

Abstract
Animal models are extremely useful tools in defining pathogenesis and treatment of human disease. For many years researchers believed that structural damage to the brain of neuropsychiatric (NP) patients lead to abnormal mental function, but this possibility was not extensively explored until recently. Imaging studies of NP-systemic lupus erythematosus (SLE) support the notion that brain cell death accounts for the emergence of neurologic and psychiatric symptoms, and evidence suggests that it is an autoimmunity-induced brain disorder characterized by profound metabolic alterations and progressive neuronal loss. While there are a number of murine models of SLE, this article reviews recent literature on the immunological connections to neurodegeneration and behavioral dysfunction in the Fas-deficient MRL model of NP-SLE. Probable links between spontaneous peripheral immune activation, the subsequent central autoimmune/inflammatory responses in MRL/MpJ-Tnfrsf6lpr (MRL–lpr) mice and the sequential mode of events leading to Fas-independent neurodegenerative autoimmune-induced encephalitis will be reviewed. The role of hormones, alternative mechanisms of cell death, the impact of central dopaminergic degeneration on behavior, and germinal layer lesions on developmental/regenerative capacity of MRL–lpr brains will also be explored. This model can provide direction for future therapeutic interventions in patients with this complex neuroimmunological syndrome.
Keywords: MRL mice, Autoimmunity, Neuroinflammation, Neurodegeneration, Impaired neurogenesis, Dopamine, Corticosteroids, Behavioral dysfunction
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune/inflammatory disease with a broad spectrum of clinical and immunological manifestations (Isenberg et al., 1989). Neurologic and psychiatric (NP) manifestations of unknown etiology are common in SLE and have been proposed to represent a more severe form of the disease, occurring in up to 75% of patients (Scolding and Joseph, 2002; Navarrete and Brey, 2000; Hanly, 2001; Bluestein, 1992; Adelman et al., 1986; Mcnicholl et al., 1994). The manifestations of NP-SLE range from diffuse CNS disorders (i.e. acute confusional state, psychosis, anxiety and depressive disorders, clinical to subclinical cognitive disorder of variable functional significance) to CNS syndromes (i.e. seizures, cerebrovascular disease, chorea and myelopathy, transverse myelitis, demyelinating syndrome and aseptic meningitis, headaches) and PNS disorders (i.e. polyneuropathies and mononeuropathies, autonomic disorders, plexopathy, myasthenia gravis) (Tincani et al., 1996). Approximately 40% of the NP-SLE manifestations develop before the onset of SLE or at the time of diagnosis and about 60% within the first year after diagnosis (van Dam, 1991). While a histologically normal brain with no specific pathognomonic brain lesions is a possible finding in NP-SLE, various abnormalities include hypoperfusion (Colamussi et al., 1995; Handa et al., 2003; Huang et al., 2002; Lopez-Longo et al., 2003) and regional metabolic abnormalities (Komatsu et al., 1999; Sibbitt and Sibbitt, 1993; Brooks et al., 1997; Volkow et al., 1988). Brain atrophy, however, is the most frequent observation on CT scans (Gonzalez-Scarano et al., 1979; Kaell et al., 1986; Miguel et al., 1994; Omdal et al., 1989; Ainiala et al., 2005; Waterloo et al., 1999) and is proposed to reflect widespread and progressive neuronal loss (Sibbitt and Sibbitt, 1993; Sibbitt et al., 1994).
Research has helped to distinguish the role of autoimmune disease as the primary factor inducing NP-SLE, apart from complications of kidney damage, infections, and steroid therapy. Autoantibodies in the serum and cerebrospinal fluid (CSF) of lupus patients have been proposed as an important factor in the etiology of CNS damage (Jennekens and Kater, 2002). Increased intrathecal synthesis (as revealed by an elevated IgG index and oligoclonal banding) in patients with CNS dysfunction (McLean et al., 1995; Hirohata et al., 1985; Winfield et al., 1983) and antigen-specific autoantibodies in the CSF (Yoshio et al., 2005) are associated with NP manifestations (Greenwood et al., 2002). For example, evidence suggests that neuronal antibodies are involved in the pathogenesis of psychiatric disease (Quismorio and Friou, 1972; Vincent et al., 2003; Diederichsen and Pyndt, 1970; Bluestein and Zvaifler, 1983), and parenchymal lesions associated with movement disorder have recently been documented in these patients (Rocca et al., 2006), supporting the link between autoimmunity, neuronal death, and neurologic manifestations. In many cases however, the correlational nature of clinical data has lead to the necessity for animal models. Using animal models, interactions between autoimmune/inflammatory phenomena and brain function can be examined in a more systematic and direct way. Several animal models develop a fatal immune complex-mediated glomerulonephritis associated with immunological abnormalities (i.e. autoantibody production) very similar to the salient features of human SLE (Theofilopoulos, 1992). The existence of murine models for this disease (both induced and spontaneous) have been extremely valuable to researchers in evaluating the various behavioral manifestations and autoimmune abnormalities which present in this syndrome.
The most commonly studied spontaneous models of lupus include the (NZB×NZW)F1(BWF1) hybrid, BXSB, and MRL mice. These strains are characterized by a wide spectrum of autoimmune manifestations (Dixon et al., 1978) and share common characteristics such as hypergammaglobulinemia and antinuclear antibodies (ANA). However, no animal model is a pure representation of an entire clinical syndrome, and each strain has distinct features which make them beneficial for examining certain aspects of disease (Dixon et al., 1978; Andrews et al., 1978). Considering that the NZB and BXSB strains of mice have a high incidence of inherited brain anomalies (Sherman et al., 1990) which can confound the assessment of autoimmunity-induced brain damage and the links between lupus-like disease and behavioral changes, the MRL model permits the examination of interrelationships between systemic autoimmunity, brain pathology, and aberrant behavior in a more controlled manner. Due to profound deficits in behavior, which appear at a high frequency during the onset of spontaneous CNS-lupus-like disease in MRL/MpJ-Tnfrsf6lpr (MRL–lpr) mice (Szechtman et al., 1997; Sakic et al., 1997), this review focuses on the validity of the MRL model in exploring organic causes of NP manifestations in SLE.
When studied together, MRL–lpr and MRL/MpJ+/+ (MRL+/+) congenic mice (sharing >99.9% of their genome) are considered to be a natural, well controlled model of NP-SLE (Sakic et al., 1997). These substrains are comparable in many respects (appearance, size and reproductive age), except in the onset of autoimmunity and neurobehavioral dysfunctions. While MRL–lpr mice have rapid onset of disease beginning around 7 weeks of age, MRL+/+ controls develop milder symptoms much later in life (Theofilopoulos, 1992). A mutation of a single autosomal recessive gene, designated lymphopoliferation (lpr), on chromosome 19 results in massive lymphoadenopathy, induced by the accumulation of abnormal T-lymphocytes in the MRL–lpr strain (Theofilopoulos, 1992). There is also a spontaneous loss-of-function mutation in Fas (Nagata, 1994) which leads to a deficit in apoptotic Fas receptor expression in MRL–lpr animals (Nagata, 1994; Singer et al., 1994). The main function of Fas is to bind to its ligand (FasL), and transduce signals leading to cell death (Nagata and Suda, 1995). Similar to various CNS cell death mechanisms and compensatory processes, the pathogenic etiology of behavioral deficits in this model of NP-SLE appears to be multi-factorial. Therefore, this review article examines the interplay between underlying genetics, autoimmunity and inflammation, hormones, and a number of secondary factors leading to nervous tissue injury, death and behavioral dysfunction in MRL–lpr mice.
2. Prelude to CNS damage: disruption of the blood–brain barrier
The blood–brain barrier (BBB) is formed by brain capillary endothelial cells that line cerebral microvessels and has an important role in maintaining a precisely regulated micro-environment for reliable neuronal signalling in the CNS (Abbott et al., 2006). Some chronic neuropathologies may involve an early phase of BBB disturbance preceding neuron damage, which suggests that vascular damage can lead to secondary neuronal disorder (Minagar and Alexander, 2003). For example, autoantibodies may have a pathogenic role should they penetrate the compromised BBB and gain access to neuronal tissues. In the case of NP-SLE, it is hypothesized that disruption of the BBB and anti-neuronal autoantibodies account for CNS manifestations of disease (Rice et al., 2005; Hanly et al., 2006). Indeed, antibodies which react with neuronal cell lines and brain tissue have been reported in the sera and CSF of patients with CNS-lupus, but they are also found in lupus patients with no clinical evidence of CNS involvement (Long et al., 1990; Denburg et al., 1988; Bluestein et al., 1981; Kelly and Denburg, 1987).
Similar to humans, evidence of this same phenomena has been found in murine models of CNS lupus (Narendran and Hoffman, 1989; Hoffman and Madsen, 1990). When the BBB is disrupted in lupus-prone mice, large molecules and cells (normally blocked from entry into the CNS), can infiltrate into the brain and lead to CNS damage. Correlational evidence in aged MRL–lpr animals (i.e. associations between ANA, dsDNA, splenomegaly, brain atrophy, and neurodegeneration) supports this notion (Sakic et al., 2000a; Ballok et al., 2003, 2006). Binding of brain-reactive autoantibodies (BRAs) to CNS tissue also appears to have detrimental consequences (Harbeck et al., 1978; Hoffman et al., 1978, 1988; Narendran and Hoffman, 1988; Sidor et al., 2005). While the mechanism by which circulating BRAs access the brain is not well understood (Hoffman and Harbeck, 1989), aberrant behavioral and emotional manifestations in human and murine forms of lupus does suggest that multiple CNS antigens and sites are targeted (Hoffman and Madsen, 1990; Hoffman et al., 1987; Crimando and Hoffman, 1992; Sakic et al., 1994; Huerta et al., 2006; DeGiorgio et al., 2001; Kowal et al., 2004). For example, autoantibody-mediated compromise of central neurotransmission is one pathogenic mechanism proposed in the etiology of NP-SLE (Bluestein and Zvaifler, 1983). Taken together, alterations in BBB permeability and BRAs appear to contribute to and mediate CNS disease in some forms of SLE (Bresnihan et al., 1979).
A dysregulated cytokine network is also thought to play a key role in NP-SLE disease pathogenesis. In general, pro-inflammatory cytokines appear to be instrumental in expanding peripheral autoimmunity to the CNS by inducing dysfunctional endothelial activation (Meroni et al., 2003; Abbott et al., 2003). For example, these cytokines have been shown to cause exaggerated intracellular adhesion molecule (ICAM)-dependent leukocyte-endothelial interactions in MRL–lpr brains (Marshall et al., 2003), which can be prevented by antibody treatment directed against pro-inflammatory cytokines or ICAM (McHale et al., 1999; Brey et al., 1997a). In addition, they can cross the BBB by specific transport systems, and can bind receptors on endothelial cells of brain vasculature to release other mediators (e.g. cytokines, nitric oxide, prostaglandins) into the CSF and brain parenchyma (Tsai et al., 1994; Svenungsson et al., 2001). Pro-inflammatory cytokines can also alter the stress hormone axis (Hayley et al., 2003; Hu et al., 1993; Rivest et al., 1992; Spangelo and Gorospe, 1995; Hansen and Krueger, 1997; Rivier and Rivest, 1993). More specifically, these cytokines are known to modulate immune responses and regulate corticosteroid levels through targeting glucocorticoid receptors in adrenal and the hypothalamic–pituitary glands (Del and Besedovsky, 2000; Lorton et al., 2003). This may ultimately contribute to the neurodegeneration and neurological dysfunction seen in NP lupus (Fig. 1) (Svenungsson et al., 2001; Jongen et al., 1990; Hafler and Weiner, 1989; Brey et al., 1997a; Shanks et al., 1999; Sakic et al., 1999; Ballok et al., 2003; Kyttaris et al., 2005). The precise regulatory mechanisms of cytokines in this dynamic autoimmune disease, however, remain unresolved.
Fig. 1.
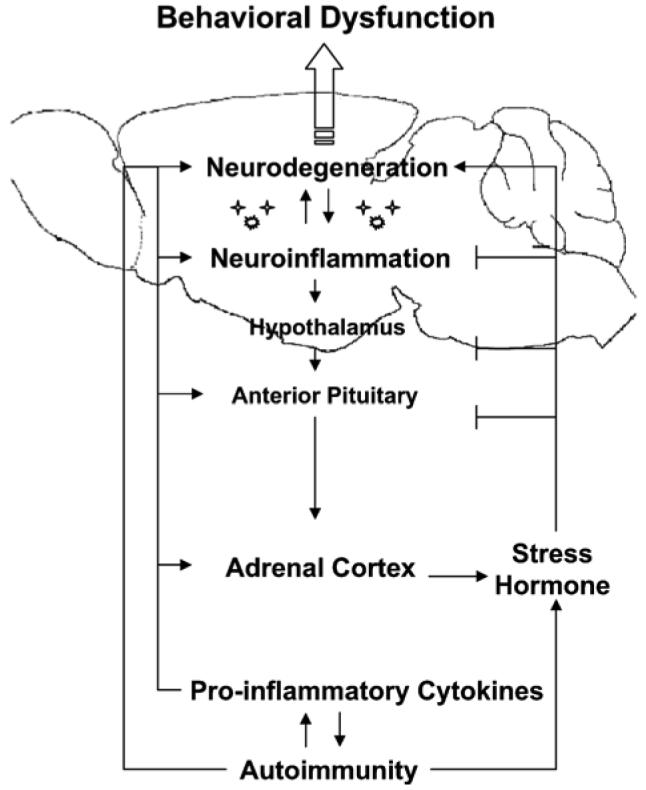
– Behavioral disorders may represent a consequence of chronic autoimmune disease-induced ‘stress’ and brain damage in neuropsychiatric lupus-like disease. The spontaneous onset of systemic autoimmunity in MRL–lpr mice includes increased levels of pro-inflammatory cytokines which mimic actions of glucocorticoids on the hypothalamic–pituitary–adrenal (HPA) axis. While these cytokines stimulate the HPA axis, glucocorticoids feedback to suppress the system at multiple levels. Due to the chronic nature of disease, however, glucocorticoids, cytokines and other autoimmune components remain elevated in lupus animals, contributing to neuronal damage and aberrant behaviors. The precise regulatory mechanisms of the dynamic interrelated systems remain largely unknown.
Abnormal cytokine production also modulates systemic autoimmunity by overactivating B-cells that differentiate into pathogenic autoantibody-forming cells. These pathogenic autoantibodies are a prelude to immune complex disease, a common feature of lupus (Peress et al., 1981). Indeed, deposition of antigen–antibody complexes (immune aggregates) in choroidal blood vessels have been associated with neuropsychiatric dysfunction, and vascular deposits within the choroid plexus (CP) are accompanied by histopathologic evidence of inflammation (Schwartz and Roberts, 1983). Therefore, autoantibodies to endothelial cells, as well as the pathogenic action of circulating–immune complexes (CIC) on microvessels, likely contribute to, if not cause, endothelial cell damage and breakdown of the BBB in lupus patients and mice (Valesini et al., 2006; Hoffman et al., 1983; Ma et al., 2006).
3. Evidence of neuroinflammation in CNS disease
Recently, the role of neuroinflammation is emerging as an important component in lupus-like disease. It has been documented that there is an age-related increase in the frequency of T-cells and perivascular leakage of IgG around brain vessels (Vogelweid et al., 1991), upregulation of adhesion molecules (McHale et al., 1999; Zameer and Hoffman, 2003), the expression of mRNA for pro-inflammatory cytokines (Tomita et al., 2001a,b), and deposition of complement proteins (Alexander et al., 2005a) within brains of MRL–lpr mice. Major histocompatibility complex (MHC) upregulation (McIntyre et al., 1990) and F4/80 microglia staining provides additional evidence for microglia-induced neuronal excitotoxicity in these animals (Ballok et al., 2006). When evaluating the global pattern of neuronal damage in diseased MRL–lpr brains (Ballok et al., 2003), the same pattern of degeneration is similarly seen in cases of hydrocephalus, meningoencephalitis, and hypoglycemic encephalopathy (Weller et al., 1978; Del Bigio, 1993; Gerber et al., 2001; Alexander and Alexander, 1983; Auer et al., 1984; Fujioka et al., 1997), all resulting in cerebritis. Supporting this notion, others have reported manifestations of hydrocephalus (Denenberg et al., 1992), meningoencephalitis (Alexander et al., 1983) and altered glucose metabolism (Alexander et al., 2005b) in the brains of these mice, with complement activation as a likely precursor to cerebral edema (Alexander et al., 2003).
In a recent study, congenic MRL+/+ animals were found to have greater cell loss, and a more aggressive, sustained microglial inflammatory response following mechanical injury and breakdown of their BBB (Hampton et al., 2004). Unlike MRL–lpr animals which develop spontaneous BBB disruption, MRL+/+ mice do not show evidence of CNS damage under normal conditions, but the MRL strain may have an inherent propensity toward exaggerated CNS inflammatory responses. For example, considering the substantia nigra of mice have more microglia than other areas (Lawson et al., 1990), one may assume that this neuroanatomical trait reflects the region-specific susceptibility of the substantia nigra in lupus mice (Ballok et al., 2004a) to inflammatory and excitotoxic metabolites produced by activated microglia.
While neuroinflammation appears to be a contributing factor to disease in MRL–lpr animals, a distinct inflammatory response is not commonly seen in brains of lupus patients. Some of the most common neuropathological findings in SLE, however, are small vessel cerebral vasculopathy and micro-infarcts. These observed features likely reflect the end result of repeated episodes of acute inflammation in the small vessels of the brain (Hess, 1997). Soluble adhesion molecules found in serum and CSF of patients with central nervous involvement (Baraczka et al., 2001) also suggests that neuroinflammatory conditions may play an understated role in some NP manifestations.
4. Evidence of neuronal death and impaired brain growth/atrophy
The MRL strain does not show a high incidence of inherited neuroanatomical abnormalities (Sherman et al., 1987) which minimize the possibility of congenital defects confounding the study of disease-induced neurodegeneration. At the onset of autoimmune symptoms in MRL–lpr mice, reports of reduced complexity of pyramidal neurons, reduced brain weights (Sakic et al., 1998b), and selectively neurotoxic CSF (Maric et al., 2001) provided indirect evidence of neuronal damage in diseased animals. Direct evidence of neuronal death, however, was first confirmed in MRL–lpr brains using the Fluoro Jade B (FJB) cytochemical stain (specific for dying neurons). A small percentage of these neurons where subsequently found to contain TdT-labeled apoptotic nuclei, and co-localized with FJB (Ballok et al., 2003) and anti-neurofilament staining (Alexander et al., 2005a). Providing further evidence of neurodegeneration, while the size of hippocampal fields and neuronal density are not reduced in young Fas-deficient lpr mice (Kovac et al., 2002), cell densities are reduced within the hippocampus, cortex (Ballok et al., 2004b) and midbrain (Ballok et al., 2004a) of aged/diseased lupus mice.
In addition to mature neurons, recent findings suggest that progenitor cells also degenerate in MRL–lpr brains. More specifically, the subventricular zone (Sakic et al., 2000b; Sidor et al., 2005), subgranual zone (Ballok et al., 2003, 2006), and substantia nigra (Ballok et al., 2004a), known to contain proliferative progenitor cells capable of neurogenesis (Yamashita et al., 2006; Suh et al., 2005; McGuire et al., 2001; Zhao et al., 2003), show signs of damage in these animals. CSF from diseased lupus mice is also cytotoxic to neurons and neuronal progenitor cells in vitro (Maric et al., 2001; Ballok et al., 2004a) supporting a link between toxic CSF IgG and neuronal/progenitor cell damage (Sidor et al., 2005; Sakic et al., 2005b). If in vitro findings are predictive of in vivo events, then autoimmune-induced lesions of germinal layers may reduce the developmental and regenerative capacity of MRL–lpr brains. An impairment in this process would likely exacerbate subsequent autoimmune/inflammatory-mediated neuronal death and behavioral deficits. For example, an impaired capacity for hippocampal neurogenesis could account for the cognitive impairments observed in these animals (Ballok et al., 2004b). Stress hormones, chronically elevated in lupus mice (Lechner et al., 2000), have also been shown to inhibit cell proliferation and neurogenesis (Mirescu and Gould, 2006), and may additionally account for impaired brain growth and regeneration along the progression of autoimmune disease.
5. Stress-like behavior is associated with autoimmune disease
The onset and progression of disease in MRL–lpr mice parallels the emergence of aberrant stress-like behaviors (Szechtman et al., 1997). The nature of this autoimmune-associated behavioral syndrome (AABS) suggests a progressive anxious- and depressive-like state (and differences in emotionality), as indicated by increased thigmotaxic behavior, impaired exploration of novel objects and spaces, performance deficits in the plus-maze and step-down tests, excessive floating in the forced swim test (FST) (Sakic et al., 1992, 1993a, 1994), reduced responsiveness to a palatable stimulus (Sakic et al., 1996a), and reduced isolation-induced inter-male fighting (Sakic et al., 1998a). Moreover, impaired “cognitive flexibility” and poor spatial learning was revealed through the Morris water maze (Sakic et al., 1993b), and spontaneous alternation behavioral test (Ballok et al., 2004b). In addition, diseased MRL–lpr animals display lower nocturnal and open-field activity, and significant deficiencies in neurological (Hess et al., 1993; Brey et al., 1997b) and psychomotor (beam-walking) tests (Sakic et al., 1993a, 1996b). Chronic social isolation stress has also been shown to exacerbate autoimmunity and reduce survival of MRL–lpr mice (Chida et al., 2005).
The causative role of autoimmunity and inflammation in the pathogenesis of AABS has been supported by studies employing the immunosuppressive drug cyclophosphamide (CY), which prevented some behavioral deficits in lupus animals (Sakic et al., 1995, 1996a; Farrell et al., 1997). More specifically, CY prevented anxiety- and depressive-like behaviors as indicated by the restoration of novel object exploration, increased responsiveness to a sweet palatable solution, and reduced floating in the FST. In addition to autoimmunity, other factors have been suggested to be involved in the emergence of AABS such as genetics (e.g. the Fas mutation), endocrine factors (e.g. corticosterone-releasing factor, glucocorticoid, prolactin), and multi-system disease (e.g. kidneys, joints, skin, eyes). Taken together, the inherited lack of anti-inflammatory-Fas-dependent mechanisms leading to unsuppressed peripheral immune activation, coincides with elevated levels of corticosteroids (Lechner et al., 2000) and the appearance of stress-like behaviors in MRL–lpr mice (Sakic et al., 1994).
6. Corticosteroids: the permissive factor for cell death?
Similar to chronic cerebral ischemia, corticosteroid therapy has been linked to cerebral atrophy (Ainiala et al., 2005) and cognitive decline often documented in NP-SLE patients (Yamauchi et al., 1994; Chinn et al., 1997). Similar to the effects of chronic stress, NP-lupus mice show brain atrophy and deficits in cognition at the onset of disease (Sakic et al., 1993b, 1998b; Ballok et al., 2004b). In addition to the peripheral dysregulation of glucocorticoids, studies have also revealed a dysfunctional hypothalamic–pituitary axis in these animals (Shanks et al., 1999; Sakic et al., 1999). Although an imbalanced neuro-immuno-endocrine network is proposed to play a key role in the etiology of brain damage, it is still not clear whether central neurons are initially damaged by an autoimmune-driven upregulation in corticosterone production.
Corticosterone-induced atrophy of neurons may be reversible, but it can also be indicative of an early stage of neurodegeneration (McEwen, 1999; Woolley et al., 1990). MRL–lpr mice normally show profound neuronal spine loss relative to asymptomatic MRL+/+ controls (Sakic et al., 1998b), but chronic corticosterone treatment further exacerbated this spontaneous process. In a recent study, while prolonged treatment with corticosterone attenuated signs of autoimmune disease, it lead to profound dendritic spine deterioration (revealed by the Golgi method) in both MRL substrains (unpublished results). Therefore, based on evidence of chronically elevated serum corticosterone levels in MRL–lpr mice (Lechner et al., 2000), sustained endogenous immunosuppression may be a precursor predisposing neurons to the degenerative autoimmune/inflammatory cascade seen at later stages of disease (i.e. following a breach in the BBB). Indeed, studies have revealed that changes in the morphology of neuronal dendrites, cerebral atrophy, and immunoreactive ubiquitin particles (denoting axon terminal degeneration) occur by 14 weeks of age in MRL–lpr brains, but progressive neurodegeneration and microglial activation does not become pronounced until terminal stages of disease (e.g. by 5 months). Few MRL–lpr mice survive beyond 6 months of age (Dixon et al., 1978), which may be attributed to profound CNS damage (Ballok et al., 2003) and brain edema (Alexander et al., 2003). Future studies examining the possibility that adrenalectomy can prevent or delay the degeneration and death of cells in these animals should be explored.
7. Behavioral consequences of CNS damage
Clinical studies have reported that NP manifestations are accompanied by cerebral atrophy (Chinn et al., 1997), progressive neuronal loss (Brooks et al., 1997; Sibbitt and Sibbitt, 1993), and parenchymal lesions associated with movement disorder (Rocca et al., 2006). In MRL–lpr mice, despite parallels between the emergence of behavioral dysfunction and systemic autoimmunity, there has been no direct evidence that brain pathology can account for aberrant behavior. Recently, the phenomena and extent of neurodegeneration in MRL–lpr brains, and the connections between brain morphology and functional damage has been explored more systematically. While it is not possible to verify causation, correlations suggest links between structural brain damage and functional/behavioral impairments in lupus animals. For example, deficits in spatial learning/memory emerged concomitant with hippocampal damage (Ballok et al., 2004b), aberrant performance in the sucrose preference test (i.e. impaired motivated behavior) coincided with lesions of the nucleus accumbens (Anderson et al., 2006), and hypoactivity (i.e. decreased locomotor capacity) accompanied the appearance of degeneration in the substantia nigra (Ballok et al., 2004a) of autoimmune mice. Although the causes and mechanisms underlying these neurological deficits are poorly understood (Hess et al., 1993; Brey et al., 1995), recent pharmacological evidence supports a link between dopaminergic circuit damage and AABS in diseased MRL–lpr animals.
In comparison to other neurotransmitters, central dopamine system activity (implicated in reward, movement, and cognitive processes) is most profoundly altered in MRL brains (Sakic et al., 2002). There is now evidence that damage to central dopaminergic circuits in MRL–lpr brains account for some behavioral deficits. For example, chronic injection with the selective D2/D3 agonist quinpirole induced self-injurious behavior in lupus mice (Sakic et al., 2002). Similarly, rotational behavior increased in MRL–lpr mice following acute injection with the selective D1/D2 dopamine agonist apomorphine (Ballok et al., 2004a). In the sucrose preference paradigm, acute injection with the indirect dopamine agonist d-amphetamine failed to alter the response rates of diseased animals to sucrose solutions (Anderson et al., 2006), while chronic treatment increased their mobility in the FST (unpublished data). Immunosuppressive treatment, suppressing autoimmunity and preventing hippocampal damage, circumvented an age-related decline in spatial memory and retrieval (Rolls and Kesner, 2006; Ballok et al., 2004b). Taken together, these results link neuropathological findings of dopaminergic cell death in nigrostriatal, mesolimbic, and mesocortical pathways to certain behavioral deficits (e.g. locomotor, motivated, and learning behaviors) in lupus-prone animals (Table 1). Although the contribution of peripheral disease manifestations on behavioral performance cannot be excluded, these pharmacological results do indicate that anhedonic- and depressive-like behaviors are a consequence of disease-driven damage to several dopamine systems in MRL–lpr brains.
Table 1.
Correlations between brain region, functional damage and dopaminergic system deficits in aged MRL–lpr mice
| Brain lesion | Behavioral task | Dopamine agonist | Behavioral consequence | Dopamine pathway |
|---|---|---|---|---|
| Hippocampus | Spontaneous alternation | n/a | Deficits in spatial learning | Mesocortical |
| Nucleus accumbens | Sucrose preference test | Amphetamine acute | Impaired motivation | Mesolimbic |
| n/a | Forced swim test | Amphetamine chronic | Increased mobility | Nigrostriatal |
| Ventral tegmental area | Activity in novel environment | n/a | Decreased exploration | Mesolimbic |
| Substantia nigra | Rotation test | Apomorphine acute | Increased movement | Nigrostriatal |
| n/a | Grooming behavior | Quinpirole chronic | Self-injurious behavior | Mesolimbic |
Major neural systems of the brain use dopamine as a principal neurotransmitter to mediate learning and memory (mesocortical system), motivated behavior (mesolimbic system), and locomotor behavior (nigrostriatal system). While young asymptomatic MRL–lpr animals do not show brain abnormalities or behavioral deficits, aged autoimmune MRL–lpr mice exhibit behavioral dysfunctions concomitant with brain damage. These behavioral impairments can be linked to specific brain structures and/or are modulated by dopamine agonists, providing pharmacological evidence that brain cell death is selective (e.g. a target of neuroactive antibodies) and accounts for some functional abnormalities in these animals. Note: ‘acute’ denotes a single injection while ‘chronic’ denotes at least 5 successive treatments with a given drug; ‘n/a’ indicates data is not available.
8. Possible mechanisms and targets of brain cell death
The well-characterized apoptotic pathway defect in MRL–lpr mice has lead to much speculation as to which compensatory processes most likely predominate in CNS disease. One consequence of impaired apoptotic mechanisms may be the accumulation of unwanted proteins which often underlie the pathogenesis of several major human neurodegenerative diseases. For example, as a result of defective ubiquitin-dependent proteolysis, neurodegeneration can occur (Layfield et al., 2001). Indeed, the ubiquitin–proteasome system appears to be overactive in MRL–lpr brains (Ballok et al., 2004b), and it is possible that autoantibodies reactive to ubiquitin in peripheral tissue of MRL–lpr mice (Elouaai et al., 1994) target these antigens in the brain. Impaired dopamine catabolism has also been suggested in the pathogenicity of CNS damage in lupus mice. Increased levels of this neurotransmitter have been reported in the tuberoinfundibular pathway of animals (Sakic et al., 2002), and may induce neurotoxicity (Blum et al., 2001; Asanuma et al., 2003) at a later stage of disease. More specifically, if impaired dopamine catabolism is a feature of other pathways (i.e. nigrostriatal, mesolimbic, and mesocortical circuits) it is possible that over time, accumulated metabolites induce the dopaminergic cell damage observed in MRL–lpr brains. Overall, impaired protein processing may be a factor contributing to progressive Fas-independent neurodegenerative autoimmune-induced encephalitis in MRL–lpr mice.
Although the Fas antigen/surface receptor (Fas/Apo-1/CD95) is not expressed in brains of MRL–lpr animals, an apoptotic mode of neuronal demise may still be mediated by mechanisms such as TNF-alpha or granzyme B receptors. Considering the massive lymphocytosis of CD8 cytotoxic T-cells (known to release granzyme B) into the third ventricles and brain parenchyma of MRL–lpr mice (Sakic et al., 2000b; Ballok et al., 2003), and TNF-alpha detected in CSF and brains of these animals (Ballok et al., 2004a; Ma et al., 2006), it is likely that both mechanisms activate terminal caspase cascades (Alexander et al., 2005a). Alternatively, lymphocytes which infiltrate MRL–lpr brains (Sakic et al., 2000b) are likely polyclonally activated and will cause substantial neuronal death in an allogeneic and syngeneic manner. More specifically, neurons have a high and selective vulnerability to T-cells in contrast to other CNS cell types (e.g. oligodendrocytes and astrocytes) which are not killed by T lymphocytes (Giuliani et al., 2003). When enough activated T-cells accumulate in the CNS of lupus mice, neuronal cytotoxicity could be mediated through cell contact-dependent mechanisms involving LFA-1 and CD40, but not FasL (due to the fas receptor deficiency).
Another pathway which could play an important role in neuronal death in MRL–lpr mice involves p53. As a well-characterized transcription factor, p53 is known to respond to DNA damage and other genotoxic stresses by the activation of downstream targets involved in repair, differentiation, senescence, growth arrest, and apoptosis (Resnick-Silverman and Manfredi, 2006). In a recent pilot study, densities of p53 dot-like particles were examined in the brain parenchyma of MRL mice. Unexpectedly, significantly reduced anti-p53 immunohistochemical staining was observed in MRL–lpr brains, relative to congenic controls (unpublished results). Consistent with this finding, cellular entry of anti-DNA antibodies into glomerular tissue of MRL–lpr mice resulted in an inhibition/reduction of p53, and suppression of apoptosis (Yanase and Madaio, 2005). When considering that elevated immunoglobulin levels are seen in the CSF and brain of lupus animals (Sidor et al., 2005; Zameer and Hoffman, 2001) one may assume that a similar autoantibody-mediated process is occurring within neurons and accessory cells of the CNS. Therefore, disrupted p53 functions may account for the morphological features and “intermediate form” of cell demise seen in MRL–lpr brains (Ballok et al., 2006), leading to cytoskeletal collapse. Future studies should examine the role of antibodies to p53 in delaying degeneration and cell death processes more closely.
There are numerous factors that can induce excitotoxic damage, and in an injured or immunologically challenged brain, cytokine-producing microglia appear to play an important role (Gwag et al., 1997). Microglia readily activate by transforming from a ramified, resting state into amoeboid cells to express MHC molecules (Lawson et al., 1990), as seen in MRL–lpr brains (McIntyre et al., 1990). Inflammatory responses are then perpetuated by both cyclooxygenase (COX) and nitric oxide (NO). In lupus mice, however, prostaglandin production abnormalities have been observed (Reilly et al., 2000), and COX inhibition is not effective in ameliorating CNS disease (Ballok et al., 2006) suggesting that inducible NO synthase (Alexander et al., 2005a) is likely the mediator of neuroinflammation in these animals. Recent ultrastructural evidence obtained from electron microscopy (Ballok et al., 2006) and a study which found significant increases in glutamine, glutamate and lactate concentrations in MRL–lpr brains (Alexander et al., 2005b) supports the notion of excitotoxic cell death in these animals. In the case of glutamate toxicity, anti-DNA antibodies may play an important role. These autoantibodies have recently been found to cross-react with central NMDA receptors in mice, producing neuronal apoptosis (DeGiorgio et al., 2001), and a similar IgG-mediated neurodegenerative event is proposed to occur in MRL–lpr brains (Sakic et al., 2005b). This causal relationship between NR2 receptor-reactive autoantibodies and brain damage has also been reported in NP-SLE patients (Omdal et al., 2005). Interestingly, the immunosuppressive drug CY has been found to abolish the infiltration of cells of the monocyte–macrophage lineage into the CP of the MRL–lpr substrain (Farrell et al., 1997) and successfully prevented atrophy of dendritic spines (Sakic et al., 2000a), and neuronal death (Ballok et al., 2004b). However, since CY affects a broad population of cells, the factors which play a predominant role in the etiology of brain damage in lupus animals is still unknown. Considering that anti-CD4 treatment (O'Sullivan et al., 1995) and a potent complement inhibitor (Alexander et al., 2005a) both ameliorated CNS disease manifestations in MRL–lpr mice, one may assume that autoimmune and inflammatory factors are both requisites for brain pathology.
9. Conclusions and future perspectives
Neurodegenerative disorders are characterized by a gradual and relentlessly progressive neuronal loss that is often selective in that it occurs in anatomically and physiologically related brain areas. Progressive neuronal death and microglial activation are concomitant with the onset and development of systemic autoimmune/inflammatory disease and behavioral deficits in the MRL–lpr model of NP-SLE. The initial role of corticosterone appears to be important in SLE-like disease given that glucocorticoids are innately elevated in MRL–lpr mice (Lechner et al., 2000), and may act similar to the iatrogenic effects of sustained corticosteroid therapy on brains of NP-SLE patients (Ainiala et al., 2005). At a later stage of disease, dopamine neurons and their progenitor cells appear to be a target of the disease process, possibly accounting for some aberrant behaviors in lupus mice. An interplay among numerous factors (i.e. activated microglia, neuroactive cytokines, autoantibodies, cytotoxic T-cells) likely leads to edema and neurodegeneration in MRL–lpr brains. Many years of clinical evidence support the therapeutic use of immunosuppressive drugs in treating SLE, with some undesirable effects (Miller, 1997). Further work elucidating precise disease factors and molecular events during neuronal death is needed to devise more effective and less toxic treatments for patients diagnosed with this complex immunological syndrome. Knowledge of the precise immune components and the biochemical profile of neurotoxic mediators in lupus mice may help identify pathogenic circuits in NP-SLE, and be beneficial when designing protocols which foster both neuroprotection and immunosuppression.
A better understanding of neuroendocrine hormones in the progression of NP-SLE may lead to novel therapeutic approaches. One hormone of recent interest is prolactin, a pituitary hormone under the control of dopamine. Elevated secretion of prolactin is commonly seen in SLE (Jara et al., 2001), and given recent results of dysfunctional dopaminergic circuits in lupus mice, one may wonder whether this endocrine imbalance is a consequence of impaired dopamine regulation in the hypothalamus (Freeman et al., 2000; Walker, 2006). Consistent with this notion, the dopamine agonist bromocriptine suppressed secretion of prolactin and ameliorated affect in SLE patients (Walker et al., 2000) and disease activity in autoimmune mice (McMurray et al., 1991; Walker, 2001). Interestingly, a recent post-mortem analysis of a NP-SLE patient's brain found evidence of hypothalamic and basal ganglia damage (Ballok et al., 2004b), suggesting that these regions may be more susceptible to disease. Additional studies, however, are needed to determine whether dopaminergic neurons die excessively in a subset of SLE patients, as seen in lupus animals. Another hormone which may be of interest in the treatment of NP-SLE is progesterone. Considering that progesterone and its precursors/metabolites have been shown to reconstitute the BBB and enhance functional and structural recovery in rodents and patients with brain edema (Shear et al., 2002; Guo et al., 2006; Sayeed et al., 2006), this hormone may prove to be an effective treatment for the edema seen in NP-SLE patients and MRL–lpr mice (van Dam, 1991; Alexander et al., 2003).
Although progenitor cells are a target in aged MRL–lpr animals (Ballok et al., 2004a), the possible effect of autoimmunity on neural stem cells may be extended into pre-natal life. For example, multipotent brain cells may be the target of an attack mounted by the immune system of autoimmune mothers. Indeed, ova-transfer experiments in mice have shown that profound deficiencies in offspring behavior are produced if embryos were reared in an autoimmune uterine environment and conversely, that the severity of behavioral dysfunction in autoimmunity-prone mice is reduced if they were transferred as embryos into a non-autoimmune uterine environment (Denenberg et al., 1991). Consistent with the possibility that the blood–placental barrier does not provide sufficient protection from mother's autoimmunity is the fact that neonatal lupus is a well-documented phenomenon in humans (Cabanas et al., 1996; Prendiville et al., 2003). Therefore, more work needs to be done to determine if conditions for progressive neurodegenerative events later in life are antedated by early neural precursor cell damage.
While the MRL–lpr strain displays many characteristics that resemble human NP-SLE, there are a number of limitations which must be considered when studying this murine model. First, human NP-SLE often has oscillating relapsing–remitting presentations of symptoms, while MRL–lpr mice show a progressive and unrelenting course of disease. This difference appears to be the most fundamental difference between human and murine SLE. Secondly, human SLE shows a strong gender preference (i.e. about nine to ten times more female patients than male). The MRL–lpr strain, however, has no gender bias, possibly suggesting a different hormonal differentiation between two sexes in human and murine lupus. Lastly, while more sophisticated and systematic assessments are administered in human SLE, affective states of MRL–lpr mice can only be assessed by behavioral tests (Scolding and Joseph, 2002; Sakic et al., 2005a), and care is required when transferring information obtained from an animal model to human disease. Despite these limitations, the MRL model of NP-SLE remains a critical tool toward understanding the complex etiopathogenesis of this neuroimmunological syndrome.
Acknowledgments
This work was supported by the doctoral research award grant MOP 38065 from the Canadian Institutes of Health Research (CIHR) awarded to D.A. Ballok.
Footnotes
Publisher's Disclaimer: This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues that you know, and providing a copy to your institution's administrator.
REFERENCES
- Abbott NJ, Mendonca LL, Dolman DE. The blood–brain barrier in systemic lupus erythematosus. Lupus. 2003;12:908–915. doi: 10.1191/0961203303lu501oa. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev., Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Adelman DC, Saltiel E, Klinenberg JR. The neuropsychiatric manifestations of systemic lupus erythematosus: an overview. Semin. Arthritis Rheum. 1986;15:185–199. doi: 10.1016/0049-0172(86)90016-8. [DOI] [PubMed] [Google Scholar]
- Ainiala H, Dastidar P, Loukkola J, Lehtimaki T, Korpela M, Peltola J, Hietaharju A. Cerebral MRI abnormalities and their association with neuropsychiatric manifestations in SLE: a population-based study. Scand. J. Rheumatol. 2005;34:376–382. doi: 10.1080/03009740510026643. [DOI] [PubMed] [Google Scholar]
- Alexander EL, Alexander GE. Aseptic meningoencephalitis in primary Sjogren's syndrome. Neurology. 1983;33:593–598. doi: 10.1212/wnl.33.5.593. [DOI] [PubMed] [Google Scholar]
- Alexander EL, Murphy ED, Roths JB, Alexander GE. Congenic autoimmune murine models of central nervous system disease in connective tissue disorders. Ann. Neurol. 1983;14:242–248. doi: 10.1002/ana.410140211. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim. Biophys. Acta. 2003;1639:169–176. doi: 10.1016/j.bbadis.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J. Immunol. 2005a;175:8312–8319. doi: 10.4049/jimmunol.175.12.8312. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Zwingmann C, Quigg R. MRL/lpr mice have alterations in brain metabolism as shown with [(1)H–(13)C] NMR spectroscopy. Neurochem. Int. 2005b;47:143–151. doi: 10.1016/j.neuint.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Anderson KK, Ballok DA, Prasad N, Szechtman H, Sakic B. Impaired response to amphetamine and neuronal degeneration in the nucleus accumbens of autoimmune MRL–lpr mice. Behav. Brain Res. 2006;166:32–38. doi: 10.1016/j.bbr.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma M, Miyazaki I, Ogawa N. Dopamine- or l-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson's disease. Neurotox. Res. 2003;5:165–176. doi: 10.1007/BF03033137. [DOI] [PubMed] [Google Scholar]
- Auer RN, Wieloch T, Olsson Y, Siesjo BK. The distribution of hypoglycemic brain damage. Acta Neuropathol. (Berl.) 1984;64:177–191. doi: 10.1007/BF00688108. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Millward JM, Sakic B. Neurodegeneration in autoimmune MRL–lpr mice as revealed by Fluoro Jade B staining. Brain Res. 2003;964:200–210. doi: 10.1016/s0006-8993(02)03980-x. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Earls AM, Krasnik C, Hoffman SA, Sakic B. Autoimmune-induced damage of the midbrain dopaminergic system in lupus-prone mice. J. Neuroimmunol. 2004a;152:83–97. doi: 10.1016/j.jneuroim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Woulfe J, Sur M, Cyr M, Sakic B. Hippocampal damage in mouse and human forms of systemic autoimmune disease. Hippocampus. 2004b;14:649–661. doi: 10.1002/hipo.10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballok DA, Ma X, Denburg JA, Arsenault L, Sakic B. Ibuprofen fails to prevent brain pathology in a model of neuropsychiatric lupus. J. Rheumatol. 2006;33:2199–2213. [PMC free article] [PubMed] [Google Scholar]
- Baraczka K, Nekam K, Pozsonyi T, Jakab L, Szongoth M, Sesztak M. Concentration of soluble adhesion molecules (sVCAM-1, sICAM-1 and sL-selectin) in the cerebrospinal fluid and serum of patients with multiple sclerosis and systemic lupus erythematosus with central nervous involvement. Neuroimmunomodulation. 2001;9:49–54. doi: 10.1159/000049007. [DOI] [PubMed] [Google Scholar]
- Bluestein HG. The central nervous system in systemic lupus erythematosus, Systemic Lupus Erythematosus. 2 ed. Churchill Livingstone; New York: 1992. pp. 639–655. [Google Scholar]
- Bluestein HG, Zvaifler NJ. Antibodies reactive with central nervous system antigens. Hum. Pathol. 1983;14:424–428. doi: 10.1016/s0046-8177(83)80287-1. [DOI] [PubMed] [Google Scholar]
- Bluestein HG, Williams GW, Steinberg AD. Cerebrospinal fluid antibodies to neuronal cells: association with neuropsychiatric manifestations of systemic lupus erythematosus. Am. J. Med. 1981;70:240–246. doi: 10.1016/0002-9343(81)90756-7. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog. Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- Bresnihan B, Hohmeister R, Cutting J, Travers RL, Waldburger M, Black C, Jones T, Hughes GR. The neuropsychiatric disorder in systemic lupus erythematosus: evidence for both vascular and immune mechanisms. Ann. Rheum. Dis. 1979;38:301–306. doi: 10.1136/ard.38.4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brey RL, Cote S, Barohn R, Jackson C, Crawley R, Teale JM. Model for the neuromuscular complications of systemic lupus erythematosus. Lupus. 1995;4:209–212. doi: 10.1177/096120339500400308. [DOI] [PubMed] [Google Scholar]
- Brey RL, Amato AA, Kagan-Hallet K, Rhine CB, Stallworth CL. Anti-intercellular adhesion molecule-1 (ICAM-1) antibody treatment prevents central and peripheral nervous system disease in autoimmune-prone mice. Lupus. 1997a;6:645–651. doi: 10.1177/096120339700600805. [DOI] [PubMed] [Google Scholar]
- Brey RL, Sakic B, Szechtman H, Denburg JA. Animal models for nervous system disease in systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 1997b;823:97–106. doi: 10.1111/j.1749-6632.1997.tb48382.x. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Sabet A, Sibbitt WL, Barker PB, van Zijl PC, Duyn JH, Moonen CT. Neurochemistry of brain lesions determined by spectroscopic imaging in systemic lupus erythematosus. J. Rheumatol. 1997;24:2323–2329. [PubMed] [Google Scholar]
- Cabanas F, Pellicer A, Valverde E, Morales C, Quero J. Central nervous system vasculopathy in neonatal lupus erythematosus. Pediatr. Neurol. 1996;15:124–126. doi: 10.1016/0887-8994(96)00159-2. [DOI] [PubMed] [Google Scholar]
- Chida Y, Sudo N, Kubo C. Social isolation stress exacerbates autoimmune disease in MRL/lpr mice. J. Neuroimmunol. 2005;158:138–144. doi: 10.1016/j.jneuroim.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Chinn RJ, Wilkinson ID, Hall-Craggs MA, Paley MN, Shortall E, Carter S, Kendall BE, Isenberg DA, Newman SP, Harrison MJ. Magnetic resonance imaging of the brain and cerebral proton spectroscopy in patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40:36–46. doi: 10.1002/art.1780400107. [DOI] [PubMed] [Google Scholar]
- Colamussi P, Giganti M, Cittanti C, Dovigo L, Trotta F, Tola MR, Tamarozzi R, Lucignani G, Piffanelli A. Brain single-photon emission tomography with Tc-99m-HMPAO in neuropsychiatric systemic lupus erythematosus: relations with EEG and MRI findings and clinical manifestations. Eur. J. Nucl. Med. 1995;22:17–24. doi: 10.1007/BF00997243. [DOI] [PubMed] [Google Scholar]
- Crimando J, Hoffman SA. Detection of brain-reactive autoantibodies in the sera of autoimmune mice using ELISA. J. Immunol. Methods. 1992;149:87–95. doi: 10.1016/s0022-1759(12)80052-4. [DOI] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- del RA, Besedovsky HO. The cytokine–HPA axis circuit contributes to prevent or moderate autoimmune processes. Z. Rheumatol. 2000;59(Suppl 2):II/31–II/35. doi: 10.1007/s003930070015. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR. Neuropathological changes caused by hydrocephalus. Acta Neuropathol. (Berl.) 1993;85:573–585. doi: 10.1007/BF00334666. [DOI] [PubMed] [Google Scholar]
- Denburg SD, Carbotte RM, Long AA, Denburg JA. Neuropsychological correlates of serum lymphocytotoxic antibodies in systemic lupus erythematosus. Brain Behav. Immun. 1988;2:222–234. doi: 10.1016/0889-1591(88)90024-4. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Mobraaten LE, Sherman GF, Morrison L, Schrott LM, Waters NS, Rosen GD, Behan PO, Galaburda AM. Effects of the autoimmune uterine/maternal environment upon cortical ectopias, behavior and autoimmunity. Brain Res. 1991;563:114–122. doi: 10.1016/0006-8993(91)91522-3. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Sherman GF, Rosen GD, Morrison L, Behan PO, Galaburda AM. A behavior profile of the MRL/Mp lpr/lpr mouse and its association with hydrocephalus. Brain Behav. Immun. 1992;6:40–49. doi: 10.1016/0889-1591(92)90058-v. [DOI] [PubMed] [Google Scholar]
- Diederichsen H, Pyndt IC. Antibodies against neurons in a patient with systemic lupus erythematosus, cerebral palsy, and epilepsy. Brain. 1970;93:407–412. doi: 10.1093/brain/93.2.407. [DOI] [PubMed] [Google Scholar]
- Dixon FJ, Andrews BS, Eisenberg RA, McConahey PJ, Theofilopoulos AN, Wilson CB. Etiology and pathogenesis of a spontaneous lupus-like syndrome in mice. Arthritis Rheum. 1978;21:S64–S67. doi: 10.1002/art.1780210909. [DOI] [PubMed] [Google Scholar]
- Elouaai F, Lule J, Benoist H, Appolinaire-Pilipenko S, Atanassov C, Muller S, Fournie GJ. Autoimmunity to histones, ubiquitin, and ubiquitinated histone H2A in NZB×NZW and MRL–lpr/lpr mice. Anti-histone antibodies are concentrated in glomerular eluates of lupus mice. Nephrol. Dial. Transplant. 1994;9:362–366. [PubMed] [Google Scholar]
- Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6:268–274. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol. Rev. 2000;80:1523–1631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Okuchi K, Hiramatsu KI, Sakaki T, Sakaguchi S, Ishii Y. Specific changes in human brain after hypoglycemic injury. Stroke. 1997;28:584–587. doi: 10.1161/01.str.28.3.584. [DOI] [PubMed] [Google Scholar]
- Gerber J, Raivich G, Wellmer A, Noeske C, Kunst T, Werner A, Bruck W, Nau R. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol. (Berl.) 2001;101:499–508. doi: 10.1007/s004010000326. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. J. Immunol. 2003;171:368–379. doi: 10.4049/jimmunol.171.1.368. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Lisak RP, Bilaniuk LT, Zimmerman RA, Atkins PC, Zweiman B. Cranial computed tomography in the diagnosis of systemic lupus erythematosus. Ann. Neurol. 1979;5:158–165. doi: 10.1002/ana.410050209. [DOI] [PubMed] [Google Scholar]
- Greenwood DL, Gitlits VM, Alderuccio F, Sentry JW, Toh BH. Autoantibodies in neuropsychiatric lupus. Autoimmunity. 2002;35:79–86. doi: 10.1080/08916930290016547. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sayeed I, Baronne LM, Hoffman SW, Guennoun R, Stein DG. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp. Neurol. 2006;198:469–478. doi: 10.1016/j.expneurol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Gwag BJ, Koh JY, Demaro JA, Ying HS, Jacquin M, Choi DW. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77:393–401. doi: 10.1016/s0306-4522(96)00473-3. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Weiner HL. MS: a CNS and systemic autoimmune disease. Immunol. Today. 1989;10:104–107. doi: 10.1016/0167-5699(89)90236-3. [DOI] [PubMed] [Google Scholar]
- Hampton DW, Seitz A, Chen P, Heber-Katz E, Fawcett JW. Altered CNS response to injury in the MRL/MpJ mouse. Neuroscience. 2004;127:821–832. doi: 10.1016/j.neuroscience.2004.05.057. [DOI] [PubMed] [Google Scholar]
- Handa R, Sahota P, Kumar M, Jagannathan NR, Bal CS, Gulati M, Tripathi BM, Wali JP. In vivo proton magnetic resonance spectroscopy (MRS) and single photon emission computerized tomography (SPECT) in systemic lupus erythematosus (SLE) Magn. Reson. Imaging. 2003;21:1033–1037. doi: 10.1016/s0730-725x(03)00200-5. [DOI] [PubMed] [Google Scholar]
- Hanly JG. Neuropsychiatric lupus. Curr. Rheumatol. Rep. 2001;3:205–212. doi: 10.1007/s11926-001-0020-7. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Robichaud J, Fisk JD. Anti-NR2 glutamate receptor antibodies and cognitive function in systemic lupus erythematosus. J. Rheumatol. 2006;33:1553–1558. [PubMed] [Google Scholar]
- Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1 beta. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 1997;42:R1246–R1253. doi: 10.1152/ajpregu.1997.273.4.R1246. [DOI] [PubMed] [Google Scholar]
- Harbeck RJ, Hoffman AA, Hoffman SA, Shucard DW, Carr RI. A naturally occurring antibody in New Zealand mice cytotoxic to dissociated cerebellar cells. Clin. Exp. Immunol. 1978;31:313–320. [PMC free article] [PubMed] [Google Scholar]
- Hayley S, Merali Z, Anisman H. Stress and cytokine-elicited neuroendocrine and neurotransmitter sensitization: implications for depressive illness. Stress. 2003;6:19–32. doi: 10.1080/1025389031000091167. [DOI] [PubMed] [Google Scholar]
- Hess DC. Cerebral lupus vasculopathy. Mechanisms and clinical relevance. Ann. N. Y. Acad. Sci. 1997;823:154–168. doi: 10.1111/j.1749-6632.1997.tb48388.x. [DOI] [PubMed] [Google Scholar]
- Hess DC, Taormina M, Thompson J, Sethi KD, Diamond B, Rao R, Feldman DS. Cognitive and neurologic deficits in the MRL/lpr mouse: a clinicopathologic study. J. Rheumatol. 1993;20:610–617. [PubMed] [Google Scholar]
- Hirohata S, Hirose S, Miyamoto T. Cerebrospinal fluid IgM, IgA, and IgG indexes in systemic lupus erythematosus. Their use as estimates of central nervous system disease activity. Arch. Intern. Med. 1985;145:1843–1846. [PubMed] [Google Scholar]
- Hoffman SA, Harbeck RJ. Implications of the Blood–Brain Barrier and Its Manipulation. Plenum Medical Book Co.; New York: 1989. CNS lupus and the blood–brain barrier; pp. 469–494. [Google Scholar]
- Hoffman SA, Madsen CS. Brain specific autoantibodies in murine models of systemic lupus erythematosus. J. Neuroimmunol. 1990;30:229–237. doi: 10.1016/0165-5728(90)90107-x. [DOI] [PubMed] [Google Scholar]
- Hoffman SA, Hoffman AA, Shucard DW, Harbeck RJ. Antibodies to dissociated cerebellar cells in New Zealand mice as demonstrated by immunofluorescence. Brain Res. 1978;142:477–486. doi: 10.1016/0006-8993(78)90910-1. [DOI] [PubMed] [Google Scholar]
- Hoffman SA, Arbogast DN, Day TT, Shucard DW, Harbeck RJ. Permeability of the blood–cerebrospinal fluid barrier during acute immune complex disease. J. Immunol. 1983;130:1695–1698. [PubMed] [Google Scholar]
- Hoffman SA, Arbogast DN, Ford PM, Shucard DW, Harbeck RJ. Brain-reactive autoantibody levels in the sera of ageing autoimmune mice. Clin. Exp. Immunol. 1987;70:74–83. [PMC free article] [PubMed] [Google Scholar]
- Hoffman SA, Narendran A, Shucard DW, Harbeck RJ. Autoantibodies, immune complexes, and behavioral disorders: neuropsychiatric involvement in systemic lupus erythematosus. Drug Dev. Res. 1988;15:237–251. [Google Scholar]
- Hu Y, Dietrich H, Herold M, Heinrich PC, Wick G. Disturbed immuno-endocrine communication via the hypothalamo–pituitary–adrenal axis in autoimmune disease. Int. Arch. Allergy Immunol. 1993;102:232–241. doi: 10.1159/000236531. [DOI] [PubMed] [Google Scholar]
- Huang WS, Chiu PY, Tsai CH, Kao A, Lee CC. Objective evidence of abnormal regional cerebral blood flow in patients with systemic lupus erythematosus on Tc-99m ECD brain SPECT. Rheumatol. Int. 2002;22:178–181. doi: 10.1007/s00296-002-0224-9. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc. Natl. Acad. Sci. U. S. A. 2006;103:678–683. doi: 10.1073/pnas.0510055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg D, Bacon P, Bombardier C, Gladman D, Goldsmith CH, Kalunian K, Liang M, Maddison P, Nived O, Richter M, et al. Criteria for assessing disease activity in systemic lupus erythematosus. J. Rheumatol. 1989;16:1395–1396. [PubMed] [Google Scholar]
- Jara LJ, Vera-Lastra O, Miranda JM, Alcala M, varez-Nemegyei J. Prolactin in human systemic lupus erythematosus. Lupus. 2001;10:748–756. doi: 10.1191/096120301717164994. [DOI] [PubMed] [Google Scholar]
- Jennekens FG, Kater L. The central nervous system in systemic lupus erythematosus: Part 2. Pathogenetic mechanisms of clinical syndromes: a literature investigation. Rheumatology (Oxford) 2002;41:619–630. doi: 10.1093/rheumatology/41.6.619. [DOI] [PubMed] [Google Scholar]
- Jongen PJ, Boerbooms AM, Lamers KJ, Raes BC, Vierwinden G. Diffuse CNS involvement in systemic lupus erythematosus: intrathecal synthesis of the 4th component of complement. Neurology. 1990;40:1593–1596. doi: 10.1212/wnl.40.10.1593. [DOI] [PubMed] [Google Scholar]
- Kaell AT, Shetty M, Lee BC, Lockshin MD. The diversity of neurologic events in systemic lupus erythematosus. Prospective clinical and computed tomographic classification of 82 events in 71 patients. Arch. Neurol. 1986;43:273–276. doi: 10.1001/archneur.1986.00520030063016. [DOI] [PubMed] [Google Scholar]
- Kelly MC, Denburg JA. Cerebrospinal fluid immunoglobulins and neuronal antibodies in neuropsychiatric systemic lupus erythematosus and related conditions. J. Rheumatol. 1987;14:740–744. [PubMed] [Google Scholar]
- Komatsu N, Kodama K, Yamanouchi N, Okada S, Noda S, Nawata Y, Takabayashi K, Iwamoto I, Saito Y, Uchida Y, Ito H, Yoshikawa K, Sato T. Decreased regional cerebral metabolic rate for glucose in systemic lupus erythematosus patients with psychiatric symptoms. Eur. Neurol. 1999;42:41–48. doi: 10.1159/000008067. [DOI] [PubMed] [Google Scholar]
- Kovac AD, Grammig J, Mahlo J, Steiner B, Roth K, Nitsch R, Bechmann I. Comparison of neuronal density and subfield sizes in the hippocampus of CD95L-deficient (gld), CD95-deficient (lpr) and nondeficient mice. Eur. J. Neurosci. 2002;16:159–163. doi: 10.1046/j.1460-9568.2002.02060.x. [DOI] [PubMed] [Google Scholar]
- Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, Volpe BT. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–188. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Kyttaris VC, Katsiari CG, Juang YT, Tsokos GC. New insights into the pathogenesis of systemic lupus erythematosus. Curr. Rheumatol. Rep. 2005;7:469–475. doi: 10.1007/s11926-005-0054-3. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Layfield R, Alban A, Mayer RJ, Lowe J. The ubiquitin protein catabolic disorders. Neuropathol. Appl. Neurobiol. 2001;27:171–179. doi: 10.1046/j.1365-2990.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- Lechner O, Dietrich H, Oliveira dos SA, Wiegers GJ, Schwarz S, Harbutz M, Herold M, Wick G. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J. Autoimmun. 2000;14:325–333. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- Long AA, Denburg SD, Carbotte RM, Singal DP, Denburg JA. Serum lymphocytotoxic antibodies and neurocognitive function in systemic lupus erythematosus. Ann. Rheum. Dis. 1990;49:249–253. doi: 10.1136/ard.49.4.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Longo FJ, Carol N, Almoguera MI, Olazaran J, onso-Farto JC, Ortega A, Monteagudo I, Gonzalez CM, Carreno L. Cerebral hypoperfusion detected by SPECT in patients with systemic lupus erythematosus is related to clinical activity and cumulative tissue damage. Lupus. 2003;12:813–819. doi: 10.1191/0961203303lu470oa. [DOI] [PubMed] [Google Scholar]
- Lorton D, Lubahn C, Bellinger DL. Potential use of drugs that target neural–immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr. Drug Targets Inflamm. Allergy. 2003;2:1–30. doi: 10.2174/1568010033344499. [DOI] [PubMed] [Google Scholar]
- Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J. Neuroimmunol. 2006;179:26–36. doi: 10.1016/j.jneuroim.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Maric D, Millward JM, Ballok DA, Szechtman H, Barker JL, Denburg JA, Sakic B. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920:183–193. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- Marshall D, Dangerfield JP, Bhatia VK, Larbi KY, Nourshargh S, Haskard DO. MRL/lpr lupus-prone mice show exaggerated ICAM-1-dependent leucocyte adhesion and transendothelial migration in response to TNF-alpha. Rheumatology (Oxford) 2003;42:929–934. doi: 10.1093/rheumatology/keg251. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp. Neurol. 2001;169:219–230. doi: 10.1006/exnr.2001.7688. [DOI] [PubMed] [Google Scholar]
- McHale JF, Harari OA, Marshall D, Haskard DO. TNF-alpha and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J. Immunol. 1999;163:3993–4000. [PubMed] [Google Scholar]
- McIntyre KR, Ayer-LeLievre C, Persson H. Class II major histocompatibility complex (MHC) gene expression in the mouse brain is elevated in the autoimmune MRL/Mp-lpr/lpr strain. J. Neuroimmunol. 1990;28:39–52. doi: 10.1016/0165-5728(90)90039-p. [DOI] [PubMed] [Google Scholar]
- McLean BN, Miller D, Thompson EJ. Oligoclonal banding of IgG in CSF, blood–brain barrier function, and MRI findings in patients with sarcoidosis, systemic lupus erythematosus, and Behcet's disease involving the nervous system. J. Neurol., Neurosurg. Psychiatry. 1995;58:548–554. doi: 10.1136/jnnp.58.5.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray R, Keisler D, Kanuckel K, Izui S, Walker SE. Prolactin influences autoimmune disease activity in the female B/W mouse. J. Immunol. 1991;147:3780–3787. [PubMed] [Google Scholar]
- Mcnicholl JM, Glynn D, Mongey AB, Hutchinson M, Bresnihan B. Prospective study of neurophysiologic, neurologic and immunologic abnormalities in systemic lupus erythematosus. J. Rheumatol. 1994;21:1061–1066. [PubMed] [Google Scholar]
- Meroni PL, Tincani A, Sepp N, Raschi E, Testoni C, Corsini E, Cavazzana I, Pellegrini S, Salmaggi A. Endothelium and the brain in CNS lupus. Lupus. 2003;12:919–928. doi: 10.1191/0961203303lu503oa. [DOI] [PubMed] [Google Scholar]
- Miguel EC, Pereira RM, Pereira CA, Baer L, Gomes RE, de Sa LC, Hirsch R, de Barros NG, de Navarro JM, Gentil V. Psychiatric manifestations of systemic lupus erythematosus: clinical features, symptoms, and signs of central nervous system activity in 43 patients. Medicine. 1994;73:224–232. doi: 10.1097/00005792-199407000-00005. [DOI] [PubMed] [Google Scholar]
- Miller E. Immunosuppression—An overview. Semin. Vet. Med. Surg. (Small Anim.) 1997;12:144–149. doi: 10.1016/s1096-2867(97)80025-4. [DOI] [PubMed] [Google Scholar]
- Minagar A, Alexander JS. Blood–brain barrier disruption in multiple sclerosis. Mult. Scler. 2003;9:540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus. 2006;16:233–238. doi: 10.1002/hipo.20155. [DOI] [PubMed] [Google Scholar]
- Nagata S. Mutations in the Fas antigen gene in lpr mice. Semin. Immunol. 1994;6:3–8. doi: 10.1006/smim.1994.1002. [DOI] [PubMed] [Google Scholar]
- Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol. Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Narendran A, Hoffman SA. Identification of autoantibody reactive integral brain membrane antigens—A two dimensional analysis. J. Immunol. Methods. 1988;114:227–234. doi: 10.1016/0022-1759(88)90178-0. [DOI] [PubMed] [Google Scholar]
- Narendran A, Hoffman SA. Characterization of brain-reactive autoantibodies in murine models of systemic lupus erythematosus. J. Neuroimmunol. 1989;24:113–123. doi: 10.1016/0165-5728(89)90105-7. [DOI] [PubMed] [Google Scholar]
- Navarrete MG, Brey RL. Neuropsychiatric systemic lupus erythematosus. Curr. Treat. Options Neurol. 2000;2:473–485. doi: 10.1007/s11940-000-0045-7. [DOI] [PubMed] [Google Scholar]
- O'Sullivan FX, Vogelweid CM, Beschwilliford CL, Walker SE. Differential effects of CD4(+) T cell depletion on inflammatory central nervous system disease, arthritis and sialadenitis in MRL/lpr mice. J. Autoimmun. 1995;8:163–175. doi: 10.1006/jaut.1995.0013. [DOI] [PubMed] [Google Scholar]
- Omdal R, Selseth B, Klow NE, Husby G, Mellgren SI. Clinical neurological, electrophysiological, and cerebral CT scan findings in systemic lupus erythematosus. Scand. J. Rheumatol. 1989;18:283–289. doi: 10.3109/03009748909095031. [DOI] [PubMed] [Google Scholar]
- Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur. J. Neurol. 2005;12:392–398. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- Peress NS, Roxburgh VA, Gelfand MC. Binding sites for immune components in human choroid plexus. Arthritis Rheum. 1981;24:520–526. doi: 10.1002/art.1780240312. [DOI] [PubMed] [Google Scholar]
- Prendiville JS, Cabral DA, Poskitt KJ, Au S, Sargent MA. Central nervous system involvement in neonatal lupus erythematosus. Pediatr. Dermatol. 2003;20:60–67. doi: 10.1046/j.1525-1470.2003.03014.x. [DOI] [PubMed] [Google Scholar]
- Quismorio FP, Friou GJ. Antibodies reactive with neurons in SLE patients with neuropsychiatric manifestations. Int. Arch. Allergy Appl. Immunol. 1972;43:740–748. doi: 10.1159/000230889. [DOI] [PubMed] [Google Scholar]
- Reilly CM, Oates JC, Cook JA, Morrow JD, Halushka PV, Gilkeson GS. Inhibition of mesangial cell nitric oxide in MRL/lpr mice by prostaglandin J2 and proliferator activation receptor-gamma agonists. J. Immunol. 2000;164:1498–1504. doi: 10.4049/jimmunol.164.3.1498. [DOI] [PubMed] [Google Scholar]
- Resnick-Silverman L, Manfredi JJ. Gene-specific mechanisms of p53 transcriptional control and prospects for cancer therapy. J. Cell. Biochem. 2006;99:679–689. doi: 10.1002/jcb.20925. [DOI] [PubMed] [Google Scholar]
- Rice JS, Kowal C, Volpe BT, DeGiorgio LA, Diamond B. Molecular mimicry: anti-DNA antibodies bind microbial and nonnucleic acid self-antigens. Curr. Top. Microbiol. Immunol. 2005;296:137–151. doi: 10.1007/3-540-30791-5_8. [DOI] [PubMed] [Google Scholar]
- Rivest S, Torres G, Rivier C. Differential effects of central and peripheral injection of interleukin-1 beta on brain c-fos expression and neuroendocrine functions. Brain Res. 1992;587:13–23. doi: 10.1016/0006-8993(92)91424-d. [DOI] [PubMed] [Google Scholar]
- Rivier C, Rivest S. Mechanisms mediating the effects of cytokines on neuroendocrine functions in the rat Corticotropin-Releasing Factor. John Wiley & Sons; Chichester: 1993. pp. 204–225. [DOI] [PubMed] [Google Scholar]
- Rocca MA, Agosta F, Mezzapesa DM, Ciboddo G, Falini A, Comi G, Filippi M. An fMRI study of the motor system in patients with neuropsychiatric systemic lupus erythematosus. NeuroImage. 2006;30:478–484. doi: 10.1016/j.neuroimage.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Kesner RP. A computational theory of hippocampal function, and empirical tests of the theory. Prog. Neurobiol. 2006;79:1–48. doi: 10.1016/j.pneurobio.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav. Immun. 1992;6:265–285. doi: 10.1016/0889-1591(92)90048-s. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Carbotte RM, Denburg JA. Brain-reactive antibodies and behavior of autoimmune MRL–lpr mice. Physiol. Behav. 1993a;54:1025–1029. doi: 10.1016/0031-9384(93)90319-b. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Carbotte RM, Denburg JA. Spatial learning during the course of autoimmune disease in MRL mice. Behav. Brain Res. 1993b;54:57–66. doi: 10.1016/0166-4328(93)90048-u. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL–lpr mice. Physiol. Behav. 1994;56:609–617. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Denburg JA. Immunosuppressive treatment prevents behavioral deficit in autoimmune MRL–lpr mice. Physiol. Behav. 1995;58:797–802. doi: 10.1016/0031-9384(95)00135-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Denburg JA, Denburg SD, Szechtman H. Blunted sensitivity to sucrose in autoimmune MRL–lpr mice: a curve-shift study. Brain Res. Bull. 1996a;41:305–311. doi: 10.1016/s0361-9230(96)00190-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Stead R, Denburg JA. Joint pathology and behavioral performance in autoimmune MRL–lpr mice. Physiol. Behav. 1996b;60:901–905. doi: 10.1016/0031-9384(96)00065-0. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA. Neurobehavioral alteration in autoimmune mice. Neurosci. Biobehav. Rev. 1997;21:327–340. doi: 10.1016/s0149-7634(96)00018-8. [DOI] [PubMed] [Google Scholar]
- Sakic B, Gurunlian L, Denburg SD. Reduced aggressiveness and low testosterone levels in autoimmune MRL–lpr males. Physiol. Behav. 1998a;63:305–309. doi: 10.1016/s0031-9384(97)00422-8. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL–lpr mice. J. Neuroimmunol. 1998b;87:162–170. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Laflamme N, Crnic LS, Szechtman H, Denburg JA, Rivest S. Reduced corticotropin-releasing factor and enhanced vasopressin gene expression in brains of mice with autoimmunity-induced behavioral dysfunction. J. Neuroimmunol. 1999;96:80–91. doi: 10.1016/s0165-5728(99)00021-1. [DOI] [PubMed] [Google Scholar]
- Sakic B, Kolb B, Whishaw IQ, Gorny G, Szechtman H, Denburg JA. Immunosuppression prevents neuronal atrophy in lupus-prone mice: evidence for brain damage induced by autoimmune disease? J. Neuroimmunol. 2000a;111:93–101. doi: 10.1016/s0165-5728(00)00364-7. [DOI] [PubMed] [Google Scholar]
- Sakic B, Maric I, Koeberle PD, Millward JM, Szechtman H, Maric D, Denburg JA. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J. Neuroimmunol. 2000b;104:147–154. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- Sakic B, Lacosta S, Denburg J, Szechtman H. Altered neurotransmission in brains of autoimmune mice: pharmacological and neurochemical evidence. J. Neuroimmunol. 2002;129:84–96. doi: 10.1016/s0165-5728(02)00171-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Hanna SE, Millward JM. Behavioral heterogeneity in an animal model of neuropsychiatric lupus. Biol. Psychiatry. 2005a;57:679–687. doi: 10.1016/j.biopsych.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakic B, Kirkham DL, Ballok DA, Mwanjewe J, Fearon IM, Macri J, Yu G, Sidor MM, Denburg JA, Szechtman H, Lau J, Ball AK, Doering LC. Proliferating brain cells are a target of neurotoxic CSF in systemic autoimmune disease. J. Neuroimmunol. 2005b;169:68–85. doi: 10.1016/j.jneuroim.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeed I, Guo Q, Hoffman SW, Stein DG. Allopregnanolone, a progesterone metabolite, is more effective than progesterone in reducing cortical infarct volume after transient middle cerebral artery occlusion. Ann. Emerg. Med. 2006;47:381–389. doi: 10.1016/j.annemergmed.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Schwartz MM, Roberts JL. Membranous and vascular choroidopathy: two patterns of immune deposits in systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1983;29:369–380. doi: 10.1016/0090-1229(83)90040-5. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Joseph FG. The neuropathology and pathogenesis of systemic lupus erythematosus. Neuropathol. Appl. Neurobiol. 2002;28:173–189. doi: 10.1046/j.1365-2990.2002.00406.x. [DOI] [PubMed] [Google Scholar]
- Shanks N, Moore PM, Perks P, Lightman SL. Alterations in hypothalamic–pituitary–adrenal function correlated with the onset of murine SLE in MRL+/+ and lpr/lpr mice. Brain Behav. Immun. 1999;13:348–360. doi: 10.1006/brbi.1998.0535. [DOI] [PubMed] [Google Scholar]
- Shear DA, Galani R, Hoffman SW, Stein DG. Progesterone protects against necrotic damage and behavioral abnormalities caused by traumatic brain injury. Exp. Neurol. 2002;178:59–67. doi: 10.1006/exnr.2002.8020. [DOI] [PubMed] [Google Scholar]
- Sherman GF, Galaburda AM, Behan PO, Rosen GD. Neuroanatomical anomalies in autoimmune mice. Acta Neuropathol. (Berl.) 1987;74:239–242. doi: 10.1007/BF00688187. [DOI] [PubMed] [Google Scholar]
- Sherman GF, Morrison L, Rosen GD, Behan PO, Galaburda AM. Brain abnormalities in immune defective mice. Brain Res. 1990;532:25–33. doi: 10.1016/0006-8993(90)91737-2. [DOI] [PubMed] [Google Scholar]
- Sibbitt WL, Sibbitt RR. Magnetic resonance spectroscopy and positron emission tomography scanning in neuropsychiatric systemic lupus erythematosus. Rheum. Dis. Clin. North Am. 1993;19:851–868. [PubMed] [Google Scholar]
- Sibbitt WL, Haseler LJ, Griffey RH, Hart BL, Sibbitt RR, Matwiyoff NA. Analysis of cerebral structural changes in systemic lupus erythematosus by proton MR spectroscopy. AJNR Am. J. Neuroradiol. 1994;15:923–928. [PMC free article] [PubMed] [Google Scholar]
- Sidor MM, Sakic B, Malinowski PM, Ballok DA, Oleschuk CJ, Macri J. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J. Neuroimmunol. 2005;165:104–113. doi: 10.1016/j.jneuroim.2005.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer GG, Carrera AC, Marshakrothstein A, Martineza C, Abbas AK. Apoptosis, fas and systemic autoimmunity: the MRL-Ipr/Ipr model. Curr. Opin. Immunol. 1994;6:913–920. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Spangelo BL, Gorospe WC. Role of the cytokines in the neuroendocrine–immune system axis. Front. Neuroendocrinol. 1995;16:1–22. doi: 10.1006/frne.1995.1001. [DOI] [PubMed] [Google Scholar]
- Suh SW, Fan Y, Hong SM, Liu Z, Matsumori Y, Weinstein PR, Swanson RA, Liu J. Hypoglycemia induces transient neurogenesis and subsequent progenitor cell loss in the rat hippocampus. Diabetes. 2005;54:500–509. doi: 10.2337/diabetes.54.2.500. [DOI] [PubMed] [Google Scholar]
- Svenungsson E, Andersson M, Brundin L, van Vollenhoven R, Khademi M, Tarkowski A, Greitz D, Dahlstrom M, Lundberg I, Klareskog L, Olsson T. Increased levels of proinflammatory cytokines and nitric oxide metabolites in neuropsychiatric lupus erythematosus. Ann. Rheum. Dis. 2001;60:372–379. doi: 10.1136/ard.60.4.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szechtman H, Sakic B, Denburg JA. Behaviour of MRL mice: an animal model of disturbed behaviour in systemic autoimmune disease. Lupus. 1997;6:223–229. doi: 10.1177/096120339700600302. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN. Murine models of lupus, Systemic Lupus Erythematosus. 2nd ed. Churchill Livingstone; New York: 1992. pp. 121–194. [Google Scholar]
- Tincani A, Brey R, Balestrieri G, Vitali C, Doria A, Galeazzi M, Meroni PL, Migliorini P, Neri R, Tavoni A, Bombardieri S. International survey on the management of patients with SLE: 2. The results of a questionnaire regarding neuropsychiatric manifestations. Clin. Exp. Rheumatol. 1996;14:S23–S29. [PubMed] [Google Scholar]
- Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci. Lett. 2001a;302:129–132. doi: 10.1016/s0304-3940(01)01679-2. [DOI] [PubMed] [Google Scholar]
- Tomita M, Holman BJ, Williams LS, Pang KC, Santoro TJ. Cerebellar dysfunction is associated with overexpression of proinflammatory cytokine genes in lupus. J. Neurosci. Res. 2001b;64:26–33. doi: 10.1002/jnr.1050. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Tsai ST, Chen KH, Thajeb P, Lin WM, Yu HS, Yu CL. Cerebrospinal fluid interleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand. J. Rheumatol. 1994;23:57–63. doi: 10.3109/03009749409103028. [DOI] [PubMed] [Google Scholar]
- Valesini G, Alessandri C, Celestino D, Conti F. Anti-endothelial antibodies and neuropsychiatric systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2006;1069:118–128. doi: 10.1196/annals.1351.010. [DOI] [PubMed] [Google Scholar]
- van Dam AP. Diagnosis and pathogenesis of CNS lupus. Rheumatol. Int. 1991;11:1–11. doi: 10.1007/BF00290244. [DOI] [PubMed] [Google Scholar]
- Vincent A, Dalton P, Clover L, Palace J, Lang B. Antibodies to neuronal targets in neurological and psychiatric diseases. Ann. N. Y. Acad. Sci. 2003;992:48–55. doi: 10.1111/j.1749-6632.2003.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J. Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Warner N, McIntyre R, Valentine A, Kulkarni M, Mullani N, Gould L. Cerebral involvement in systemic lupus erythematosus. Am. J. Physiol. Imaging. 1988;3:91–98. [PubMed] [Google Scholar]
- Walker SE. Bromocriptine treatment of systemic lupus erythematosus. Lupus. 2001;10:762–768. doi: 10.1191/096120301717165010. [DOI] [PubMed] [Google Scholar]
- Walker SE. Impaired hypothalamic function, prolactinomas, and autoimmune diseases. J. Rheumatol. 2006;33:1036–1037. [PubMed] [Google Scholar]
- Walker SE, Smarr KL, Parker JC, Weidensaul DN, Nelson W, McMurray RW. Mood states and disease activity in patients with systemic lupus erythematosus treated with bromocriptine. Lupus. 2000;9:527–533. doi: 10.1177/096120330000900709. [DOI] [PubMed] [Google Scholar]
- Waterloo K, Omdal R, Jacobsen EA, Klow NE, Husby G, Torbergsen T, Mellgren SI. Cerebral computed tomography and electroencephalography compared with neuropsychological findings in systemic lupus erythematosus. J. Neurol. 1999;246:706–711. doi: 10.1007/s004150050436. [DOI] [PubMed] [Google Scholar]
- Weller RO, Mitchell J, Griffin RL, Gardner MJ. The effects of hydrocephalus upon the developing brain. Histological and quantitative studies of the ependyma and subependyma in hydrocephalic rats. J. Neurol. Sci. 1978;36:383–402. doi: 10.1016/0022-510x(78)90046-1. [DOI] [PubMed] [Google Scholar]
- Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA, III, Koffler D. Intrathecal IgG synthesis and blood–brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am. J. Med. 1983;74:837–844. doi: 10.1016/0002-9343(83)91075-6. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–231. doi: 10.1016/0006-8993(90)90778-a. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernandez AP, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi H, Fukuyama H, Ogawa M, Ouchi Y, Kimura J. Callosal atrophy in patients with lacunar infarction and extensive leukoaraiosis. An indicator of cognitive impairment. Stroke. 1994;25:1788–1793. doi: 10.1161/01.str.25.9.1788. [DOI] [PubMed] [Google Scholar]
- Yanase K, Madaio MP. Nuclear localizing anti-DNA antibodies enter cells via caveoli and modulate expression of caveolin and p53. J. Autoimmun. 2005;24:145–151. doi: 10.1016/j.jaut.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Yoshio T, Hirata D, Onda K, Nara H, Minota S. Antiribosomal P protein antibodies in cerebrospinal fluid are associated with neuropsychiatric systemic lupus erythematosus. J. Rheumatol. 2005;32:34–39. [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. Immunoglobulin binding to brain in autoimmune mice. J. Neuroimmunol. 2001;120:10–18. doi: 10.1016/s0165-5728(01)00412-x. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J. Neuroimmunol. 2003;142:67–74. doi: 10.1016/s0165-5728(03)00262-5. [DOI] [PubMed] [Google Scholar]
- Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7925–7930. doi: 10.1073/pnas.1131955100. [DOI] [PMC free article] [PubMed] [Google Scholar]