. GRP-75 is known to be induced under conditions of low glucose, nutritional and other environmental stress [40]. Increased levels of GRP-75 were also observed in ischemic brain which suggests that it may protect the tissue from metabolic stress such as oxygen/glucose deprivation [38]. It was reported to be present at multiple sub cellular sites including endoplasmic reticulum, cytoplasmic vesicles, and cytosol while residing predominantly in mitochondria [40]. GRP-75 performs multiple functions including mitochondrial import, intracellular trafficking, receptor internalization, and is also implicated in antigen recognition and cell proliferation. Although, GRP-75 contains a mitochondrial translocation sequence, following its phosphorylation in the cytoplasm, we did not observe an increase in translocation to the mitochondria. Thus, phosphorylation of GRP-75 and its translocation to mitochondria appear to be independent events. The exclusive localization of phosphorylated GRP-75 to the cytoplasm suggests that it may regulate cytoplasmic protein functions through protein:protein interactions. In this context, it is important to note that GRP-75 was shown to bind to multiple proteins and alter their functional activity [40]. It was shown to interact with mevolanate pyrophosphate decarboxylase (MPD), a protein known to furnish prenylation of many proteins including p21Ras and over expression of GRP-75 resulted in reduced level of Ras activity and p42 MAP Kinase [46]. In transformed human tumor cells, GRP-75 was shown to bind to tumor suppressor protein p53 and this resulted in cytoplasmic sequestration of p53 and inhibition of its transcriptional inactivation function [40, 47]. Additional GRP-75 interacting proteins include GRP-94, interleukin-1 receptor and fibroblast growth factor-1 (FGF-1) [40, 48, 49]. It was shown that tyrosine phosphorylation of GRP-75 is required for its interactions with FGF-1 [49]; however, currently no information is available if tyrosine/serine phosphorylation affects its interactions with MPD, p53 or GRP-94. Since phosphorylation is a key regulatory event in signal transduction pathways, our finding that Ang II rapidly induces GRP-75 phosphorylation suggest that it may alter the functional activity of proteins during Ang II signaling.
In a previous report [35] we showed in α-thrombin-treated cells that a phospo-specific anti-Stat3 antibody raised against a phospho-peptide of Stat3 specifically cross-reacted with phosphorylated GRP-75. In this study, we demonstrated in Ang II-treated WB rat liver cells that phospho-specific anti-caveolin-1 antibody cross-reacted with GRP-75. Although these are structurally and functionally unrelated proteins, sequence comparison of GRP-75 revealed two separate short regions of homology, one to caveolin-1 phosphopeptide (G-LY, aa 158-161 in GRP-75) (Fig. 7), and another to Stat3 phospho-peptide (PYL aa 330–332 in GRP-75) [35]. Both homology regions contain a single tyrosine residue (Y161 and Y331) and it is likely that, anti-phospho-Stat3 and anti-caveolin-1 antibodies recognize these sites following tyrosine phosphorylation by Ang II. Antibodies to epitopes outside the phosphorylation domains are removed by extensive pre-adsorption with un-phosphorylated peptides during preparation of phospho-specific antibodies, and therefore, they are bound to recognize sites with short sequences. In separate reciprocal immunoprecipitation experiments, we showed that Ang II induces GRP-75 phosphorylation at both Y161 and Y331 residues. Further, phosphoamino acid analysis showed the ability of Ang II to induce phosphorylation at both tyrosine and serine residues. Although the location of serine phosphorylation site(s) on GRP-75 is not clear, it appears to have a sequence (LYSP, aa 160–163) similar to the p42/44 MAP kinase consensus site. The higher levels of serine phosphorylation may also be due to multiple sites of serine phosphorylation on GRP-75 following Ang II stimulation. Identification of all Ang II-induced serine/tyrosine phosphorylation sites on GRP-75 requires additional study.
The observation that pervanadate, which is a known inducer of oxidative stress in cells causes phosphorylation of GRP-75 suggests that similar mechanism may be involved in Ang II-induced GRP-75 phosphorylation. Our observation that pretreatment of cells with the DPI, an inhibitor of NADH/NADPH oxidase, completely abolished the ability of Ang II to induce tyrosine phosphorylation of GRP-75 further suggests that ROS may be involved in this process. Although Ang II induced p42/p44 MAP kinases, it appears that MAP kinase pathway is not involved in Ang II-mediated tyrosine phosphorylation of GRP-75. A number of studies have documented the generation of ROS during Ang II induced signaling (reviewed in ref. 2). Evidence suggests that cardiovascular diseases are associated with increased oxidative stress in blood vessels [50, 51]. ROS such as superoxide and H2O2 causes blood vessels to thicken, produce inflammation in the vessel wall, and therefore, are “risk factors” for vascular disease, where as ROS also acts as signaling molecule in many aspects of growth factor-mediated physiological responses [50]. ROS act as second messengers that are capable of activating diverse intracellular signaling pathways [51] which mediate growth, migration, cellular apoptosis and survival [52]. In VSMCs, Ang II stimulates ROS generation through NADH/NADPH oxidase located in the cell membrane [2, 50, 53]. ROS activate p38MAP kinase and Akt leading to protein synthesis, hyperplasia and hypertrophy in VSMCs [53]. ROS are also implicated in the activation of p42/44 MAP kinase and PYK2 [54] and stimulates the expression of endothelial vascular cell adhesion molecule-I, important in cell-cell interactions and in process associated with atherosclerosis [55]. Evidences also indicate that Ang II contributes to liver fibrosis though activation of NADPH oxidase and production of ROS in hepatic stellate cells [56, 57]. One possible scenario is that phosphorylated GRP-75 may protect cellular proteins from the damaging effect of ROS through protein:protein interactions. Multiple sites of phosphorylation may confer GRP-75 the ability to bind with diverse group of proteins through site specific interactions. Interestingly, the ability of Ang II to induce GRP-75 phosphorylation is also shared by α-thrombin which binds to the G-protein coupled receptor, PAR1 [35], and similar to Ang II, also induces oxidative stress [58]. The identification of proteins, whose functions are modulated by GRP-75 through its phosphorylation and interaction, represents an important area of research and may provide therapeutic strategies to control oxidative stress-induced cardiovascular diseases.
Acknowledgments
This work was supported by National Institutes of Health RO1 grant # HL66000 to G.J.B. We thank Dr. Srivenugopal for useful discussions and providing reagents for phosphoamino acid analysis, and Dr. Richard Cook, Baylor College of Medicine, Houston, for Mass Spectrometric analysis.
Footnotes
Richard G. Cook, Personal communication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim S, Iwao H. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 2.Touz RM, Schiffrin EL. Pharmcol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 3.Marshal RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, McAnulty RJ, Laurent GJ. Am J Phyiol Lung Cell Mol Physiol. 2004;286:156–164. doi: 10.1152/ajplung.00313.2002. [DOI] [PubMed] [Google Scholar]
- 4.Guo G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S. Am J Physiol Renal Physiol. 2001;280:F777–F785. doi: 10.1152/ajprenal.2001.280.5.F777. [DOI] [PubMed] [Google Scholar]
- 5.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, Brenner DA. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng Y-I, Shukla SD. Biochem Biophys Acta. 2002;1589:285–297. doi: 10.1016/s0167-4889(02)00189-1. [DOI] [PubMed] [Google Scholar]
- 7.Dinh DT, Frauman AG, Johnston CI, Fabiani ME. Clin Sci. 2001;100:481–492. [PubMed] [Google Scholar]
- 8.Booz G, Day JNE, Baker KM. J Mol Cell Cardiol. 2002;34:1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 9.Touyz RM, Berry C. Braz J Med and Biol Res. 2002;35:1001–1015. doi: 10.1590/s0100-879x2002000900001. [DOI] [PubMed] [Google Scholar]
- 10.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T. J Biol Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 11.Murasawa S, Mori Y, Nozawa Y, Gotoh N, Shibuya M, Masaki H, Maruyama K, Tsutsumi Y, Moriguchi Y, Shibazaki Y, Tanaka Y, Iwasaka T, Inada M, Matsubara M. Circ Res. 1998;82:1338–1348. doi: 10.1161/01.res.82.12.1338. [DOI] [PubMed] [Google Scholar]
- 12.Schafer B, Marg B, Gschwind A, Ullrich A. J Biol Chem. 2004;279:47929–47938. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- 13.Zuo L, Ushio-Fukai M, Hilenski LL, Alexander RW. Arterioscler Thromb Vasc Biol. 2004;24:1223–1228. doi: 10.1161/01.ATV.0000132400.25045.2a. [DOI] [PubMed] [Google Scholar]
- 14.Ushio-Fukai M, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling KK, Alexander RW. J Biol Chem. 2001;276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- 15.Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, Marumo F, Hirata Y, Inagami T. J Biol Chem. 1999;274:36483–36851. doi: 10.1074/jbc.274.52.36843. [DOI] [PubMed] [Google Scholar]
- 16.Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. J Biol Chem. 2001;276:7957–7962. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- 17.Ushio-Fukai M, Zuo L, Ikeda S, Tojo T, Patrushev NA, Alexander RW. Circ Res. 2005;97:829–836. doi: 10.1161/01.RES.0000185322.46009.F5. [DOI] [PubMed] [Google Scholar]
- 18.Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Arterioscler Thromb Vasc Biol. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]
- 19.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 20.Olivares-Reyes JA, Shah BH, Hernandez-Aranda J, Gracia-Caballero A, Farshori MP, Garcia-Sainz JA, Catt KJ. Mol Pharmacol. 2005;68:356–364. doi: 10.1124/mol.104.010637. [DOI] [PubMed] [Google Scholar]
- 21.Ishizaka N, Griendling KK, Lassegue B, Alexander RW. Hypertension. 1998;32:459–466. doi: 10.1161/01.hyp.32.3.459. [DOI] [PubMed] [Google Scholar]
- 22.Wyse BD, Prior IA, Qian H, Morrow IC, Nixon S, Muncke C, Kurzchalia TV, Thomas WG, Parton RG. J Biol Chem. 2003;278:23738–23746. doi: 10.1074/jbc.M212892200. [DOI] [PubMed] [Google Scholar]
- 23.Shah BH, Yesilkaya A, Olivares-Reyes JA, Chen HD, Hunyady L, Catt KJ. Mol Endocrinol. 2004;18:2035–2048. doi: 10.1210/me.2003-0476. [DOI] [PubMed] [Google Scholar]
- 24.Andresen BT, Linnoila JJ, Jackson EK, Romero GG. Hypertension. 2003;41(part2):781–786. doi: 10.1161/01.HYP.0000049426.61176.DF. [DOI] [PubMed] [Google Scholar]
- 25.Earp HS, Huckle WR, Dawson TL, Li X, Graves LM, Dy R. J Biol Chem. 1995;270:28440–28447. doi: 10.1074/jbc.270.47.28440. [DOI] [PubMed] [Google Scholar]
- 26.Zohn IE, Yu H, Li X, Cox AD, Earp HS. Mol Cell Biol. 1995;15:6160–6168. doi: 10.1128/mcb.15.11.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maloney JA, Tsygankova O, Szot A, Yang L, Li Q, Williamson JR. Am J Physiol Cell Physiol. 1998;274:974–982. doi: 10.1152/ajpcell.1998.274.4.C974. [DOI] [PubMed] [Google Scholar]
- 28.Maloney J, Tsyganova OM, Yang L, Li Q, Szot A, Baysal K, Williamson JR. Am J Physiol. 1999;276:C221–C230. doi: 10.1152/ajpcell.1999.276.1.C221. (Cell Physiol. 45) [DOI] [PubMed] [Google Scholar]
- 29.Graves LM, He Y, Lambert J, Hunter D, Li X, Earp HS. J Biol Chem. 1997;272:1920–1928. doi: 10.1074/jbc.272.3.1920. [DOI] [PubMed] [Google Scholar]
- 30.Yu H, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, Earp HS. J Biol Chem. 1996;271:29993–29998. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Earp HS. J Biol Chem. 1997;272:14341–14348. doi: 10.1074/jbc.272.22.14341. [DOI] [PubMed] [Google Scholar]
- 32.Bokkala S, Joseph SK. J Biol Chem. 1997;272:12454–12461. doi: 10.1074/jbc.272.19.12454. [DOI] [PubMed] [Google Scholar]
- 33.Thekkumakara TJ, Linas SL. Biochem J. 2003;370:631–639. doi: 10.1042/BJ20020960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang LJ, Guo YL, Tsygankova O, Li QY, Malony JA, Steinhauer M, Williamson JR. Biochemical Pharmacol. 1999;57:425–432. doi: 10.1016/s0006-2952(98)00308-6. [DOI] [PubMed] [Google Scholar]
- 35.Bhat GJ, Samikkannu T, Thomas JJ, Thekkumkara TJ. J Biol Chem. 2004;279:48915–48922. doi: 10.1074/jbc.M409043200. [DOI] [PubMed] [Google Scholar]
- 36.Bhat GJ, Baker KM. Mol Cell Biochem. 1997;170:171–176. doi: 10.1023/a:1006865721939. [DOI] [PubMed] [Google Scholar]
- 37.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Arterioscler Thromb Vasc Biol. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 38.Massa SM, Longo FM, Zuo J, Wang S, Chen J, Sharp FR. J Neurosci Res. 1995;40:807–819. doi: 10.1002/jnr.490400612. [DOI] [PubMed] [Google Scholar]
- 39.Singh B, Soltys BJ, Wu ZC, Patel HV, Freeman KB, Gupta RS. Exp Cell Res. 1997;243:205–216. doi: 10.1006/excr.1997.3609. [DOI] [PubMed] [Google Scholar]
- 40.Wadhwa R, Taira K, Kaul SC. Cancer Therapy. 2003;1:173–178. [Google Scholar]
- 41.Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers RN, Bick RJ, Smith JR, Pereira-Smith OM. Biochem Biophys Res Commun. 2000;275:174–179. doi: 10.1006/bbrc.2000.3237. [DOI] [PubMed] [Google Scholar]
- 42.Mizzen LA, Chang C, Garrels JI, Welch WJ. J Biol Chem. 1989;264:20664–20675. [PubMed] [Google Scholar]
- 43.Carpenter G, Cohen S. J Biol Chem. 1990;265:7709–7712. [PubMed] [Google Scholar]
- 44.Downward J, Parker P, Waterfield MD. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- 45.Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardai G. Pharmacol Therapeutics. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 46.Wadhwa R, Yaguchi T, Hassan MK, Taira K, Kaul SC. Biochem Biophys Res Comm. 2003;302:735–742. doi: 10.1016/s0006-291x(03)00226-2. [DOI] [PubMed] [Google Scholar]
- 47.Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel R, Mitsui Y, Kaul SC. J Biol Chem. 1998;273:29586–29591. doi: 10.1074/jbc.273.45.29586. [DOI] [PubMed] [Google Scholar]
- 48.Sacht G, Brigelius-Flohe R, Kiess M, Sztajer H, Flohe L. Biofactors. 1999;9:49–60. doi: 10.1002/biof.5520090107. [DOI] [PubMed] [Google Scholar]
- 49.Mizukoshi E, Suzuki M, Misono T, Loupatov A, Munekata E, Kaul SC, Wadhwa R, Imamura T. Biochem Biophys Res Commun. 2001;280:1203–1209. doi: 10.1006/bbrc.2001.4225. [DOI] [PubMed] [Google Scholar]
- 50.Ushio-Fukai M, Alexander RW. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 51.Berk BC. Thrombosis Haemostasis. 1999;82:810–817. [PubMed] [Google Scholar]
- 52.Irani K. Cir Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 53.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 54.Frank GD, Eguchi S, Inagami T, Motley ED. Biochem Biophys Res Comm. 2001;280:1116–1119. doi: 10.1006/bbrc.2001.4251. [DOI] [PubMed] [Google Scholar]
- 55.Pueo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Arteriosclr Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 56.Bataller R, Sancho—Bru P, Gines P, Brenner DA. AntioxidRedox Signal. 2005;7:1346–1355. doi: 10.1089/ars.2005.7.1346. [DOI] [PubMed] [Google Scholar]
- 57.Arteel GE. Hepatology. 2004;40:263–265. doi: 10.1002/hep.20296. [DOI] [PubMed] [Google Scholar]
- 58.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]