

Abstract
Trypanosomatid RNA editing is a unique process and essential for these organisms. It therefore represents a drug target for a group of protozoa that includes the causative agents for African sleeping sickness and other devastating tropical and subtropical diseases. Here, we present drug-like inhibitors of a key enzyme in the editing machinery, RNA-editing ligase 1 (REL1). These inhibitors were identified through a strategy employing molecular dynamics to account for protein flexibility. A virtual screen of the REL1 crystal structure against the National Cancer Institute Diversity Set was performed by using AutoDock4. The top 30 compounds, predicted to interact with REL1's ATP-binding pocket, were further refined by using the relaxed complex scheme (RCS), which redocks the compounds to receptor structures extracted from an explicitly solvated molecular dynamics trajectory. The resulting reordering of the ligands and filtering based on drug-like properties resulted in an initial recommended set of 8 ligands, 2 of which exhibited micromolar activity against REL1. A subsequent hierarchical similarity search with the most active compound over the full National Cancer Institute database and RCS rescoring resulted in an additional set of 6 ligands, 2 of which were confirmed as REL1 inhibitors with IC50 values of ≈1 μM. Tests of the 3 most promising compounds against the most closely related bacteriophage T4 RNA ligase 2, as well as against human DNA ligase IIIβ, indicated a considerable degree of selectivity for RNA ligases. These compounds are promising scaffolds for future drug design and discovery efforts against these important pathogens.
Keywords: molecular dynamics, relaxed complex scheme, RNA ligase, African sleeping sickness, receptor flexibility
Many advances in our understanding of the fundamental biology of trypanosomatids (order Kinetoplastida) have occurred over the past few decades, including the sequencing of their genomes (1). Yet, the development of new and effective drugs to treat the diseases caused by these protozoan parasites has been relatively nonexistent. Each year, millions of people in the poorest countries in the world suffer from the infectious tropical diseases caused by these pathogens, including human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis. These “orphan diseases” cause not only death, but also contribute to a crippling cycle of poverty within the Americas, Asia, and Africa, which collectively harbor a disproportionate amount of the neglected disease burden (2). Existing drugs are costly and difficult to deliver, induce debilitating or fatal side effects, and are showing increased signs of resistance (3). As telling as it is unfortunate, only 1 new drug against HAT has been registered within the past 50 years (4).
A unique aspect of the biology of these protozoan parasites is that most of their mitochondrial mRNAs undergo extensive editing in the form of internal uridine insertion and removal, catalyzed by large, multiprotein complexes known as editosomes and directed by guide RNAs (gRNAs) (5). Although all of the components of the editosome are not yet fully characterized, decades of study have allowed many intriguing details to emerge, in particular, with respect to the 20S editosomes (5). These complexes exist in at least 3 different forms with respect to their endonuclease component. Each of the 3 complexes comprises 14–15 proteins and appears to be highly dynamic (6).
The editing process is initiated by endonucleolytic cleavage at a site of mismatch identified when a premRNA is bound by its cognate gRNA, a 50- to 60-nt transcript complementary to a so-called anchor region in the premRNA as well as to the edited version of that RNA. The type of RNA mismatch determines which 20S editosome catalyzes the cleavage reaction. As specified by the gRNA sequence, U's are then either added, by the terminal uridylyl transferase (TUTase) RET2, or deleted by a U-specific 3′-exoribonuclease (ExoUase). The processed RNA fragments are then religated by 1 of 2 RNA ligases, RNA editing ligase 1 (REL1) or 2 (REL2). REL1 has been shown to play a key role in the viability of Trypanosoma brucei, as it is required for survival of both the insect and bloodstream forms of the pathogen (7, 8). In addition, it is a particularly attractive drug target because there are no known close human homologs.
Virtual screening (VS) is a widely used computational method to identify inhibitors out of a large database of compounds (9). The treatment of receptor flexibility within the scope of VS is still in its infancy and a very active area of research, because it is widely accepted that receptor flexibility plays an important role in molecular recognition (10). A promising approach is the relaxed complex scheme (RCS), a hybrid computational method that combines the advantages of docking algorithms with dynamic structural information provided by molecular dynamics (MD) simulations (11, 12). The use of structural information provided from all-atom MD simulations allows us to incorporate ensemble-based information into the drug discovery and design process, whereas conventional VS techniques typically consider only 1 or a few static receptor structures. The incorporation of dynamic receptor information, although computationally more intensive, can discover and take advantage of new binding pockets (13, 14) and improve the ranking of predicted compounds. To our knowledge, the RCS is the only computational technique that exploits full-receptor main-chain flexibility, although techniques allowing side-chain and limited receptor backbone flexibility have been developed (15–17). An important example of the success of the RCS was demonstrated with raltegravir, the first FDA-approved drug-targeting HIV integrase (18, 19).
In this work, we present 5 drug-like inhibitors of T. brucei REL1, which we discovered through an improved RCS, integrated within a VS approach. The high-resolution crystal structure of TbREL1 (20) provides an excellent platform for rational drug design as well as for MD simulations. A 20-ns simulation was used to investigate the dynamics of TbREL1 (21), and the resulting structures were used in conjunction with the RCS to predict the most promising compounds. Through the use of QR factorization, we are able to distill the structural ensemble generated through the MD simulations to a nonredundant set, thus significantly reducing the computational expense. In vitro inhibition assays of the first step in the ligation reaction and tests for nonspecific inhibition through aggregation confirmed at least 3 inhibitory compounds with IC50 values in the low-micromolar range. The top compounds were also tested against 2 related and 1 unrelated ATP-binding protein to investigate off-target activity. Ultimately, we show that the use of receptor flexibility in the VS process provided an important enrichment of the recommended set of compounds, resulting in the discovery of several promising scaffolds that may aid in the development of new drugs against several devastating tropical diseases.
Results and Discussion
Initial Set of Recommended Compounds.
The initial VS of the National Cancer Institute (NCI) Diversity Set used the static TbREL1 crystal structure to dock and rank ≈1,800 compounds according to their predicted binding affinity. Typical VSs use a single crystal structure to predict the binding affinity of the compounds in the screening set. To take receptor flexibility into account and to validate and refine the top hits, the top 2% of the screening hits (corresponding to the top 30 compounds), all of which were predicted to interact with the ATP-binding pocket, were redocked into the full holo MD ensemble (Fig. 1). This RCS rescoring created a so-called binding spectrum for each compound and the mean of this binding spectrum energy (i.e., RC energy) was used to rank-order the compounds. The RC energy ranking reordered the top compounds such that several compounds that ranked poorly in the crystal structure screen were subsequently ranked as the best compounds (Table 1). An additional filtering of the compounds according to Lipinski's rules was performed, allowing us to focus our search on the most promising drug-like candidate compounds.
Fig. 1.

An overview of the relaxed complex scheme with virtual screening procedure is shown. The first screen is performed with the crystal structure and the NCI diversity set with AutoDock4 (AD4). MD simulations are performed with the solvated crystal structure, generating the receptor ensemble. The compounds can be docked into the full ensemble (400 structures) or a reduced set, here determined with QR factorization (QR ensemble, 33 structures). The reordered compounds are then selected for experimental testing.
Table 1.
First set of recommended compounds
| Compound ID | NSC No. | Crystal structure BE | RC-mean BE | RC-QR-mean BE | Rank from crystal structure screen | % Activity at 10 μM, with TX-100 |
|---|---|---|---|---|---|---|
| V1 | 45208 | −9.92 | −12.13 | −12.06 | 16 | 43.1 |
| V2 | 7223 | −10.97 | −11.98 | −11.89 | 8 | N.I.* |
| V3 | 125908 | −10.76 | −11.73 | −11.74 | 15 | 87.2 |
| V4 | 117079 | −11.59 | −11.39 | −11.25 | 2 | 68.5 |
| V5 | 601364 | −11.07 | −11.01 | −10.97 | 7 | N.I. |
| V6 | 45544 | −11.25 | −10.89 | −10.73 | 5 | N.I. |
| V7 | 9600 | −12.61 | −10.33 | −10.48 | 1 | N.I. |
| V8 | 117269 | −11.32 | −8.09 | −8.04 | 4 | N.I. |
The compound ID, NSC number, predicted binding energy (BE) based on the single static crystal structure (Crystal Structure BE), mean binding energy based on the full receptor ensemble (RC-mean BE), reduced representative ensemble (RC-QR-mean BE), rank from the crystal structure screen, and percent TbREL1 activity at 10 μM concentration of inhibitor and 0.1% Triton X-100 (TX-100) (% Activity at 10 μM; N.I. denotes ″not inhibiting″;
*, denotes compound is an aggregator in our assays).
To improve the efficiency of the RCS, we reduced the structural ensemble used for redocking with the QR factorization algorithm (22). This technique was originally developed to remove inherent bias in structure databases and distill, from a vast quantity of redundant information, a minimal basis set of protein structures that accurately spans the evolutionary conformation space of a particular protein. Here, we incorporate the QR factorization to generate a nonredundant and representative set of structures spanning the configurational space sampled in the MD simulations. This algorithm reduced the initial set of 400 structures to 33 (supporting information (SI) Fig. S1). Notably, a comparison of the mean RC energy and the mean QR-RC energy for the top 30 compounds was very close (R2 = 0.9932), indicating that redocking into the QR-reduced set is a more efficient way to capture the effects of receptor flexibility without loss of binding spectrum information (Table 1). The integration of QR to the RCS resulted in 90% decrease in computational cost for the redocking calculations, providing an order of magnitude speedup. The top 8 compounds were recommended for experimental testing (Table 1 and Table S1).
Low-Micromolar Inhibitors Found in Round 1.
The first step in the reaction pathway of RNA and eukaryotic DNA ligases is the reaction of a lysine residue with ATP to form a covalent enzyme-AMP intermediate (adenylylation reaction). Measuring formation of TbREL1-[32P]AMP by SDS/PAGE and autoradiography, we tested the initial recommended set for their ability to inhibit the adenylylation reaction (Fig. S2A). The assay was carried out at 10 μM compound concentration in the presence of 0.1% Triton X-100 as a first measure to select against any promiscuous, aggregate-based inhibitors (23). Two compounds, V1 [4,5-dihydroxy-3-(1-naphthyldiazenyl)-2,7-naphthalenedisulfonic acid] and V4 [1-amino-4-(3-(aminosulfonyl)anilino)-9,10-dioxo-9,10-dihydro-2-anthracenesulfonic acid] (Fig. 2) inhibited TbREL1 adenylylation by ≈57% and ≈31%, respectively (Table 1 and Fig. S2A). Another compound, V2 [3-hydroxy-4-((1-hydroxy-2-naphthyl)diazenyl)-7-(hydroxy(oxido)amino)-1-naphthalenesulfonic acid], showed complete inhibition at 10 μM in the absence of detergent (data not shown). Addition of 0.1% Triton X-100 to the assay completely restored activity and, therefore, that inhibitor was categorized as aggregate-based and not analyzed further. Interestingly, some of the compounds appear to up-regulate adenylylation activity (Fig. S2A), possibly because of small amounts of denaturing agent facilitating structural transitions (24). Subsequent studies focused on the best hit, compound V1.
Fig. 2.

Structures of the 5 inhibitors discovered. The chemical structures of the 5 drug-like, low-micromolar inhibiting compounds presented in this work.
Inhibition by V1 was not significantly influenced by the presence of 0.1 mg/mL BSA (Fig. S3A), again ruling out nonspecific, aggregate-based inhibition (25). We determined an IC50 for V1 of 1.95 ± 0.33 μM (Fig. 3). Comparison with bacteriophage T4 RNA ligase 2 (T4Rnl2; refs. 26 and 27) and human DNA ligase IIIβ (HsLigIIIβ; ref. 28) indicated considerable selectivity for RNA vs. DNA ligases: V1 inhibited adenylylation of T4Rnl2 and HsLigIIIβ with IC50s of 3.53 ± 1.17 μM and 27.49 ± 6.40 μM, respectively. Luciferase, an unrelated but also ATP-dependent enzyme, was not affected up to 3 mM, the highest concentration tested (data not shown).
Fig. 3.
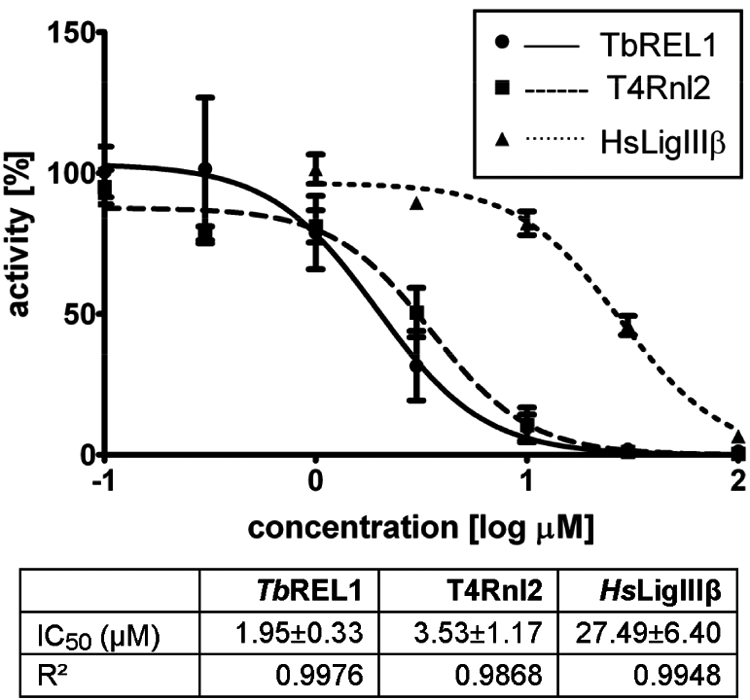
First round of inhibitor testing. Dose–response curve for V1 vs. TbREL1 (solid line, circles), T4Rnl2 (dashed line, squares), and HsLigIIIβ (dotted line, triangles). Corresponding IC50 and R2 values are listed beneath.
Hierarchical Search over the Full NCI Database.
Hierarchical screening is an efficient strategy that allows an initial broad search over a chemically and pharmacologically diverse set of compounds, followed by a focused search over a much larger database to find molecules related to potential lead compounds (29). A similarity search over the full NCI database with V1 resulted in 117 compounds, which we docked to the TbREL1 crystal structure and ranked according to predicted binding energy. The top compounds based on a binding energy cutoff of −12 kcal/mol (corresponding to the top 10% of the similarity set) were then redocked into the QR ensemble. The resulting QR binding spectrum energy was used for the final ranking and the top 6 compounds with the highest affinity for TbREL1 were selected for experimental testing (Table 2).
Table 2.
Second set of compounds based on similarity to V1
| Compound ID | NSC No. | Crystal structure BE | RC-QR-mean BE | % Activity at 10 μM, with TX-100 |
|---|---|---|---|---|
| S1 | 100234 | −14.05 | −11.85 | 8.6 |
| S2 | 45207 | −12.99 | −11.47 | N.I. |
| S3 | 7829 | −12.88 | −10.95 | N.I. |
| S4 | 86033 | −12.55 | −10.02 | N.I. |
| S5 | 16209 | −12.73 | −9.95 | 4.1 |
| S6 | 45201 | −12.79 | −9.84 | 98.6 |
The compound ID, NSC number, predicted binding energy (BE) based on the single static crystal structure (Crystal Structure BE), mean binding energy based on the full receptor ensemble (RC-mean BE), reduced representative ensemble (RC-QR-mean BE), rank from the crystal structure screen, and percent TbREL1 activity at 10 μM concentration of inhibitor and 0.1% Triton X-100 (TX-100) (% Activity at 10 μM; N.I. denotes ″not inhibiting″).
Related Compound S5 Showed Increased Inhibition.
The 6 best compounds identified in the similarity screen were tested at 10 μM concentration in the adenylylation assay, as above, carried out in the presence of 0.1% Triton X-100 (Fig. S2B and Table S2). Two compounds, S5 [3-((4-(ethylamino)phenyl)diazenyl)-4,5-dihydroxy-2,7-naphthalenedisulfonic acid] and S1 [3-((5-chloro-2-hydroxyphenyl)diazenyl)-4,5-dihydroxy-2,7-naphthalenedisulfonic acid] (Fig. 2, Fig. S2, and Table 2) strongly inhibited TbREL1 activity.
Addition of 0.1 mg/mL BSA had no significant effect on inhibition for either of these 2 compounds, confirming that they do not act through aggregation (Fig. S3B and data not shown). Dose–response curves established IC50 values of 1.01 ± 0.16 μM and 1.95 ± 0.61 μM for S5 and S1, respectively (Fig. 4). For S5, this reflects an approximately 2-fold decrease compared with V1. Interestingly, IC50 values for T4Rnl2 and HsLigIIIβ decreased ≈3.5- and 4.2-fold, respectively. For S1, IC50 values for all 3 ligases were not significantly different from V1. The control enzyme luciferase was not affected up to 1 mM, the highest concentration tested for both S5 and S1 (data not shown).
Fig. 4.
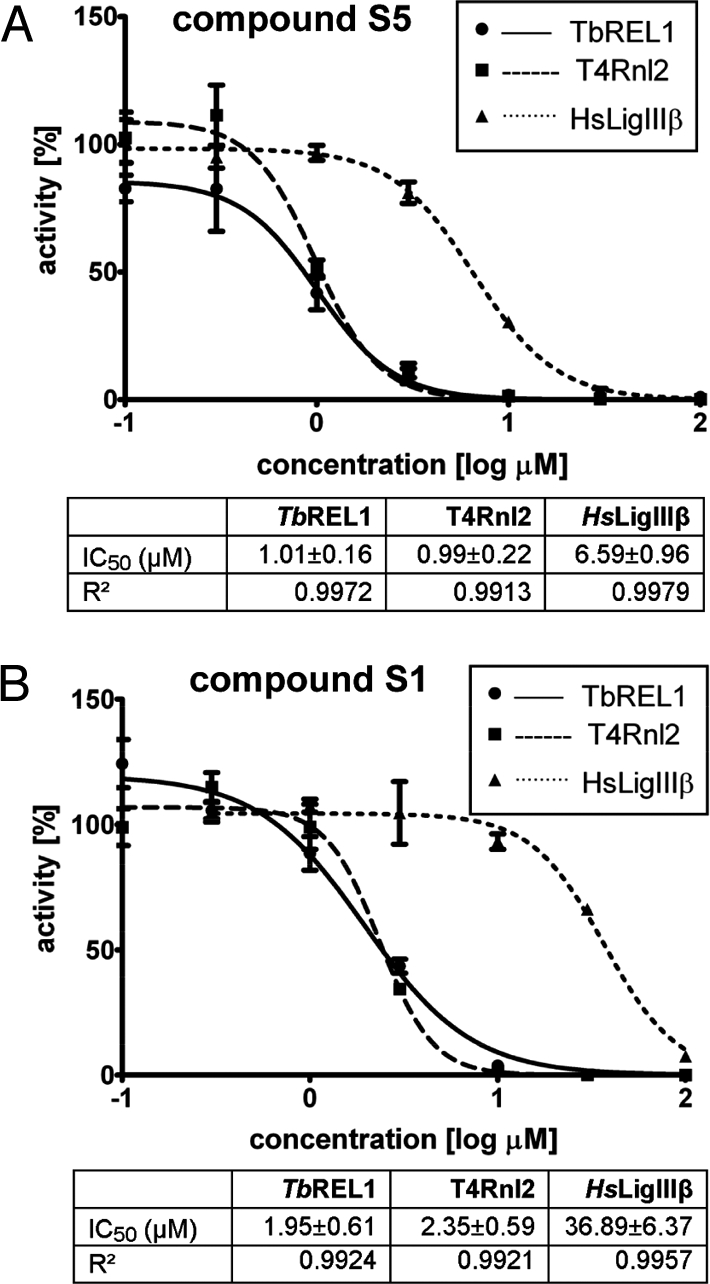
Second round of inhibitor testing. (A) Dose–response curve for S5 vs. TbREL1 (solid line, circles), T4Rnl2 (dashed line, squares), and HsLigIIIβ (dotted line, triangles). Corresponding IC50 and R2 values are listed beneath. (B) Same for S1.
Predicted Modes of Binding for the Top 3 Compounds.
The 3 micromolar drug-like inhibitors of TbREL1 that we have identified, V1, S5, and S1, all share a core 4,5-dihydroxy-2,7-naphthalenedisulfonic acid scaffold, which is predicted to bind in the deep ATP-binding cleft (Fig. 5). Based on our RCS calculations, we expect S3 and S6, which share the core scaffold, to also inhibit TbREL1, albeit at higher concentrations than tested here.
Fig. 5.

Predicted binding mode of S5. The most populated and lowest energy pose is shown for S5 docked into the TbREL1 crystal structure. S5 is shown in ball and stick, with carbons in green, nitrogens in blue, sulfur in yellow, oxygen in red, and hydrogens in white. Hydrogen bonds and salt-bridge interactions are shown with black lines. The protein residues are shown in licorice, with the same coloring except for carbons, which are cyan.
Comparison of V1 with S2 suggests that maintaining both hydroxyl groups may be an important feature, because the compounds are identical with the exception of the hydroxyl group on position 5 (Tables S1 and S2). The core scaffold, including several hydrogen-bonding interactions with the backbone groups of V88, E86, and the side chain of R111, conserves many protein–ligand interactions previously occupied by ATP (Fig. 5). The naphthalene group conserves aromatic pi-stacking interactions between the adenosine ring and F209. On the opposite side of the ring, hydrophobic interactions are maintained with the side chain of V286. Interestingly, the C7 sulfonic acid group in all 3 compounds is predicted to replace a critical water molecule bridging a hydrogen bond network between R288, D210, the backbone carbonyl of F209, Y58, and the N1 atom of the adenosine ring (Fig. S4). Toward the periphery of the binding site, the aromatic rings in all of the compounds are in position to form cation–pi interactions with R309 or R111.
Because of their varying chemical structure, compounds V1, S1, and S5 are predicted to differ in their receptor interactions near the periphery of the binding site. The top compound, S5, may benefit from an additional hydrogen bond formed between the amino group in its ethylamino-phenyl group and the backbone carbonyl of G90, as well as hydrophobic interactions between T91 and the ethyl group. Compared with S6, the differences on the para-substituent account for a large difference in activity (Table S2 and Fig. S2B). In the lowest-energy and most populated predicted binding modes the para-substituent is placed on the periphery of the binding site, and we speculate that the dynamics of residues G90, T91, and N193 may account for these differences (Fig. S1). Further optimizations of the core scaffold may increase the affinity of binding, such as extensions of hydrogen-bonding groups on the C8 atom of the naphthalene moiety or a small hydrophobic group on the C1 atom.
Receptor Flexibility in Computer-Aided Drug Design.
The development of methodologies for computer-aided drug design depends on the critical compromise between accuracy and computational costs. Ideally, the most reliable prediction of molecular affinity can be obtained through rigorous free-energy calculations of the ligand-binding process (ref. 30 and references within); however, these techniques are prohibitively expensive for use in VS experiments. In this work, we combined VS with all-atom MD simulations in an approach we call the relaxed complex scheme (Fig. 1). The use of QR factorization to reduce structural redundancy in our MD-generated receptor ensemble enabled us to accommodate full receptor flexibility in a computationally feasible approach.
Two of the best inhibiting compounds from the first round, V1 and V3, were initially ranked 16th and 15th in the crystal structure screen and therefore would not have been selected for testing based on the crystal structure screen alone. Remarkably, V1 was correctly predicted to be the best inhibitor, and the other inhibiting compounds identified in the first round of testing were reordered to the top of the list based on their predicted rankings by the RC-QR mean binding energy (Table 1). We note, however, that they are not rank-ordered correctly. Still, the success of these predictions underscores the importance of incorporating receptor flexibility in docking and scoring methods. Further refinement of the predicted compounds based on the RCS reranking provided an important enrichment of the final ranked set, resulting in a hit rate (at 10 μM concentration) of 38%. The secondary hierarchical screening, the result of a similarity search for compound V1, resulted in a hit rate of 33%.
Potential Off-Target Activity.
As an example for a related host protein, we experimentally tested the identified inhibitors against the human DNA ligase IIIβ (to date, no human RNA ligases have been identified) and showed that these compounds have 6- to 36-fold selectivity for the trypanosomal target (Figs. 3 and 4). Docking of all 3 compounds to human DNA ligase I (1X9N; ref. 31) indicates that this may be due, in part, to differences in the depth of the ATP-binding pocket between the human and trypanosomal ligase and in residues on the periphery of the active site (data not shown). Efforts to reduce off-target activity can be pursued by strategically modifying these compounds in ways that exploit structural differences between the ATP-binding sites of DNA and RNA ligases, in particular, REL1, that have been identified through a structural phylogenetic analysis of the superfamily (21). Although 8 residues are strictly conserved within the 5 core motifs, residues flanking the core motifs vary among the superfamily members. For example, several key ATP-binding residues flanking motifs I, IV, and V, namely C85, N92, R288, and I305 (TbREL1 numbering), create a binding site topology in TbREL1 distinct from the human DNA ligases. Modifications to the core scaffolds that take advantage of these differences may provide additional selectivity for TbREL1 over the human enzymes.
Dye-Like Compounds as Antitrypanosomal Therapeutics.
The use of dyes as therapeutics has a rich history, beginning in 1891 with the reporting of methylene blue as a potent antimalarial by Guttman and Ehrlich (32). Ehrlich also had interest in developing compounds against HAT and discovered antitrypanosomal activity in mice with the dyes Trypan Red and Trypan Blue. Although these compounds were not active in humans, chemical modifications of the azo linkage to reduce the compounds' tissue-staining capacity led to the discovery of suramin, which is still used today (33).
The 3 best inhibitors we have identified are all azo dyes and similar to suramin in that they are polysulfonated naphthyl compounds. Although suramin's mode of trypanocidal action is still a matter of debate, it is known to be quite promiscuous in terms of inhibitory activity. TbREL1 does not appear to be a target of this drug, however, because dyskinetoplastic trypanosomes, which do not rely on RNA editing (34), are usually susceptible to treatment with suramin (35). Suramin binds to plasma proteins through its charged sulfonate groups (36). Although such groups are shared between suramin and our top compounds, the lack of an effect of BSA on their activity suggests that they do not have high affinity for this plasma protein (Fig. S3). This may indicate a generally lower promiscuity relative to suramin, although more extensive biochemical assays with glycolytic enzymes, dehydrogenases, and kinases are warranted. The latter 2 groups of enzymes are known to bind sulfonated aromatic groups (37), albeit with different chemical structures than the compounds presented here.
The discovery of these smaller, soluble compounds as new antitrypanosomal drug leads has important pharmacological implications. The potential to develop orally available drugs and therapeutics that do not rely on membrane transporter proteins is a key challenge facing HAT therapeutics, because reduced drug uptake has been shown to be a mechanism of drug resistance in African trypanosomes (38). These smaller and significantly less charged (compared with suramin) drug-like compounds, which were strictly chosen to obey all of the Lipinski rules, may be able to passively diffuse through membranes or use other transport mechanisms.
Conclusions
Despite many great advances in the understanding of trypanosomal biology over the past few decades—including the sequencing of the genomes of representatives for the 3 major trypanosomid pathogen groups—little of this knowledge has so far been exploited to develop more effective drugs against these parasites. To address this pharmaceutical need, a cooperative effort between academia and the pharmaceutical industry is urgently needed. In this work, we present 5 compounds that are active against an editosome enzyme required for the viability of T. brucei, which we hope will provide promising scaffolds for future drug discovery and design efforts against these parasites. The fact that 3 of the compounds are dye derivatives may bode well for future development, both in terms of the cost of manufacturing and bioavailability. Importantly, these compounds were discovered through a computational scheme that combines VS experiments with all-atom MD simulations in an ensemble-based approach to drug discovery. The method we present here, which takes advantage of advances in computer hardware and software algorithms, provides a flexible receptor drug discovery scheme that is significantly reduced in computational expense. This approach is widely applicable to nearly any biological target and provides encouraging evidence for the advantages of moving to ensemble-based approaches in scientific discovery.
Materials and Methods
Target and Compound Preparation and Screening Protocol.
A VS of the NCI Diversity Set (NCIDS) database was performed with the original crystal structure by using AutoDock4 (AD4) (39, 40). The 1.2-Å resolution 1XDN crystal structure of TbREL1 was used to optimize the AD4 parameters as a control case (20). In this benchmark, ATP and its coordinated magnesium ion were redocked into the crystal structure. For the receptor, protonation states of all residues were assigned according to a pH of 7.0 by using the PDB 2PQR web site (41). The resulting PDB was processed with the AD4 receptor preparation script. The C4 atom in the bound ATP was chosen as the grid center and the grid contained 60 × 60 × 60 grid points with 0.375-Å spacing. See SI Text for a detailed description of the AD4 parameter optimization.
The optimized AD4 parameters were used to screen the NCIDS (42, 43); 1,823 compounds were screened. The ligand files were processed with AutoDockTools v1.4.5. All torsions were allowed to rotate through the AutoTors program. The initial position and conformation were randomly assigned and 100 dockings were performed. Top hits were filtered for drug-likeness by their adherence to Lipinski's “rule of fives” (44), because it has been recommended that compounds conform to 2 or more of these rules (45). We applied a more strict criterion, selecting compounds that conformed to all 4 rules.
Hierarchical Similarity Search.
The top compound identified from the experimental assays, V1, was used in a similarity search (i.e., hierarchical search) over the full NCI database. A Tanimoto similarity index of 80% was used to identify compounds with 80% or greater chemical similarity (46). These compounds were then docked into the static receptor by using a similar procedure as described above and used in the RCS as described below.
The Relaxed Complex Scheme.
The top 30 compounds (corresponding to an energy cutoff of −10.0 kcal/mol) were redocked to 400 snapshots extracted from the ATP bound MD simulations at 50-ps intervals. The MD preparation, details, and results are described elsewhere (21). New receptor grid files were generated for each of the receptor structures. The ligand-docking parameters were identical to those used for the VS, except that 20 docking runs were performed for each ligand. The lowest docked energy poses were extracted for each frame and the mean of the docking energies is reported for each as RC-mean binding energy (BE).
Generating a Representative Ensemble from MD.
To reduce the redundancy of the MD-generated structures, a QR factorization method was used as implemented in VMD 1.8.6 (47). The integration of this technique into the RCS has been fully described in ref. 12. Use of a QH threshold of 0.86 to the REL1 MD structures reduced the initial set of 400 structures to 33 (reducing the number of dockings from 11,200 to 924), with essentially no loss of binding spectrum information (Table 1).
Compounds and Reagents.
Compounds for biochemical screens were obtained from the Developmental Therapeutics Program at the NCI, National Institutes of Health, and dissolved in DMSO. Other reagents were from Sigma, unless noted otherwise.
Recombinant TbREL1 Expression and Purification.
See SI Text for a detailed description. In brief, full-length TbREL1 was expressed in insect cells by using the baculovirus system and purified via a C-terminal tandem affinity purification (TAP) tag (48).
Enzymatic Assays and Curve Fitting.
See SI Text for a detailed description including buffer conditions. Adenylylation reactions with TbREL1 were performed, essentially as described in ref. 20, in a volume of 20 μL with 0.1 pmol of protein and 1.8 μCi (30 nM) [α-32P]ATP. Triton X-100 (0.1% wt/vol) or BSA (0.1 mg/mL) were included as indicated. Adenylylation reactions with T4 phage RNA ligase 2 (T4Rnl2, New England Biolabs) and with human DNA ligase IIIβ were performed with 1.8 μCi (30 nM) [α-32P]ATP in 20-μL reactions containing 0.1 pmol and 1.2 pmol of protein, respectively.
Formation of enzyme–[32P]AMP complexes was analyzed by SDS/PAGE and phosphorimaging (Storm, Molecular Dynamics). Inhibitor candidates, dissolved in DMSO, were included at the concentrations indicated and parallel reactions with DMSO alone served as controls. All reactions were done in at least triplicate. IC50 values were determined through nonlinear regression analysis with the GraphPad Prism 5 software.
Supplementary Material
Acknowledgments.
We thank Tom Ellenberger and In-Kwon Kim (Washington University, St. Louis) for HsLigIIIβ; Jim Champoux (University of Washington, Seattle) for access to his insect cell facility; Art Olson, William Lindstrom, and Garrett Morris (Scripps Research Institute, La Jolla) for early access to AutoDock4 and helpful discussions; and Maria Kontoyianni for critically reading the manuscript. This work was supported by National Institutes of Health Grant F32-GM077729 and National Science Foundation Grant MRAC CHE060073N (to R.E.A.); National Institutes of Health Grants AI069057 and NS061733 (to A.S.) and GM42188 (to K.S.); and National Institutes of Health Grant GM31749 and National Science Foundation Grants MCB-0506593 and MCA93S013 (to J.A.M.). This work was also supported by the Howard Hughes Medical Institute, San Diego Supercomputing Center, the W.M. Keck Foundation, Accelrys, Inc., the National Biomedical Computational Resource, and the Center for Theoretical Biological Physics.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0805820105/DCSupplemental.
References
- 1.Cross GA. Trypanosomes at the gates. Science. 2005;309(5733):355. doi: 10.1126/science.1116055. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. WHO Report on Global Surveillance of Epidemic-Prone Infectious Diseases. Geneva, Switzerland: World Health Organization; 2003. [Google Scholar]
- 3.Croft SL, Barrett MP, Urbina JA. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005;21(11):508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 4.Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: Pharmacological re-engagement with a neglected disease. Br J Pharmacol. 2007;152(8):1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. Complex management: RNA editing in trypanosomes. Trends Biochem Sci. 2005;30(2):97–105. doi: 10.1016/j.tibs.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol. 2008;28(1):122–130. doi: 10.1128/MCB.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schnaufer A, et al. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science. 2001;291(5511):2159–2162. doi: 10.1126/science.1058955. [DOI] [PubMed] [Google Scholar]
- 8.Rusche LN, et al. The two RNA ligases of the Trypanosoma brucei RNA editing complex: Cloning the essential band IV gene and identifying the band V gene. Mol Cell Biol. 2001;21(4):979–989. doi: 10.1128/MCB.21.4.979-989.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oprea TI, Matter H. Integrating virtual screening in lead discovery. Curr Opin Chem Biol. 2004;8(4):349–358. doi: 10.1016/j.cbpa.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Carlson HA. Protein flexibility and drug design: How to hit a moving target. Curr Opin Chem Biol. 2002;6(4):447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 11.Lin JH, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J Am Chem Soc. 2002;124(20):5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 12.Amaro R, Baron R, McCammon JA. An improved relaxed complex scheme for incorporating receptor flexibility in computer-aided drug design. J Comput Aided Mol Des. 2008 doi: 10.1007/s10822-10007-19159-10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schames JR, et al. Discovery of a novel binding trench in HIV integrase. J Med Chem. 2004;47(8):1879–1881. doi: 10.1021/jm0341913. [DOI] [PubMed] [Google Scholar]
- 14.Cheng LS, et al. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J Med Chem. 2008;51:3878–3894. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337(1):209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49(2):534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 17.Claussen H, Buning C, Rarey M, Lengauer T. FlexE: Efficient molecular docking considering protein structure variations. J Mol Biol. 2001;308(2):377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz M, et al. Potent antiviral effect of MK-0518, novel HIV-1 integrase inhibitor, as part of combination ART in treatment-naive HIV-1 infected patients. XVI International AIDS Conference; August 13–18, 2008; Toronto, Canada. Geneva, Switzerland: International AIDS Society; 2006. Available at: www.thebody.com/content/treat/art38247.html#3. [Google Scholar]
- 19.Hazuda DJ, et al. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc Natl Acad Sci USA. 2004;101:11233–11238. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng J, Schnaufer A, Salavati R, Stuart KD, Hol WG. High resolution crystal structure of a key editosome enzyme from Trypanosoma brucei: RNA editing ligase 1. J Mol Biol. 2004;343(3):601–613. doi: 10.1016/j.jmb.2004.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Amaro R, Swift R, McCammon JA. Functional and structural insights revealed by molecular dynamics simulations of an essential RNA editing ligase in Trypanosoma brucei. PLoS Negl Trop Dis. 2007;1(2):e68. doi: 10.1371/journal.pntd.0000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67(4):550–573. doi: 10.1128/MMBR.67.4.550-573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryan AJ, Gray NM, Lowe PN, Chung CW. Effect of detergent on “promiscuous” inhibitors. J Med Chem. 2003;46(16):3448–3451. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 24.Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46(20):4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 26.Ho CK, Wang LK, Lima CD, Shuman S. Structure and mechanism of RNA ligase. Structure. 2004;12(2):327–339. doi: 10.1016/j.str.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Yin S, Ho CK, Shuman S. Structure-function analysis of T4 RNA ligase 2. J Biol Chem. 2003;278(20):17601–17608. doi: 10.1074/jbc.M300817200. [DOI] [PubMed] [Google Scholar]
- 28.Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: Structure, reaction mechanism, and function. Chem Rev. 2006;106(2):687–699. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J Med Chem. 2004;47(27):6681–6690. doi: 10.1021/jm049504o. [DOI] [PubMed] [Google Scholar]
- 30.Chipot C, Pohorille A. Free Energy Calculations. New York: Springer; 2007. [Google Scholar]
- 31.Pascal JM, O'Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432(7016):473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 32.Guttman P, Ehrlich P. Uber die wirkung des Methylenblau bei Malaria. Berl Klin Wochenschr. 1891;39:953–956. [Google Scholar]
- 33.Wainwright M. Dyes in the development of drugs and pharmaceuticals. Dyes Pigm. 2007;76:582–589. [Google Scholar]
- 34.Schnaufer A, Domingo GJ, Stuart K. Natural and induced dyskinetoplastic trypanosomatids: How to live without mitochondrial DNA. Int J Parasitol. 2002;32(9):1071–1084. doi: 10.1016/s0020-7519(02)00020-6. [DOI] [PubMed] [Google Scholar]
- 35.Brun R, Lun ZR. Drug sensitivity of Chinese Trypanosoma evansi and Trypanosoma equiperdum isolates. Vet Parasitol. 1994;52(1–2):37–46. doi: 10.1016/0304-4017(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 36.Wang CC. Molecular mechanisms and therapeutic approaches to the treatment of African trypanosomiasis. Annu Rev Pharmacol Toxicol. 1995;35:93–127. doi: 10.1146/annurev.pa.35.040195.000521. [DOI] [PubMed] [Google Scholar]
- 37.Kopperschläger G, Böhme H-J, Hofmann E. Cibacron blue F3G-A and Related Dyes as Ligands in Affinity Chromatography. Berlin: Springer; 1982. pp. 101–138. [Google Scholar]
- 38.Maser P, Luscher A, Kaminsky R. Drug transport and drug resistance in African trypanosomes. Drug Resist Updat. 2003;6(5):281–290. doi: 10.1016/j.drup.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. [Google Scholar]
- 40.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 41.Dolinsky T, Nielsen J, McCammon J, Baker N. PDB 2PQR: An automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi LM, et al. Mining and visualizing large anticancer drug discovery databases. J Chem Inf Comput Sci. 2000;40(2):367–379. doi: 10.1021/ci990087b. [DOI] [PubMed] [Google Scholar]
- 43.Voigt JH, Bienfait B, Wang S, Nicklaus MC. Comparison of the NCI open database with seven large chemical structural databases. J Chem Inf Comput Sci. 2001;41(3):702–712. doi: 10.1021/ci000150t. [DOI] [PubMed] [Google Scholar]
- 44.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 45.Leach AR, Hann MM, Burrows JN, Griffen EJ. Fragment screening: An introduction. In: Hubbard RE, editor. Structure-Based Drug Discovery: An Overview. Vol 2. Cambridge, UK: Royal Society of Chemistry; 2006. pp. 429–446. [Google Scholar]
- 46.Butina D. Unsupervised data base clustering based on daylight's fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J Chem Inf Comput Sci. 1999;39:747–750. [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Schnaufer A, et al. Separate insertion and deletion subcomplexes of the Trypanosoma brucei RNA editing complex. Mol Cell. 2003;12(2):307–319. doi: 10.1016/s1097-2765(03)00286-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.