

Abstract
Cytochrome P450 1B1 (Cyp1b1) metabolism contributes to physiological functions during embryogenesis, but also to carcinogenic activation of polycyclic aromatic hydrocarbons (PAH). We generated Cyp1b1-deficient mice carrying the Min allele of the Adenomatous polyposis coli gene. These Cyp1b1-deficient Min mice developed twice as many tumors as Min controls, which, however, remained similar in size and histology. Tumors from older (130 day) Cyp1b1-deficient Min mice exhibited focal areas of nuclear atypia associated with less organized epithelia. The metabolism of endogenous substrates by Cyp1b1, therefore, suppresses tumor initiation, but also affects progression. Treatment of Min mice with 7,12-dimethylbenzanthracene (DMBA) doubled both tumor multiplicity and size within 20 days, but not when mice lacked Cyp1b1. This was paralleled by an abnormal staining of crypts with β catenin, phospho-IKK, and ReIA, which may represent an early stage of tumorigenesis similar to aberrant crypt formation. Cyp1b1 deletion did not affect circulating DMBA and metabolites. Cyp1b1 expression was higher in the tumors compared to normal small intestines. Increased tumorigenesis may, therefore, arise from generation of DMBA metabolites by Cyp1b1 in the developing tumors. Benzo(a)pyrene (BP), which is similarly activated by Cyp1b1 in vitro, did not affect tumorigenesis in Min mice. By contrast, BP and DMBA each suppressed tumor multiplicity in absence of Cyp1b1. Cyp1b1 metabolism of DMBA and endogenous oxygenation products may each impact a tumor promoting NF-κB. activation, whereas Ah receptor activation by PAH effects suppression. Tumorigenesis may, therefore, depend on activation of PAH by Cyp1b1, and on off-setting suppression by Cyp1b1 of endogenous tumor-enhancing substrates.
Keywords: Cytochrome P450 1B1 (Cyp1b1, mouse; CYP1B1, human), Adenomatous polyposis coli (Apc), Polycyclic aromatic hydrocarbons (PAH), DMBA, and Intestinal neoplasia
Introduction
Colorectal cancer is the second leading cause of cancer death in the United States. This disease is the culmination of epigenetic and genetic alterations that initiate tumorigenesis and drive progression. An early molecular event is the inactivation of the Adenomatous polyposis coli (Apc) gene (1). The role of Apc in maintaining homeostasis is best exemplified by the existence of familial adenomatous polyposis (FAP). These patients with this hereditary form of the disease develop hundreds to thousands of colonic tumors by the second decade of life.
Apc maintains homeostasis in the intestine as an integral component of Wnt signaling (2), which disrupts the cytoplasmic complex of Apc with β-catenin, Axin, and GSK3β. This prevents GSK-initiated proteolosis of β-catenin, enhances translocation to the nucleus, and stimulates transcription of target genes, including c-Myc, Cyclin D, MMP-9, PPARδ, and Sox-9. β-catenin also directly contributes to epithelial cell adhesion. Loss of Apc activity and elevated β-catenin drastically alters the intestinal epithelium; including expanding the stem cell population. c-Myc is a key mediator of these dramatic biological changes (3). Array analysis of several colon cancer models demonstrates that mouse and human tumors recapitulate embryonic developmental signatures, irrespective of etiology (3).
Several mouse models of FAP have been developed. Mice carrying the Min allele of Apc express an altered form of Apc protein, lacking the C-terminal domains that bind Axin, β-catenin, and GSK3β (2). C57BL6/J (B6) mice carrying Min develop on average 100 tumors along the entire length of the intestinal tract. Several genes involved in polyunsaturated fatty acid signaling, including secreted phospholipase A2, Cox1, Cox2, LpI, PPARδ, and PPARγ, affect the multiplicity of intestinal tumors in Min mice (4–6).
Carcinogens also affect intestinal tumorigenesis in Min mice. Some carcinogens affect tumorigenesis in specific regions of the intestine. Treatment of Min mice with ethylnitrosourea (ENU), a direct acting alkylating agent, increased the multiplicity of small intestinal tumors by a factor of 3 (7). The tumors from these mice often carried a point mutation that inactivated the wildtype allele of Apc. Similarly, treatment of Min mice with 2-amino-1-methyl-6-phenylimidazol [4,5-B] pyridine (PhIP) increased the multiplicity of small intestinal tumors, by inactivating the wildtype allele through a point mutation, and in part through inducing genetic instability (8). Other carcinogens specifically affect tumorigenesis in the colon. Treatment of Min mice with azoxymethane (AOM) increased the incidence and multiplicity of colonic tumors (9). The majority of tumors from these mice exhibited loss of heterozygosity (LOH) at the Apc locus. Thus, exogenous chemicals can affect intestinal tumorigenesis in Min mice by promoting the inactivation of the wildtype allele of Apc through variety of distinct mechanisms. Most carcinogens, including PhIP and AOM, must be metabolically activated by cytochrome P450s and other metabolizing enzymes before exerting their effect on tumorigenesis (10). In this paper, we treat Min mice with polycyclic aromatic hydrocarbons (PAHs) for the first time. These chemicals, to which humans are extensively exposed, cause many cancers through their conversion to reactive dihydrodiol epoxides (11).
CYP1B1 is often more highly expressed in neoplasms, notably those of the bladder, breast, colon, kidney, ovaries, and prostate than in the adjacent normal epithelium (12–14). Cytochrome P450 1B1 (CYP1B1)-mediated conversion of estradiol to 4-OH estradiol has been linked to a variety of human endocrine cancers (13–14). Certain allelic variants of CYP1B1 have been further associated with increased estradiol 4-hydroxylation and increased risk for colon cancer, and elevated CYP1B1 expression is a marker for more aggressive colon tumors (15). Mouse Cyp1b1, which differs from human CYP1B1 by 20 percent (amino acid level), demonstrates appreciably different PAH metabolism and much less estradiol metabolism (16).
The CYP1B1 gene is scarcely expressed in liver, but is frequently found in extra-hepatic tissues (17). Cyp1b1 was initially purified from mouse embryonic fibroblasts (17) was first cloned from human keratinocytes (18). Cloning from the rodent tissue established that the same form exhibited very diverse expression (17). The gene has a very unusual structure, containing a promoter with multiple hormonal control elements, only two introns, and a highly extended 3′ UTR (19). Cyp1b1 is regulated by a highly conserved 200 bp enhancer that responds to the Ah receptor, to cAMP through a far upstream enhancer, and also through a miRNA that targets the 3′UTR. This latter mechanism has been linked to the elevated expression in tumors (20).
The effect of Cyp1b1 on carcinogenesis in mice is tissue specific (21). Treatment of wildtype mice with 7,12-dimethylbenzanthracene (DMBA) induces tumors in several organs, including ovary, skin, uterus, and lung, which occur less frequently in Cyp1b1-deficient mice. DMBA and benzo(a)pyrene (BP) bind with high affinity to recombinant mouse Cyp1b1 forming typical substrate complexes (16). DMBA and BP are converted to dihydrodiol epoxides by Cyp1b1 (22). Formation of tumors parallels formation of DNA-DMBA dihydrodiol adducts in the responsive tissues (21). By contrast, in these same studies, Cyp1b1 suppressed DMBA-induced lung tumors. This tissue specificity, therefore, may reflect a cancer-inducing role for endogenous substrates of Cyp1b1. Physiological substrates are implicated in the highly conserved expression during early development in mice, which plays a role in embryonic patterning (23–24). A lack of Cyp1b1 activity in mice and humans causes aberrant eye development (23,25).
We describe the multiplicity, size, and pathology of tumors throughout the intestinal tract of Cyp1b1-deficient Min mice and Min controls, as well as the effects of treating both groups of mice with DMBA or BP, which are similarly mutagenic through dihydrodiol epoxides in vitro (22). We use two intraperitoneal (IP) injections at times that correspond to ongoing intestinal development. This mode of administration provides a sustained systemic exposure that can be compared to the previous studies. The reported experiments provide evidence for opposing effects of Cyp1b1 on intestinal tumorigenesis, derived from the metabolism of endogenous substrates and the activation of PAH. The role of endogenous substrates is particularly notable, owing to our recent finding that a lack of Cyp1b1 also substantially affects adiposity (Larsen and Jefcoate, unpublished) and the manner in which fatty acid metabolism impacts intestinal tumorigenesis in the Min mouse. A simple model is developed to account for these paradoxical findings that involves opposing contributions of Cyp1b1 to the tumorigenic process through metabolism of the PAH and endogenous substrates.
Methods
Mouse breeding and maintenance
Mice were maintained in the AAALAC accredited University of Wisconsin School of Medicine and Public Health Animal Care Facility. All experiments were carried out in accordance with protocols approved by the Animal Care and Use Committee in accordance to the NIH Guide for the Care and Use of Laboratory Animals. All mice were provided food and water ad libitum and were maintained on a 12 hour light/dark cycle. The mice were fed 2019 Teklad Global 19% Protein Extruded Rodent Diet, which is essentially free of flavonoids. The initial C57BL/6 (B6) ApcMin/+ founders were obtained from Dr. Amy Moser at the University of Wisconsin (26). A colony was maintained by breeding wildtype B6 females with B6 ApcMin/+ males. The initial B6 CYP1B1−/− founders were obtained from Dr. Frank Gonzalez (21). A colony was maintained by intercrossing CYP1B1−/− offspring. To produce B6 ApcMin/+ CYP1B1−/− mice, B6 CYP1B1−/− females were first bred to B6 ApcMin/+ males and then B6 CYP1B1−/− females were bred to B6 CYP1B1+/− ApcMin/+ males. Offspring were genotyped prior to 21 days of age, in accordance to previously published protocols (7,21).
Treatments
BP, DMBA, and olive oil were purchased from either Sigma Chemical Company (St. Louis, MO) or Accustandard, Inc. (New Haven, CT). Female mice were injected IP on days 28 and 35 with 0.1 mg of either BP or DMBA (or other doses as indicated) in olive oil (200 uL, 0.5 mg/ml) or with vehicle alone. Eight to ten mice from three to four litters were treated over an extended period of time. In one experiment, DMBA was injected as a single dose of 0.2 mg on day 28. Mice were sacrificed on days 55, 85, or 130, as specified.
Tumor analysis
Mice were sacrificed by CO2 asphyxiation. The intestinal tissues were removed and rinsed in PBS. Four centimeter segments of proximal, medial, and distal small intestine, as well as the entire colon were placed on Whatman filter paper. Each tissue sample was opened longitudinally, rinsed with 1× PBS to wash away luminal contents, and fixed overnight in 4% paraformaldehyde solution at room temperature. After 16 hours, the tissue samples were carefully removed from the filter paper and stored in 75% ethanol at room temperature. Tumor multiplicity was determined by two investigators, who were both blinded to the genotype of the animal and treatment, using an Olympus dissecting microscope at 10× magnification. The maximum diameter of each tumor was measured using a calibrated eyepiece reticule.
Statistical Analysis
Tumor multiplicity and size were compared among different groups of mice using the Wilcoxon Rank Sum Test (4).
Histology
Intestinal tumors and adjacent normal tissue were analyzed by immunohistochemistry. The tissues were embedded in paraffin and serially cut into a sequential series of 10 micrometer sections. Sections were stained with hemetoxylin and eosin, β-catenin (27), phospho-IKK (Cell Signaling Technologies) (28), and p65 (ReIA) (Cell Signaling Technologies) (29), following the manufacturer’s protocol. A minimum of four tumors from each treatment were subjected to detailed pathological analysis.
In situ Hybridization
Paraffin sections of intestinal tissue were hybridized with either a sense or antisense 35S-radiolabeled Cyp1a1 and Cyp1b1 riboprobe, as described previously (30). The Cyp1a1 probe spanned nucleotides 5562 to 6212 and Cyp1b1 spanned nucleotides 752 to 1489. After development, sections were counter-stained with propidium iodide and images were documented under darkfield and epifluorescent illumination. Using this method, strong expression of Cyp1a1 was observed in the liver and lung of DMBA-treated mice, as described previously (30).
Results
CYP1B1 is overexpressed in many types of neoplasms, including human colorectal tumors. We, therefore, sought to test whether Cyp1b1 activity contributes to the development of intestinal tumors in Min mice. Cyp1b1-deficient Min mice and Min controls were sacrificed at 55 days of age and the intestinal tract was removed. Tumors were scored in three segments of the small intestine corresponding to the first 4 cm nearest the stomach (proximal), 4 cm in the middle (medial), and the last 4 cm nearest the cecum (distal), as well as the entire colon. A lack of Cyp1b1 dramatically affected tumor multiplicity along the entire length of the intestinal tract in Min mice (Figure 1A–C and Table 1). Cyp1b1-deficient Min developed on average 37 ± 18 intestinal tumors, whereas Min controls developed on average of 15 ± 9. This difference is statistically significant (p=0.02; two-sided Wilcoxon analysis). Thus, a lack of Cyp1b1 metabolism of endogenous substrates clearly enhances the early stages of tumorigenesis. Tumors in Cyp1b1-deficient Min mice were similar in size to those in Min controls, either at 55 or 85 days of age (Figure 2A). Many tumors from 85 day old mice exceeded 1 mm in maximum diameter, a size that requires angiogenesis (5). Thus, a lack of Cyp1b1 activity does not affect tumor expansion.
Figure 1. A deficiency of Cyp1b1 increases tumorigenesis throughout the entire intestinal tract of Min mice, but leads to a suppression of tumorigenesis by DMBA and BP.
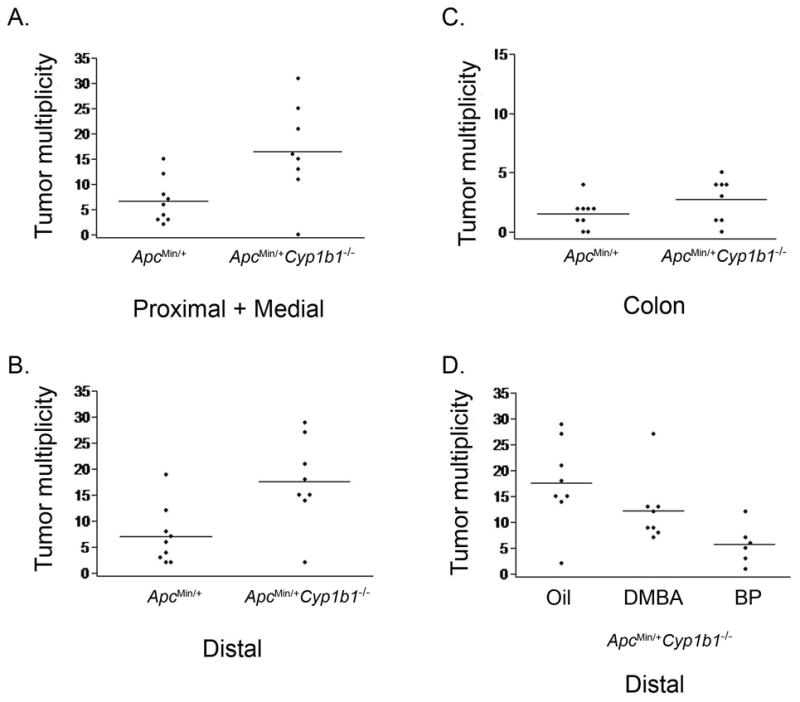
Tumors were scored in the proximal, medial, and distal regions of the small intestine and the entire colon of Cyp1b1-deficient Min mice and Min controls (panels A–C) or the distal region of Cyp1b1-deficient mice treated with either oil, DMBA or BP (panel D).
Table 1.
Tumor multiplicity in ApcMin/+and ApcMin/+Cyp1b1−/− mice.
| Mean tumor count
|
|||||||
|---|---|---|---|---|---|---|---|
| Genotype | Treatment | N of mice | Proximal SI | Medial SI | Distal SI | Colon | Total tumor count, mean ± SD |
| ApcMin/+ | Oil | 9 | 3 | 4 | 7 | 2 | 15±10 |
| DMBA | 10 | 3 | 7† | 12† | 1 | 23±11 | |
| BP | 10 | 3 | 4 | 8 | 2 | 17±11 | |
| ApcMin/+ Cyp1b1−/− | Oil | 8 | 6 | 11 | 18 | 3 | 37*±18 |
| DMBA | 8 | 3 | 7‡ | 12‡ | 2 | 24±8 | |
| BP | 6 | 5 | 8 | 6 | 1 | 20±8 | |
Significantly different from oil-treated ApcMin/+ mice (p=0.02).
Significantly different from oil-treated ApcMin/+ mice (p=0.03)
Significantly different from oil-treated ApcMin/+ Cyp1b1−/− mice (p<0.05).
Mice received PAH injections (0.1 mg each) on days 28 and 35 of age and were sacrificed at 55 days of age. SI, small intestine.
Figure 2. The size of intestinal tumors is increased by treating Min mice with DMBA.

The maximum diameter was determined for tumors in the distal region of the small intestine from several different genetic backgrounds and treatment groups. A lack of Cyp1b1 activity did not affect tumor size in Min mice at 55 and 85 days of age (panel A), whereas treatment with DMBA (D) increased tumor size in Min mice at 55 days of age, relative to oil-treated controls (C), with the significant percentage having a maximum diameter between 0.5 and 1 mm (panels B and C; ▲ DMBA, ■ BP, ◆ oil-treated control).
Min tumors have been extensively examined in many laboratories and are uniformly adenomas, which exhibit a relatively uniform progression (4–5,7) that was seen here in each treatment group. We used antibody staining for β catenin (Figure 3, left), which is increased by Apc LOH, and for phospho-IKK (Figure 3, center) and ReIA (Figure 3, panel B, right), which each mediate NF-κB activation (28), a key participant in intestinal cancer (31). β catenin is also a component of E-Cadherin cell-cell adhesion complexes, and strong inter-epithelial β catenin complexes are prevalent in the luminal epithelia of normal villi and also on the surface of the tumors (brown laddering feature, panel B). β catenin is low within these day 55 Min tumors, but increases in Cyp1b1−/− tumors (panel A). There was a general trend for increased levels of phospho-IKK in the day 55 Cyp1b1−/− tumors, both in the normal luminal borders and tumor epithelia (Figure 3, panel A (center) and enlarged inserts (right)). Surprisingly, ReIA, which is released from NF-κB complexes by IKK, was low in the border villi epithelia, irrespective of Cyp1b1 status (panel B). We observed abundant punctuate, nuclear ReIA in the vascular region of the normal villi (not shown).
Figure 3. Effects of Cyp1b1 deletion and DMBA treatment on the histology of Min tumors at days 55 and 130.
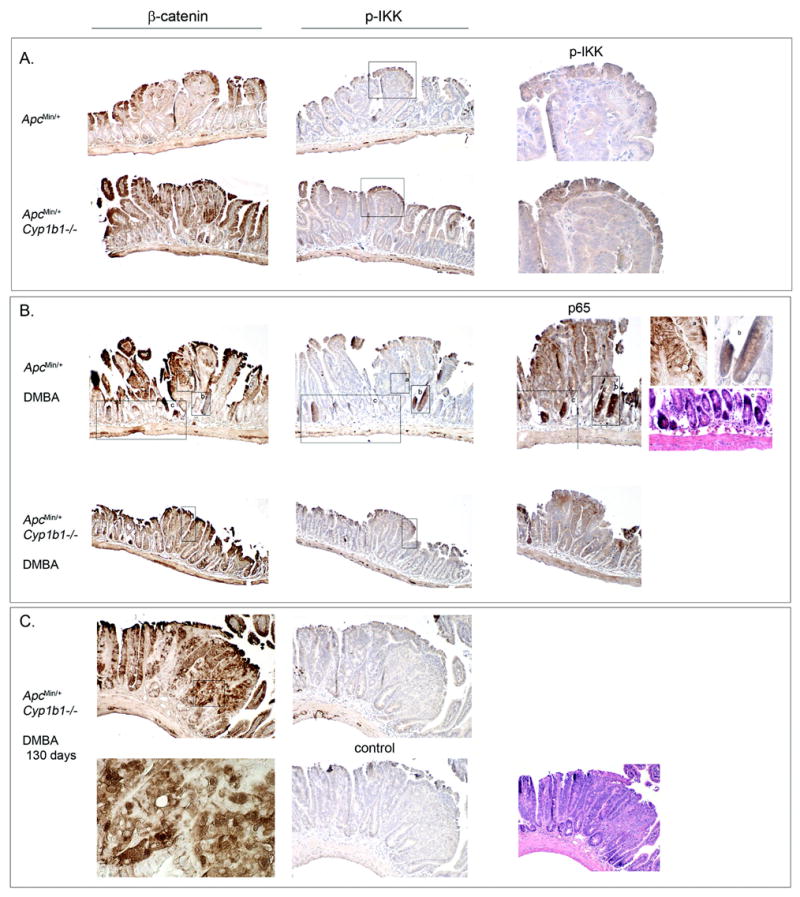
Mice were sacrificed at either 55 days of age (panels A and B) or 130 days of age (panel C). Tumors were isolated from day 55 oil-treated Min and Cyp1b1-deficient Min control mice (panel A), day 55 DMBA-treated Min and Cyp1b1-deficient Min mice (panel B), and 130 day old DMBA-treated Cyp1B1-deficient Min mice (panel C). Sections were stained with hematoxylin and eosin (column 3 inserts) or β catenin (left), phospho-IKK (center), or RelA (right insert) (each dark brown), as indicated. In panel A (right), pIKK staining is shown in the enlarged regions (4x; boxes shown). In panel B (right) 4x enlargements are shown for boxed regions a-c stained with anti-p65, anti-β catenin, anti-pIKK, and H/E, as indicated. Panel C (lower row) also shows 4x enlargement of the anti-β catenin staining (left, box shown), an antibody-deleted control (center) and an H/E stained section (right). Panel B shows anti-p65 (RelA) staining of DMBA-treated ApcMin/+ (upper) and DMBA-treated ApcMin/+Cyp1b1−/− (lower).
β catenin stained luminal surface epithelia as a laddering of cell junctional E-cadherin/b catenin complexes (panel B, left images of DMBA-treated ApcMin/+ and ApcMin/+ Cyp1b1−/−tissue). Typically, the proportion of β catenin stained cells within tumors (indicative of Apc-LOH) increased with tumor enlargement, but was also higher in tumors of similar size from Cyp1b1−/− Min mice. DMBA treatment of Min mice produced many enlarged tumors at day 55 (panel B) that were typical of Min tumors seen at about day 100. Tumors from DMBA treatment of Cyp1b1-deficent Min mice, again, were mostly typical of normal Min tumors at day 55 (panel B). Tumors from DMBA treatment of Min mice showed strong staining for phospho-IKK, β catenin, ReIA, and hemotoxylin in about a third of the crypts, although not specifically in the tumors and with only partial overlap of these responses. The larger tumors seen at day 130 commonly showed regions of disorganized growth, exhibiting nuclear atypia, intense β catenin staining with common nuclear localization (enlarged in panel C), and absence of phospho-IKK. These atypia regions were also seen in tumors from these Cyp1b1-deficient mice without DMBA treatment (not shown), but not in the equivalent day 55 tumors or in Cyp1b1+/+ Min mice.
We next tested whether Cyp1b1 metabolism of DMBA, affects intestinal tumorigenesis in Min mice. Moser and her colleagues found that a high dose of DMBA was toxic to Min mice, presumably because of the high tumor burden in the intestine (26). We determined the toxicity of DMBA across a range of doses at days 28 and 35 of age. When mice were administered a dose of 0.1 mg of DMBA at 28 and 35 days of age, the majority of Min mice (10/11) survived to 55 days of age, but typically failed to survive beyond 75 days, even with lower doses (0.02 mg/Kg). The mice typically exhibited extensive peritoneal inflammation. Importantly, all Cyp1b1-deficient Min mice survived to 100 days of age, regardless of treatment.
Treatment with DMBA (0.1.mg, days 28 and 35) substantially affected both the multiplicity (Figure 1D and Table 1) and size (Figure 2B and 2C) of intestinal tumors, even within a mere 20 days of the second administration. DMBA stimulated twice as many tumors in the medial and distal regions of the small intestine as oil-treated Min animals, wildtype for Cyp1b1 (Min controls) (19 ± 10 versus 11 ± 9), but was ineffective in the proximal segment. The difference is statistically significant (p=0.03; one-sided Wilcoxon analysis). DMBA doubled the average maximum tumor diameter at 55 days of age and greatly increased the proportion of large tumors (Figures 2B and 2C). In addition, over 20% of the tumors in DMBA-treated Min mice exceeded 1 mm in maximum diameter, whereas none reached this size in the Min controls (p<0.0001). When the same total amount of DMBA (0.2 mg) was administered as a single dose on day 28, the number and size of tumors in the most responsive distal region was the same as in the control-treated Min mice (Figure 2B). Consequently, a second DMBA exposure appears to be an essential part of the carcinogenesis process. Min mice similarly treated with BP showed no changes in multiplicity (Table 1) or size (Figure 2C) of the tumors. BP and DMBA are similarly toxic to a variety of cells in vitro. BP, however, is similarly less toxic than DMBA in bone marrow (30).
We compared the effect of DMBA on the multiplicity and size distribution of intestinal tumors in Cyp1b1-deficient Min mice. DMBA-treated Cyp1b1-deficient mice actually developed significantly fewer tumors in the medial and distal region than vehicle-treated mice (19 ± 8 versus 29 ± 15 tumors; Figure 1A and B and Table 1). The size of the tumors in the distal region was also unchanged (diameter 0.54 ± 0.23 mm versus 0.63 ± 0.32 mm; Figure 2B). Together, these observations indicate that the stimulatory effect of DMBA on intestinal tumor size in Min mice is eliminated when Cyp1b1 activity is removed. The appreciable decrease in tumor multiplicity produced by DMBA in Cyp1b1-deficient Min mice indicates a strong DMBA suppression effect in absence of CYP1B1 metabolism. There was no net effect of Cyp1b1 deficiency in the presence of DMBA (Table 1, line 2 versus 5). This arises because the effect of Cyp1b1 deficiency on endogenous regulation (line 4 versus 1) is opposite to the effect of Cyp1b1 deficiency on DMBA activity (line 5 versus 4). DMBA, in absence of metabolism by Cyp1b1, appears to prevent the stimulation produced by loss of endogenous metabolism. An equivalent BP treatment did not affect Min tumorigenesis, but, surprisingly, BP was at least as effective as DMBA in decreasing tumor multiplicity in Cyp1b1-deficient Min mice (Figure 1D and Table 1). This suppression by DMBA and BP may derive from a tumor suppression effect resulting from activation of the Ah receptor (AhR), which is appreciable in PAH-treated mouse colon and small intestine (32). BP is a much more effective activator of AhR than DMBA (30).
β catenin staining was elevated in the larger Min tumors produced by the DMBA treatment, but not when Cyp1b1 was deleted, thus paralleling tumor numbers and size (Figure 3, panels B and C). β catenin was strikingly evident in about a third of the crypts after DMBA-treatment (panel B, boxes b and c), which also stained more strongly with hemotoxylin,. These distinctive crypts, which were present in clusters, likewise stained strongly for phospho-IKK and ReIA. These activated crypts were present outside the tumors. They were greatly diminished when Cyp1b1 was deleted (panel B), even at 130 days (panel C). The increases in IKK phosphorylation and ReIA and β catenin expression only partially overlapped. This process may be related to the formation of aberrant crypt foci, which have been recognized as clusters of hemotoxylin and methylene blue stained crypts in the colon of chemically treated mice and are implicated as an early step towards tumor formation (33).
At day 55, the punctate nuclear β catenin was most visible following DMBA treatment, consistent with the increased tumor growth (panel B, right insert box a), but was particularly evident in the disorganized atypia of the 130 day DMBA-treated Cyp1b1−/− Min mice (panel C). This loss of cell organization, which was particularly evident with H/E staining (panel C, right), was also seen in equivalent tumors from untreated Cyp1b1−/− mice (not shown), but was not seen when Cyp1b1 was present. The enhanced β catenin staining seen at day 55 tumors with Cyp1b1 deletion (panel B) corresponded mostly to epithelial adherens complexes with little punctate nuclear staining.
Unlike the tumor epithelia of day 55 Cyp1b1−/− min mice, the atypia seen after 130 days were completely devoid of phospho IKK. Low phospho-IKK appeared to be typical of the larger Min tumors, irrespective of conditions (Figure 3, panel C). Surprisingly, ReIA expression was low in the border epithelia, even though they exhibited high phospho-IKK (Figure 3, panel B). While the typical IKK phosphorylation of the inhibitory regulator (IκB) releases ReIA to the nucleus, phosphorylation by IKKα leads to proteolysis of ReIA (31). The antibody recognizes both IKKα/β forms. By contrast, atypia expressed robust ReIA expression, although rarely with punctate nuclear distribution (not shown).
We examined whether Cyp1b1 deletion had a systemic effect on DMBA and its metabolism. In Cyp1b1-deficient Min mice, the blood concentrations of DMBA and the active 3,4 dihydrodiol metabolite were 6.3 ± 3.1 pmol/ml and 2.1 ± 0.9 pmol/ml, respectively, compared to 5.3 ±2.1 and 2.9 ± 1.0 in the wild type mice. Since Cyp1b1 deletion does not affect circulating DMBA or metabolites, the affect on DMBA-induced tumorigenesis derives from enhanced metabolism in the small intestine.
CYP1B1 is expressed at a very low level in the normal intestinal epithelium of humans, but is elevated in tumors (15). Cyp1b1 expression was very low in microsomes isolated from normal mouse intestine and was only detected after high exposures to PAHs (34–35). In situ hybridization shows that Cyp1b1 expression was clearly elevated in intestinal tumors relative to adjacent normal tissue, particularly in the region of the crypts and associated stroma (Figure 4A). By contrast, Cyp1a1 was not expressed (Figure 4B). Previous work has demonstrated the expression of multiple CYPs in the mouse intestine (35). Microarray analysis of mRNA isolated from three Min tumors and their adjacent epithelia confirmed that Cyp1b1 expression was substantially elevated in each tumor (Figure 4C). These changes were very selective since only three other Cyps among over 30 represented on the array increased. Two additional Cyps, Cyp2B9 and Cyp2B10, which are commonly found in liver, increased 5- to 10-fold (not shown). Importantly, Cyp26b1, which removes retinoic acid in embryos, was substantially elevated in all three tumors. The highly expressed Cyp26a1 was elevated in two of three tumors, consistent with modest increases previously reported for Min tumors (36).
Figure 4. Intestinal tumors from Min mice exhibit elevated expression of Cyp1b1 and Cyp26 forms relative to surrounding tissues, but no Cyp1a1.

Tumor sections were hybridized with antisense or sense probes against Cyp1b1 (A and B) or Cyp1a1 (B). Microarray analyses of three Min tumors relative to normal intestinal tissue. The arrays confirm increased expression of Cyp1b1, and also show increases in Cyp26a1 (2 out of 3 tumors) and Cyp26b1.
Discussion
We have demonstrated that removal of Cyp1b1 more than doubles intestinal tumorigenesis by 55 days of age in the Min mouse cancer (Figure 1 and Table 1), but with only minor affects on tumor size, histology, or distribution (Figures 2 and 3, Table 1). This exclusive affect on tumor multiplicity indicates that the metabolism of endogenous substrates by Cyp1b1 suppresses early stages of intestinal tumorigenesis. This expands on previous work, which has demonstrated over-expression of Cyp1b1 in many tumors (12), including human colon cancers (13). Paradoxically, we show that Cyp1b1 mediates a rapid stimulation of these adenomas by DMBA within 3 weeks, at exposure levels that are only moderately higher than can be obtained from environmental PAH exposures (37). We report a large difference according to the PAH (DMBA≫BP) that parallels differences that we have characterized for bone marrow toxicity (30).
Cyp1b1 metabolism of endogenous substrates can limit tumorigenesis by either generating a suppressor or removing an activator. Participation of Cyp1b1 in the generation of retinoic acid, a known tumor suppressor, plays a conserved functional role in early embryogenesis as a contributor to retinoic acid synthesis (24). Cyp1b1 is expressed at substantially higher levels in intestinal tumors than in surrounding normal tissue (Figure 4), notably in the region that is associated with stroma and the crypts. Cyp26b1 and, to a lesser extent, Cyp26a1, which specifically remove retinoic acid are also over-expressed in Min tumors (Figure 4). Thus, deletion of the Cyp1b1 may cooperate with enhanced Cyp26 expression to lower retinoic acid restraints on early tumor development. Human colonic tumors have elevated CYP3A4, CYP2S1, CYP2U1, and CYP51 and also express CYP26A1 (13). Suppression of local retinoic acid, including increases in Cyp26a1 mRNA in Min tumors in human familial adematous polyposis (FAP) adenomas, consistent with β-catenin regulation have previously been demonstrated (36). Human CYP1B1 also metabolizes estradiol to genotoxic quinones, but much more effectively than rodent Cyp1b1 (16,38). The estradiol in these female Min mice appreciably suppresses Min tumors (39) and, therefore, any elevation by removal of Cyp1b1 should lower tumorigenesis.
Min tumors and human colonic tumors each exhibit high clusterin expression, consistent with activation of NF-κB (27). Activation of the NF-κB complex by IKKβ releases dimeric NF-κB transcription factors, including ReIA, which generate anti-apoptotic and pro-inflammatory transcripts that enhance colon tumorigenesis (29,31,40). In Cyp1b1−/− Min small intestines, phospho-IKK increased modestly in the normal villi and tumor epithelia (Figure 3, panels A and B), consistent with NF-κB activation. ReIA also increased, but without specifc nuclear localization associated with IKK activation. A similar constitutive activation of NF-κB in the lungs of Cyp1b1-deficient mice could explain the elevation of DMBA-induced lung tumors in these mice (21).
Linoleic and arachidonic oxygenation products are plausible Cyp1b1 substrates. Thus they modulate NF-κB activities, including inflammatory recruitment of macrophage (40), and can be activators (20-HETE, leukotriene B4) (41). Prostaglandins formed by Cox1 and Cox 2 also stimulate Min tumorigenesis by mediating angiogenesis when tumors exceed a critical size of 1mm (5). However, Cyp1b1 deletion increased Min tumor initiation rather than enlargement.
Loss of Apc activates the Min tumorigenesis process by enhancing β-catenin signaling. This results in enhanced expansion of a mutated stem cell population at the bottom of the crypts. Adenomas in Min mice chimeric for ROSA26 expression demonstrate distinct transformation events in adjacent crypts, suggesting that initiation in one crypt facilitates initiation in an adjacent crypt (42). Stimulation of NF-κB activity commonly increases the release of chemokines, which may enhance this inter-crypt proximity effect. Adenomas also expand through a crypt fission process, which starts when stem cells reach a threshold level (43). This process is enhanced by loss of Apc activity and by damage to the crypts, including the effects of reactive chemicals.
The metabolism of DMBA by Cyp1b1 doubles the number and size of intestinal tumors in Min mice in a mere 20 days, as evidence by the reversal with Cyp1b1 deletion (Table 1 and Figure 2B). BP was ineffective despite effective activation in vitro (17,22). We have reported this difference for Cyp1b1-mediated bone marrow toxicity, apparently due to activation of AhR-dependent protective mechanisms by this potent AhR ligand. The stimulation was selective to the medial and distal segments and accompanied by a novel large stimulation of β catenin, ReIA, and phosho-IKK in the about a third of intestinal crypts, including outside the tumors (Figure 3). This crypt activation may also be associated with the generation of aberrant crypt foci, an early step in colon cancer that can derive from chemical activation of crypt fission (33,43). The staining of adjacent crypts (Figure 3, panel B enlargements) may arise in this way.
Tumor multiplicities in mice lacking Cyp1b1 were actually decreased by DMBA and still more by BP (Figure 1D). This paradoxical reversal of the Cyp1b1-deficient phenotype by DMBA and BP can be captured by a simple model in which Cyp1b1 deficiency not only increases endogenous chemicals that activate NF-κB transcription (shown in Figure 5), but uncovers an opposing suppression through stimulation of AhR by PAHs that can predominate in absence of their metabolic activation. AhR is substantially activated in the mouse colon and small intestine by PAHs (32), and BP is a much more effective activator than DMBA (30). AhR inhibition is realized through direct interaction with the Rel components of the NF-κB transcription complex and has been reported for lung responses to cigarette condensate in vivo (44). AhR activity in the crypts of mouse small intestine is also demonstrated by the AhR-Cre recombinase method for Apc deletion (45–46). The pro-carcinogenic effect produced by PAHs may, therefore, depend on the balance between metabolite activation (high for DMBA) and AhR suppression (high for BP). The absence of alfalfa and soybean meal from the mouse diet thereby excludes phytochemicals, such as flavonoids (47) and indole derivatives, which are potential Cyp1b1 substrates (48) and AhR inducers (46).
Figure 5. A model for participation of Cyp1b1 metabolism of both endogenous substrates and PAHs in Min tumorigenesis.

Increased NF-κB activation increases tumorigenesis through anti-apoptotic and pro-inflammatory effects. By contrast, Cyp1b1 activates PAHs to mutatgenic dihydrodiol epoxides. In absence of Cyp1b1, PAHs become elevated in the intestine and exhibit enhanced AhR activity. This suppresses the NF-κB activity, thus reversing the effect of endogenous reactive oxygen species (ROS), which can also increase in Cyp1b1 deficient cells. Cyp1b1 may, additionally, suppress tumorigenesis by converting retinaldehyde to retinoic acid, an active Min tumor suppressor (36).
Cyp1b1 metabolism of endogenous substrates or PAHs may contribute to loss of the wildtype Apc allele, which arises by three principal mechanisms: somatic recombination, epigenetic silencing, or mutation through DMA damage (49). The spontaneous loss of heterozygosity (LOH) for Apc occurs through somatic recombination in these B6 Min mice and is much higher in the distal intestine, whereas tumorigenesis from allelic silencing predominates at the ileocecal junction (49). The increased multiplicity produced by Cyp1b1 deletion occurs equally in each region, suggesting enhanced somatic recombination. DMBA, however, is most effective in the distal segment (Table 1). Cyp1b1 is preferentially expressed in the small intestine relative to the colon (35).
Cyp1b1 deletion does not affect circulating levels of PAHs or their dihydrodiol metabolites. The net effect of Cyp1b1 on tumorigenesis in the mouse intestine should, therefore, depend on local metabolism of PAHs or the dihydrodiol precursors by Cyp1b1 (Figure 5). The elevation of expression of Cyp1b1 seen here (Figure 4) and in many human tumors, including in the colon (13–14), may occur early in the transformation process. This is likely since DMBA metabolism is completed in a moderately short time period after each administration. By contrast, the effects of Cyp1b1 on the endogenous substrates are continuously present during the tumor development. Single IP doses of DMBA (but not BP) produce major changes in bone marrow hematopoiesis that are eventually transmitted to other sites (30). Importantly, these cells derived from bone marrow migrate to the intestine and participate in tumorigenesis (40). The finding that repeat administration is necessary is consistent with previous studies with AOM and ENU (7,10). This is consistent with a model in which an activated crypt, such as may be indicated by phospho-IKK and ReIA staining, facilitates activation of an adjacent crypt. This clustering of transformed crypts was evident in the histology following DMBA treatment.
Although a lack of Cyp1b1 activity increased the number of tumors in Min mice, these mice persisted beyond 130 days of age. The atypia that are evident in these large Cyp1b1-deficient tumors indicates that Cyp1b1 additionally affects tumor progression (Figure 3). These regions of atypia strongly express nuclear β catenin, but not the inter-membrane adhesion complexes typical of organized epithelia (39). The absence of phospho-IKK contrasts with the elevation in early tumors associated with Cyp1b1 deletion (Figure 3), but also reflects a trend in other larger tumors. The tumor enhancement provided by Cyp1b1 deletion has no noticeable effect on mortality despite many very large tumors. This altered development of Min tumors in absence of Cyp1b1, demonstrated by prevalent atypia, may contribute to particularly benign tumors.
The link established here between Cyp1b1 and tumorigenesis is particularly relevant to human colon cancer (12). CYP1B1, in combination with nuclear β catenin, provides an effective biomarkers to screen for colorectal carcinoma (15), and the common allelic variants may represent risk factors (13–14).
Acknowledgments
We thank Dr. William Dove for his critical reading of this manuscript and Joe Hardin for his contribution to tumor histology.
This work was supported by NIH grants R01 CA016265 (CRJ) and R01 CA87609 (CRJ). University of Wisconsin Clinical Cancer Grant 5P30-CA014520-34 (Director, G. Wilding/C.R. Jefcoate, member)
Footnotes
Dedicated to Margaret Thorn, whose valiant fight with colon cancer was an added motivation for this research.
References
- 1.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 2.Clarke AR. Wnt signalling in the mouse intestine. Oncogene. 2006;25:7512–21. doi: 10.1038/sj.onc.1210065. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser S, Park YK, Franklin JL, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cormier RT, Hong KH, Halberg RB, et al. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet. 1997;17:88–91. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- 5.Takeda H, Sonoshita M, Oshima H, et al. Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003;63:4872–7. [PubMed] [Google Scholar]
- 6.Reed KR, Sansom OJ, Hayes AJ, et al. PPARdelta status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene. 2004;23:8992–6. doi: 10.1038/sj.onc.1208143. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker AR, Moser AR, Dove WF. N-ethyl-N-nitrosourea treatment of multiple intestinal neoplasia (Min) mice: age-related effects on the formation of intestinal adenomas, cystic crypts, and epidermoid cysts. Cancer Res. 1995;55:4479–85. [PubMed] [Google Scholar]
- 8.Andreassen A, Vikse R, Mikalsen A, et al. 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) induces genetic changes in murine intestinal tumours and cells with ApcMin mutation. Mutat Res. 2006;604:60–70. doi: 10.1016/j.mrgentox.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Mollersen L, Paulsen JE, Alexander J. Loss of heterozygosity and nonsense mutation in Apc in azoxymethane-induced colonic tumours in min mice. Anticancer Res. 2004;24:2595–9. [PubMed] [Google Scholar]
- 10.Sohn OS, Faila ES, Requeijo SP, et al. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001;61:8435–40. [PubMed] [Google Scholar]
- 11.Kleiner HE, Vulimiri SV, Hatten WB, et al. Role of cytochrome p4501 family members in the metabolic activation of polycyclic aromatic hydrocarbons in mouse epidermis. Chem Res Toxicol. 2004;17:1667–74. doi: 10.1021/tx049919c. [DOI] [PubMed] [Google Scholar]
- 12.Murray GI, Taylor MC, McFadyen MCE, et al. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997;57:3026–31. [PubMed] [Google Scholar]
- 13.Kumarakulasingham M, Rooney PH, Dundas SR, et al. Cytochrome P450 profile of colorectal cancer: identification of markers of prognosis. Clin Cancer Res. 2005;11:3758–65. doi: 10.1158/1078-0432.CCR-04-1848. [DOI] [PubMed] [Google Scholar]
- 14.Sissung TM, Price DK, Sparreboom A, et al. Pharmacogenetics and regulation of human cytochrome P450 1B1: implications in hormone-mediated tumor metabolism and a novel target for therapcutic intervention. Mol Cancer Res. 2006;4:135–50. doi: 10.1158/1541-7786.MCR-05-0101. [DOI] [PubMed] [Google Scholar]
- 15.Chang H, Su JM, Huang CC, et al. Using a combination of cytochrome P450 1B1 and β-catenin for early diagnosis and prevention of colorectal cancer. Cancer Detection and Prevention. 2005;29:562–9. doi: 10.1016/j.cdp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Savas U, Carstens CP, Jefcoate CR. Recombinant mouse CYP1B1 expressed in Escherichia coli exhibits selective binding by polycyclic hydrocarbons and metabolism which parallels C3H10T1/2 cell microsomes, but differs from human recombinantCYP1B1. Arch Biochem Biophys. 1997;347:181–92. doi: 10.1006/abbi.1997.0339. [DOI] [PubMed] [Google Scholar]
- 17.Savas U, Bhattacharyya KK, Christou M, et al. Mouse cytochrome P-450EF, representative of a new 1B subfamily of cytochrome P-450s. Cloning, sequence determination, and tissue expression. J Biol Chem. 1994;269:14905–11. [PubMed] [Google Scholar]
- 18.Sutter TR, Yang YM, Hayes CL, et al. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem. 1994;269:13092–9. [PubMed] [Google Scholar]
- 19.Tang YM, Wo YY, Stewart J, et al. Isolation and characterization of the human cytochrome P450 CYP1B1 gene. J Biol Chem. 1996;271:28324–30. doi: 10.1074/jbc.271.45.28324. [DOI] [PubMed] [Google Scholar]
- 20.Tsuchiya Y, Nakajima M, Takagi S, et al. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 2006;66:9090–8. doi: 10.1158/0008-5472.CAN-06-1403. [DOI] [PubMed] [Google Scholar]
- 21.Buters J, Quintanilla-Martinez J, Schober W, et al. CYP1B1 determines susceptibility to low doses of 7,12-dimethylbenz[a]anthracene-induced ovarian cancers in mice: correlation of CYP1B1-mediated DMA adducts with carcinogenicity. Carcinogenesis. 2003;24:327–34. doi: 10.1093/carcin/24.2.327. [DOI] [PubMed] [Google Scholar]
- 22.Shimada T, Murayama N, Okada K, et al. Different mechanisms for inhibition of human cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic inhibitors. Chem Res Toxicol. 2007;20:489–96. doi: 10.1021/tx600299p. [DOI] [PubMed] [Google Scholar]
- 23.Choudhary D, Jansson I, Sarfarazi M, et al. Physiological significance and expression of P450s in the developing eye. Drug Metab Rev. 2006;38:337–52. doi: 10.1080/03602530600570149. [DOI] [PubMed] [Google Scholar]
- 24.Chambers D, Wilson L, Maden M, et al. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development. 2007;134:1369–83. doi: 10.1242/dev.02815. [DOI] [PubMed] [Google Scholar]
- 25.Libby RT, Smith RS, Savinova OV, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003;299:1578–81. doi: 10.1126/science.1080095. [DOI] [PubMed] [Google Scholar]
- 26.Moser AR, Mattes EM, Dove WF, et al. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc Natl Acad Sci USA. 1993;90:8977–81. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Halberg RB, Ehrhardt WM, et al. Clusterin as a biomarker in murine and human intestinal neoplasia. Proc Natl Acad Sci. 2003;100:9530–5. doi: 10.1073/pnas.1233633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata W, Maeda S, Hikiba Y, et al. The IKb kinase inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol. 2007;179:2681–5. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- 29.Steinbrecher KA, Harmel-Laws E, Sitcheran R, et al. Loss of epithelial ReIA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–99. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 30.Galvan N, Teske DE, Zhou G, et al. Induction of CYP1A1 and CYP1B1 in liver and lung by benzo(a)pyrene and 7,12-dimethylbenz[a]anthracene do not affect distribution of polycyclic hydrocarbons to target tissue: role of AhR and CYP1B1 in bone marrow cytotoxicity. Toxicol Appl Pharmacol. 2005;202:244–57. doi: 10.1016/j.taap.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Karin M, Greten FR. NF-kB: linking inflammation and immunity to cancer development and progression. Nature Reviews. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 32.Campbell SJ, Carlotti F, Hall PA, et al. Regulation of the CYP1A1 promoter in transgenic mice: an exquisitely sensitive on-off system for cell specific gene regulation. J Cell Sci. 1996;109:2619–25. doi: 10.1242/jcs.109.11.2619. [DOI] [PubMed] [Google Scholar]
- 33.Papanikolaou A, Wang QS, Papanikolaou D, et al. Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane-induced colon carcinogenesis. Carcinogenesis. 2000;21:1567–72. [PubMed] [Google Scholar]
- 34.Zhang QY, Dunbar D, Kaminsky LS. Characterization of mouse small intestinal cytochrome P450 expression. Drug Metabol Dispos. 2003;31:1346–51. doi: 10.1124/dmd.31.11.1346. [DOI] [PubMed] [Google Scholar]
- 35.Uno S, Dragin N, Miller ML, et al. Basal and inducible CYP1 mRNA quantitation and protein localization throughout the mouse gastrointestinal tract. Free Rad Biol Med. 2008;44:570–83. doi: 10.1016/j.freeradbiomed.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shelton DN, Sandoval IT, Eisinger A, et al. Up-regulation of CYP26A1 in adenomatous polyposis coli-deficient vertebrates via a Wnt-dependent mechanism: implications for intestinal cell differentiation and colon tumor development. Cancer Res. 2006;66:7571–7. doi: 10.1158/0008-5472.CAN-06-1067. [DOI] [PubMed] [Google Scholar]
- 37.Phillips DH. Polycyclic aromatic hydrocarbons in the diet. Mutat Res. 1999;443:139–47. doi: 10.1016/s1383-5742(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 38.Rahman M, Hayes Sutter C, Emmert GL, et al. Regioselective 2-hydroxylation of 17 beta-estradiol by rat cytochrome P4501B1. Toxicol Appl Pharmacol. 2006;216:469–78. doi: 10.1016/j.taap.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Javid SH, Moran AE, Carothers AM, et al. Modulation of tumor formation and intestinal cell migration by estrogens in the ApcMin/+ mouse model of colorectal cancer. Carcinogenesis. 2005;26:587–95. doi: 10.1093/carcin/bgh346. [DOI] [PubMed] [Google Scholar]
- 40.Greten FR, Eckman L, Greten TF, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Pidgeon GP, Lysaght J, Krishnamoorthy S, et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–24. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- 42.Thliveris AT, Halberg RB, Clipson L, et al. Polyclonality of familial murine adenomas: analysis of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc Natl Acad Sci USA. 2005;102:6960–5. doi: 10.1073/pnas.0502662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev. 2006;2:203–12. doi: 10.1007/s12015-006-0048-1. [DOI] [PubMed] [Google Scholar]
- 44.Thatcher TH, Sanjay BM, Baglole CJ, et al. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component ReIB. Am J Pathol. 2007;170:855–64. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sansom OJ, Reed KR, Hayes AJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–90. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito S, Chen C, Satoh J, et al. Dietary phytochemicals regulate whole-body CYP1A1 expression through an arylhydrocarbon receptor nuclear translocator-dependent system in gut. J Clin Invest. 2007;117:1940–50. doi: 10.1172/JCI31647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang TKH, Chen J, Yeung EYH. Effect of ginko biloba extract on procarcinogen-bioactivating human CYP1 enzymes: identification of isorhamnetin, kaempferol, and quercetin as potent inhibitors of CYP1B1. Toxicol Appl Pharmacol. 2006;213:18–26. doi: 10.1016/j.taap.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Prince M, Campbell CT, Robertson TA, et al. Naturally occurring coumarins inhibit 7,12-dimethylbenz[a]anthracene DMA adduct formation in mouse mammary gland. Carcinogenesis. 2006;6:1204–13. doi: 10.1093/carcin/bgi303. [DOI] [PubMed] [Google Scholar]
- 49.Haigis KM, Hoff PD, White A, et al. Tumor regionality in the mouse intestine reflects the mechanism of loss of Apc function. Proc Natl Acad Sci USA. 2004;101:9769–73. doi: 10.1073/pnas.0403338101. [DOI] [PMC free article] [PubMed] [Google Scholar]